
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microfluidic platform for 3D cell culture with live imaging and clone retrieval†
Carla
Mulas‡
 *a,
Andrew C.
Hodgson‡
a,
Timo N.
Kohler
b,
Chibeza C.
Agley
a,
Peter
Humphreys
a,
Hans
Kleine-Brüggeney
b,
Florian
Hollfelder
b,
Austin
Smith
abc and
Kevin J.
Chalut
*ad
*a,
Andrew C.
Hodgson‡
a,
Timo N.
Kohler
b,
Chibeza C.
Agley
a,
Peter
Humphreys
a,
Hans
Kleine-Brüggeney
b,
Florian
Hollfelder
b,
Austin
Smith
abc and
Kevin J.
Chalut
*ad
aWellcome-MRC Cambridge Stem Cell Institute, Jeffrey Cheah Biomedical Centre, University of Cambridge, Puddicombe Way, Cambridge Biomedical Campus, Cambridge, CB2 0AW, UK. E-mail: cm623@cam.ac.uk; kc370@cam.ac.uk
bDepartment of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge, CB2 1GA, UK
cLiving Systems Institute, University of Exeter, Exeter, EX4 4QD, UK
dDepartment of Physics, University of Cambridge, 19 J J Thomson Avenue, Cambridge, CB3 0HE, UK
First published on 23rd June 2020
Abstract
Combining live imaging with the ability to retrieve individual cells of interest remains a technical challenge. Combining imaging with precise cell retrieval is of particular interest when studying highly dynamic or transient, asynchronous, or heterogeneous cell biological and developmental processes. Here, we present a method to encapsulate live cells in a 3D hydrogel matrix, via hydrogel bead compartmentalisation. Using a small-scale screen, we optimised matrix conditions for the culture and multilineage differentiation of mouse embryonic stem cells. Moreover, we designed a custom microfluidic platform that is compatible with live imaging. With this platform we can long-term culture and subsequently extract individual cells-in-beads by media flow only, obviating the need for enzymatic cell removal from the platform. Specific beads may be extracted from the platform in isolation, without disrupting the adjacent beads. We show that we can differentiate mouse embryonic stem cells, monitor reporter expression by live imaging, and retrieve individual beads for functional assays, correlating reporter expression with functional response. Overall, we present a highly flexible 3D cell encapsulation and microfluidic platform that enables both monitoring of cellular dynamics and retrieval for molecular and functional assays.
Introduction
Asynchrony and heterogeneity are key challenges when studying dynamic processes in biology. These challenges can be overcome by following individual cells over time using live imaging systems. Live imaging has proven successful in studying dynamic processes across systems, from embryonic stem cells1,2 or hematopoietic stem cell differentiation,3 to large-scale in vivo morphogenesis across multiple organisms (for example4–6). However, the number of fluorescent channels available, as well as the phototoxicity associated with imaging, often limit the number of parameters (e.g. reporters or dyes) that can be observed at the same time. We can infer morphological parameters, but determining cell state often requires reporter systems. We can more accurately infer cell state, and significantly increase the number of parameters, by using single cell RNA sequencing. Yet, in transcriptomic analysis, the history of a cell is typically lost, and we can only place cells in ‘pseudo-time’ by making significant and hard-to-test assumptions about the trajectories cells have followed.7 On the other hand, microfluidic platforms have emerged which promise to overcome the limitations mentioned above and integrate live imaging with mRNA sequencing (for example8,9). These systems, however, often lack versatility, so that it is not possible to recover cells for functional assays, such as testing differentiation potential. Furthermore, given the need for most cells to grow in adherent conditions, enzymatic methods, which require inactivation steps, would generally need to be used to extract cells from the device.Combining live imaging with cell retrieval for functional and molecular analysis could greatly help us better understand dynamic cellular process in controlled conditions. For this purpose, we have designed a highly flexible cell encapsulation system and microfluidic platform, and validated it using embryonic stem (ES) cell differentiation as a model system. Single ES cells possess the remarkable ability to divide indefinitely while maintaining the capacity to give rise to all cell types of the adult organism.10 Exit from the ES cell state and initiation of differentiation is a key step. However, the study of this transition is hindered by a large degree of temporal heterogeneity.11–13 Indeed, at any given point in time during exit from the ES cell state, cells can be in different stages of the differentiation process and respond very differently to differentiation cues.14 This asynchrony has confounded our understanding of how ES cells change state and initiate multilineage differentiation.
With these challenges in mind, we set out to design a platform that would enable: (i) live imaging a dynamic and asynchronous process using reporter systems; (ii) isolating individual events for downstream molecular and functional characterisation at specific timepoints. The platform had to fulfil two requirements: cells had to be cultured in contact with extracellular matrix (ECM), without attaching to the microfluidic device directly; and channels had to be controlled individually so that specific cells or cell aggregates of interest could be extracted independently.
Hydrogel beads provide an ideal system to keep cells in contact with ECM, without direct attachment to a dish or surface. Previously, it was shown that ES cells can self-renew and initiate the process of differentiation in agarose-only beads.15 Here, we optimise conditions for encapsulating cells in 3D agarose–fibrin hydrogel beads. We also develop a microfluidic platform that enabled us to retain individual beads in culture, or extract them for downstream analysis, using media flow, without the need to enzymatically dissociate the matrix. Finally, we show that this platform can be used to follow the asynchronous exit from the ES cell state over time, and determine the functional properties of cells at specific points during that process.
Results and discussion
3D agarose–fibrin matrix mini-screen
The foundation for our encapsulation strategy is agarose beads. However, agarose is biologically inert and does not provide specific cell adhesion or ECM retention. Therefore, we sought to optimise the cell encapsulation platform by providing a biologically active scaffold component. Thrombin can polymerise fibrinogen molecules to form fibrin scaffolds that are highly biocompatible and able to bind various ECM proteins, including laminin.16,17 Moreover, fibrin can be remodelled and digested by cells over time providing a system to potentially escape compression associated with cell division,17 while its stability can be increased by covalent modification.18 It should be noted, however, that excess remodelling can eventually lead to disintegration of the scaffold. To strike a balance between biocompatibility, degradability and stability, we made use of agarose–fibrin composite matrices, where agarose provides the non-degradable scaffold.To identify the optimal agarose/fibrin scaffold composition for ES cell differentiation in hydrogel beads, we designed a 3D matrix screen (adaptable template provided in Table S1†). ES cells harbouring a Sox1::GFP (a neural marker) knock-in reporter were embedded in 96-well plates containing matrices of varying composition, and differentiated towards the neural lineage (Fig. 1A). We varied the concentration of agarose, fibrinogen, thrombin and laminin in combination (Table 1), and examined the effect on differentiation efficiency. Some of the combinations led to scaffold disintegration over time so that cells grew adherent at the bottom of the dish and were, therefore, poor candidates for stable encapsulation. In other conditions, however, the scaffold was stable, and cells grew in 3D, embedded in the hydrogels. Using visual inspection, we assigned each well a matrix-integrity score of 1–4. A matrix integrity scores of 1 indicated scaffolds that disintegrated and therefore were unsuitable for our current purposes. Integrity scores of 2 indicated scaffolds that largely disintegrated but remained partly intact. Integrity scores of 3 indicated scaffolds that were mostly intact, but partly disintegrated. Wells with a matrix integrity score of 4 contained only embedded aggregates, suggesting the gels were stable and would not disintegrate in the final platform, making these conditions good candidates for our purposes. After 5 days, we dissolved scaffolds with agarase and dissociated aggregates into single cells using trypsin. The differentiation efficiency was analysed by flow cytometry (Fig. 1A). All conditions resulted in Sox1 upregulation in >70% of cells (Fig. 1B). Conditions where cells escaped and grew adherent to the culture plates (matrix-integrity score of 1–2) showed higher cell numbers, which is likely due to the slightly higher proliferation rate of ES cells in 2D versus 3D.15 Nevertheless, harvesting cells that escape from their microenvironment and attach to the microfluidic chip would necessitate enzymatic methods; thus, we discarded such conditions.
