
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExcitation energy transfer between monomolecular layers of light harvesting LH2 and LH1-reaction centre complexes printed on a glass substrate†
Xia
Huang‡
ab,
Cvetelin
Vasilev‡
 *a and
C. Neil
Hunter
a
*a and
C. Neil
Hunter
a
aDepartment of Molecular Biology and Biotechnology, University of Sheffield, Sheffield S10 2TN, UK. E-mail: c.vasilev@sheffield.ac.uk
bCAS Key Lab of Bio-Medical Diagnostics, Suzhou Institute of Biomedical Engineering and Technology, Chinese Academy of Sciences, 215163, Suzhou, China
First published on 11th June 2020
Abstract
Light-harvesting 2 (LH2) and light-harvesting 1 – reaction centre (RCLH1) complexes purified from the photosynthetic bacterium Rhodobacter (Rba.) sphaeroides were cross-patterned on glass surfaces for energy transfer studies. Atomic force microscopy (AFM) images of the RCLH1 and LH2 patterns show the deposition of monomolecular layers of complexes on the glass substrate. Spectral imaging and fluorescence life-time imaging microscopy (FLIM) revealed that RCLH1 and LH2 complexes, sealed under physiological conditions, retained their native light-harvesting and energy transfer functions. Measurements of the amplitude and lifetime decay of fluorescence emission from LH2 complexes, the energy transfer donors, and gain of fluorescence emission from acceptor RCLH1 complexes, provide evidence for excitation energy transfer from LH2 to RCLH1. Directional energy transfer on the glass substrate was unequivocally established by using LH2-carotenoid complexes and RCLH1 complexes with genetically removed carotenoids. Specific excitation of carotenoids in donor LH2 complexes elicited fluorescence emission from RCLH1 acceptors. To explore the longevity of this novel nanoprinted photosynthetic unit, RCLH1 and LH2 complexes were cross-patterned on a glass surface and sealed under a protective argon atmosphere. The results show that both complexes retained their individual and collective functions and are capable of directional excitation energy transfer for at least 60 days.
Introduction
Photosynthesis involves harvesting solar energy by light-harvesting (LH) antenna complexes, then transfer to a specialized, membrane-bound complex called the reaction centre (RC) where this energy is converted to charge current. The LH function can be performed either by large pigment–protein complexes such as chlorosomes or phycobilisomes, which lie on the membrane surface and deliver harvested energy to an underlying RC, or by a series of repeating LH units that sit alongside the RC within the membrane bilayer.1 The latter case comprises the photosynthetic apparatus of plants and, at a simpler level, the photosynthetic membranes found in purple phototrophic bacteria. The structural and functional characterisation for the phototrophic bacterium Rhodobacter (Rba.) sphaeroides has reached a level where all of the steps of photosynthesis, from absorption of solar energy, through to trapping at the RC, generation of a proton motive force and the production of adenosine triphosphate (ATP), have been combined into an in silico model of the photosynthetic membrane.2–4 The detail of this membrane model is such that it can account for the doubling time of the bacterium,5 and it also encourages the design and ‘bottom up’ fabrication of bio-hybrid energy trapping systems that capture, convert and store solar energy.New bio-hybrid energy transfer and trapping assemblies take many forms, and range from incorporating new chromophores into native6–14 and de novo-designed15,16 proteins, to using a variety of lithographic patterning methods to precisely position a single type of photosynthetic complex.17–22 In this case, the assembly of extensive two-dimensional architectures for energy harvesting, transfer and trapping requires the ability to direct the relative positions of two or more types of photosynthetic complex on the same surface. The two complexes chosen for this two-protein patterning work are the LH2 antenna of Rhodobacter sphaeroides, and its native energy acceptor, the RCLH1 complex.2,23 Both are membrane-intrinsic, multisubunit proteins in which transmembrane polypeptides bind light-absorbing bacteriochlorophyll and carotenoid pigments.24 The short distances between pigments ensure the rapid delocalization of excited states and their transfer within and between complexes in the native membrane.24–26 Here we use a simple, robust lithographic procedure to construct intersecting domains of LH2 antenna and RCLH1 complexes, deposited as monomolecular layers, effectively creating a new micron-scale ‘photosynthetic unit’. A two-stage micro-contact printing method was used to fabricate a two-dimensional grid of cross-patterned LH2 and RCLH1 proteins, which was interrogated by AFM and fluorescence microscopy. Spectral and lifetime imaging shows that light absorbed by the LH2 antenna is transferred to RCLH1 complexes; thus, these arrays contain functionally coupled components for absorbing and transferring excitation energy, thereby performing the first two steps of photosynthesis.
Methods
Protein purification
Wild type LH2, RCLH1 and ΔcrtB RCLH1 proteins were purified as described previously.27,28 Briefly, semi-aerobically grown cell were harvested and disrupted in a French pressure cell at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 psi. After centrifugation, the supernatant was loaded onto a sucrose gradient in order to isolate the intracytoplasmic membranes (ICM). After harvesting, the ICMs were solubilised in 3% (v/v) β-DDM for RCLH1 and ΔcrtB RCLH1, and in 4% N,N-dimethyldodecylamine-N-oxide (LDAO) for LH2 by stirring in the dark at 4 °C for 45 min. The solubilized membrane solution was diluted at least three-fold in working buffer and centrifuged for 1 hour in a Beckman Ti70.1 rotor at 48000 rpm (160000 × g) at 4 °C to remove unsolubilized material. The supernatant was further purified by using ion-exchange chromatography.
000 psi. After centrifugation, the supernatant was loaded onto a sucrose gradient in order to isolate the intracytoplasmic membranes (ICM). After harvesting, the ICMs were solubilised in 3% (v/v) β-DDM for RCLH1 and ΔcrtB RCLH1, and in 4% N,N-dimethyldodecylamine-N-oxide (LDAO) for LH2 by stirring in the dark at 4 °C for 45 min. The solubilized membrane solution was diluted at least three-fold in working buffer and centrifuged for 1 hour in a Beckman Ti70.1 rotor at 48000 rpm (160000 × g) at 4 °C to remove unsolubilized material. The supernatant was further purified by using ion-exchange chromatography.
Sample preparation
The Si master template (Mikromasch, TGZ11) with linear arrays of 5 μm width, 10 μm pitch and 1.35 μm step height was used as master to replicate a polydimethylsiloxane (PDMS) stamp.29 Prior to casting of the PDMS mixture, the Si master template was treated in trichlorosilane (Sigma-Aldrich) vapour under vacuum (20 mbar) for 16 hours. The PDMS mixture were prepared by mixing Sylgard184 silicon elastomer base (Dow Corning) and Sylgard184 silicon elastomer curing agent (Dow Corning) at a ratio of 10:1. The PDMS mixture was stirred for five minutes to reach uniformity and centrifuged at 3000 × g for 15 min to remove air bubbles. Then the PDMS mixture was cast onto the Si master and cured at 74 °C for 8 h, before being carefully detached as a PDMS replica stamp.
The PDMS soft-patterning was performed on a poly-L-lysine coated substrate, either a glass coverslip (Fisher Scientific, Corning BioCoat, REF 354085). The substrate was treated with 20 mM dimethyl suberimidate (DMS) (Thermo Fisher Scientific, Prod #20700) for 40 min at pH 8.5 to activate lysine as the cross-linker for amine attachment.
The PDMS stamp was covered with solution of LH2 complexes (15 μM protein in buffer consisting of 20 mM HEPES, 0.03% β-DDM, pH 7.8) for 5 min, then blown dry with argon to form a surface layer of LH2. The LH2 inked stamp was gently placed onto the substrate to print LH2 arrays and left for 5 minutes before being gently lifted away. Samples with cross-patterned LH2 and RCLH1 arrays were made using the same printing steps performed for the RCLH1 complexes (either ΔcrtB RCLH1 or WT RCLH1) with the second printing orientation at roughly 90 degrees to the previous LH2 arrays. AFM was used to image samples prepared on silicon substrates in air. Samples prepared on glass coverslips were sealed either in a dry argon atmosphere or in a 20 mM HEPES buffer, pH 7.8 with protocatechuate-dioxygenase (50 nM)/3,4-dihydroxybenzoic acid (2.5 mM) enzymatic oxygen scavenging system30 before being imaged by fluorescence life-time microscopy for energy transfer studies.
