
A microfluidic approach for experimentally modelling the intercellular coupling system of a mammalian circadian clock at single-cell level†
 ‡§bc
Yanyi
Huang
*af
‡§bc
Yanyi
Huang
*af
Abstract
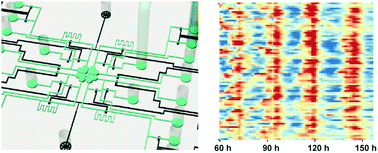
In mammals, it is believed that the intercellular coupling mechanism between neurons in the suprachiasmatic nucleus (SCN) confers robustness and distinguishes the central clock from peripheral circadian oscillators. Current in vitro culturing methods used in Petri dishes to study intercellular coupling by exogenous factors invariably cause perturbations, such as simple media changes. Here, we design a microfluidic device to quantitatively study the intercellular coupling mechanism of circadian clock at the single cell level, and demonstrate that vasoactive intestinal peptide (VIP) induced coupling in clock mutant Cry1-/- mouse adult fibroblasts engineered to express the VIP receptor, VPAC2, is sufficient to synchronize and maintain robust circadian oscillations. Our study provides a proof-of-concept platform to reconstitute the intercellular coupling system of the central clock using uncoupled, single fibroblast cells in vitro, to mimic SCN slice cultures ex vivo and mouse behavior in vivo phenotypically. Such a versatile microfluidic platform may greatly facilitate the studies of intercellular regulation networks, and provide new insights into the coupling mechanisms of the circadian clock.
 Please wait while we load your content...
Please wait while we load your content...