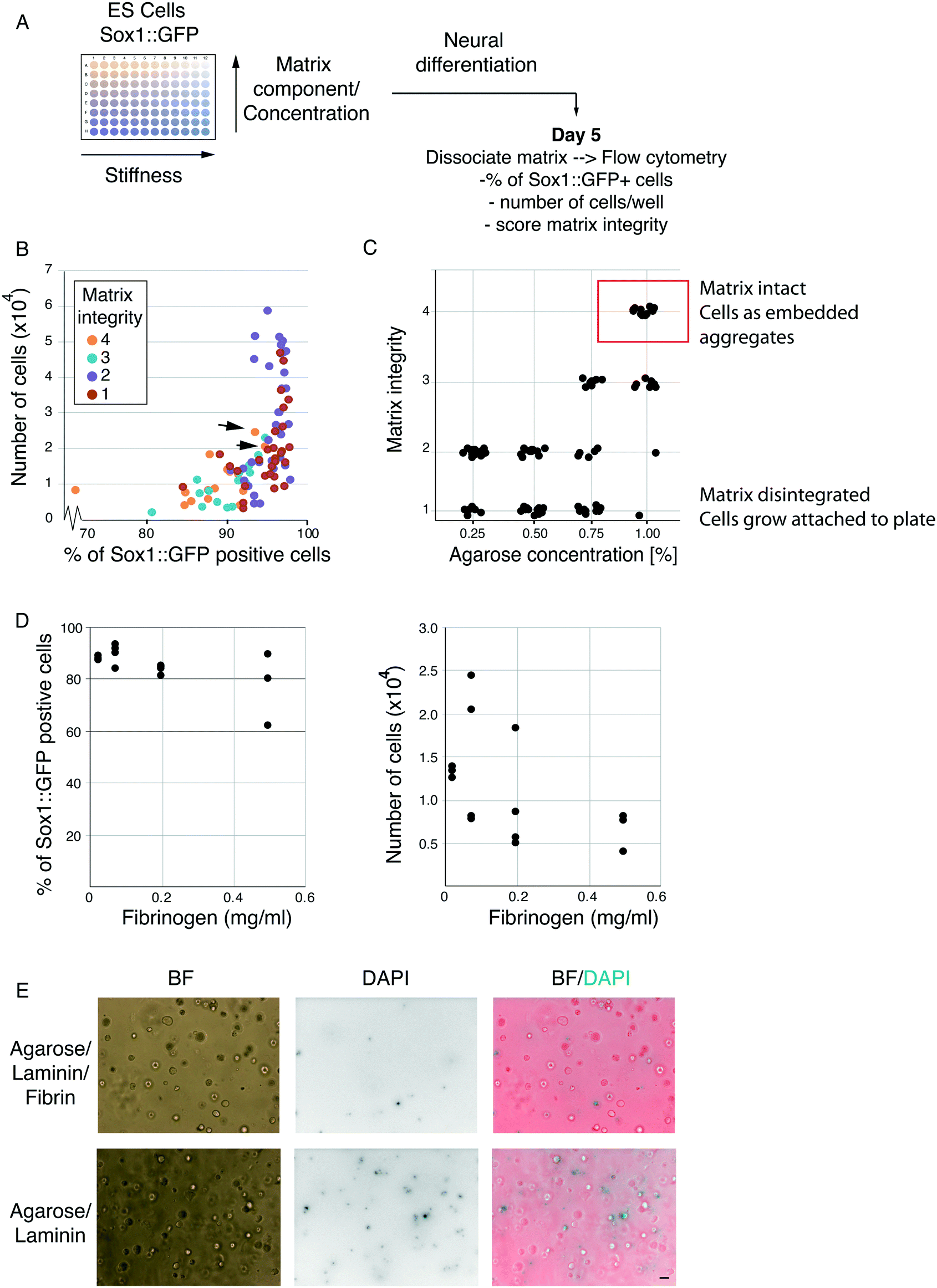 | ||
| Fig. 1 3D matrix mini-screen. A. Schematic of 3D matrix screen to determine hydrogel composition for efficient neural differentiation. B. Percentage Sox1::GFP positive cells and number of cells on day 5 of neural differentiation in beads. Each dot represents a different matrix condition, coloured according to a matrix integrity score. The matrix integrity score was assigned based on visual inspection of each well under a light microscope. Scores were assigned as follows: 4 = all cells in aggregates embedded in hydrogel, 3 = most cells in aggregates embedded in hydrogel but some cells seen growing adherent at the bottom of the dish, 2 = most cells growing adherent to the bottom of the well but a few 3D aggregates embedded in the matrix visible, 1 = all cells growing adherent to the bottom of the well and no matrix visible. Black arrows indicate the conditions selected for further characterisation, as they resulted in a large number of Sox1 positive cells, while maintaining maximum matrix integrity (score of 4). C. Effect of agarose concentration on matrix integrity. D. Effect of fibrinogen concentration on percentage of Sox1::GFP cells (linear regression showing negative correlation, R2 = 0.934, p = 0.022) and number of cells (E) after 5 days of differentiation. E. Cell survival in agarose/fibrinogen/laminin composite gels vs. agarose/laminin-only matrices on day 3. DAPI is able to enter only dead or dying cells. Scale bar 100 μm. | ||
| Agarose% | Fibrinogen mg ml−1 | Thrombin U ml−1 | Laminin μg ml−1 |
|---|---|---|---|
| 1 | 0.5 | 0.25 | 0.6 |
| 0.85 | 0.2 | 0.5 | 0.3 |
| 0.70 | 0.075 | 1 | |
| 0.50 | 0.025 |
As expected, agarose concentration was the biggest determining factor for matrix integrity (Fig. 1C). Notably, however, in the gels with the highest matrix integrity, increased fibrinogen concentration showed a slight negative correlation with the percentage of Sox1::GFP positive cells and cell numbers (Fig. 1D). Laminin and thrombin concentration did not show significant trends on their own (not shown). Taken together, our results indicate that the optimal conditions for ES cell differentiation were in hydrogel beads formed under the higher agarose concentrations and lower fibrin. However, fibrin was still necessary, as omitting fibrin altogether resulted in increased cell death after 3 days in culture (Fig. 1E). We presume this is because fibrin co-polymerises with agarose and allows gels to bind and retain laminin, and because, at low density, cells require matrix attachment to efficiently survive and differentiate. Thus, we chose to carry forward the condition which resulted in the highest matrix stability score, cell proliferation and differentiation efficiency [1% agarose, 0.075 mg ml−1 fibrinogen, 0.5 U ml−1 thrombin and 3 ng ml−1 laminin, Fig. 1B, black arrows].
Functionalised hydrogel beads support multilineage differentiation of mES cells
Next, we examined whether we could generate usable hydrogel beads with the optimised matrix conditions. To this end, we used a flow-focusing microfluidic encapsulation device.15 Low-melt agarose was dissolved and cooled to 37 °C as previously described,15 before adding fibrinogen and laminin. This solution was fed through one inlet of the encapsulation device. Newly formed agarose beads were collected in tubes on ice. Basal media supplemented with thrombin was added to the collection tube, and agarose–fibrin beads were de-emulsified by addition of perfluorooctanol (PFO) (Fig. 2A). Thrombin addition led to fibrin/fibrinogen incorporation and retention in the hydrogel beads (Fig. 2B). The beads remained stable and did not attach to the dish.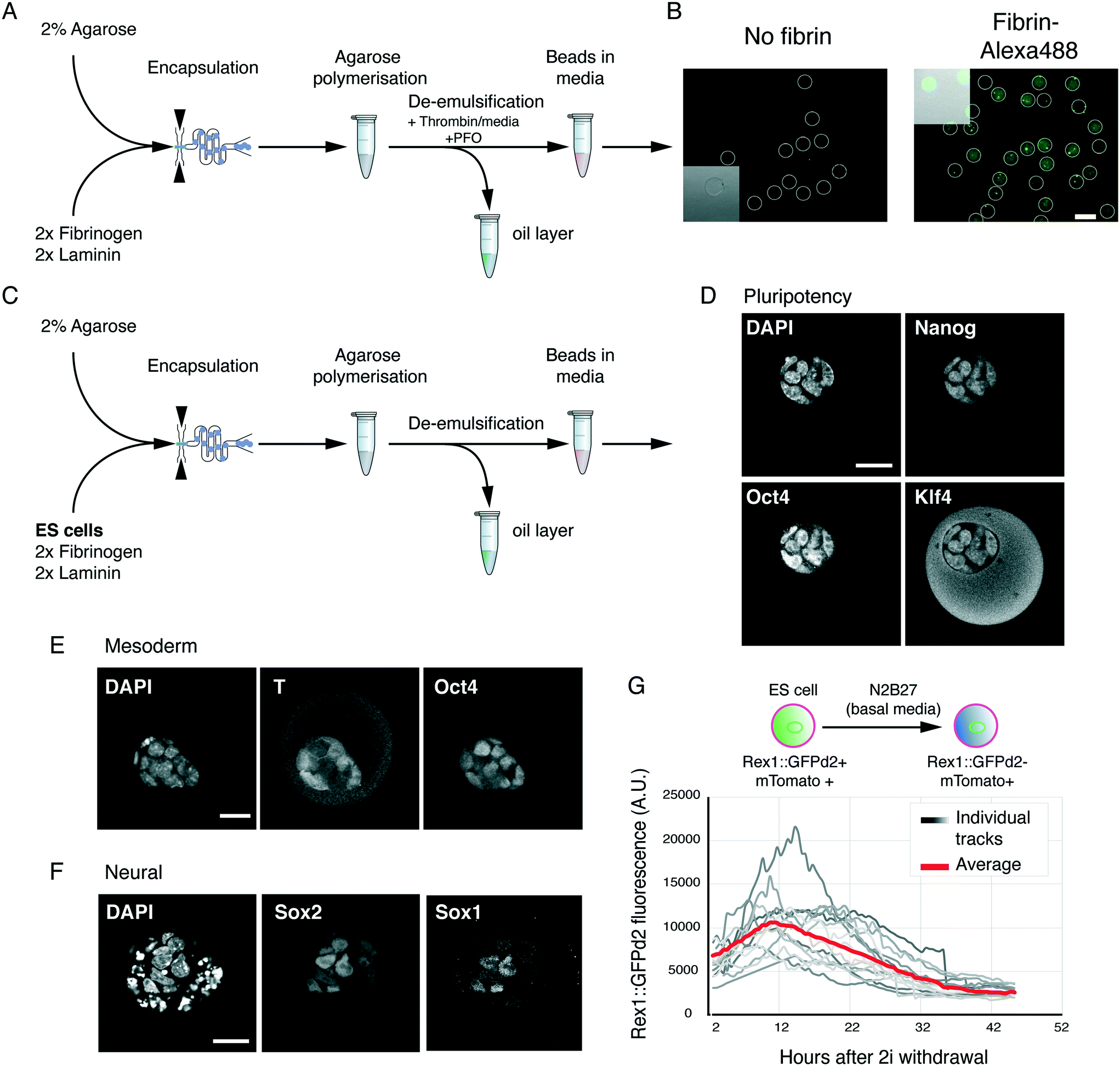 | ||
| Fig. 2 ES cell differentiation in functionalised hydrogel beads. A. A flow-focusing device is used to generate agarose/fibrin/laminin hydrogel beads in oil/carrier solution. After incubating on ice to allow agarose polymerisation, fresh media with thrombin is added, as well as PFO, to demulsify the beads. The aqueous fraction containing agarose beads is used for further experiments. B. Fluorescently labelled fibrin is successfully retained in microgel beads. White outlines show beads, identified from the brightfield channel. Insert shows overlay of brightfield and fluorescent channel for a section of the image. C. Strategy for encapsulating ES cells in hydrogel beads. D. Encapsulated ES cells maintained in ES cell media and fixed for 2 days for immunostaining show expression of pluripotency-associated transcription factors Nanog, Oct4 and Klf4. Nuclei were counterstained with DAPI. Scale bar 20 μm. E. Encapsulated ES cells were placed in mesoderm-inducing media and fixed for immunostaining on day 3. Oct4/T double positive cells are early mesoderm progenitors, while Oct4+/T-cells are as-yet uncommitted cells. Scale bar 20 μm. F. Encapsulated ES cells were directed towards the neural lineage and analysed on day 4 for the expression of transcription factors Sox1 and Sox2. Punctuated DAPI staining shows dead or dying cells. Scale bar 20 μm. G. Live imaging of differentiating ES cells encapsulated in 3D hydrogels. Membrane tomato (mTomato) expression was used to segment aggregates in 3D. The average aggregate level was determined for each bead and plotted against the time after withdrawal of self-renewal signals (2i). Individual tracks and average are shown. | ||
Next, we tested if the agarose–fibrin beads could sustain ES cell self-renewal and differentiation. 2% low-melt agarose solution was heated and subsequently cooled to 37 °C. ES cells were resuspended in basal media (N2B27) supplemented with 2× fibrinogen and laminin solution. The agarose and cell suspension were mixed, and quickly passed through the cell encapsulation microfluidic device (Fig. 2C). Encapsulated cells were de-emulsified as shown previously,15 in a media containing thrombin.
In ES cell culture conditions (2i/LIF), encapsulated cells proliferated at a rate similar to what was previously reported in 3D15 and maintained expression of pluripotency markers Nanog, Klf4 and Oct4 (Fig. 2D). When placed in mesoderm-inducing conditions, we could detect expression of the pan-primitive streak marker T(Bra) (Fig. 2E), while in neural conditions we could detect Sox1 and Sox2 double positive neural precursor cells (Fig. 2F). Finally, we tested whether the hydrogel beads were amenable to live imaging. To follow exit from the ES cell state, we used the Rex1::GFPd2 reporter, known to mark undifferentiated ES cells in vitro, and the naïve epiblast in vivo.11 Rex1::GFPd2 is lost in cells that have irreversibly exited the ES cell state, thereby providing a high-fidelity reporter for the ES cell state. ES cells harbouring both Rex1::GFPd2 and a constitutive membrane marker (mTomato) were encapsulated and driven to exit the ES cell state in basal media (N2B27). 3D stacks of individual beads were acquired every 30 min and automatically segmented using the mTomato membrane marker in order to measure the total GFP level. This allowed us to track the downregulation of Rex1::GFPd2 as ES cells exited the naïve, undifferentiated, ES cell state (Fig. 2G). In agreement with previous reports,12,14 the downregulation of Rex1::GFPd2 was asynchronous, with cells in some gel beads downregulating the reporter before others.
Overall, the data demonstrate that agarose and fibrin can be successfully co-polymerised in hydrogel beads, enabling support for both self-renewal and multi-lineage differentiation. Moreover, the system is compatible with live imaging. We noted that in some conditions, especially after 5–6 days of culture, cells towards the outside of the aggregate showed signs of cell death (e.g. fragmented nuclei), possibly due to those cells being highly compressed. Since pluripotency transition experiments take place within 48 h of encapsulation, we do not anticipate this being a limitation. Moreover, the matrix conditions could be further optimised for longer term experiments. Finally, the hydrogel beads can easily be dissolved following agarase treatment (not shown). Since agarase is not cytotoxic it can be added to culture media, without a need to deactivate or remove it for subsequent culture (we found no excess cell death).