Characterisation of printed photosynthetic complexes by atomic force microscopy
The AFM data was collected on a Multimode 8 instrument equipped with a 15 μm scanner (E-scanner) coupled to a NanoScope V controller (Bruker). NanoScope software (v9.2, Bruker) was used for data collection and Gwyddion (v2.52, open source software covered by GNU general public license, www.gwyddion.net) and OriginPro (v8.5.1, OriginLab Corp.) software packages were used for data processing and analysis. The patterned surfaces with the immobilized protein molecules on them were imaged in PeakForce Tapping mode at nearly-physiological conditions in buffer (20 mM HEPES, pH 7.8), at room temperature using BL-AC40TS probes (Olympus). In this case, the Z-modulation amplitude was adjusted to values in the range 20–24 nm, while the Z-modulation frequency was 1 kHz and the contact tip–sample force was kept in the range 80–100 pN.Fluorescence life-time imaging microscopy (FLIM)
The fluorescence emission properties of samples were measured on a home-built time-resolved fluorescence microscope. The microscope is equipped with 2 sets of light sources: a 470 nm LED light (Thorlabs, M470L2) for wide field fluorescence images; and a 485 nm picosecond diode laser (PicoQuant, PDL 828) for spectral and lifetime measurements. The excitation light is focused by a 100× objective (PlaneFluorite, NA = 1.4, oil immersion, Olympus) and the fluorescence emission is collected from the same focal spot on the sample. The collected light is then filtered by dichroic beam-splitters to remove the background excitation light: using the 458 nm dichroic beam-splitter (Semrock) when exciting with the 420 nm LED or the 425 nm laser; or using the 495 nm dichroic beam-splitter (Semrock) when exciting with the 470 nm LED or the 485 nm laser. A filter wheel was equipped with 6 filters to allow filter selecting as each measurement requires. A spectrometer (Acton SP2558, Princeton Instruments) was equipped for wavelength selecting, an electron-multiplying charge-coupled device (EMCCD) detector (ProEM 512, Princeton Instruments) was equipped for photon collecting and a hybrid detector (HPM-100-50, Becker & Hickl) was equipped for photon counting. The modulation of the laser was synchronized with a time correlated single-photon counting (TCSPC) module (SPC-150, Becker & Hickl) for the lifetime decay measurement. The objective is equipped with a piezo scanner (nPoint) to allow laser scanning of the sample for acquiring fluorescence spectral images and fluorescence lifetime images. Samples were excited by the 420/485 nm pulsed laser at 1 MHz repetition rate and fluence of ∼2 × 1014 photons per pulse per cm2. Time-correlated single-photon counting (TCPSC) was applied for triggering the laser and counting the photon arrival time. TCPSC is a well-established and a common technique for fluorescence lifetime measurements. It detects single photons and measures their arrival times in respect to the light source. During the measurement in this work, the entrance slit of the spectrometer was closed to 100 μm. A grating with 150 lines per mm was used to select the wavelength. A band-pass filter and a secondary exit slit on the spectrometer were used to narrow the recording wavelength range to 3 nm. Fluorescence lifetime images were recorded by scanning the excitation laser over the sample using the piezo scanner.Wide field fluorescence images were analysed by ImageJ, the spectral data were analysed in OriginPro, and the fluorescence decay curves were analysed in OriginPro and TRI2 (open source), with fitting using the multi-exponential decay function:
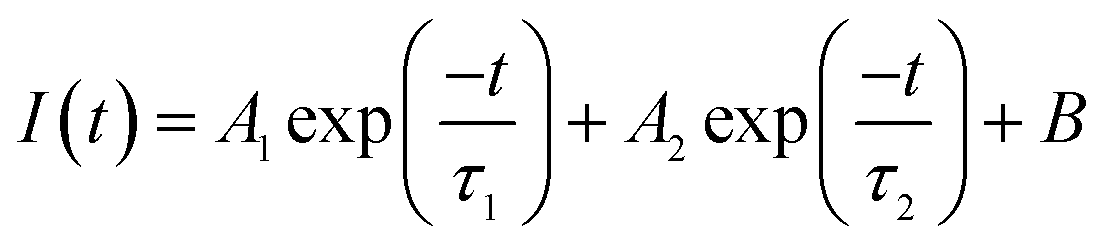
where tk is the time point k, I(tk) is the data at the time point k, Ic(tk) is the fit at the time point k, n is the number of the data points and p is the number of the variable fit parameters (n − p = degrees of freedom).
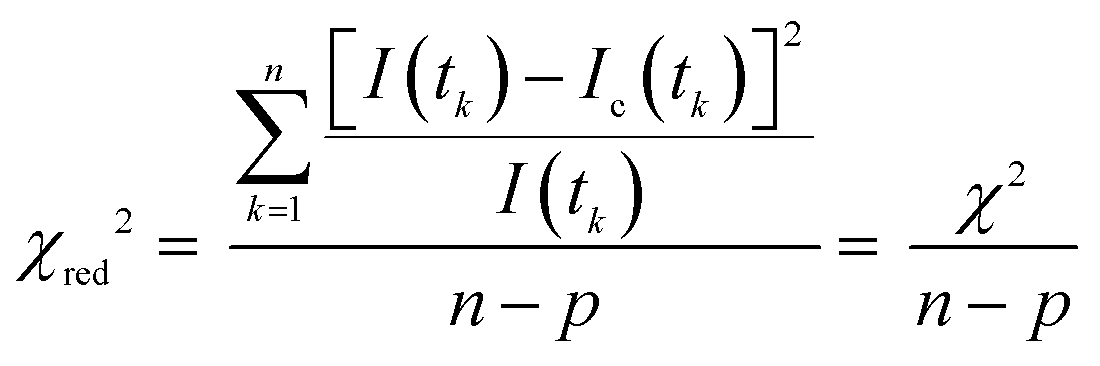
Using a mirror to replace the sample, the time delay of the laser from the pulse starting point to the instrument responding point was measured. Such time delay was defined as the instrument response function (IRF), which was approximately 130 ps on the home-built fluorescence microscope. The IRF was taken into account when the fitting was performed for the decay curves.
Results and discussion
Directed formation of crossed-patterned LH2 and RCLH1 complexes on glass and silicon
We created artificial light-harvesting networks using a very simple and low cost soft-lithographic technique, based on the micro-contact printing approach,31–33 schematically represented in Fig. 1. Optically transparent glass functionalized with poly-L-lysine (PLL) was chosen as a substrate, to facilitate characterisation of immobilized protein complexes by fluorescence microscopy. As a first step, the LH2 complexes (in green, Fig. 1G and H) were printed onto the substrate using a soft PDMS stamp (Fig. 1B and C) inked with the protein solution (Fig. 1D, see M&M), followed by the printing of RCLH1 complexes (in red, Fig. 1I), performed in a similar way but at a 90° angle to the LH2 lines.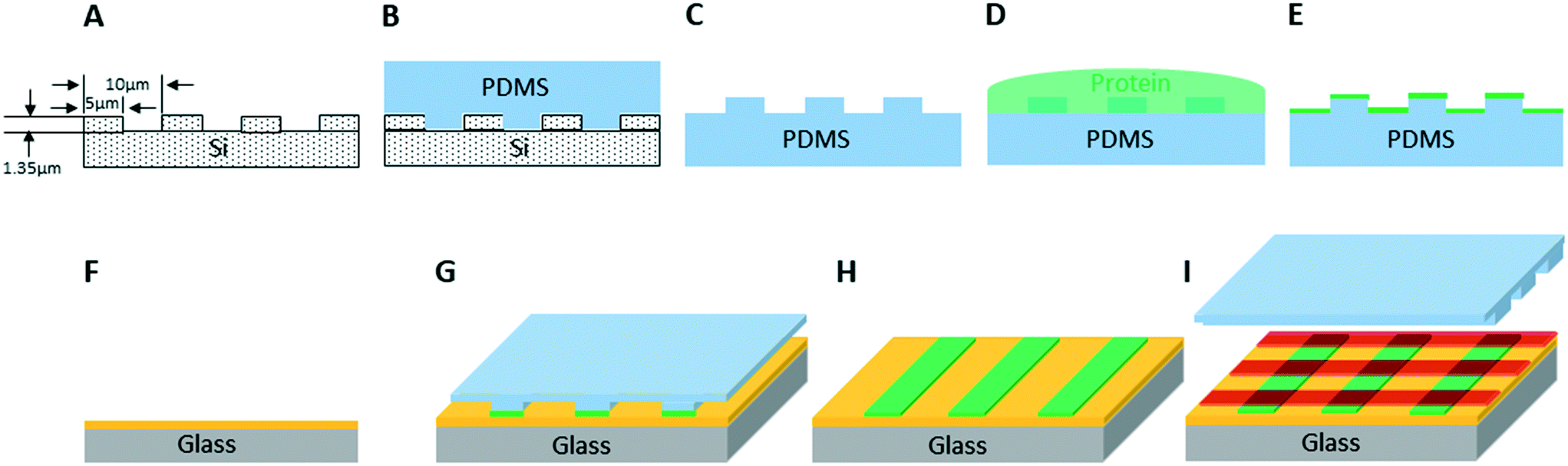 | ||
| Fig. 1 Schematic diagram of the micro-contact printing method used to fabricate the cross-patterned LH2 and RCLH1 protein arrays. A – Si master with rectangular arrays of 5 μm width, 10 μm pitch and 1.35 μm step height. B and C – Casting a PDMS replica of the master. D and E – Inking the stamp with LH2 (green). F – PLL coated glass reacted with DMS (yellow). G and H – Printing LH2 on glass; I – printing RCLH1 complexes (red) on glass orthogonally to LH2 arrays. | ||
As a first step we investigated the surface density, orientation and surface coverage of the immobilized protein complexes on the substrate by using AFM. Fig. 2 shows an AFM topographic image of protein complexes cross-patterned on a PLL-coated glass under near-physiological conditions, in imaging buffer. The LH2 complexes were printed first and, subsequently, the RCLH1 complexes were cross-printed at approximately 90°, forming a zone of intersection as shown in Fig. 2A. The cross-section across the LH2 line reveals heights in the range 6 to 7 nm (Fig. 2B, green), while the cross-section across the RCLH1 line (Fig. 2B, red) reveals an average height of around 10 nm. Both values conform with the known sizes of the two complexes and with previous AFM measurements34 and indicate that the patterned protein domains have a single-molecule thickness. At the intersection of the LH2 and RCLH1 lines the average height increases to around 15 nm (Fig. 2B, purple). The height of this profile (Fig. 2B) indicates that, during the second printing step, some of the RCLH1 complexes in the cross-over area are likely immobilized on top of the existing LH2 molecules. This arrangement creates the conditions for energy transfer perpendicular to the plane of the glass support, rather than the lateral transfer process found in nature. We also assume that, in areas where the LH2 layer is incomplete, the RCLH1 complexes are immobilized directly onto the functionalised glass surface, thus intermixing with the LH2 monolayer and creating the conditions for energy transfer in the plane of the glass support, similar to their relative arrangement in the native biological energy transfer network. The diagram in Fig. 2C summarises the likely arrangement of complexes in these cross-patterns.
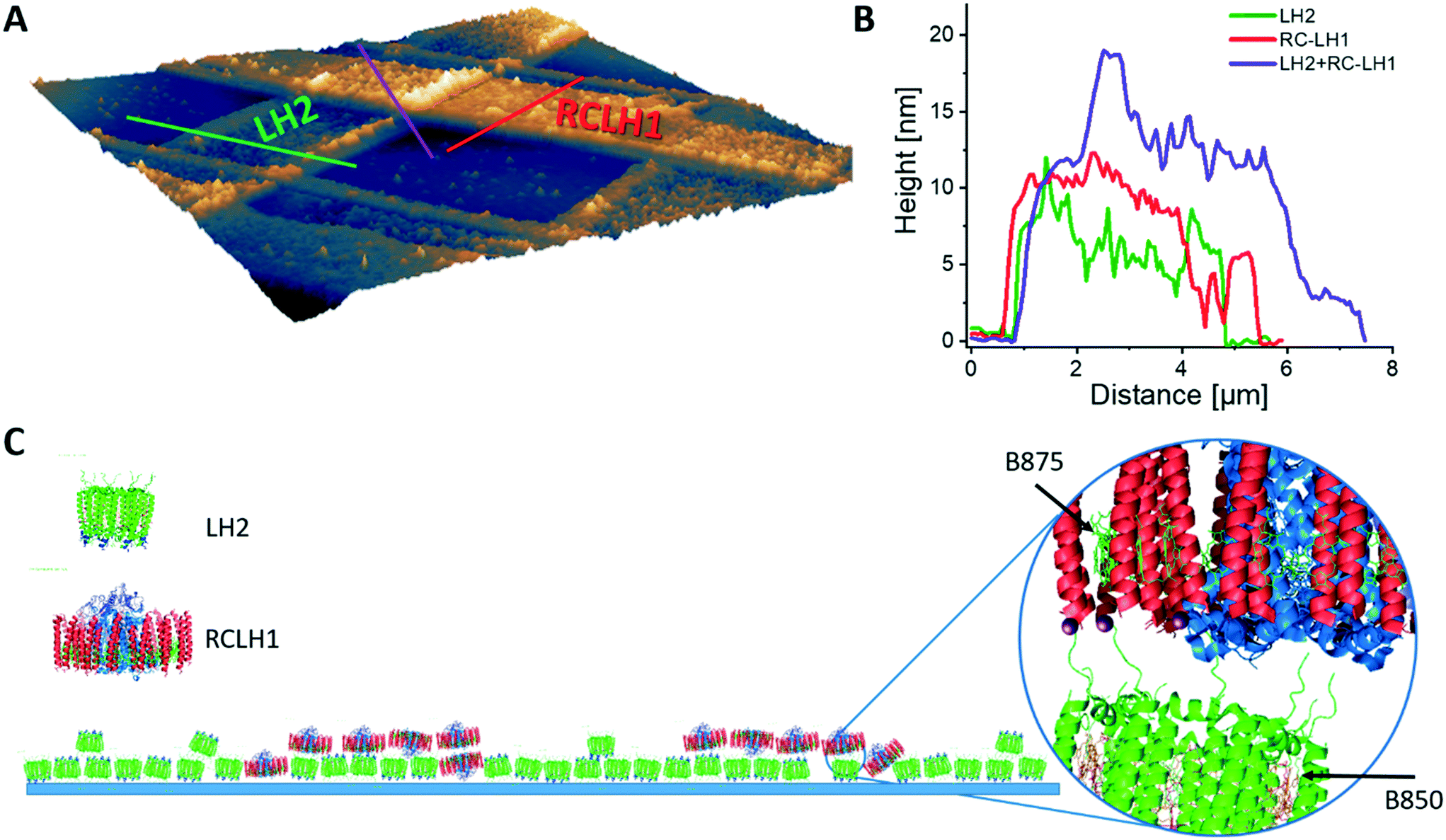 | ||
| Fig. 2 Topography of the cross-patterned LH2/RCLH1 complexes on a PLL-coated glass substrate. A – AFM topography image acquired under physiological conditions (in imaging buffer), with three cross-sections indicated: green – LH2; red – RCLH1; purple intersection. B – Cross-section across the LH2 domains (green), RCLH1 domains (red), and across the LH2/RCLH1 intersecting area (purple), with typical heights of 6 nm, 10 nm and 15 nm, respectively. C – Depiction of the likely arrangement of LH2 (green) and RCLH1 (blue/red) complexes in imprinted cross-patterns. | ||
Excitation energy transfer between LH2 and RCLH1 under physiological conditions
Native biological light-harvesting networks are stabilized in membrane bilayers and operate under tightly controlled physiological conditions, so the complexes and energy transfer assemblies studied here could be hindered by their removal from their in vivo context. In order to overcome this limitation, the surface-patterned LH complexes were sealed in argon-sparged imaging buffer. In bacterial photosynthetic systems the LH2 antenna complexes harvest light energy and transfer it to the RCLH1 core complex, where the excitation energy is stabilized as a photo-chemical charge separation. In order to reproduce the processes of excitation energy transfer in an artificial LH network, it is important to show that the protein complexes have retained their optical and structural properties following their immobilization on the substrate. Thus, the artificial microarrays of cross-printed LH2 and RCLH1 complexes were characterized by fluorescence lifetime and spectral imaging in a home-built FLIM setup. The samples were excited either at 485 nm or at 470 nm in the absorption band of the carotenoids present in both the LH2 and RCLH1 complexes (Fig. 3). The excitation energy is absorbed by carotenoids and transferred to B800 and B850 bacteriochlorophylls (BChls) within the LH2 complexes, where energy is partly emitted as fluorescence and partly transferred to the B875 pigments in the RCLH1 complexes. In turn, LH1 B875 BChls can either emit some of the excitation energy as fluorescence or transfer it to the RC, where it is trapped as a charge separation. The LH2 and RCLH1 complexes we used in this work exhibit fluorescence emission peaks at 862 nm and 893 nm, respectively. Analysis of emission spectra of immobilized LH complexes, in terms of peak position and shape, show that their structural integrity is retained after patterning on the glass substrate.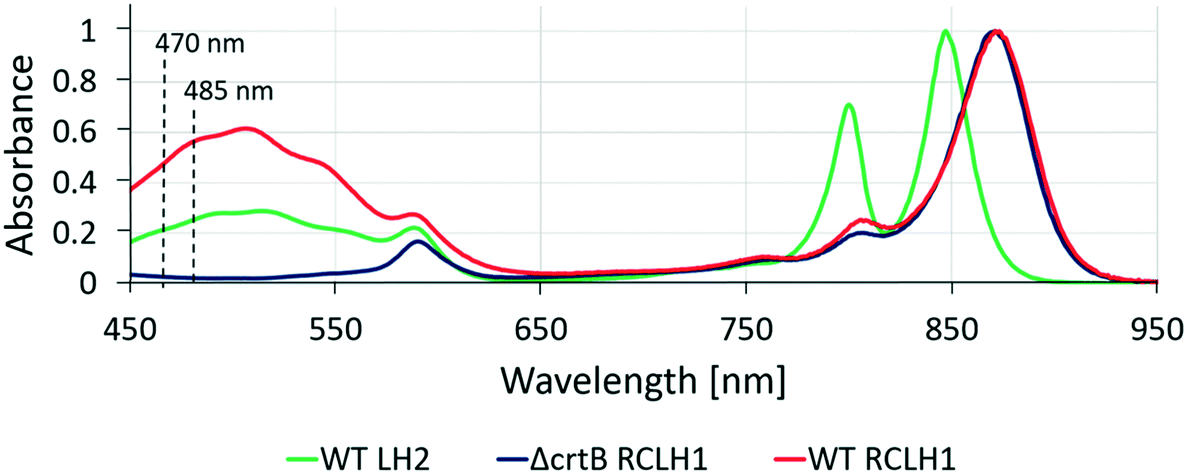 | ||
| Fig. 3 Absorption spectra of purified complexes used in this study. LH complexes were excited either at 470 nm or 485 nm for fluorescent emission. At 485 nm, the absorbance of RCLH1, LH2 and ΔcrtB RCLH1 are 0.57, 0.26 and 0.02, respectively. At 470 nm, the absorption of RCLH1, LH2 and ΔcrtB RCLH1 are 0.50, 0.22 and 0.02, respectively. | ||
Fig. 4 shows fluorescence data acquired from a sample cross-printed with LH2 and RCLH1 complexes on PLL-coated glass, sealed in imaging buffer (20 mM HEPES, argon-sparged). The false-color fluorescence image (Fig. 4A) of the sample, acquired in epi-fluorescence mode and illuminated by the 470 nm LED source, shows the distribution of the LH complexes on the surface; the green regions correspond to LH2 complexes (857/30 nm bandpass filter), and those in red are from RCLH1 complexes (900/32 nm bandpass filter). When switching to scanning confocal mode and using the 485 nm pulsed laser as excitation light source, we were able to record the spectral map of fluorescence emission of the sample (Fig. 4B–D). The fluorescence intensity maps acquired at 860 nm and 890 nm (Fig. 4B and C, respectively), confirm the immobilization of the LH2 complexes along the near-horizontal lines and immobilization of the RCLH1 complexes along the near-vertical lines. A striking observation in the cross-over area (marked with number 2 in Fig. 4A and B), where LH2 and RCLH1 complexes are in very close proximity, is the decrease in the LH2 emission intensity, accompanied by a comparable increase in RCLH1 emission. This observation is confirmed by the deconvolution of the spectra extracted from the pixel marked with the number 2 (cross-over area), orange curve in Fig. 4D, and its comparison with the individual spectra, green and red in Fig. 4D, extracted from the pixels marked with the numbers 1 and 3 in panel B (LH2-only and RCLH1-only areas, respectively). We interpret these changes in the emission intensities as an indication of excitation energy transfer (EET) between the LH2 and RCLH1 complexes.
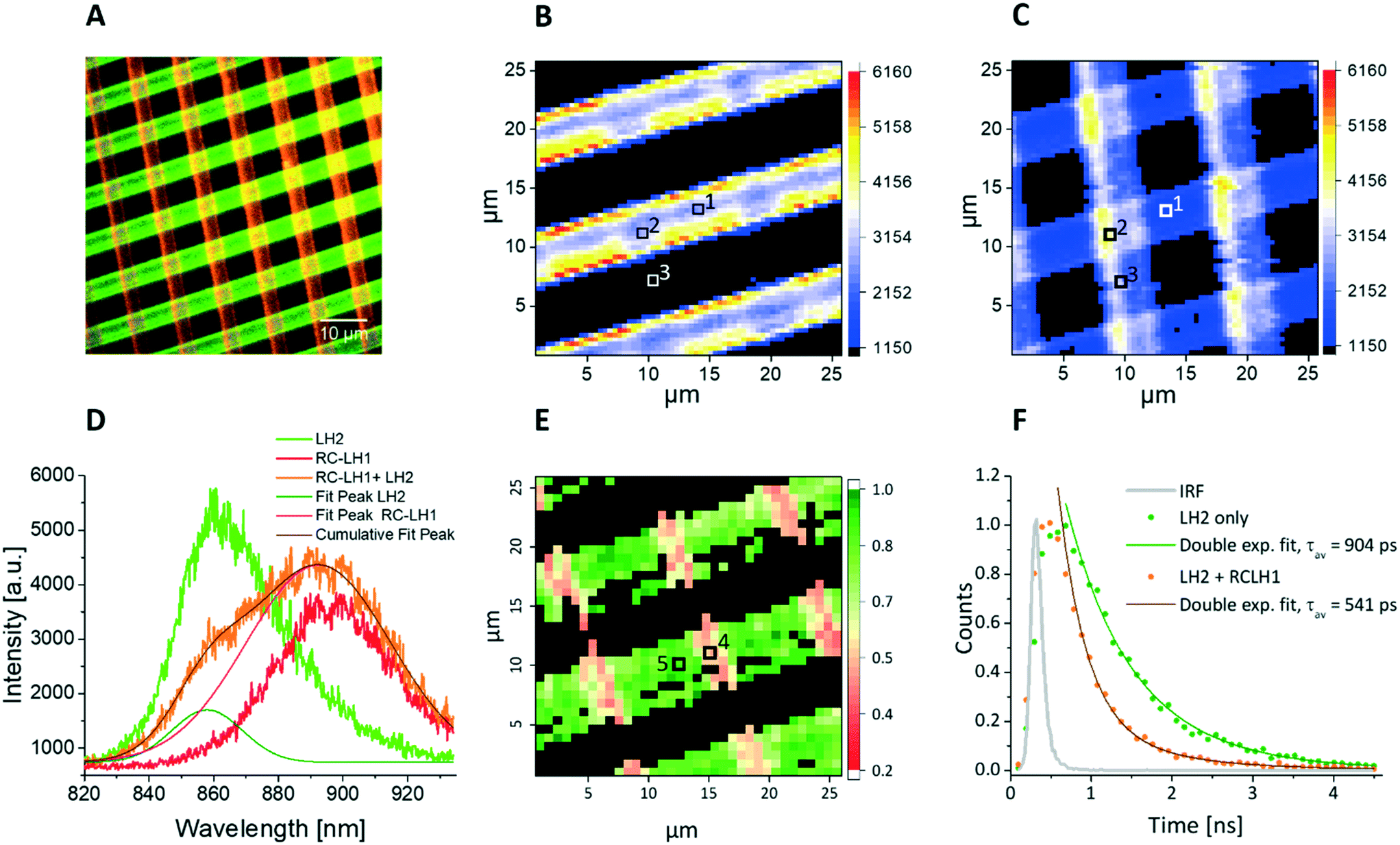 | ||
| Fig. 4 Spectral and lifetime fluorescence obtained from cross-patterned LH2 and RCLH1 complexes under physiological conditions (in imaging buffer) on a functionalised glass substrate. A – False colour fluorescence image (wide field excitation at 470 nm), showing the LH2 (green) and RCLH1 (red) lines in a grid-like pattern with a period of 10 μm and line width of 5 μm. B – Spectral map showing the emission intensity at 860 nm (LH2 emission peak), with excitation at 485 nm from a pulsed laser; scan size 26 μm. The LH2 emission intensity drops in the areas of intersection with RCLH1. C – Simultaneously acquired spectral map showing the emission intensity at 890 nm (RCLH1 emission peak), scan size 26 μm. D – Individual emission spectra recorded in the pixels of the images in panels B and C marked with 1 (LH2 only, green), 2 (LH2/RCLH1 intersection, orange) and 3 (RCLH1 only, red). The spectral deconvolution in D shows a clear drop in the LH2 emission (olive peak fit) compared to the LH2 emission outside the cross-over area, accompanied with a corresponding increase in the RCLH1 emission (pink peak fit) compared to the RCLH1-only zone outside the cross-over area. E – Amplitude weighted average lifetime image obtained at 485 nm excitation, and 860 nm emission (LH2 complex emission peak), clearly showing a decrease in the lifetime in the cross-over areas, where the two complexes are in close proximity, scan size 26 μm. F – Individual decay curves recorded in the pixels of the lifetime image in panel E marked with 4 (orange, average lifetime of 541 ps) and 5 (green, average lifetime of 904 ps), respectively. | ||
In order to study EET between surface-immobilized complexes in more detail we recorded, simultaneously with the spectral map, a fluorescence lifetime map of the LH2 complexes on the surface. The photon fluence for all lifetime measurements was about 2.0 × 1014 photons per pulse per cm2, which is sufficiently low to minimise excitonic annihilation in the LH complexes. An amplitude-averaged lifetime image of the cross-patterned sample, recorded at 860 nm (LH2 peak emission wavelength) is shown in Fig. 4E with two individual fluorescence decay curves shown in Fig. 4F. From Fig. 4E, the LH2-only areas (green) generally show longer lifetimes in the 750–900 ps range, compared with the LH2–RCLH1 intersection area (pink) where the LH2 lifetimes are in the 400–550 ps range. In Fig. 4F, the green decay curve was extracted from the pixel marked 5 (Fig. 4E) corresponding to the LH2-only area, and the bi-exponential decay function fitting result shows an amplitude-averaged lifetime τav = 904 ± 85 ps, with components A1 = 0.39 ± 0.08, τ1 = 1119 ± 97 ps and A2 = 0.61 ± 0.11, τ2 = 678 ± 77 ps; the orange curve corresponds to a part of the LH2–RCLH1 cross-patterned zone (marked 4, Fig. 4E), and the bi-exponential decay function fitting result shows an amplitude-averaged lifetime τav = 541 ± 47 ps, with components A1 = 0.09 ± 0.01, τ1 = 1138 ± 112 ps and A2 = 0.91 ± 0.21, τ2 = 348 ± 38 ps. This reduction of the LH2 fluorescence lifetime indicates EET from the LH2 complex to RCLH1 complex in the cross-over areas where the two protein molecules are in very close proximity.
Energy transfer from LH2 to ΔcrtB RCLH1 in a protective environment
The LH2 and RCLH1 complexes studied in Fig. 2 both contained carotenoids and therefore were both excited by the 485 nm light source, which complicates the assignment of energy transfer between the complexes. In order to overcome this problem, and to specifically excite only the LH2 energy transfer donor, we purified RCLH1 complexes from the ΔcrtB RCLH1 strain,35 which has no carotenoids and therefore has negligible absorption at the excitation wavelength of 485 nm (Fig. 3). Due to the lack of carotenoids ΔcrtB RCLH1 in particular is expected to be less photostable compared to WT-RCLH1, so the surface-patterned LH complexes were partially dehydrated, and then sealed under a protective argon atmosphere. It is worth noting that the same combination of proteins was also investigated under hydrated conditions, as used for experiments in Fig. 2, and as described in the ESI† (Fig. S1). Fig. 5A shows a false colour epifluorescence image of cross-patterned LH2 (green) and ΔcrtB RCLH1 (red) complexes, with wide field excitation at 470 nm and the emission signals filtered by 857/30 nm and 900/32 nm bandpass filters, respectively. Fig. 5A shows the precision of patterning of the two types of complex, delineating their positions, and the differing emission bands in panel D provide evidence that the immobilized complexes have retained their properties and structural integrity under the protective conditions of our experiment. Spectral imaging of the co-patterned complexes (Fig. 5B) shows lines arising from LH2 emission at 860 nm. Orthogonal RCLH1 lines are absent because absorption of the 485 nm excitation light is very weak due to the absence of carotenoids (see Fig. 2), so fluorescence emission was barely detectable over the background in the areas where the ΔcrtB RCLH1 complexes were immobilized on their own. As expected, the spectral intensity map shows that the lines of LH2 emission were not uniform; individual emission spectra, corresponding to the pixels in Fig. 5B and C and marked with the numbers 1(LH2-only area, green), 2 (LH2/RCLH1 intersection area, orange), and 3 (RCLH1 area, red), are shown in Fig. 5D. Spectral deconvolution shows a clear drop in the LH2 emission (olive peak fit) at the LH2/RCLH1 intersection and a large increase in the ΔcrtB RCLH1 emission (pink peak fit), compared to the ΔcrtB RCLH1 emission outside the intersecting area (pixel 3, red). A simultaneously acquired spectral map of emission intensity at 890 nm from ΔcrtB RCLH1 complexes (Fig. 5C) shows major signals at the LH2/RCLH1 intersections; given that excitation is specific for LH2, this RCLH1 emission must have arisen from excitation energy transfer from neighbouring LH2 complexes.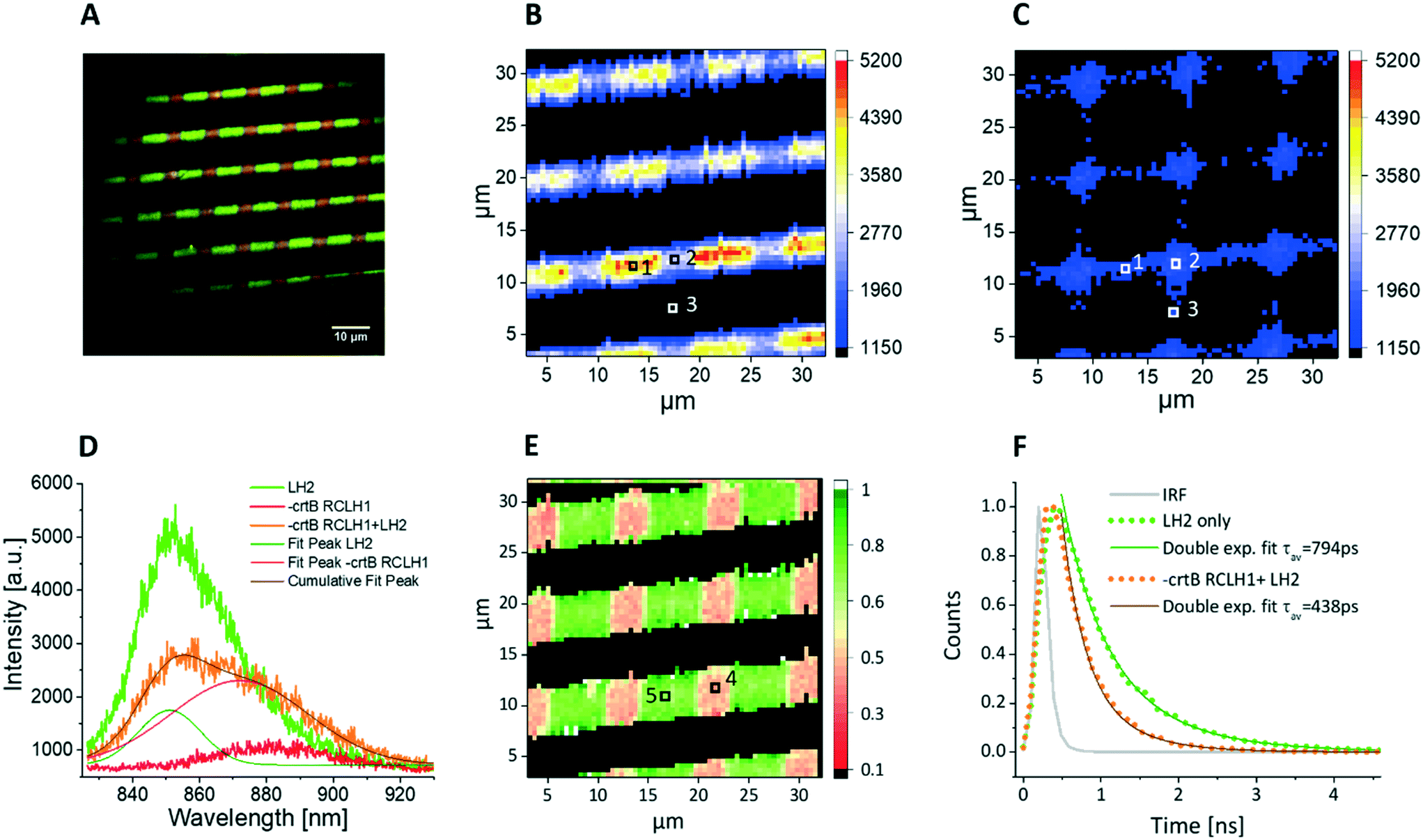 | ||
| Fig. 5 Spectral and lifetime fluorescence data of cross-patterned LH2 and ΔcrtB RCLH1 complexes on a functionalised glass substrate imaged in protective atmosphere of argon. A – False colour fluorescence image (wide field excitation at 470 nm), showing a grid-like pattern of LH2 and ΔcrtB RCLH1 complexes with a period of 10 μm and line width of 5 μm. The LH2 emission is in green, while the areas where ΔcrtB RCLH1 complexes were present are shown in red, filtered by 857/30 nm and 900/32 nm bandpass filters, respectively. B – Spectral map showing the emission intensity at 860 nm (LH2 emission), with excitation at 485 nm from a pulsed laser, scan size 32 μm. The positions of three pixels used for acquiring spectral data are marked. C – Simultaneously acquired spectral map showing the emission intensity at 890 nm (ΔcrtB RCLH1 emission), scan size 32 μm, with increased emission in the areas where LH2 is present as the energy transfer donor. D – Individual emission spectra recorded in the pixels of the images in panels B and C marked with 1 (LH2 only, green line), 2 (intersecting area, orange line) and 3 (ΔcrtB RCLH1 only, red line), respectively. The spectral deconvolution shows a clear drop in the LH2 emission (olive peak fit) and an increase in the ΔcrtB RCLH1 emission (pink peak fit), compared to the LH2-only emission and the ΔcrtB RCLH1-only emission outside the intersecting area. E – Amplitude weighted average lifetime image obtained with 485 nm excitation and recording 857 nm emission (LH2 complex emission peak), clearly showing a decrease in the lifetime in the intersecting areas, where the two complexes are in close proximity, scan size 32 μm. F – Individual decay curves recorded in the pixels of the lifetime image in panel E marked with 4 (orange, average lifetime of 438 ps) and 5 (green, average lifetime of 794 ps), respectively. | ||
Fig. 5E shows an amplitude-weighted fluorescence lifetime map of surface-attached LH2 and RCLH1 complexes, with excitation at 485 nm and emission monitored at 857 nm, the peak emission wavelength of LH2. Individual decay curves corresponding to pixels 4 and 5 of the lifetime image are shown in Fig. 5F. From Fig. 5E, the LH2 only areas (green) generally show longer lifetimes of between 700–900 ps, compared with the LH2–ΔcrtB RCLH1 intersecting area (pink) of about between 400–500 ps. In Fig. 5F, the green decay curve was extracted from the pixel marked 5 (Fig. 5E) corresponding to the LH2-only area, and the bi-exponential decay function fitting shows an amplitude-averaged lifetime τav = 794 ± 67 ps, with components A1 = 0.45 ± 0.06, τ1 = 974 ± 83 ps and A2 = 0.55 ± 0.08, τ2 = 512 ± 55 ps; the orange curve represents the fluorescence decay in the intersecting area (marked 4, Fig. 5E), and the bi-exponential decay function fitting result shows an amplitude-averaged lifetime τav = 438 ± 48 ps, with components A1 = 0.25 ± 0.04, τ1 = 645 ± 71 ps and A2 = 0.75 ± 0.12, τ2 = 284 ± 39 ps. Again, the lowered LH2 fluorescence lifetime confirms the EET from the LH2 complex to ΔcrtB RCLH1 complex in the intersecting areas where the two protein molecules are in close proximity.
It is worth noting that our experiment allows for the presence of some inactivated RCLH1 complexes. When using wild type RCLH1 (Fig. 4), the 485 nm light excites the carotenoids in both LH2 and RCLH1 complexes, whereas when using ΔcrtB RCLH1 (Fig. 5) the excitation light targets only LH2, and not the carotenoidless ΔcrtB RCLH1. Looking at Fig. 4D (red line), we can see a larger amplitude of LH1 fluorescence than in Fig. 5D (red line), likely because of direct excitation of the carotenoids in RCLH1, thus turning over RCs photochemistry and generating a ‘closed’ (inactive) state. There is far less chance of this occurring in carotenoidless ΔcrtB RCLH1 complexes, and in this case, more RCs will be ‘open’, i.e. active. Despite the likely presence of ‘closed’ RCs in Fig. 4, we still see the characteristics of directional energy transfer from LH2 to LH1, namely a shortened fluorescence lifetime and greatly lowered fluorescence intensity in LH2.
Long-term stability of cross-patterned LH2 and ΔcrtB RCLH1 complexes on glass surfaces
In order to test the stability and “shelf life” of these artificial light-harvesting/energy transfer systems, the samples were sealed in argon atmosphere and stored at 4 °C in the dark. Regular FLIM measurements showed that both LH2 and ΔcrtB RCLH1 are quite stable under these conditions. From days 1 to 60, there is a small variation in the fluorescence lifetime of LH2 complex (measured in an LH2 only area) with an average lifetime of 740 ± 90 ps (Table 1). At the same time, the average lifetime of the LH2 complex measured in the LH2 + ΔcrtB RCLH1 intersecting area remains relatively constant at 400 ± 50 ps (Table 1). Furthermore, based on the observed quenching of the LH2 fluorescence, our fluorescence lifetime images (Fig. S2C†) clearly show excitation energy transfer between LH2 and ΔcrtB RCLH1 60 days after sample preparation. The fluorescence intensity images at 857 nm (Fig. S2A†) and 890 nm (Fig. S2B†), respectively, show that the pattern is well preserved on the functionalised glass substrate with no diffusion of protein complexes, while the emission spectra (Fig. S2D†) indicate that both LH2 and ΔcrtB RCLH1 complexes remain undamaged.| Storage time | τ 1 [ps] LH2 + ΔcrtB RCLH1 | τ 1 [ps] LH2 | τ 2 [ps] LH2 + ΔcrtB RCLH1 | τ 2 [ps] LH2 | τ av [ps] LH2 + ΔcrtB RCLH1 | τ av [ps] LH2 |
|---|---|---|---|---|---|---|
| Day 1 | 640 | 1040 | 280 | 520 | 430 | 770 |
| Day 5 | 480 | 1040 | 290 | 500 | 380 | 740 |
| Day 10 | 510 | 950 | 280 | 430 | 380 | 680 |
| Day 15 | 600 | 1100 | 360 | 600 | 450 | 840 |
| Day 20 | 490 | 1010 | 280 | 500 | 370 | 770 |
| Day 30 | 450 | 950 | 290 | 470 | 350 | 710 |
| Day 60 | 540 | 840 | 320 | 430 | 410 | 650 |
Conclusions
Previous work using reconstituted membranes has shown that energy transfer can be observed in mixed assemblies of LH2 and RCLH1 complexes,36–38 but controlling the relative stoichiometries and two-dimensional organization of energy donor/acceptor photosynthetic complexes on a nanometer scale, and the locations where excitation energy can migrate between them, presents new challenges. For some time, it has been possible to construct nanoarrays of single types of photosynthetic complex, starting with the light-harvesting LH2 complex of Rba. sphaeroides,18,19,21 and later the RCLH1 complex20 and the LHCII complex of plants.22 In each case the function of the complex, in terms of fluorescence emission, was retained; for LHCII it was possible to directly image the ability of immobilized molecules of LHCII to switch between fluorescent and quenched states. For LH2, there were indications of long-range excitation energy transfer; 80 nm-wide nanolines of LH2 complexes exhibited energy propagation on micron length scales, which greatly exceed the natural energy propagation lengths found in in native photosynthetic membranes.17 Excitation energy is generally trapped before it migrates for more than 50–100 nm within natural energy transfer and trapping networks such as the chromatophore vesicles of purple bacteria,3,4 or the thylakoids of cyanobacteria, algae and plants.39Nanoarrays of two or more types of photosynthetic complex would be valuable tools to investigate LH antenna-to-RC trap ratios, to explore length scales and geometries of energy migration and trapping that lie beyond those found in biology. However, progress with these aims relies on the ability to co-pattern two or more types of complex on the same surface and on the application of spectral and time-resolved microscopies to assess the functional state of immobilized assemblies. Recent developments in surface chemistries do allow multiprotein patterning,40 and an alternating linear LHCII/EGFP pattern has been reported.22 Here, we have used a simple lithographic method to cross-print LH and energy trapping complexes, and at the points of intersection we demonstrate collection of light by one complex, LH2, and its subsequent transfer to the RCLH1 complex. Thus, this assembly can be regarded as a fabricated ‘photosynthetic unit’, where the complexes adopt a predetermined, geometric configuration. Energy transfer requires separations of 5 nm or less, so nanoscale arrangements between complexes must be established within the intersecting LH2/RCLH1 domains.
Although the stability of surface-immobilized proteins is a potential problem, many studies have shown that photosystem complexes are stable on a variety of substrates. Purified bacterial RCs, stabilised using peptide surfactants, retain their function when deposited on indium-tin oxide (ITO)-coated glass,41 gold electrodes,42–44 or gallium arsenide.45 Stable coatings of RCLH1, photosystem II46–48 and photosystem I49–51 complexes on electrodes have been reported. To our knowledge, there has been no systematic study of the long-term stability of surface-attached complexes on a functionalised glass substrate, so we undertook a 60 day test with the cross-patterned arrays of photosynthetic complexes sealed in argon and stored at 4 °C in the dark. These artificial light-harvesting systems proved to be extremely stable under these conditions, and further work will examine the stability of other natural and de novo designed maquette complexes.
Studies of LH2-only bacterial membranes (where the packing of protein complexes is optimal) reveal an average fluorescence lifetime of around 500 ps,52 while solubilized LH2 complexes exhibit a fluorescence lifetime of around 1 ns. In our experiments, the LH2-only areas of the patterns exhibit an average lifetime in between those two values (800–900 ps). This indicates that the packing of the LH2 complexes on the surface is not as dense as in the biological membranes, however, the LH2 complexes are close enough to observe partial quenching of the fluorescence lifetime.
It is worth noting that although some of the RCLH1 complexes (in the cross-over area) are immobilized on top of the existing LH2 layer (the rest are likely attached directly onto the substrate thus intermixing with the LH2 complexes), this stacked arrangement still allows efficient energy transfer to occur between the LH2 and RCLH1 molecules despite the fact that both complexes evolved for the ‘lateral’ energy transfer occurring in biological membranes.
The means to control the relative positions of two or more types of molecule on the same surface allows construction of ‘mix and match’ combinations of molecules that could not be created through genetic means, such as arrays comprising mixed bacterial/plant, or plant/artificial maquette complexes. Further functional tests will include measuring the nanoelectrical properties of RC traps within native, biohybrid and bioinspired photosynthetic arrays, deposited on conducting substrates. This development would add a third function of charge separation to the absorption and transfer of energy demonstrated in the present work.
Author contributions
Conceptualization: X. H., C. V. and C. N. H. Data curation and formal analysis: C. V. and X. H. Funding acquisition: C. N. H. Investigation: X. H. and C. V. Methodology: C. V. and C. N. H. Project administration and resources: C. N. H. Supervision: C. V. and C. N. H. Writing-original draft: X. H., C. V. and C. N. H. Writing-review and editing: C. V. and C. N. H.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported as part of the Photosynthetic Antenna Research Center (PARC), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001035. PARC supported the AFM, spectral imaging and fluorescence lifetime studies, provided a doctoral studentship for X. H., supported C. V. and provided partial support for C. N. H. C. N. H. also acknowledges financial support from the Biotechnology and Biological Sciences Research Council (BBSRC UK), award number BB/M000265/1.References
- R. G. Saer and R. E. Blankenship, Biochem. J., 2017, 474, 2107–2131 CrossRef CAS PubMed.
- M. L. Cartron, J. D. Olsen, M. Sener, P. J. Jackson, A. A. Brindley, P. Qian, M. J. Dickman, G. J. Leggett, K. Schulten and C. N. Hunter, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 1769–1780 CrossRef CAS PubMed.
- M. Sener, J. Strumpfer, A. Singharoy, C. N. Hunter and K. Schulten, eLife, 2016, 5, e09541 CrossRef PubMed.
- M. Şener, J. Strümpfer, J. A. Timney, A. Freiberg, C. N. Hunter and K. Schulten, Biophys. J., 2010, 99, 67–75 CrossRef PubMed.
- A. Hitchcock, C. N. Hunter and M. Sener, J. Phys. Chem. B, 2017, 121, 3787–3797 CrossRef CAS.
- P. K. Dutta, S. Levenberg, A. Loskutov, D. Jun, R. Saer, J. T. Beatty, S. Lin, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2014, 136, 16618–16625 CrossRef CAS PubMed.
- P. K. Dutta, S. Lin, A. Loskutov, S. Levenberg, D. Jun, R. Saer, J. T. Beatty, Y. Liu, H. Yan and N. W. Woodbury, J. Am. Chem. Soc., 2014, 136, 4599–4604 CrossRef CAS PubMed.
- K. Gundlach, M. Werwie, S. Wiegand and H. Paulsen, Biochim. Biophys. Acta, Bioenerg., 2009, 1787, 1499–1504 CrossRef CAS PubMed.
- M. A. Harris, J. Jiang, D. M. Niedzwiedzki, J. Jiao, M. Taniguchi, C. Kirmaier, P. A. Loach, D. F. Bocian, J. S. Lindsey, D. Holten and P. S. Parkes-Loach, Photosynth. Res., 2014, 121, 35–48 CrossRef CAS PubMed.
- M. A. Harris, P. S. Parkes-Loach, J. W. Springer, J. Jiang, E. C. Martin, P. Qian, J. Jiao, D. M. Niedzwiedzki, C. Kirmaier, J. D. Olsen, D. F. Bocian, D. Holten, C. N. Hunter, J. S. Lindsey and P. A. Loach, Chem. Sci., 2013, 4, 3924–3933 RSC.
- M. A. Harris, T. Sahin, J. Jiang, P. Vairaprakash, P. S. Parkes-Loach, D. M. Niedzwiedzki, C. Kirmaier, P. A. Loach, D. F. Bocian, D. Holten and J. S. Lindsey, Photochem. Photobiol., 2014, 90, 1264–1276 CrossRef CAS PubMed.
- K. A. Meadows, K. Iida, K. Tsuda, P. A. Recchia, B. A. Heller, B. Antonio, M. Nango and P. A. Loach, Biochemistry, 1995, 34, 1559–1574 CrossRef CAS PubMed.
- J. W. Springer, P. S. Parkes-Loach, K. R. Reddy, M. Krayer, J. Jiao, G. M. Lee, D. M. Niedzwiedzki, M. A. Harris, C. Kirmaier, D. F. Bocian, J. S. Lindsey, D. Holten and P. A. Loach, J. Am. Chem. Soc., 2012, 134, 4589–4599 CrossRef CAS PubMed.
- Y. Yoneda, T. Noji, T. Katayama, N. Mizutani, D. Komori, M. Nango, H. Miyasaka, S. Itoh, Y. Nagasawa and T. Dewa, J. Am. Chem. Soc., 2015, 137, 13121–13129 CrossRef CAS.
- G. Kodali, J. A. Mancini, L. A. Solomon, T. V. Episova, N. Roach, C. J. Hobbs, P. Wagner, O. A. Mass, K. Aravindu, J. E. Barnsley, K. C. Gordon, D. L. Officer, P. L. Dutton and C. C. Moser, Chem. Sci., 2017, 8, 316–324 RSC.
- J. A. Mancini, G. Kodali, J. Jiang, K. R. Reddy, J. S. Lindsey, D. A. Bryant, P. L. Dutton and C. C. Moser, J. R. Soc., Interface, 2017, 14, 20160896 CrossRef PubMed.
- M. Escalante, A. Lenferink, Y. Zhao, N. Tas, J. Huskens, C. N. Hunter, V. Subramaniam and C. Otto, Nano Lett., 2010, 10, 1450–1457 CrossRef CAS.
- M. Escalante, P. Maury, C. M. Bruinink, K. van der Werf, J. D. Olsen, J. A. Timney, J. Huskens, C. N. Hunter, V. Subramaniam and C. Otto, Nanotechnology, 2008, 19, 025101 CrossRef PubMed.
- M. Escalante, Y. Zhao, M. J. W. Ludden, R. Vermeij, J. D. Olsen, E. Berenschot, C. N. Hunter, J. Huskens, V. Subramaniam and C. Otto, J. Am. Chem. Soc., 2008, 130, 8892–8893 CrossRef CAS PubMed.
- S. Patole, C. Vasilev, O. El-Zubir, L. Wang, M. P. Johnson, A. J. Cadby, G. J. Leggett and C. N. Hunter, Interface Focus, 2015, 5, 20150005 CrossRef PubMed.
- N. P. Reynolds, S. Janusz, M. Escalante-Marun, J. Timney, R. E. Ducker, J. D. Olsen, C. Otto, V. Subramaniam, G. J. Leggett and C. N. Hunter, J. Am. Chem. Soc., 2007, 129, 14625–14631 CrossRef CAS PubMed.
- C. Vasilev, M. P. Johnson, E. Gonzales, L. Wang, A. V. Ruban, G. Montano, A. J. Cadby and C. N. Hunter, Langmuir, 2014, 30, 8481–8490 CrossRef CAS PubMed.
- P. Qian, M. Z. Papiz, P. J. Jackson, A. A. Brindley, I. W. Ng, J. D. Olsen, M. J. Dickman, P. A. Bullough and C. N. Hunter, Biochemistry, 2013, 52, 7575–7585 CrossRef CAS PubMed.
- A. Freer, S. Prince, K. Sauer, M. Papiz, A. Hawthornthwaite-Lawless, G. McDermott, R. Cogdell and N. W. Isaacs, Structure, 1996, 4, 449–462 CrossRef CAS PubMed.
- D. Noy, C. C. Moser and P. L. Dutton, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 90–105 CrossRef CAS PubMed.
- M. Şener, J. Hsin, L. G. Trabuco, E. Villa, P. Qian, C. N. Hunter and K. Schulten, Chem. Phys., 2009, 357, 188–197 CrossRef PubMed.
- T. Walz, S. J. Jamieson, C. M. Bowers, P. A. Bullough and C. N. Hunter, J. Mol. Biol., 1998, 282, 833–845 CrossRef CAS PubMed.
- P. Qian, C. N. Hunter and P. A. Bullough, J. Mol. Biol., 2005, 349, 948–960 CrossRef CAS PubMed.
- X.-M. Zhao, Y. Xia and G. M. Whitesides, J. Mater. Chem., 1997, 7, 1069–1074 RSC.
- M. Swoboda, J. Henig, H.-M. Cheng, D. Brugger, D. Haltrich, N. Plumeré and M. Schlierf, ACS Nano, 2012, 6, 6364–6369 CrossRef CAS.
- S. A. Ruiz and C. S. Chen, Soft Matter, 2007, 3, 168–177 RSC.
- A. Bernard, J. P. Renault, B. Michel, H. R. Bosshard and E. Delamarche, Adv. Mater., 2000, 12, 1067–1070 CrossRef CAS.
- Y. Xia and G. M. Whitesides, Annu. Rev. Mater. Sci., 1998, 28, 153–184 CrossRef CAS.
- J. D. Tucker, C. A. Siebert, M. Escalante, P. Adams, J. D. Olsen, C. Otto, D. L. Stokes and C. N. Hunter, Mol. Microbiol., 2010, 76, 833–847 CrossRef CAS PubMed.
- K. J. Grayson, K. M. Faries, X. Huang, P. Qian, P. Dilbeck, E. C. Martin, A. Hitchcock, C. Vasilev, J. Yuen, D. M. Niedzwiedzki, G. J. Leggett, D. Holten, C. Kirmaier and C. N. Hunter, Nat. Commun., 2017, 8, 13972 CrossRef CAS PubMed.
- C. N. Hunter, R. Van Grondelle, N. G. Holmes and O. T. Jones, Biochim. Biophys. Acta, Bioenerg., 1979, 548, 458–470 CrossRef CAS.
- A. Sumino, T. Dewa, M. Kondo, T. Morii, H. Hashimoto, A. T. Gardiner, R. J. Cogdell and M. Nango, Langmuir, 2011, 27, 1092–1099 CrossRef CAS PubMed.
- C. Uragami, Y. Sugai, K. Hanjo, A. Sumino, R. Fujii, T. Nishioka, I. Kinoshita, T. Dewa, M. Nango, A. T. Gardiner, R. J. Cogdell and H. Hashimoto, J. Photochem. Photobiol., A, 2015, 313, 60–71 CrossRef CAS.
- K. Amarnatha, D. I. G. Bennett, A. R. Schneider and G. R. Fleming, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1156–1161 CrossRef PubMed.
- O. El Zubir, S. Xia, R. E. Ducker, L. Wang, N. Mullin, M. L. Cartron, A. J. Cadby, J. K. Hobbs, C. N. Hunter and G. J. Leggett, Langmuir, 2017, 33, 8829–8837 CrossRef CAS PubMed.
- R. Das, P. J. Kiley, M. Segal, J. Norville, A. A. Yu, L. Wang, S. A. Trammell, L. E. Reddick, R. Kumar, F. Stellacci, N. Lebedev, J. Schnur, B. D. Bruce, S. Zhang and M. Baldo, Nano Lett., 2004, 4, 1079–1083 CrossRef CAS.
- M.-H. Ham, J. H. Choi, A. A. Boghossian, E. S. Jeng, R. A. Graff, D. A. Heller, A. C. Chang, A. Mattis, T. H. Bayburt, Y. V. Grinkova, A. S. Zeiger, K. J. Van Vliet, E. K. Hobbie, S. G. Sligar, C. A. Wraight and M. S. Strano, Nat. Chem., 2010, 2, 929–936 CrossRef CAS PubMed.
- S. A. Trammell, I. Griva, A. Spano, S. Tsoi, L. M. Tender, J. Schnur and N. Lebedev, J. Phys. Chem. C, 2007, 111, 17122–17130 CrossRef CAS.
- J. Zhao, Y. Zou, B. Liu, C. Xu and J. Kong, Biosens. Bioelectron., 2002, 17, 711–718 CrossRef CAS PubMed.
- L. Frolov, Y. Rosenwaks, S. Richter, C. Carmeli and I. Carmeli, J. Phys. Chem. C, 2008, 112, 13426–13430 CrossRef CAS.
- A. Badura, B. Esper, K. Ataka, C. Grunwald, C. Wöll, J. Kuhlmann, J. Heberle and M. Rögner, Photochem. Photobiol., 2006, 82, 1385–1390 CrossRef CAS PubMed.
- M.-J. den Hollander, J. G. Magis, P. Fuchsenberger, T. J. Aartsma, M. R. Jones and R. N. Frese, Langmuir, 2011, 27, 10282–10294 CrossRef CAS PubMed.
- N. Terasaki, M. Iwai, N. Yamamoto, T. Hiraga, S. Yamada and Y. Inoue, Thin Solid Films, 2008, 516, 2553–2557 CrossRef CAS.
- C. J. Faulkner, S. Lees, P. N. Ciesielski, D. E. Cliffel and G. K. Jennings, Langmuir, 2008, 24, 8409–8412 CrossRef CAS PubMed.
- H. Krassen, A. Schwarze, Br. Friedrich, K. Ataka, O. Lenz and J. Heberle, ACS Nano, 2009, 3, 4055–4061 CrossRef CAS PubMed.
- O. Yehezkeli, O. I. Wilner, R. Tel-Vered, D. Roizman-Sade, R. Nechushtai and I. Willner, J. Phys. Chem. B, 2010, 114, 14383–14388 CrossRef CAS PubMed.
- K. Timpmann, M. Chenchiliyan, E. Jalviste, J. A. Timney, C. N. Hunter and A. Freiberg, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 1835–1846 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00156b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |