
Nanoplasmon-enhanced drop-screen for high throughput single-cell nucleocytoplasmic miRNA profiling†

Abstract
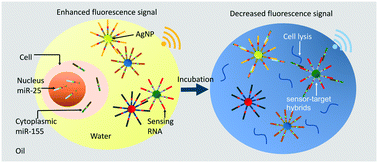
Cell nucleocytoplasmic profiles of microRNAs (miRNAs) are critical to determining a single cell's essential functionalities, such as cellular transcription, nucleus export and degradation, which gives a comprehensive view of cellular processes. Despite the importance of addressing nucleocytoplasmic heterogeneity, the challenge of high-throughput screening remains. Although a droplet-based approach was developed for single-cell miRNA assays, the challenge of quantifying miRNA with high sensitivity to indicate nucleocytoplasmic heterogeneity remains. In this study, a nanoplasmon-enhanced droplet screening platform was developed to quantify single-cell nucleocytoplasmic heterogeneity with the high sensitivity of 0.1 nM. Droplet screening and multiplexed plasmonic assays are synergistic: droplet screening is used to isolate single cells for high-throughput screening, while enhanced nanoplasmonic assays are conducted to precisely determine different types of miRNAs, addressing the cell nucleocytoplasmic profile. Here, two nucleic acid-functionalized plasmonic nanosensors, silver nanoparticles functionalized with designed sequences to target miRNAs, are synthesized. After the targets are bound, competitive formation of sensor-target hybrids interferes with plasmonic coupling between the nanoparticles, decreasing a fluorescence signal and thus enabling high-sensitivity single-cell miRNA quantification. Using the fluorescence signal change as a readout allows continuous-flow measurement to provide a single-cell nucleocytoplasmic profile in a high-throughput manner (∼100 cells per minute) for effective quantitative cell biology.
- This article is part of the themed collection: Single Cell Analysis
 Please wait while we load your content...
Please wait while we load your content...