Microfluidics for single hydrogel bead culture and retrieval
Next, we designed a microfluidic platform that would allow culturing cells in beads under live imaging, and extracting specific beads one at a time. To achieve single-bead control we designed a 2-layer microfluidic platform using photolithography. In this design, a figure-8 channel layer contains individual bead traps (Fig. 3A, insert [i]), inlets and outlets, and an auxiliary module contains an array of bead traps (Fig. 3A, insert [ii] and C) and multiple independent inlets/outlets for immunofluorescence (to allow for fixing, washes and staining). A second layer was superimposed onto the channel layer to control media flow by means of PDMS pneumatic (or Quake) valves (Fig. 3B).19 To achieve complete channel closure upon valve actuation, the mould of the channel layer was constructed using a two-step process. First, non-compressible features were generated with SU8 2100. Then, features containing the channels under pneumatic control were superimposed onto the SU8 features, in AZ 40 XT and re-flown by reheating to generate rounded profiles (Fig. 3A top insert and S1†20).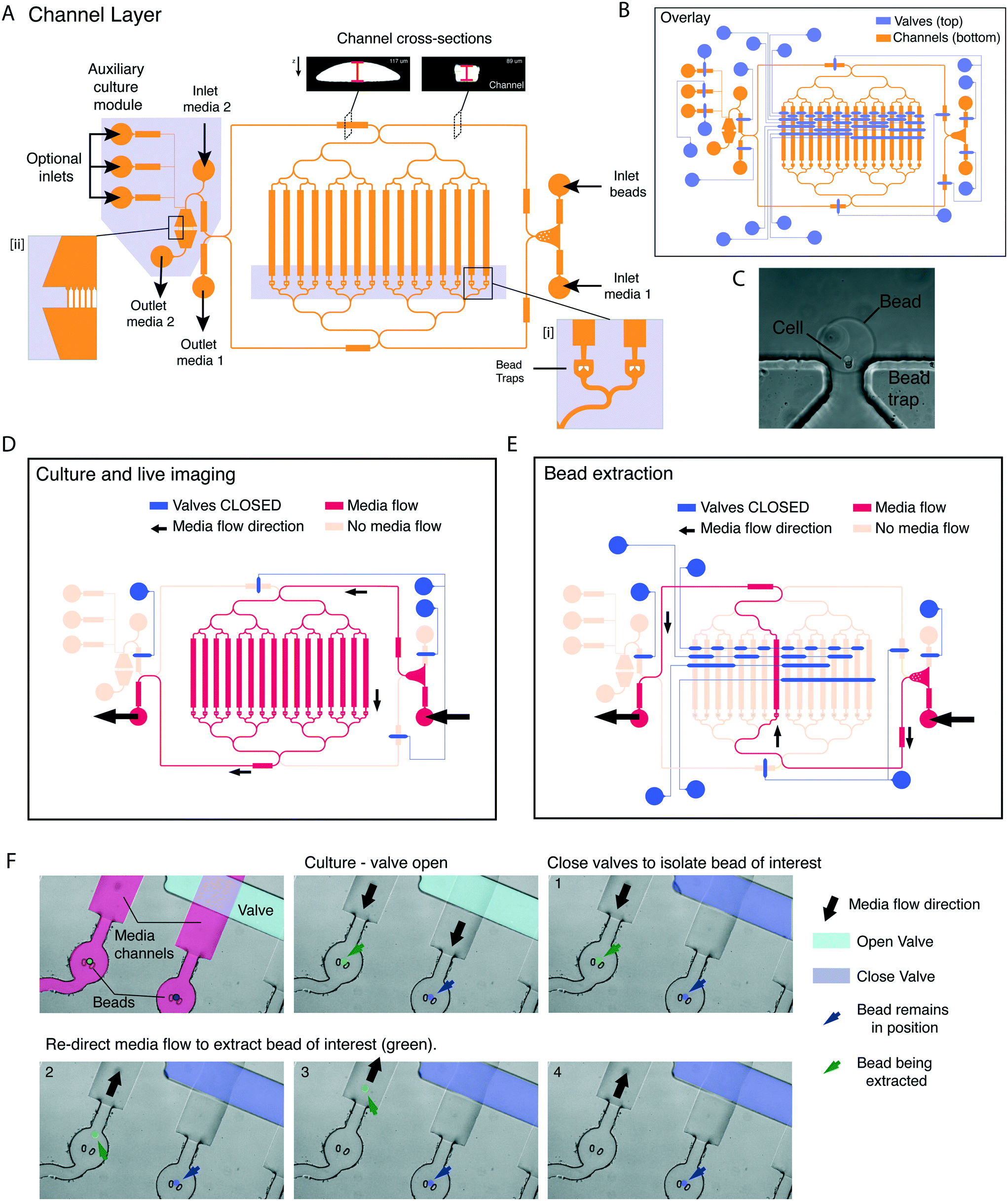 | ||
| Fig. 3 Microfluidic platform for independent capture and release of aggregates. A. Schematic of the channel (bottom) layer of the microfluidic platform showing individual components and modules. (i) Shows individual bead traps, (ii) shows a magnified view of a secondary culture module. Insert shows cross-section of different channel regions. Thicker segments can be closed by valves. B. Schematic of the overlay of the valve and channel layer in the final design. C. Brightfield image of a cell, encapsulated in an agarose/fibrin/laminin bead, on the bead trap. D. Valve actuation and media flow for culture and live imaging. Media flow keeps all beads in position in the traps. E. By actuating a combination of valves (example shows extraction of bead in trap 8), the media flow along a single channel can be reversed, those allowing for the extraction of a bead of interest. Media flow is blocked along all other channels, so that all remaining beads remain in the traps. F. Snapshots of live imaging showing valve actuation (1), and re-direction of media by valve actuation as shown in (E) to removal of a single bead (false colour in green – steps 2–4). While a neighbouring bead (false colour blue) remains in position (4). | ||
In the design, a number of valves control inlet/outlet access. These are actuated to prevent backflow and direct the media flow to specific outlets and/or auxiliary modules. Two valves control the direction of media flow into the trap module, while eight valves provide single channel control. Table S2† describes the combination of valves and inlets used for bead loading and culture. Activating different sets of valves enables controlling the direction of media flow during culture (Fig. 3D), and removing individual beads (Fig. 3E). Valve actuation is achieved through a pressure increase in the valve layer, which is controlled by a MUX microfluidic flow switch matrix (Elveflow), which can apply constant pressure over any combination of valves. For the bead removal to be effective, the sequence and timing of valve actuations is controlled by a custom MATLAB graphical user interface (GUI), for which we provide the code (see Methods). Within the GUI, each combination of valve actuations was mapped to a specific button relating to a corresponding bead trap. The user can dictate the flow direction, channel of interest and destination (outlet vs. auxiliary module) through single mouse clicks on the GUI window.
We tested whether individual beads could be removed without displacing the remaining beads (Fig. 3F). When the media flow was directed towards the trap (Fig. 3F, panel 1) hydrogel beads are held in position. To extract an individual bead, the media flow was initially redirected towards the desired channel by closing off all the alternative channels (Fig. 3F, panel 2). Next, the direction of media flow was reversed to extract the bead of interest into a well of a 96-well plate (Fig. 3F, panels 3 and 4). Notably, the neighbouring bead (Fig. 3F, false colour blue) remained in position.
Therefore, our platform allows for cell culturing under live imaging conditions of individual cells/beads, which can be independently extracted. The platform, moreover, provides a very high degree of flexibility and scalability. The number of channels and beads analysed can be doubled by adding two extra control valves. Moreover, the independent inlets can be used to carry out media change or drug administration at precise intervals of time.
Live imaging of differentiation and single cell retrieval for functional studies
We then designed an experiment to validate the platform. ES cells can be maintained in an undifferentiated, relatively homogenous, state in N2B27 basal medium supplemented with GSK3 and MEK1/2 inhibitors (collectively termed 2i), with the optional addition of LIF21 (2i/LIF medium). In N2B27 alone (no inhibitors of cytokines), cells initiate differentiation.11,21 As cells exit the ES cell state, they lose the ability to self-renew/survive in ES cell maintenance media (2i/LIF).11 The ability to self-renew correlates with the expression of Rex1::GFPd2; cells that have extinguished Rex1::GFPd2 expression can no longer self-renew in selective ES cell culture conditions.12,14 The phenotype of cells with intermediate levels of Rex1::GFPd2, in part due to asynchrony in the differentiating population (Fig. 2G and ref. 12), is as yet unknown.Our microfluidic platform allows us to image cells live as they begin to differentiate, as shown by loss of Rex1::GFPd2 expression. To determine if cells with intermediate levels of Rex1::GFPd2 could self-renew or had irreversibly lost ES cell identity, we encapsulated Rex1::GFPd2 cells harbouring the constitutive mTomato membrane marker into hydrogel beads functionalised with laminin. Individual cell/beads were captured using the microfluidic platform and the media was switched from ES cell-maintenance medium to N2B27 differentiation medium (no supplements). This allowed cells to initiate differentiation as monitored by Rex1::GFPd2 downregulation. We imaged cells every 30 min for 27 h, at which point they displayed a wide distribution of Rex1::GFPd2 expression levels.12 Next, we extracted individual cells/beads from the platform and placed each cell/bead in a well of a 96 well plate containing selective ES cell culture conditions (2i/LIF) (Fig. 4A). The capacity to form self-renewing colonies was determined after 5 days. We found that cells that retained detectable expression of Rex1::GFPd2, even if significantly lower than ES cell-controls, could still self-renew in 2i/LIF (Fig. 4B and C). This shows that the ability to self-renew in 2i/LIF conditions is lost only after cells have fully downregulated Rex1::GFPd2 expression. Therefore, our platform allows us to directly correlate reporter expression over time with the functional response to signalling pathways/culture conditions.
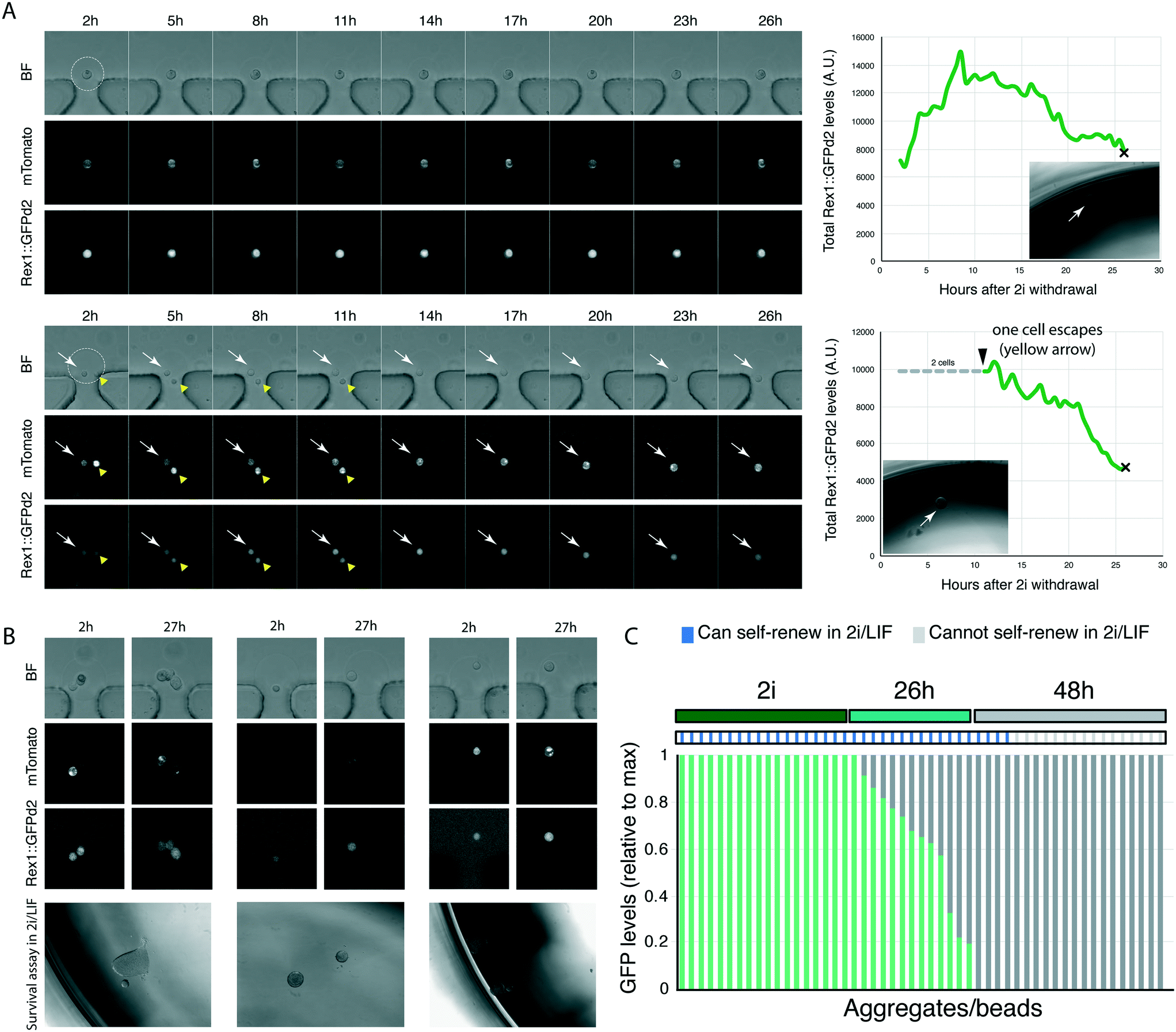 | ||
| Fig. 4 Combined platform for live imaging and retrieval of live cells for functional studies. A. Example tracks of single encapsulated cells, tracked over 27 h during exit from the ES cell state. Cells were automatically segmented using mTomato and the total GFP level determined. Graphs on the right show total Rex1::GFP levels, normalised to cell/aggregate volume. ‘×’ denotes point of extraction and insert figure shows colony arising after 5 days in ES cell culture conditions (2i/LIF). On the bottom panel, two cells are initially encapsulated together (white and yellow arrows), but one cell (yellow arrow) escapes confinement after 11 h. Graph shows quantification of remaining cell. B. Images show examples of beads on traps; bright field, Rex1::GFPd2 and mTomato marker expression of start and end point of live imaging. Bottom panels show the result of the self-renewal assay in 2i/LIF. C. Quantification of the total level of GFP, normalised to peak, expression, and the outcome of the self-renewal assay in 2i/LIF. | ||
Conclusions
In this study we present a versatile platform for integrating live imaging with individual cell retrieval. To achieve this, we combine a hydrogel 3D encapsulation system with a microfluidic platform that is compatible with live imaging and single clone retrieval. The platform offers the potential to link the cell's history with cell state, characterised at the molecular or functional level.To generate biocompatible and tuneable hydrogel beads, we developed an agarose-based encapsulation strategy. Agarose scaffolds are non-toxic and stable but do not allow for ECM or cell attachment. Indeed, they have been extensively used for anchorage-independent growth, for example to screen for transformed cells.22 Thus, we co-polymerized agarose with fibrin to obtain a composite matrix capable of binding any ECM protein, such as laminin. Agarose–fibrin gels have been previously used in combination with bioprinting to generate hollow scaffolds for modelling renal proximal tubules on chip.23 Agarose–fibrinogen–hydroxyapatite aggregates were also used for co-culture of HUVEC and fibroblast cells.24 In that report, however, fibrinogen was used as a cell-adhesive component rather than as a scaffold.24 Also, the use of hydroxyapatite was essential for the incorporation of fibrinogen. This is likely due to the absence of thrombin24 which allows for fibrinogen polymerisation into fibrin.16 By co-polymerising agarose and fibrin, we have a versatile system that can bind different ECM proteins, where we can readily customise the stiffness and degree of matrix degradation/remodelling by changing the proportion of agarose and fibrin. Furthermore, the polymerisation technique that was used in the previous report resulted in a large degree of size variation in the aggregates, making them unsuitable for microfluidics.24 Therefore, to our knowledge, this is the first report of customisable ECM-functionalised hydrogel beads compatible with single cell culture and microfluidic handling. Importantly, our protocol allows for hydrogel beads to be formed in mild gelation/polymerisation conditions, in the absence of harsh chemicals or UV light. The hydrogel matrix screen approach, for which we provide a template (Table S1†), as well as the use of functionalised hydrogel beads, could both provide useful platforms for culture for different cell types across a number of different applications.
Many microfluidic devices have now been reported to overcome technical limitations of large-scale cell culture, single clone retrieval and live imaging. Droplet phenotyping devices can screen many droplets and/or retrieving droplets with a particular phenotype (e.g. positive staining for a dye or protein expression).25–29 This can allow identifying rare or transient events if enough droplets are screened. However, all cells must be screened/retrieved simultaneously, and the history of the cell is lost. A key requirement for our experiments is to end the experiment for different cells at different times in order to analyse intermediate steps within dynamic processes, while retaining a record of the biological process of interest (i.e. how the cells got to that particular stage). Alternative cell culture devices, while allowing for small-scale culture and live imaging of cells,30,31 require cells to grow adhering to the microfluidic device. Therefore, cells must be extracted by coring or detaching the chip, high pressure media flow,32 or flowing enzymatic solutions.33 These set ups, while retaining the history of cells, also often require all samples to be extracted at once. Our device, on the other hand, through combining elements of microfluidic valve technology,34 multiplexing35 and microfluidic droplet cell encapsulation,36 allows following dynamic biological processes in real time and easily retrieving specific, transient, events. We can capture individual hydrogel beads, culture or differentiate them while performing imaging, and remove individual beads at specific times while leaving the rest undisturbed. Since the cells do not adhere to the microfluidic chip, individual beads can be extracted by changing the direction of media flow, without using high pressure, having to flow through cell dissociation reagents (e.g. trypsin), or destroying the chip.
We have introduced a number of design elements that make the microfluidic chip scalable beyond 16-channels. The valve multiplexing design allows doubling the number of captured hydrogel beads for each two control valves that are added.37 By using a high working-distance objecting and imaging systems capable of fast z-stack acquisition, we could obtain 3D images compatible with segmentation pipelines. Finally, the system can be integrated with previously-validated reporter systems used to infer parameters such as differentiation status, signalling pathway activity, cell cycle phase, or many other cellular features. Coupling dynamic readouts of reporters with end-point functional or molecular analysis could provide unparalleled information on cell state. The ability to isolate individual beads while leaving the remainder undisturbed could prove a useful system for isolating specific stages in processes that occur asynchronously or are of transient duration. Overall, we expect this platform to be adaptable to a wide range of applications for assessing dynamics of cellular processes.
Methods
Mouse ES cell culture and differentiation
Mouse ES cell culture and differentiation was performed following previously described protocols.14,38 Briefly, cells carrying a constitutive tomato marker and a destabilised form of GFP under the control of the Rex1 promoter were routinely grown in 2i or 2iLIF [1 μM PD0325901, 3 μM CHIR99021 with or without 100 U ml−1 leukaemia inhibitor factor (LIF)] in N2B27 basal media [1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 DMEM/F12 (Sigma-Aldrich D6421), 1:2 Neurobasal (Thermo Fisher 21103049), 0.5X B27 (Invitrogen 17504044), 1X N2.BV (see below), 50 μM b-mercaptoethanol (Thermo Fisher 31350), 2 mM L-glutamine (Thermo Fisher 25030081), 12.5 μg ml−1 insulin human recombinant zinc solution (Thermo Fisher 1258014)]. 100X N2.BV was made in house [8.791 mg ml−1 apotransferrin (Sigma Aldrich T1147), 1X DMEM/F12, 0.66% BSA fraction V, 3 μM sodium selenite (Sigma Aldrich S5261), 1.688 mg ml−1 putrescine (Sigma Aldrich P5780), 2.08 μg ml−1 progesterone (Sigma Aldrich P8783)]. Cells were split every two days and plated at a density of 1.5 × 104 cells per cm2. Differentiation in aggregates was carried out by placing hydrogel beads in N2B27 (without 2i or LIF).
:2 DMEM/F12 (Sigma-Aldrich D6421), 1:2 Neurobasal (Thermo Fisher 21103049), 0.5X B27 (Invitrogen 17504044), 1X N2.BV (see below), 50 μM b-mercaptoethanol (Thermo Fisher 31350), 2 mM L-glutamine (Thermo Fisher 25030081), 12.5 μg ml−1 insulin human recombinant zinc solution (Thermo Fisher 1258014)]. 100X N2.BV was made in house [8.791 mg ml−1 apotransferrin (Sigma Aldrich T1147), 1X DMEM/F12, 0.66% BSA fraction V, 3 μM sodium selenite (Sigma Aldrich S5261), 1.688 mg ml−1 putrescine (Sigma Aldrich P5780), 2.08 μg ml−1 progesterone (Sigma Aldrich P8783)]. Cells were split every two days and plated at a density of 1.5 × 104 cells per cm2. Differentiation in aggregates was carried out by placing hydrogel beads in N2B27 (without 2i or LIF).
Matrix screen
Low-melt agarose (Lonza SeaPlaque 50105) was dissolved at twice the concentration required and kept at 37 °C to prevent polymerisation. 2× thrombin solution was added to each tube containing agarose. 50 μl of agarose/thrombin were added to each well of a 96 well plate kept on a hot plate set to 37 °C to prevent polymerisation. On a separate 96 well plate, basal media was supplemented with laminin (final concentration 2×) and mixed with cells (final cell concentration per well 3500). 50 μl of the media/laminin/fibrinogen and cell mixture was added to each well containing agarose/thrombin and mixed by pipetting while keeping the plate at 37 °C. Once sufficiently mixed, the plate was placed at 4 °C for 15 min to allow agarose to polymerise. 100 μl of N2B27 was added once the hydrogel was polymerised. Cells were differentiated for 5 days, before assigning each well a matrix integrity score based on visual inspection: 4 = all cells in aggregates embedded in hydrogel, 3 = most cells in aggregates embedded in hydrogel but some cells adhering to the culture plate, 2 = most cells growing adherent to the bottom of the well but a few 3D aggregates embedded in the matrix visible, 1 = all cells growing adherent to the bottom of the well and no matrix visible. Next, media was carefully removed and replaced with 50 μl of fresh N2B27 supplemented with agarase (Thermo Fisher Scientific EO0461) and trypsin. Hydrogels were dissolved after 15 min, and gentle pipetting resulted in a single cell suspension. Differentiation efficiency was analysed by flow cytometry. A linear regression model on the average percentage of Sox1 positive cells at each fibrin concentration was used to determine the level of negative correlation. A template for customising the matrix screen, alongside detailed instructions and calculations, is provided in Table S1.†Immunofluorescence
Hydrogel beads were fixed in 4% PFA for 30 min at room temperature, before washing twice with PBS for 5 min each time. Aggregates were permeabilised and blocked for 2–4 h in block solution (PBS, 0.3% Triton 100X (Sigma Aldrich T8787) and 3% Donkey serum (Sigma Aldrich D9663)). Primary antibodies were diluted in block solution and incubated overnight at 4 °C with gentle rocking. Beads were washed three times for 15 min with PBST (PBS, 0.3% Triton 100X) before incubating with secondary antibodies diluted in block solution. After 3 washes of PBST for 15 min each, beads were stored at 4 °C in PBS to reduce background before mounting in VECTASHIELD antifade mounting media (Vector Laboratories H-1000). Images were acquired on a confocal laser scanning microscope (Leica SP5). The following primary antibodies were used: Nanog (1:200, eBioscience 14-5761-80), Oct4 C-10 (1:400, Santa Cruz sc5279), Klf4 (1:400, R&D AF3158), T(Bra) (1:300, R&D AF2085), Sox2 (1:200, eBioscience 14-9811-82), Sox1 (1:200, Cell Signalling 4194). Alexa Fluor-conjugated secondary antibodies were used (Life Technologies). Nuclei were stained with DAPI.
Live imaging and analysis
Live imaging of agarose drops outside and inside the microfluidic device was done using a Zeiss 710 confocal microscope using an 20× 0.5 EC Epiplan-Neofluar objective. A custom made-heated black box was used to surround the microscope and syringe pumps and avoid large temperature gradients. Ten z-stacks were acquired every 30 min for GFP, mTomato and bright field. Image analysis outside the microfluidic chip (Fig. 2G) was carried out using Imaris. The membrane fluorescent protein (mTomato) was used to segment cells using Imaris Surfaces. For each region of interest, the total GFP level and volume was determined. At each timepoint, GFP levels were normalised to total aggregate volume. To quantify GFP levels in beads in the microfluidic device, we used a custom Matlab code (https://github.com/CMulas/Beads_3D_segmentation).Hydrogel bead generation
Low melt agarose (Lonza SeaPlaque 50105) was dissolved at twice the required concentration (2%) in PBS by heating at 80 °C, before reducing the temperature to 37 °C. Cells were dissociated to obtain a single cell suspension at a concentration of 5–10 × 106 cells per ml in 2i supplemented with 0.15 mg ml−1 (2×) fibrinogen (Sigma Aldrich F4883) and 6 ng ml−1 (2×) laminin (Mouse purified, Merck Millipore CC095). The agarose solution was mixed 1:1 with the cells/fibrinogen/laminin in 2i media and kept at 37 °C briefly to avoid polymerisation. The encapsulation was carried out as previously described (with a few modifications) using tested parameters to generate 80 μm diameter beads.15 Briefly, chips were plasma-bonded to glass coverslips and the inlets were immediately flushed with a solution of 1% PFOTS (trichloro(1H,1H,2H,2H-perfluorooctyl)silane, Sigma Aldrich 448931) in HFE-7500 (3M Novec Engineered Fluid, Fluorochem 051243). For hydrogel bead generation, one inlet was used for the agarose/cell suspension and one for the oil/carrier solution [HFE-7500 supplemented with 0.3% Pico-Surf 1 (Sphere Fluidics)] which were flown through at 6 μl min−1 and 30 μl min−1 respectively. Beads were collected for ∼15 min. Newly formed hydrogel beads were collected on ice and polymerised for 15 min. Cell culture media (either 2i or N2B27 alone) was supplemented with 0.5 U ml−1 thrombin (Sigma Aldrich T4393) and added to the bead/oil emulsion. Deemulsification was carried out by adding 1H,1H,2H,2H-perfluorooctanol (PFO) (Alfa Aesar, B20156) directly to the oil layer, quickly vortexing and extracting the top aqueous layer containing the hydrogel beads (∼100 beads per μl). These were temporarily stored in an Eppendorf tube inside a tissue culture incubator until loading the platform.
Microfluidic platform generation
For the channel layer mould, a silicon wafer was spin-coated with SU8 2100 (MicroChem, UK) at 3100 RPM to achieve a 90 μm layer across the wafer surface before baking for 5 min at 65 °C and 30 minutes at 95 °C. To create the SU8 features within the 90 μm layer, shown in Fig. S1,† a photomask (printed by JD Photo Data, Hitchin, UK) was used with a MJB-4 contact mask aligner (Karl Suss, Munich, Germany) to selectively expose the SU8 coated wafer for 18 s (365–405 nm, 20 mW cm−2). Exposed regions in the 90 μm layer of SU8 crosslink, becoming insoluble. Following a subsequent post-expose bake of 15 minutes at 95 °C, the wafer is developed in propylene-glycol-mono-methyl-ether-acetate (PGMEA) (Merk Millipore) for 7 minutes. This removes all non-crosslinked areas of SU8, leaving 90 μm high channels on the wafers surface. Any residue is removed with isopropanol and the wafer is baked at 200 °C to complete the SU8-layer fabrication.
The same wafer was then spin coated with AZ 40 XT (MicroChem, UK) at 900 RPM. A slow-ramped bake from 65–126 °C was performed over 20 minutes before the spin-and-bake process was repeated to achieve a double thickness layer. A photomask with the features marked ‘AZ’ on Fig. S1† was used to generate the channel segments that can be closed by valves. The wafer is carefully aligned using the MJB-4 so the channels of the mask fit between the SU8 channels which are visible though the AZ layer. Exposure consists of two 90 second rounds using 365–405 nm light at 20 mW cm−2. The wafer is post-exposure baked at 105 °C, and then immersed in AZ 726 developer (MicroChemicals GmbH, Ulm, Germany) with occasional water rinses until the features are revealed. To achieve the rounding of the valve sections, the wafer is then finally baked for 5 minutes at 110 °C to reflow to a maximum height of 115–120 μm (measured with stylus profilometer, Dektak, Bruker, MA, USA). To produce a mould for the valve layer, SU8 2025 was spun across a silicon wafer at 1700 rpm to produce a feature height of 35 μm. Bake and exposure protocols followed the manufacturer's guidelines.
:1 (base:curing agent) PDMS (Sylgard 184, Dow Corning) over the channel layer mould described above. This is sufficient to cover the SU8 and features to create a thin PDMS membrane. We found that 450 RPM is sufficient to produce a robust membrane while being thin enough to deform as a valve. This layer is cured at 60 °C for 6 hours.
The valve layer was formed using 5:1 (base:curing agent) PDMS poured over the valve layer mould to a thickness of 1–1.5 cm and cured for 1 hour at 60 °C. A scalpel is used to cut the PDMS chip around the outside of the features to free from the mould. A 1.5 mm biopsy punch is then used to create holes (indicated by the circular sections of Fig. 3B and S1†) for fluidic access to the valve channels. The valve and channel layers are then exposed, feature side up, to oxygen plasma for 20 s at 100 W (Diener Electronic GmbH and Co. KG, Germany). Using a stereo microscope, the two layers are aligned as in Fig. 3B and brought into contact to bond together. Following a further 1 hour bake at 60 °C, a scalpel is used to extract the PDMS chip from the silicon wafer. Fluidic access is enabled through a 1.5 mm biopsy punch at the circular sites of Fig. 3B and S1.† To complete the fabrication process, the PDMS device is bonded to a glass slide (76 × 52 × 1.2 mm, Fisher Scientific) using the oxygen plasma protocol described above before baking at 60 °C for one hour.
For fluidic control, each of the valve inlets are connected via FEP tubing to the MUX multiplexing unit (Elveflow, France). The MUX unit can distribute pressure to any combination of the on-chip microfluidic valves with tuneable pressure. For fluidic delivery, the channel inlets are connected via FEP tubing to a syringe pump (Nemesys, Cetoni). All equipment to control valves, flow and pressure are housed within a purpose-built cabinet containing a heater set to 37 °C. The operating conditions and valve set-up for equilibration, loading, culture and extraction are provided in Table S2.† Several steps, outlined in Table S3,† were taken to reduce experimental failures.
The microfluidic device uses 14 of the available MUX channels (excluding the valves for the auxiliary module). A pressure source, in this case a Fluigent MFCS-EZ microfluidic flow control system, with a maximum pressure of 1.3 bar, connects to the MUX. As the valves within the microfluidic device are a closed system with the MUX, any combination of ‘open’ or ‘closed’ results in the same pressure in any actuated valve. The pressure is calibrated at the start of each experiment to effectively close the on-chip valve when the corresponding MUX outlet is opened.
Each valve on the microfluidic device is connected to the MUX channels as shown in Fig. S1.† To deliver beads to the on-chip traps, a step-by-step protocol is described in Table S2.† At step 12, the combination of valves directs the bead in position 16 to either be extracted or delivered to the secondary culture array by actuating valve C4 or D1 respectively. To specify an individual bead for removal or relocation, a combination of valves A1–4 and B1–4 are actuated to deliver fluid flow only to the specified channel. The various combinations required to remove each bead are programmed into a MATLAB graphical user interface (https://github.com/achodgson/mulashodgson2020/blob/master/Bead_Control_GUI.txt), enabling the user to direct the bead with a single click.
Author contributions
Conceptualisation, CM, ACH, AS and KC; methodology, CM, ACH, CCA, PH; formal analysis, CM; investigation, CM, ACH, TNK, HKB; writing, CM, ACH, KJC; supervision, FH, AS, KJC.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We would like to thank Liisa Van Vliet and Fabrice Gielen for useful discussions. CM and ACH were supported by a Leverhulme Trust Research Grant (RPG-2016-418) and ERC grant (772798), TNK was supported by an AstraZeneca Graduate Studentship, CCA was supported by an MRC grant (MR/M011089/1), FH is a H2020 ERC Advanced Investigator [695669]. The Cambridge Stem Cell Institute receives core funding from the Wellcome Trust and the Medical Research Council. AS is a Medical Research Council Professor. KC was supported by a Royal Society University Research Fellowship.References
- K. Brown, K. M. Loh and R. Nusse, Live Imaging Reveals that the First Division of Differentiating Human Embryonic Stem Cells Often Yields Asymmetric Fates, Cell Rep., 2017, 21, 301–307 CrossRef CAS PubMed.
- A. Filipczyk, et al. Network plasticity of pluripotency transcription factors in embryonic stem cells, Nat. Cell Biol., 2015, 17, 1235–1246 CrossRef CAS PubMed.
- H. M. Eilken, S.-I. Nishikawa and T. Schroeder, Continuous single-cell imaging of blood generation from haemogenic endothelium, Nature, 2009, 457, 896–900 CrossRef CAS PubMed.
- P. J. Keller, A. D. Schmidt, J. Wittbrodt and E. H. K. Stelzer, Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy, Science, 2008, 322, 1065–1069 CrossRef CAS PubMed.
- F. Bosveld, et al. Mechanical control of morphogenesis by Fat/Dachsous/Four-jointed planar cell polarity pathway, Science, 2012, 336, 724–727 CrossRef CAS PubMed.
- K. McDole, et al. In Toto Imaging and Reconstruction of Post-Implantation Mouse Development at the Single-Cell Level, Cell, 2018, 175, 859–876 CrossRef CAS PubMed.
- C. Weinreb, A. Rodriguez-Fraticelli, F. D. Camargo and A. M. Klein, Lineage tracing on transcriptional landscapes links state to fate during differentiation, Science, 2020, 367(6479), eaaw3381 CrossRef CAS PubMed.
- J. Yuan, J. Sheng and P. A. Sims, SCOPE-Seq: a scalable technology for linking live cell imaging and single-cell RNA sequencing, Genome Biol., 2018, 19, 5 CrossRef PubMed.
- R. J. Kimmerling, et al. A microfluidic platform enabling single-cell RNA-seq of multigenerational lineages, Nat. Commun., 2016, 7, 7 Search PubMed.
- G. Martello, P. Bertone and A. Smith, Identification of the missing pluripotency mediator downstream of leukaemia inhibitory factor, EMBO J., 2013, 32, 2561–2574 CrossRef CAS PubMed.
- T. Kalkan and A. Smith, Mapping the route from naive pluripotency to lineage specification, Philos. Trans. R. Soc., B, 2014, 369(1657), 20130540 CrossRef PubMed.
- T. Kalkan, et al. Tracking the embryonic stem cell transition from ground state pluripotency, Development, 2017, 142711–142756, DOI:10.1242/dev.142711.
- S. Semrau, et al. Dynamics of lineage commitment revealed by single-cell transcriptomics of differentiating embryonic stem cells, Nat. Commun., 2017, 1–16, DOI:10.1038/s41467-017-01076-4.
- C. Mulas, T. Kalkan and A. Smith, NODAL Secures Pluripotency upon Embryonic Stem Cell Progression from the Ground State, Stem Cell Rep., 2017, 9, 77–91 CrossRef PubMed.
- H. Kleine-Bruggeney, et al. Long-Term Perfusion Culture of Monoclonal Embryonic Stem Cells in 3D Hydrogel Beads for Continuous Optical Analysis of Differentiation, Small, 2019, 15(5), e1804576 CrossRef PubMed.
- P. A. Janmey, J. P. Winer and J. W. Weisel, Fibrin gels and their clinical and bioengineering applications, J. R. Soc., Interface, 2009, 6, 1–10 CrossRef CAS PubMed.
- S. Jockenhoevel and T. C. Flanagan, in Tissue Engineering for Tissue and Organ Regeneration, IntechOpen, 2011, DOI:10.5772/19761.
- J. W. Weisel, Fibrinogen and fibrin, Adv. Protein Chem., 2005, 70, 247–299 CrossRef CAS PubMed.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Monolithic microfabricated valves and pumps by multilayer soft lithography, Science, 2000, 288, 113–116 CrossRef CAS PubMed.
- P. M. Fordyce, C. A. Diaz-Botia, J. L. DeRisi and R. Gomez-Sjoberg, Systematic characterization of feature dimensions and closing pressures for microfluidic valves produced via photoresist reflow, Lab Chip, 2012, 12, 4287–4295 RSC.
- Q. Ying, et al. The ground state of embryonic stem cell self-renewal, Nature, 2008, 453, 519–523 CrossRef CAS PubMed.
- A. W. Hamburger and S. E. Salmon, Primary bioassay of human tumor stem cells, Science, 1977, 197, 461–463 CrossRef CAS PubMed.
- K. A. Homan, et al. Bioprinting of 3D Convoluted Renal Proximal Tubules on Perfusable Chips, Sci. Rep., 2016, 6, 1–13 CrossRef PubMed.
- A. Y. Rioja, E. L. H. Daley, J. C. Habif, A. J. Putnam and J. P. Stegemann, Distributed vasculogenesis from modular agarose-hydroxyapatite-fibrinogen microbeads, Acta Biomater., 2017, 55, 144–152 CrossRef CAS PubMed.
- E. Brouzes, et al. Droplet microfluidic technology for single-cell high-throughput screening, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14195–14200 CrossRef CAS PubMed.
- M. T. Guo, A. Rotem, J. A. Heyman and D. A. Weitz, Droplet microfluidics for high-throughput biological assays, Lab Chip, 2012, 12(12), 2146–2155 RSC.
- L. Mazutis, et al. Single-cell analysis and sorting using droplet-based microfluidics, Nat. Protoc., 2013, 8, 870–891 CrossRef CAS PubMed.
- J.-C. Baret, et al. Fluorescence-activated droplet sorting (FADS): efficient microfluidic cell sorting based on enzymatic activity, Lab Chip, 2009, 9(13), 1850–1858 RSC.
- M. M. Wang, et al. Microfluidic sorting of mammalian cells by optical force switching, Nat. Biotechnol., 2004, 23, 83–87 CrossRef PubMed.
- P. J. Hung, P. J. Lee, P. Sabounchi, R. Lin and L. P. Lee, Continuous perfusion microfluidic cell culture array for high-throughput cell-based assays, Biotechnol. Bioeng., 2004, 89, 1–8 CrossRef PubMed.
- R. Gómez-Sjöberg, A. A. Leyrat, D. M. Pirone, C. S. Chen and S. R. Quake, Versatile, fully automated, microfluidic cell culture system, Anal. Chem., 2007, 79, 8557–8563 CrossRef PubMed.
- C. Luni, et al. High-efficiency cellular reprogramming with microfluidics, Nat. Methods, 2016, 13, 446–452 CrossRef CAS PubMed.
- S. Giulitti, et al. Direct generation of human naive induced pluripotent stem cells from somatic cells in microfluidics, Nat. Cell Biol., 2019, 21, 275–286 CrossRef CAS PubMed.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Monolithic microfabricated valves and pumps by multilayer soft lithography, Science, 2000, 288, 113–116 CrossRef CAS PubMed.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Microfluidic large-scale integration, Science, 2002, 298, 580–584 CrossRef CAS PubMed.
- S.-Y. Teh, R. Lin, L.-H. Hung and A. P. Lee, Droplet microfluidics, Lab Chip, 2008, 8(2), 198–220 RSC.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Microfluidic large-scale integration, Science, 2002, 298, 580–584 CrossRef CAS PubMed.
- C. Mulas, et al. Defined conditions for propagation and manipulation of mouse embryonic stem cells, Development, 2019, 146, dev173146 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00165a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |