
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A flexible 3-dimensional microelectrode array for in vitro brain models†
David A.
Soscia
 a,
Doris
Lam
b,
Angela C.
Tooker
a,
Heather A.
Enright
b,
Michael
Triplett
a,
Piyush
Karande
a,
Sandra K. G.
Peters
b,
Ana Paula
Sales
a,
Elizabeth K.
Wheeler
a and
Nicholas O.
Fischer
*b
a,
Doris
Lam
b,
Angela C.
Tooker
a,
Heather A.
Enright
b,
Michael
Triplett
a,
Piyush
Karande
a,
Sandra K. G.
Peters
b,
Ana Paula
Sales
a,
Elizabeth K.
Wheeler
a and
Nicholas O.
Fischer
*b
aEngineering Directorate, Lawrence Livermore National Laboratory, 7000 East Ave, Livermore, CA 94550, USA
bPhysical and Life Sciences Directorate, Lawrence Livermore National Laboratory, 7000 East Ave, Livermore, CA 94550, USA. E-mail: fischer29@llnl.gov
First published on 23rd January 2020
Abstract
Three-dimensional (3D) in vitro models have become increasingly popular as systems to study cell–cell and cell–ECM interactions dependent on the spatial, mechanical, and chemical cues within the environment of the tissue, which is limited in traditional two-dimensional (2D) models. Although electrophysiological recordings of neuronal action potentials through 2D microelectrode arrays (MEAs) are a common and trusted method of evaluating neuronal function, network communication, and response to chemicals and biologicals, there are currently limited options for measuring electrophysiological activity from many locations simultaneously throughout a 3D network of neurons in vitro. Here, we have developed a thin-film, 3D flexible microelectrode array (3DMEA) that non-invasively interrogates a 3D culture of neurons and can accommodate 256 channels of recording or stimulation. Importantly, the 3DMEA is straightforward to fabricate and integrates with standard commercially available electrophysiology hardware. Polyimide probe arrays were microfabricated on glass substrates and mechanically actuated to collectively lift the arrays into a vertical position, relying solely on plastic deformation of their base hinge regions to maintain vertical alignment. Human induced pluripotent stem cell (hiPSC)-derived neurons and astrocytes were entrapped in a collagen-based hydrogel and seeded onto the 3DMEA, enabling growth of suspended cells in the matrix and the formation and maturation of a neural network around the 3DMEA probes. The 3DMEA supported the growth of functional neurons in 3D with action potential spike and burst activity recorded over 45 days in vitro. This platform is an important step in facilitating noninvasive electrophysiological characterization of 3D networks of electroactive cells in vitro.
Introduction
Traditionally, in vitro systems serve as a reductionist approach to study complex brain function. Isolated from brain tissue, dissociated neurons are typically grown on a 2-dimensional (2D) planar substrate to study cell health and function. In a controlled environment, these systems serve as testbeds to assess the electrical activity of neurons and their networks in response to chemical and biological exposures. However, 2D neuronal monocultures lack other cell types within the brain (e.g. astrocytes, oligodendrocytes and microglia), as well as mechanical and chemical cues present in three-dimensional (3D) in vivo tissue. In recent years, there has been an impetus to develop models that more accurately represent the function and physiology of the in vivo brain, by increasing the complexity of cellular composition and extracellular milieu. Currently, there are an increasing number of studies in the organ-on-a-chip field that aim to recapitulate brain function on 2D devices by incorporating extracellular matrix components,1 co-culturing support cells,2 and compartmentalizing multiple brain regions.3–5 Mirroring the 3D architecture of the brain has also been a significant goal in recent years, with a number of model systems developed such as brain organoids6–9 or the use of natural and/or synthetic 3D matrices to encapsulate neurons.10–14 Several of these studies have shown that there are significant differences in the way neurons organize, develop, and communicate when cultured in a 3D matrix as compared to those grown on 2D substrates, and that 3D models more closely resemble the functional behavior of in vivo tissue.15–17Multi-electrode arrays (MEAs) provide a non-invasive method to monitor the electrophysiological activity of neurons in vitro over time. Action potentials, the electrical impulses from neurons, can be detected using these electrodes by measuring a change in transmembrane or extracellular voltage of a cell or cluster of cells. In an interconnected network of neurons, this can be observed as organized bursts of action potentials and/or synchronized activity that is detected across multiple electrodes on the array. The temporal and spatial organization and synchronization of action potentials can provide crucial insight into the health of and communication throughout a population of neurons.3–5 For neurons cultured on a 2D surface, MEAs consisting of several thin-film metal electrodes patterned on the cell growth surface are utilized.18–24 As such, MEAs can record from many localized sites in parallel over long periods of time, providing a means to correlate activity across large populations of neurons and evaluate the system as an interconnected network. Thus, MEAs complement approaches that provide true intracellular recordings, such as patch clamping, that provide a greater level of detail regarding neuronal electrophysiological signalling.
There are currently very few options to assess the electrical activity of 3D cell culture models, and in particular, devices that allow for interrogating many locations simultaneously throughout a 3D network of neurons in vitro. Traditional single-electrode probes like glass25–28 and tungsten13,29,30 have been most commonly utilized by inserting the probe into the cell-containing matrix after maturation of the network. In other studies, 2D arrays of electrodes have been extended from substrates to measure a single plane of neurons; these generally are in the form of mechanically stiff “spike” electrode arrays similar to the in vivo Utah Array.31,32 An intriguing new style of MEA has also been introduced, intended to minimize mechanical impact of the array on the cell matrix by utilizing a thin, flexible polymer “mesh” containing either nanowire or thin-film electrodes.33–35 This approach has been focused on in vivo or ex vivo tissue-based applications, and it is limited in its ability to be integrated with an in vitro system.
In order to significantly advance the utility of 3D neuronal organ models, a new MEA design is needed that can capture network-wide electrophysiology of neurons in a 3D in vitro matrix. This requires the development of an ordered, integrated 3D array of microelectrodes that is straightforward to fabricate, biocompatible, minimally disruptive to the 3D biological system, amenable to facile cell culturing, and readily integrated into existing commercial recording instrumentation. We have developed an integrated in vitro platform that addresses all the aforementioned challenges for measuring the neuronal activity of 3D in vitro cultures, leveraging knowledge from implantable in vivo MEAs developed by our group.36–38 The 3D Microelectrode Array (3DMEA) is comprised of 80 thin-film electrodes distributed across 10 flexible polymer probes, with three independent 3DMEAs integrated on each device. While the arrays are fabricated using traditional 2D microfabrication, the array probes can be collectively actuated for vertical alignment using a custom apparatus. Plastic deformation of the probe hinge region ensures that the probes maintain vertical positioning, especially in a 3D biological system that is cultured long term.
To demonstrate the utility of the 3DMEA, we non-invasively monitored the electrophysiological activity of a human 3D neuronal culture. The 3D culture consisted of human iPSC-derived neurons and astrocytes entrapped in a collagen hydrogel supplemented with extracellular matrix (ECM) proteins (ECM-collagen) around the actuated 3DMEA probes. Detailed device characterization, 3D culture analysis and visualization, as well as electrophysiological recordings from the 3DMEA, will be discussed.
Methods
3D probe device fabrication
Devices were fabricated using wafer-level cleanroom processing. First, a chrome release layer was patterned on 6′′ Borofloat-33 glass wafers (University Wafer, South Boston, MA) using wet etching. 8 μm of polyimide (HD Microsystems, Parlin, NJ) was then deposited and cured. Next, the first metal layer of 20 nm Ti/250 nm Au/20 nm Ti was patterned via wet etching followed by deposition of 2 μm of polyimide. After defining interconnection vias, this process was repeated for a second trace metal layer. After an additional interconnection layer, an electrode metal layer of 20 nm Ti/250 nm Au was then patterned using a wet etch process. A final 2 μm layer of polyimide was added, connection pad and electrode vias were defined, and a device via etch to the substrate was performed on the polyimide. All polyimide etching was done using dry oxygen plasma. A detailed cross-sectional process flow is shown in Fig. S1.† The wafers were then diced into individual chips and immersed in CR-7 chrome etchant (Transene, Danvers, MA) for 5 hours at room temperature until the release layer was fully dissolved away.Actuation shank design and fabrication
To vertically actuate the flexible polyimide probes, an approach was developed to flex the probe bodies off the surface and subsequently lift them upright using rigid structures, which we call “buckling shanks” and “lifting shanks,” respectively (Fig. 2). Briefly, shank designs were lithographically patterned on 250 μm-thick Si wafers for buckling shanks, and silicon-on-insulator (SOI) wafers with 100 μm-thick device layers for lifting shanks (University Wafer, South Boston, MA). The silicon was then etched using a DRIE process. For buckling shanks, the shanks were released from a backing wafer using PRS2000 resist stripper (Fischer Scientific, Hampton, NH). For lifting shanks, the shanks were immersed in 49% hydrofluoric acid for 3 days for release. To limit probe damage during actuation, all silicon shanks were uniformly coated with a 3.5 μm-thick Parylene C layer by vapor deposition.Actuation apparatus design and assembly
An actuation apparatus was developed to precisely position the shanks relative to the probe arrays (Fig. 2). The rig was assembled using optical stages and microdrives, including a rotation mount to define the buckling shank angle relative to the substrate (Newport, Irvine, CA). Given the small size and spacing of the probes in each array, the components of the actuation apparatus needed to be precision manufactured, therefore a solid model of the rig was designed using SOLIDWORKS (Dassault Systèmes, Waltham, MA). All 4 buckling shanks were held by a custom computer numerical control (CNC)-machined aluminum clamp. Shim stock was used to precisely space the 4 shanks relative to one another (McMaster-Carr, Santa Fe Springs, CA). The lifting shanks are adhered to a separate custom CNC-machined aluminum piece with double-sided Kapton tape.Post-cleanroom processing and characterization
Actuation angle characterization
To characterize the angle of probe actuation once the cell culture wells had been affixed, the intrinsic autofluorescence of the polyimide was utilized to image the probes via confocal fluorescence microscopy. Arrays were imaged using green fluorescence emission settings (488 nm excitation, 523 nm emission) at 2.5× magnification (Zeiss Fluar 2.5x/0.12 M27 objective) using vertical steps of 20 μm on a Zeiss LSM700 confocal microscope (Carl Zeiss Microscopy, Thornwood, NY). Orthogonal views of the stacks were prepared using Zeiss ZenBlue software and exported as images, then imported into ImageJ and measured using the angle annotation tool (Fig. S2†).3D human neuron and astrocyte co-cultures
As previously reported,39 human iPSC-derived neurons and astrocytes (Neucyte) were encapsulated in ECM-collagen hydrogel and were grown on the 3DMEA devices. All reagents were from Thermo Fisher Scientific (Franklin, MA) unless otherwise stated. ECM-Collagen gel solution, with suspended cells, was prepared at a volume of 75 μL with a final concentration of collagen at 3 mg mL−1. Briefly, concentrated rat tail collagen Type 1 (9.41 mg mL−1, Corning, Bedford, MA) was diluted in seeding neuronal media (Neucyte, San Jose, CA) adapted from Iordan et al.,40 and the ECM mixture, 200 μg ml−1 of MaxGel (Sigma-Millipore, St. Louis, MO) for a volume of 50 μL. The ECM-collagen gel solution was neutralized to ∼pH 7.4 with 0.2 N of NaOH and chilled on ice, to prevent fibrillogenesis of the collagen, before the addition of the cell suspension (25 μL). The cell suspension contained human iPSC-derived glutamatergic and GABAergic neurons, two major neuronal classes that are critical for proper neural network function, and astrocytes, a supporting glial cell type shown to enhance the development and synchronization of neuronal networks from hiPSC-derived neurons.41–43 Cells were encapsulated at ratios recommended by the vendor: 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 glutamatergic to GABAergic neurons, and 75:25 for neurons to astrocytes. To obtain these ratios, purified and concentrated stocks of the respective cell types were thawed and diluted (1:2) in DMEM before sampling the cell suspension to obtain a live cell count using the Countess Automated Cell Counter (Thermo Fisher Scientific). At the vendor's recommended ratio, the volume of the live cell population for each cell type was taken and pooled for a total of 375000 or 500000 cells. The mixed cell suspension was centrifuged (to remove the cyropreservative) and resuspended in seeding neuronal media before added to the ECM-collagen gel solution for a seeding density of 5 × 106 cells per mL and 6.67 × 106 cells per mL, respectively. The cell containing gel solution flooded each array, and collagen fibrillogenesis occurred in a humidified incubator (37 °C) with 5% CO2 regulation for 2 hours. After 2 hours, seeding neuronal media (Neucyte) was added to wells and maintained in a humidified incubator (37 °C, 5% CO2). Then, 24 h later cells were maintained in short term media for 1 week before being switched to long term media for the duration of the experiment. For cell maintenance, 50% of media was replaced every 2–3 days.
:30 glutamatergic to GABAergic neurons, and 75:25 for neurons to astrocytes. To obtain these ratios, purified and concentrated stocks of the respective cell types were thawed and diluted (1:2) in DMEM before sampling the cell suspension to obtain a live cell count using the Countess Automated Cell Counter (Thermo Fisher Scientific). At the vendor's recommended ratio, the volume of the live cell population for each cell type was taken and pooled for a total of 375000 or 500000 cells. The mixed cell suspension was centrifuged (to remove the cyropreservative) and resuspended in seeding neuronal media before added to the ECM-collagen gel solution for a seeding density of 5 × 106 cells per mL and 6.67 × 106 cells per mL, respectively. The cell containing gel solution flooded each array, and collagen fibrillogenesis occurred in a humidified incubator (37 °C) with 5% CO2 regulation for 2 hours. After 2 hours, seeding neuronal media (Neucyte) was added to wells and maintained in a humidified incubator (37 °C, 5% CO2). Then, 24 h later cells were maintained in short term media for 1 week before being switched to long term media for the duration of the experiment. For cell maintenance, 50% of media was replaced every 2–3 days.
Immunocytochemistry
The 3D cultures were fixed with 4% paraformaldehyde (PFA, 1 h), washed in PBS (5 min, 3×), permeabilized with 0.2% Triton-×100 (10 min), and blocked with 5% BSA (4 °C, overnight). Cultures were washed in PBS (1 h, 3×) before labeling with the primary antibody for anti-class III beta-tubulin (Tuj1, chicken, 1:200, Neuromics, Edina, MN). Cultures were then washed with PBS (2× for 1 h, 1× overnight) before incubating with the secondary antibody, Alexa Fluor 647-conjugated goat anti-chicken (1:200, Life Technologies, Eugene, OR). For nuclear staining, cultures were incubated (5 h) in 4′-6-diamidino-2-phenylindole (DAPI; 1:3000; ThermoFisher). Samples were stored in PBS (dark, 4 °C) before imaging using a LSM700 confocal microscope (Carl Zeiss Microscopy, Thornwood, NY).
Imaging acquisition and processing of cell-containing 3D cultures
Optimal camera exposure and microscope settings were fixed and remained the same for all replicates of ECM-Collagen gel samples on the device. 2D images (1024 × 1025 pixels) in the XY plane at 2.5×, and 10× magnifications (Zeiss Fluar 2.5×/0.12 M27, and EC PlanNeo 10×/0.30, respectively) were acquired at 10 μm intervals from the bottom of the gel (no fluorescence) to the top of the gel (disappearance of fluorescent signal), or at 0.5 μm intervals near the bottom of the gel (e.g., 150 μm). Sampling of 3D cultures included 1–2 regions of interest from each of the three replicates. To visualize cells and probes of the 3DMEA device, Z-stack images of ECM-Collagen gel samples were compiled in ZEN Blue (Carl Zeiss Microscopy), filtered using the binomial filtered method, and rendered into a 3D object in Orthogonal viewer with maximal intensity projection selected.Electrophysiology and data analysis
A 256-channel Multichannel Systems MEA2100 electrophysiology system (Multichannel Systems, Reutlingen, Germany) was used to record electrophysiology activity from 3DMEA devices for 30 min at a sampling frequency of 10 kHz and bandpass filtered between 4–4000 Hz. Devices were placed within a 5% CO2-regulated chamber on a heated stage at 37 °C; recordings started after a 5 min equilibration time. An action potential spike was defined by a lower limit threshold, set at 6.5× the standard deviation of baseline noise, for each electrode. Data was exported as a hdf5 file and analyzed using an in-house custom R package to remove silent electrodes (<2 spikes) and to calculate features such as number of spikes and bursts. Burst parameters, defined previously,44,45 include: maximum beginning ISI of 0.1 s, maximum end ISI of 0.2 s, minimum IBI of 0.5 s, minimum burst duration of 0.05 s, and minimum number of spikes per burst of 6. Features were calculated per channel for each device. Mean and standard error of the mean (SEM) values per device were calculated for each feature.3D electrophysiological activity mapping and visualization
Using the as-designed location of the electrodes within each array prior to actuation, combined with angle quantification data of the actuated probes, a unique electrode map in 3D space was generated for each cultured well. Each actuated probe in the array was modeled using a combination of two rectangular mesh surfaces. These surfaces defined the section before the hinge on the X–Y plane and the section containing the electrodes; raised during the actuation procedure. The location of each electrode on the actuated probe was calculated using its X–Y position on the surface and the actuation angle obtained using the orthogonal views of the stacked images and ImageJ. The mesh points were colored gray on the surface of the probe and black on the edges of the probe and boundaries of the electrodes. Within the circular region of each electrode on the probe, the points were assigned a color that varied with the features recorded on that electrode. A logarithmic scale was used for the number of spikes to facilitate a better visualization of its large range observed across the 3D cell culture (Fig. 5D). A high-resolution 3D rendering was created using the Python interface of Plotly.46 A suitable viewing position (X, Y, Z) was then selected to capture a 2D projection of the visualization.Results and discussion
3D probe device design
The 3DMEA was designed for performance, ease of use, and compatibility with existing commercial electrophysiology instrumentation. Hence, the device contained the same substrate dimensions and interconnection pad locations as the Multichannel Systems MEA2100 electrophysiological recording system which allows recording of up to 256 electrodes. The chip, having dimensions of 49.25 mm × 49.25 mm × 0.5 mm, consists of three independent probe arrays each contained within a separate cell culturing well (Fig. 1A–C), allowing up to three parallel experiments to be conducted (and simultaneously recorded) on each device. Each array contains ten flexible polyimide probes of length 1100 μm and width 90 μm (Fig. 1C). The 10 probes are arranged into 4 “columns” containing either 2 or 3 probes (Fig. 1D). To control the precise location at which the probe would bend during actuation, a hinge region was incorporated into the design (Fig. 1E). This region features a void in the polyimide, resulting in an inherently flexible region compared to the rest of the probe body. The hinges are 20 μm wide and either 20 μm or 40 μm in length. Each probe contains eight 50 μm-diameter electrodes along the body of individual probes (Fig. 1E). The electrode diameter was chosen based on a previous report from our group using a 2D planar MEA,5 although smaller electrodes could be readily fabricated if necessary. Electrodes have an edge-to-edge separation distance of 75 μm along the body of each probe (Z-plane distance once actuated) and 440 μm between probes (X–Y plane distance once actuated). The electrodes in each array are thus reproducibly distributed across a 3D volume of 2.38 cubic mm.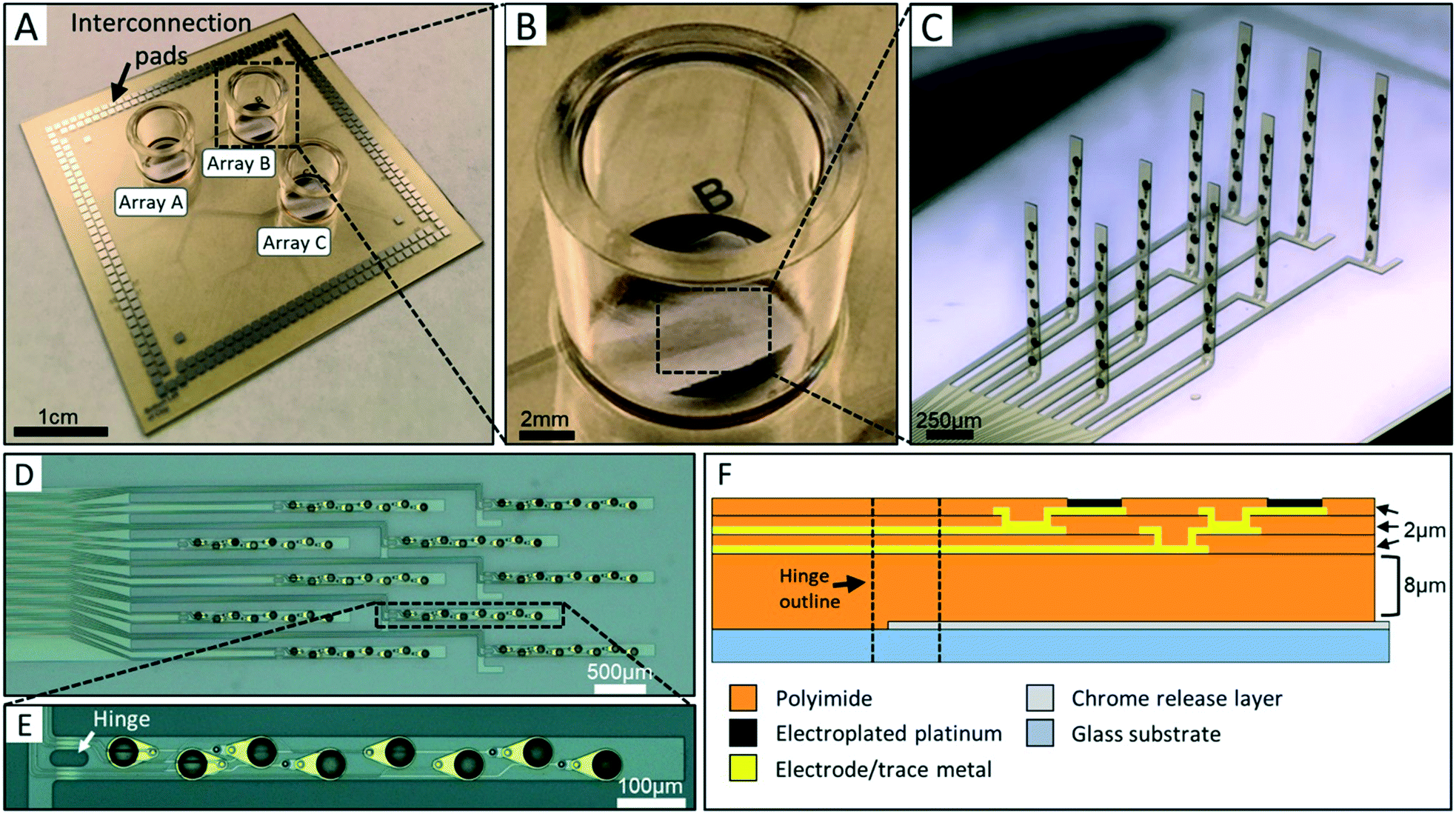 | ||
| Fig. 1 3DMEA device prior to actuation. A) A completed device. The overall length and width, as well as pad locations are identical to a commercial multichannel systems brand device, allowing for seamless integration into existing electrophysiology electronics. B) Close-up image of a single cell culture well. The large dark metal features at the top and bottom of each cell culture well are ground electrodes, which are all electrically shorted to each other. C) Light micrograph of a single 3DMEA post-actuation. The hinge regions are plastically deformed and allow the probes to stand upright without additional supports. D) Brightfield image of one MEA array containing 10 probes and 80 total electrodes prior to actuation. Moving from left to right, columns containing 2, 3, 2, and 3 probes, respectively, can be seen. E) Detail of one probe containing 8 electrodes, all electroplated with platinum black. The hinge region to the left is plastically deformed during the actuation process. F) Cross-sectional cartoon showing material stack of microfabricated probe and relative location of hinge (not to scale). | ||
A cross-sectional stack of the probes is shown in Fig. 1F. The final probe thickness is ∼15 μm. Polyimide was chosen as the main structural component of the probes due to its flexibility and biocompatibility. The 8 μm base polyimide layer thickness was chosen to prevent damage to subsequent functional metal layers during the “lifting” phase of actuation of the probes (see “Mechanical actuation component design” section of results). Additionally, this thickness allows both of the subsequent trace metal layers running through the hinge region to be in mechanical compression during actuation since they are located above the neutral plane at ∼7.5 μm, thus limiting the risk of trace breakage during this step.
Mechanical actuation component design
To ensure both rapid and reproducible actuation of the array probes, an actuation apparatus was developed (Fig. 2A). This apparatus provided precise control over probe actuation, and allowed all 10 probes to be actuated simultaneously. The actuation process, enabled by the apparatus, employed the flexible properties of the polyimide probe arrays. First, the buckling shanks contact the distal end of each probe at an acute angle off the glass surface (Fig. 2Bi–ii). A compressive load is applied in the longitudinal direction of the probe as the shank arms are driven in the direction of the hinge using microdrives, and a controlled buckling of the probes occurs, arching the middle region of the probes off the glass substrate (Fig. 2Bii). With the buckling shanks held in place, a “lifting shank” is carefully positioned under the buckled probes (Fig. 2Biii). The lifting shank is parallel to and has clearance of ∼25 μm from the substrate during this step to avoid contact with the polyimide-passivated metal traces on the surface connecting the electrodes with the contact pads. Once the lifting shank is in place, the buckling shanks are drawn away from the probes allowing them to relax back to a straight conformation. Lastly, the lifting shank is moved upward then toward the hinge region of the probes, to vertically actuate the probes off the surface (Fig. 2Biv).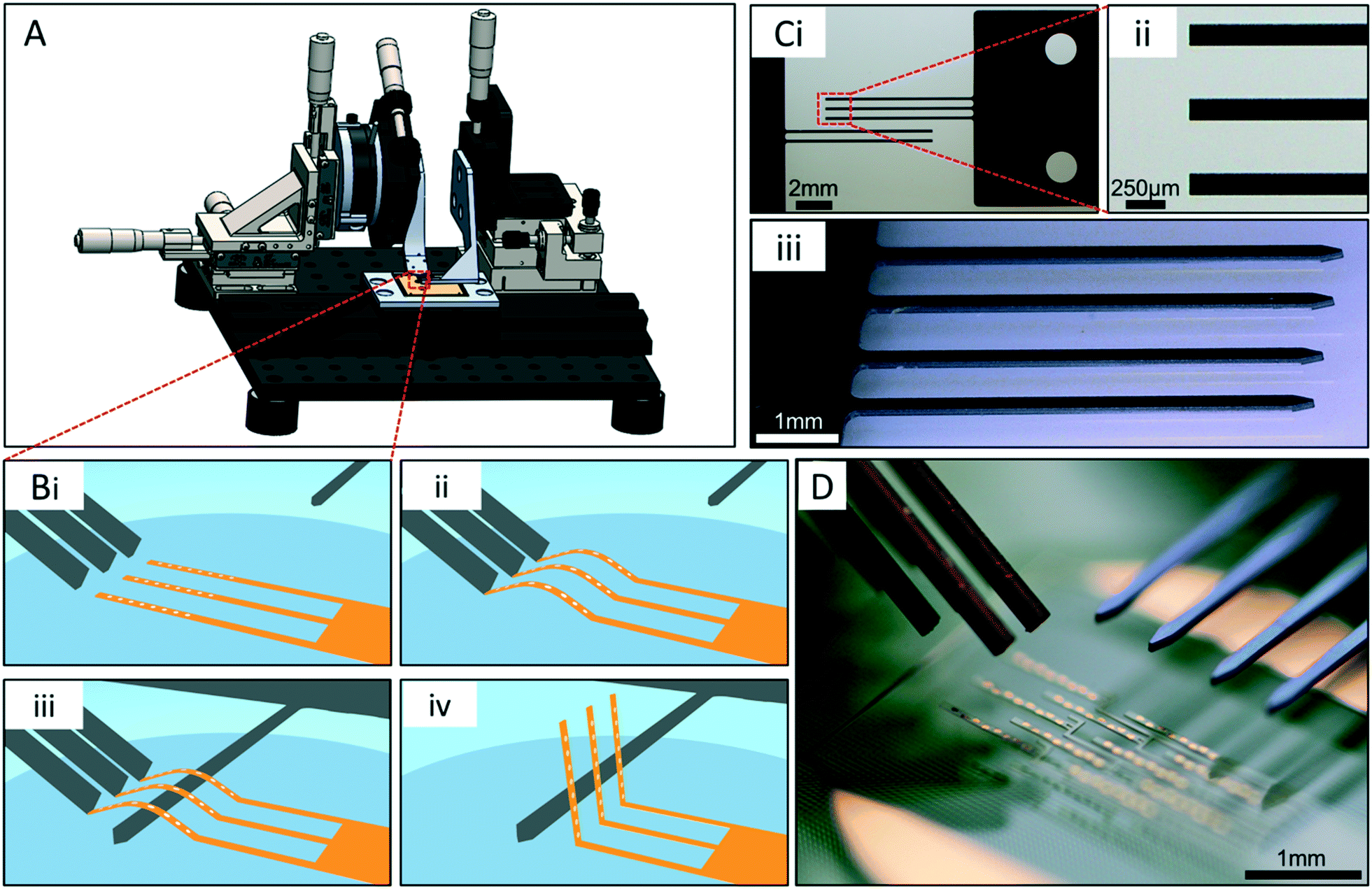 | ||
| Fig. 2 Components and process of mechanical actuation of 3DMEA probes. A) Solid model of actuation apparatus. The orange element in the center is the 3DMEA device. B) Conceptualization of actuation approach using silicon shanks and flexible probes; i) angled buckling shanks contact the substrate, then are moved toward the distal end of the probes. ii) The flexible polyimide probe bodies buckle off the surface and arch as the buckling shanks are moved longitudinally relative to the probes. iii) A lifting shank is guided under the buckled probes. iv) After retraction of the buckling shanks, the lifting shank is moved upward, then toward the hinge region of the probes to deform it and finalize the vertical actuation process. C) Actuation shanks. i) 2- and 3-arm buckling shanks, ii) detail of the flat ends of the arms. iii) Angled view of a lifting shank containing arms with pointed tips. D) Photograph of the buckling and lifting shanks relative to a probe array before performing the actuation process. There are 4 buckling shanks stacked on one another to buckle all 4 “columns” of probes at once. | ||
The silicon buckling shanks have either 2 or 3 arms per shank (Fig. 2Ci) since the “columns” of probes within an array contain either 2 or 3 probes each. The tips of the arms are flat such that they make flush contact with the distal end of each probe, helping to stabilize the probes during buckling (Fig. 2Bii). The lifting shank has 4 arms for simultaneous lifting of all 4 columns of probes and pointed tips to help guide them under the buckled probes (Fig. 2Ciii). The buckling shank arms are 140 μm wide by 7.5 mm long, and the lifting shanks are 150 μm wide by 5 mm long. These dimensions allowed the shanks to be sufficiently stiff relative to the flexible probes.
The shanks are aligned relative to one another using alignment pins in the holding clamp, inserted through holes in the shank tab region (two holes are visible in Fig. 2Ci). The force vector in the longitudinal direction of the probes during the buckling process was maximized by keeping the buckling angle as low as possible relative to the substrate. An angle of 32° was optimal for the buckling shanks to achieve this and allow enough space under them for the lifting shank to slide. With this configuration, the lifting shanks were arranged approximately 25 μm from the glass substrate to fit under the buckled probes (Fig. 2D).
The mechanical approach described in this work provides benefits over previously published methods for actuating flexible MEAs. In 2003, Takeuchi et al. described a method for creating a 3D in vivo MEA whereby an embedded layer of a ferromagnetic metal (e.g. nickel) was deposited within layers of polyimide probes,47 and an external magnetic field of 380 mT generated by an industrial electromagnet was used to actuate the probes to ∼90°. Similarly, Chen et al. published a study in 2011 in which an external electrostatic field of 8 kV was used to actuate flexible SU8- and parylene C- based MEA probes for in vivo implantation.48 In both cases, external energized equipment was required to perform actuation, which introduces potential hazards. Additionally, the magnetic actuation approach requires the use of ferromagnetic metals like nickel, which are generally cytotoxic.49 Thus, if the polymer sheath enclosing the material is damaged during manufacture or use, the exposed metals would be detrimental to nearby cells or tissue. The actuation approach used in our study does not require any hazardous equipment, and the probes described in our study contain no cytotoxic components.
Actuation process and physical and functional characterization of 3DMEA
There were three key considerations when designing the 3DMEA probes. First, it was desired that they be flexible to more closely match the low stiffness of the hydrogel. Second, they needed to be amenable to the actuation process, such that the electrodes and traces remain intact and undamaged after hinge bending. Third, the probes needed to remain vertically actuated upon removal of the actuation shanks.We first evaluated the ability to buckle all probes simultaneously and uniformly. It was found that the buckling shanks were successful in achieving this, such that all lifting shank arms could be inserted under the buckled probes without contacting them (Fig. S3†). To eliminate damage to the electrode region of the probes, it was critical to have the final deformation of the probe body contained within the hinge region. Additionally, it was important to not use any external fixing elements to hold the probes upright once actuated, which would add additional fabrication effort and time, and add materials that could compromise biocompatibility. Immediately after actuating the probes vertically using the lifting shank, these characteristics were evaluated using brightfield microscopy. It was determined that the approach was successful in actuating the probes arrays vertically from the surface, and negligible damage was seen on the individual probes. In addition, the plastic deformation in the probe body occurred entirely in the hinge region, and the plastic deformation of the hinge alone was able to hold the probes upright (Fig. 1C). Comparatively, the electrostatic actuation paper by Chen et al. required a secondary “fixing” element made from injecting polyethylene glycol (PEG) at the hinge and subsequent curing it to hold the probes in their actuated position, rather than employing the much simpler method of plastic deformation of the polymer for mechanical stability.48 The simplicity of our approach also results in time savings during preparation, as each device containing three 3DMEAs could be easily actuated within 5 min.
To evaluate electrode integrity before and after actuation, impedance measurements were taken across devices. Individual electrode impedances pre-actuation ranged from 58 ± 6 kΩ to 69 ± 6 kΩ for bare gold electrodes and 37 ± 7 kΩ to 48 ± 6 kΩ for Pt-plated electrodes. The decrease in impedance from electroplating was less than previously reported due to significantly lower voltage applied and time used for electroplating to reduce the risk of the plated Pt film cracking or delaminating during actuation. These values did not significantly change post-actuation and testing for open circuits indicated >95% of electrodes that were functional before actuation were also functional after the process (data not shown).
To maximize the 3DMEA vertical alignment and thus extend the recording capability of the electrodes in the Z-dimension, it was desired to actuate the probes to an angle of approximately 90° relative to the substrate. Immediately after actuation, angles of the actuated probes were assessed. The probe-to-probe variations within arrays were rather consistent, having ranges of 8.8° (80.0–88.8°) to 12.7° (81.9–94.6°). Between arrays, the average actuation angles varied from 81.9° ± 3.9° to 95.6° ± 3.6° (Fig. 3A). To evaluate whether the actuation angle changes over time absent of any additional handling, a device was actuated and left dry for 4 months post-actuation. The average actuation angles of the device decreased by 13.8°, less than 1 degree per week (Fig. S4†), indicating that, while some minor settling of the probes occurs over time, the probes retain significant vertical alignment over weeks of storage.
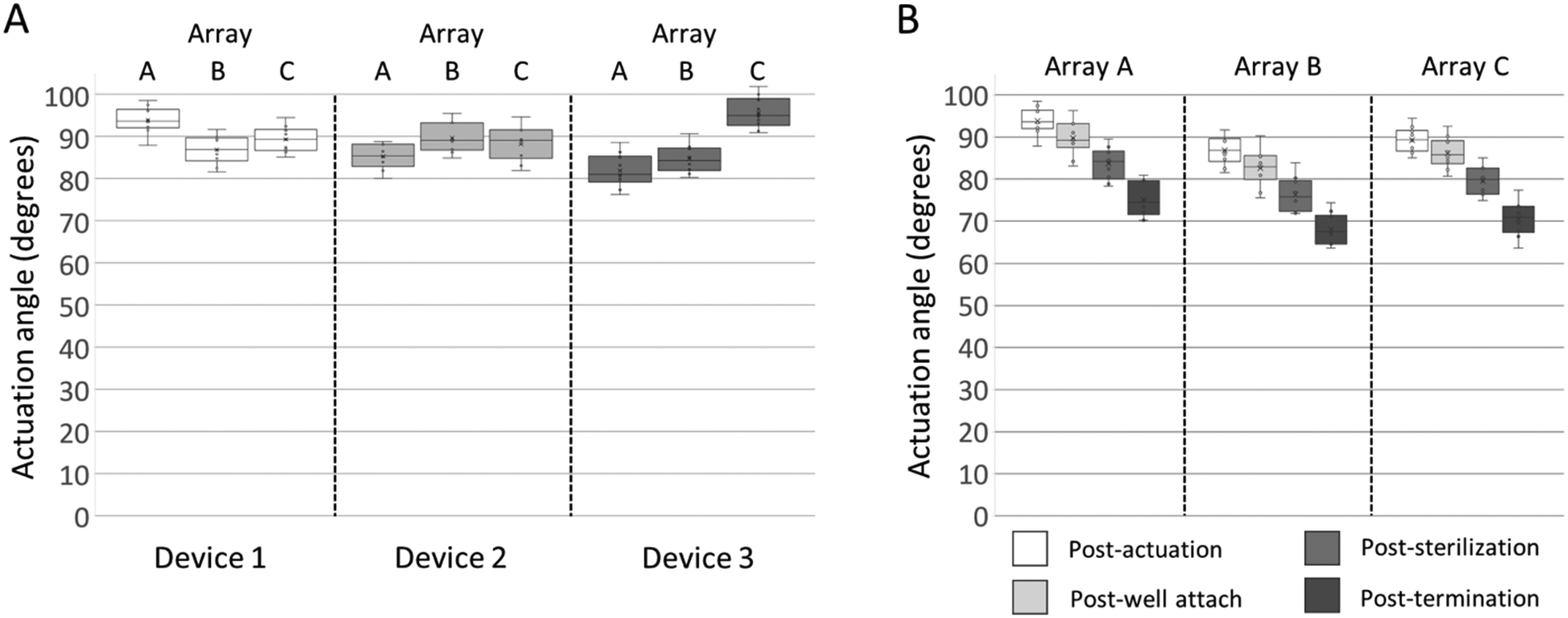 | ||
| Fig. 3 3DMEA probe actuation quantification. A) Comparison of probe angles of individual arrays from three devices immediately after actuation. B) Average actuation angles from individual arrays at 4 key points during device usage: immediately post-actuation, post-well attachment, after ethanol sterilization and rinse steps, and immediately after termination of the cell culture. Lines in boxes indicate median value. Boxes denote upper/lower quartiles, whiskers denote max/min values. | ||
To evaluate whether handling of the devices during the steps required for preparation for in vitro 3D cultures changes the probe actuation angles, they were measured at four key points in the device preparation process: 1) immediately post-actuation; 2) after well attachment with epoxy; 3) prior to seeding after the arrays have undergone multiple liquid treatments, including ethanol sterilization and water rinsing; and 4) “post-termination” after hydrogel/cell addition, media changes, culture maturation over several weeks, and incubation/rinsing steps associated with immunocytochemistry. After each handling step, the probe actuation angle decreased (Fig. 3B). When comparing the angle of the probes post-actuation versus post-termination of the cell culture, the averages per array decreased between 9.9° to 20.0°. The data indicated that moderate changes in probe actuation angles occur as a result of preparation and handling. Since the decreases were relatively consistent, in future studies the probes can be actuated to defined angles over 90° and allowed to relax to their final angles after cell seeding.
3D neuronal culture growth and electrophysiological activity in 3DMEA array
In order for the 3DMEA device to be suitable for use as a 3D in vitro platform, it is critical for the devices to be biocompatible and support the growth of neurons entrapped in a 3D ECM-collagen gel. After flooding the actuated 3DMEA wells with human iPSC-derived neurons and astrocytes suspended in ECM-collagen hydrogel and allowing the gel to polymerize, a representative neuronal culture was evaluated after 24 days in vitro (DIV). As shown in Fig. 4A and B, a dense network of cells and neuronal processes are observed that stained positive for TUJ-1, a common marker for neuronal labeling. This indicated that the 3DMEA supported a healthy neuronal culture that was not adversely affected by any materials present in the 3DMEA arrays (Fig. 4A and B), underscoring the biocompatibility of the device and fabrication process. The polyimide in the probes exhibited autofluorescence, facilitating the visualization of the probes within the 3D culture. Qualitatively, there was no preference of cell body distribution and process growth as a function of distance from the probes (Fig. 4B), supporting the theory that the flexibility and small form factor of the probes creates a minimally invasive approach to measuring electrophysiological activity in 3D hydrogel cultures. Additionally, X–Z orthogonal views of the TUJ-1-labeled cultures reveal a distribution of cells throughout the height of the hydrogel (i.e., 750–1000 μm thick) while also visualizing the location of the actuated probes (Fig. 4C and D).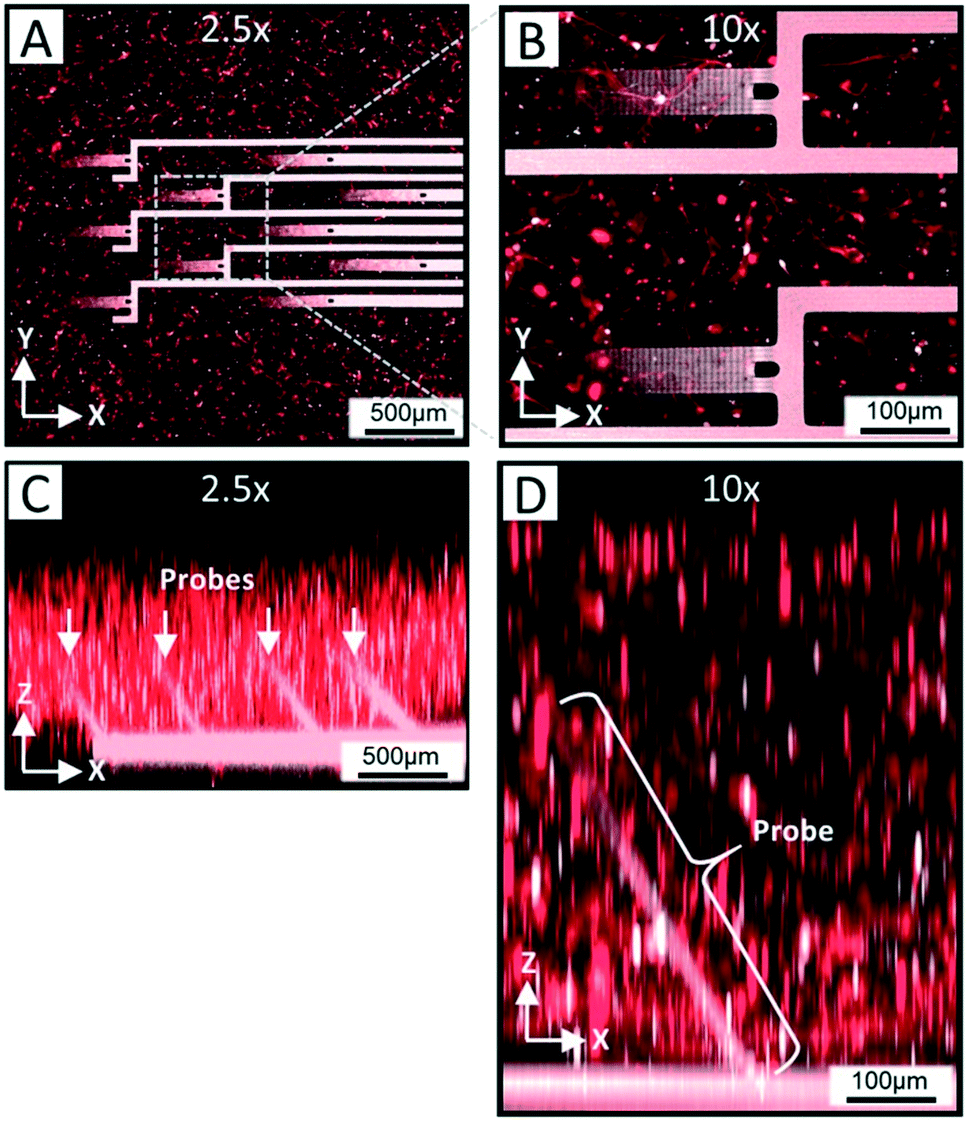 | ||
| Fig. 4 Confocal images of TUJ-1 labeled neurons within a 3DMEA at DIV 24. A) 2.5× maximum projection in the X–Y planar view of entire array illustrating neuronal network formation throughout 3DMEA. B) 10× maximum projection X–Y planar view of two 3D probes within array detailing the networks of neuronal soma and processes surrounding probes. C) and D) 2.5× and 10× X–Z planar view with maximum projections of 3DMEA/3D neuronal culture showing distribution of suspended neurons relative to 3D probes. | ||
To assess the ability of the 3DMEA device to monitor the electrical activity of a 3D neuronal culture system, recordings were performed on human iPSC-derived neuronal cultures over 38 DIV. By DIV 15 electrodes began detecting electrophysiological activity from the cells, which was consistent with prior studies involving rat hippocampal30 and cortical27 neurons in 3D hydrogels. Across the 3 arrays from the device examined, there were 12, 31, and 27 active electrodes out of the total 80 electrodes in arrays A, B, and C, respectively that exhibited activity at DIV 38. An example overlay of action potential waveforms captured by one electrode is shown in Fig. 5B. Bursting activity was also present on 58.3%, 48.4%, and 33.3% of active electrodes for each array, respectively, which is expected for functional and mature networks from GABAergic and Glutamatergic neurons (Fig. 5A and C).50,51 A summary of neuronal activity at 38 DIV from three arrays on a single device is shown in Table 1. We observed neuronal activity up to 45 DIV before the culture was terminated for immunocytochemistry (data not shown). Electrophysiological activity was distributed throughout the 3DMEA and was able to be visually represented in 3D plots based on the location of the electrodes calculated from the probe actuation angles measured at termination of the cell culture (Fig. 5D). These visualizations, along with the potential for future spatial and temporal mapping of electrophysiology data across the 3D volume, are enabled by the integration of the 3DMEA within the culture substrate, as microfabrication of the probes facilitates identification of electrode locations in space via the quantified actuation angles. By comparison to the 3DMEA, non-integrated flexible “mesh” arrays like those described in Kireev, Tian and Zhou's papers are initially in the form of an extremely flexible sheet which requires additional support structures like electrospun nanofibers to allow handling,33–35 and recording from multiple planes in the Z-direction requires meshes to be folded or rolled, further complicating handling and limiting precise electrode positioning.
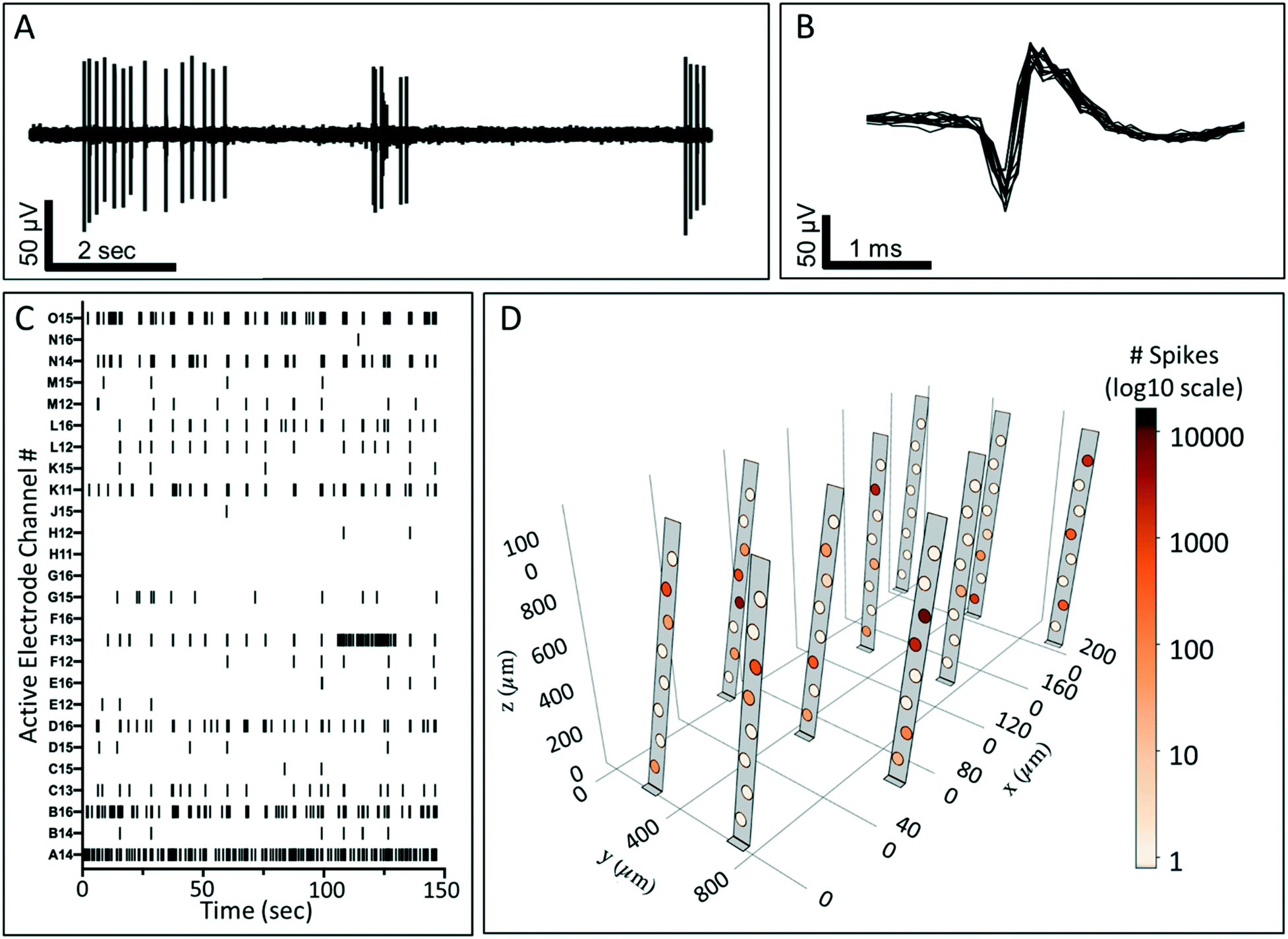 | ||
| Fig. 5 Electrophysiology recording from 3DMEA/3D human iPSC-derived neuronal cultures at DIV 38. A) Example of raw neuronal spiking data, demonstrating the detection of action potential spikes and bursts from the 3DMEA. B) Waveform overlay of spikes from (A) detected from a single electrode during a recording experiment. The amplitude of the detected signal is ∼±65 mV. C) Example raster plot from a single array of the 3DMEA device showing spiking and bursting activity. In this plot, each hash mark represents a detected spiking event. D) 3D visualization plot showing spike activity from a single array of the 3DMEA device throughout 3D space over a 30-min recording. The number of spikes recorded from each electrode is represented by shading of electrode on a log10 scale. | ||
| Features of neural activity at 38 DIV (mean ± SEM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| # of active electrodes | # of spikes | Firing rate (Hz) | ISI (s) | % of active electrodes bursting | # of bursts | # spikes in bursts | Burst duration (s) | IBI (s) | |
| Array A | 12 | 1385.5 ± 540.0 | 0.77 ± 0.30 | 24.2 ± 15.3 | 58.3 | 120 ± 48.2 | 9.1 ± 1.6 | 0.4 ± 0.07 | 189.3 ± 118.49 |
| Array B | 31 | 1362.4 ± 9311.4 | 0.75 ± 0.18 | 62.7 ± 27.7 | 48.4 | 153.1 ± 20.3 | 12.1 ± 1.4 | 0.5 ± 0.004 | 46.9 ± 35.34 |
| Array C | 27 | 823.0 ± 299.4 | 0.46 ± 0.17 | 42.6 ± 18.3 | 33.3 | 106.9 ± 27.9 | 11.1 ± 2.8 | 0.4 ± 0.05 | 51.3 ± 34.66 |
The 3DMEA device was designed to be minimally invasive to the 3D biological system. Here, we entrapped human cells within an ECM-collagen hydrogel. The advantage of the entrapment method is that the cells are suspended in a liquid gel solution, which then undergoes collagen fibrillogenesis or polymerization with cells suspended in 3D space. Over time, neuronal processes grow within the hydrogel and around the actuated probes. The addition of astrocytes in the cultured system promoted and improved neuronal synapse formation, maturation, and supported electrophysiological activity in vitro, which requires astrocyte contact and/or astrocyte-derived molecules.52–55 We have recently shown that a greater cell density was needed for long-term maintenance of primary rat and human iPSC-derived neurons and astrocytes co-cultured in 3D ECM-collagen hydrogel,39 as compared to published studies using primary rat neurons30,56–58 which were encapsulated at a lower cell density. In using this high density co-cultured system, we have demonstrated that the materials used to fabricate the novel 3DMEA are biocompatible with human cells This was shown by the electrophysiological spiking and bursting data recorded on our 3DMEA at over several weeks in vitro, as previously reported on a 2D MEA device.1,5 In the future, low-pass filtering could be applied to capture local field potentials for evaluating summated activity from neurons further from electrodes.59
We have demonstrated the capability to visualize spatial and temporal mapping of electrophysiological data across a 3D volume. This will help future studies examine whether the mapping of activity correlates to the distribution of cells within the hydrogel. Recently, we found that cell concentration and atmospheric CO2 contribute to cell distribution in ECM-collagen gel.39 It is not clear whether cell distribution affects features of network activity. Future directions will aim to optimize the method of 3D biological systems to be able to accelerate the onset of neuronal activity and increase the percentage of active electrodes within each 3DMEA. It is likely that a higher percentage of channels will exhibit activity by further increasing cell density near the probes.
Conclusions
The objective of this study was to develop and test a device that provides a means to electrically interrogate 3D in vitro cultures of electroactive cells, such as neurons, using integrated 3D microelectrode arrays. It was important that the device be biocompatible, relatively easy to fabricate, able to be integrated with existing commercial electrophysiology hardware, and straightforward to use for those preparing 3D cultures. Lastly, it was desirable to have the 3DMEA positioned in 3D space prior to cell–matrix addition, such that the cellular network could form naturally rather than inserting MEA probes into established neuronal cultures and thereby damaging the intricate neuronal network. The flexibility and resilience of the probes was beneficial for the mechanical actuation method, allowing hands-free actuation that could be automated using electronically driven microdrives in the future. Although we did not investigate the mechanical effect of the flexible probes on the 3D neuronal culture in this study, it is worth evaluating in future studies whether the lower-stiffness polyimide-based probes reduce damage to the culture during micromovements caused by handling, similar to published in vivo studies demonstrating this benefit over stiff (e.g. silicon-based) probes.60,61The 3DMEA was able to record electrophysiological activity from human iPSC-derived neurons co-cultured with astrocytes in a 3D ECM-collagen hydrogel. To our knowledge, this demonstrates the first functional recording of human iPSC-derived neurons entrapped in a 3D gel by a microelectrode array, and the first in vitro device containing a 3D microelectrode array integrated into the substrate to achieve electrophysiological recordings simultaneously in all three dimensions. In the future, network analysis across 3D matrices of electroactive cells can be assessed using healthy and/or diseased phenotypes, and upon addition of chemicals of interest. Additionally, 3D ECM-based neuronal cultures will be optimized via cell/ECM concentration and the incorporation of other support cells (e.g. microglia, oligodendrocytes) to increase the number of electrically active cells per unit volume.
The device design used for this study represents one possible arrangement of a 3DMEA, however in the future, the platform can be modified to, for example: 1) increase the electrode density on one or both sides of the probes to provide greater resolution or coverage of the 3D neuronal network; 2) decrease electrode size to better capture single-unit spiking events; and 3) accommodate 3D spheroids or organoids. The number of electrodes per probe, probe spacing, probe length, etc. can easily be tailored by adjusting array and shank designs in future versions to best accommodate the cell population to be interrogated. Therefore, the 3DMEA has wide-ranging applications for drug development, countermeasure validation, and disease research for any organ system containing electroactive cells. This work represents an important step in the advancement of human-relevant in vitro systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by LDRD Award 17-SI-002 under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344 Lawrence Livermore National Security, LLC. We would like to acknowledge Laura Forde and Jeenali Shah for their contributions to this project. IM Release #: LLNL-JRNL-795358.References
- D. Lam, H. A. Enright, J. Cadena, S. K. Peters, A. P. Sales, J. J. Osburn, D. A. Soscia, K. S. Kulp, E. K. Wheeler and N. O. Fischer, Sci. Rep., 2019, 9, 4159 CrossRef PubMed.
- M. Geissler and A. Faissner, J. Neurosci. Methods, 2012, 204, 262–272 CrossRef PubMed.
- S. Dauth, B. M. Maoz, S. P. Sheehy, M. A. Hemphill, T. Murty, M. K. Macedonia, A. M. Greer, B. Budnik and K. K. Parker, J. Neurophysiol., 2017, 117, 1320–1341 CrossRef CAS PubMed.
- M. Garcia-Munoz, E. Taillefer, R. Pnini, C. Vickers, J. Miller and G. W. Arbuthnott, Front. Syst. Neurosci., 2015, 9, 63 CrossRef PubMed.
- D. Soscia, A. Belle, N. Fischer, H. Enright, A. Sales, J. Osburn, W. Benett, E. Mukerjee, K. Kulp and S. Pannu, PLoS One, 2017, 12, e0188146 CrossRef CAS PubMed.
- Y.-T. L. Dingle, M. E. Boutin, A. M. Chirila, L. L. Livi, N. R. Labriola, L. M. Jakubek, J. R. Morgan, E. M. Darling, J. A. Kauer and D. Hoffman-Kim, Tissue Eng., Part C, 2015, 21, 1274–1283 CrossRef CAS PubMed.
- A. M. Paşca, S. A. Sloan, L. E. Clarke, Y. Tian, C. D. Makinson, N. Huber, C. H. Kim, J.-Y. Park, N. A. O'Rourke, K. D. Nguyen, S. J. Smith, J. R. Huguenard, D. H. Geschwind, B. A. Barres and S. P. Paşca, Nat. Methods, 2015, 12, 671 CrossRef PubMed.
- D. Pamies, P. Barreras, K. Block, G. Makri, A. Kumar, D. Wiersma, L. Smirnova, C. Zang, J. Bressler, K. M. Christian, G. Harris, G.-L. Ming, C. J. Berlinicke, K. Kyro, H. Song, C. A. Pardo, T. Hartung and H. T. Hogberg, ALTEX, 2017, 34, 362–376 CrossRef PubMed.
- S. Velasco, A. J. Kedaigle, S. K. Simmons, A. Nash, M. Rocha, G. Quadrato, B. Paulsen, L. Nguyen, X. Adiconis and A. Regev, Nature, 2019, 1 Search PubMed.
- D. Zhang, M. Pekkanen-Mattila, M. Shahsavani, A. Falk, A. I. Teixeira and A. Herland, Biomaterials, 2014, 35, 1420–1428 CrossRef CAS PubMed.
- L. D'Aiuto, J. Naciri, N. Radio, S. Tekur, D. Clayton, G. Apodaca, R. Di Maio, Y. Zhi, P. Dimitrion and P. Piazza, Stem Cell Res. Ther., 2018, 9, 134 CrossRef PubMed.
- D. Sood, K. Chwalek, E. Stuntz, D. Pouli, C. Du, M. Tang-Schomer, I. Georgakoudi, L. D. Black and D. L. Kaplan, ACS Biomater. Sci. Eng., 2016, 2, 131–140 CrossRef CAS PubMed.
- M. D. Tang-Schomer, J. D. White, L. W. Tien, L. I. Schmitt, T. M. Valentin, D. J. Graziano, A. M. Hopkins, F. G. Omenetto, P. G. Haydon and D. L. Kaplan, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13811–13816 CrossRef CAS PubMed.
- M. D. Tang-Schomer, W. Wu, D. Kaplan and M. Bookland, J. Tissue Eng. Regener. Med., 2018, 12, 1247–1260 CrossRef CAS PubMed.
- J. L. Bourke, A. F. Quigley, S. Duchi, C. D. O'Connell, J. M. Crook, G. G. Wallace, M. J. Cook and R. M. Kapsa, J. Tissue Eng. Regener. Med., 2018, 12, 490–493 CrossRef CAS PubMed.
- Y. Lai, K. Cheng and W. Kisaalita, PLoS One, 2012, 7, e45074 CrossRef CAS PubMed.
- S. P. Paşca, Nature, 2018, 553, 437 CrossRef PubMed.
- M. E. J. Obien, K. Deligkaris, T. Bullmann, D. J. Bakkum and U. Frey, Front. Neurosci., 2014, 8, 423 Search PubMed.
- P. Charlesworth, E. Cotterill, A. Morton, S. G. Grant and S. J. Eglen, Neural Dev., 2015, 10, 1 CrossRef PubMed.
- D. A. Wagenaar, J. Pine and S. M. Potter, BMC Neurosci, 2006, 7, 11 CrossRef PubMed.
- S. I. Morefield, E. W. Keefer, K. D. Chapman and G. W. Gross, Biosens. Bioelectron., 2005, 15, 383–396 CrossRef.
- E. Defranchi, A. Novellino, M. Whelan, S. Vogel, T. Ramirez, B. Van Ravenzwaay and R. Landsiedel, Front. Neuroeng., 2011, 4, 6 CAS.
- B. J. Whalley and A. Constanti, Neuroscience, 2006, 140, 939–956 CrossRef CAS PubMed.
- H. A. Enright, S. H. Felix, N. O. Fischer, E. V. Mukerjee, D. Soscia, M. McNerney, K. Kulp, J. Zhang, G. Page, P. Miller, A. Ghetti, E. K. Wheeler and S. Pannu, Analyst, 2016, 141, 5346–5357 RSC.
- W. L. Cantley, C. Du, S. Lomoio, T. DePalma, E. Peirent, D. Kleinknecht, M. Hunter, M. D. Tang-Schomer, G. Tesco and D. L. Kaplan, ACS Biomater. Sci. Eng., 2018, 4, 4278–4288 CrossRef CAS.
- P. W. Coates, B. Fermini, J. C. Strahlendorf and H. K. Strahlendorf, Dev. Neurosci., 1992, 14, 35–43 CrossRef CAS PubMed.
- H. R. Irons, D. K. Cullen, N. P. Shapiro, N. A. Lambert, R. H. Lee and M. C. LaPlaca, J Neural Eng., 2008, 5, 333 CrossRef PubMed.
- Z.-N. Zhang, B. C. Freitas, H. Qian, J. Lux, A. Acab, C. A. Trujillo, R. H. Herai, V. A. Nguyen Huu, J. H. Wen, S. Joshi-Barr, J. V. Karpiak, A. J. Engler, X.-D. Fu, A. R. Muotri and A. Almutairi, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3185 CrossRef CAS PubMed.
- M. Frega, M. Tedesco, P. Massobrio, M. Pesce and S. Martinoia, Sci. Rep., 2014, 4, 5489 CrossRef CAS PubMed.
- T. Xu, P. Molnar, C. Gregory, M. Das, T. Boland and J. J. Hickman, Biomaterials, 2009, 30, 4377–4383 CrossRef CAS PubMed.
- G. Charvet, L. Rousseau, O. Billoint, S. Gharbi, J.-P. Rostaing, S. Joucla, M. Trevisiol, A. Bourgerette, P. Chauvet and C. Moulin, Biosens. Bioelectron., 2010, 25, 1889–1896 CrossRef CAS PubMed.
- M. O. Heuschkel, M. Fejtl, M. Raggenbass, D. Bertrand and P. Renaud, J. Neurosci. Methods, 2002, 114, 135–148 CrossRef.
- D. Kireev, V. Rincón Montes, J. Stevanovic, K. Srikantharajah and A. Offenhäusser, Front. Neurosci., 2019, 13, 320 CrossRef PubMed.
- B. Tian, J. Liu, T. Dvir, L. Jin, J. H. Tsui, Q. Qing, Z. Suo, R. Langer, D. S. Kohane and C. M. Lieber, Nat. Mater., 2012, 11, 986 CrossRef CAS PubMed.
- T. Zhou, G. Hong, T.-M. Fu, X. Yang, T. G. Schuhmann, R. D. Viveros and C. M. Lieber, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5894–5899 CrossRef CAS PubMed.
- A. Tooker, D. Liu, E. B. Anderson, S. Felix, K. G. Shah, K. Y. Lee, J. E. Chung, S. Pannu, L. Frank and V. Tolosa, Conf. Proc. IEEE Eng. Med. Biol. Soc., 2014, 2014, 6830–6833 Search PubMed.
- A. Tooker, T. E. Madsen, A. Yorita, A. Crowell, K. G. Shah, S. Felix, H. S. Mayberg, S. Pannu, D. G. Rainnie and V. Tolosa, Conf. Proc. IEEE Eng. Med. Biol. Soc., 2013, 2013, 5159–5162 Search PubMed.
- A. Tooker, V. Tolosa, K. G. Shah, H. Sheth, S. Felix, T. Delima and S. Pannu, Conf. Proc. IEEE Eng. Med. Biol. Soc., 2012, 2012, 5999–6002 Search PubMed.
- D. Lam, H. A. Enright, S. K. Peters, M. L. Moya, D. A. Soscia, J. Cadena, J. A. Alvarado, K. S. Kulp, E. K. Wheeler and N. O. Fischer, J. Neurosci. Methods, 2019, 108460 Search PubMed.
- A. Iordan, A. Duperray, A. Gerard, A. Grichine and C. Verdier, Biorheology, 2010, 47, 277–295 CAS.
- M. N. Ishii, K. Yamamoto, M. Shoji, A. Asami and Y. Kawamata, Toxicology, 2017, 389, 130–138 CrossRef CAS PubMed.
- J. Kuijlaars, T. Oyelami, A. Diels, J. Rohrbacher, S. Versweyveld, G. Meneghello, M. Tuefferd, P. Verstraelen, J. R. Detrez and M. Verschuuren, Sci. Rep., 2016, 6, 36529 CrossRef CAS PubMed.
- A. M. Tukker, F. M. Wijnolts, A. de Groot and R. H. Westerink, Neurotoxicology, 2018, 67, 215–225 CrossRef CAS PubMed.
- A. M. Belle, H. A. Enright, A. P. Sales, K. Kulp, J. Osburn, E. A. Kuhn, N. O. Fischer and E. K. Wheeler, Sci. Rep., 2018, 8, 10820 CrossRef PubMed.
- D. Soscia, A. Belle, N. Fischer, H. Enright, A. Sales, J. Osburn, W. Benett, E. Mukerjee, K. Kulp, S. Pannu and E. Wheeler, PLoS One, 2017, 12, e0188146 CrossRef CAS PubMed.
- Collaborative data science, Plotly Technologies Inc., Montréal, QC, 2015, https://plot.ly Search PubMed.
- S. Takeuchi, T. Suzuki, K. Mabuchi and H. Fujita, J. Micromech. Microeng., 2003, 14, 104 CrossRef.
- C.-H. Chen, S.-C. Chuang, H.-C. Su, W.-L. Hsu, T.-R. Yew, Y.-C. Chang, S.-R. Yeh and D.-J. Yao, Lab Chip, 2011, 11, 1647–1655 RSC.
- K. K. Das, S. N. Das and S. A. Dhundasi, Indian J. Med. Res., 2008, 128, 412–425 CAS.
- J. Suresh, M. Radojicic, L. L. Pesce, A. Bhansali, J. Wang, A. K. Tryba, J. D. Marks and W. van Drongelen, J. Neurophysiol., 2016, 115, 3073–3089 CrossRef CAS PubMed.
- A. Odawara, H. Katoh, N. Matsuda and I. Suzuki, Sci. Rep., 2016, 6, 26181 CrossRef CAS PubMed.
- M. Pyka, C. Wetzel, A. Aguado, M. Geissler, H. Hatt and A. Faissner, Eur. J. Neurosci, 2011, 33, 2187–2202 CrossRef PubMed.
- M. Geissler and A. Faissner, J. Neurosci. Methods, 2012, 204, 262–272 CrossRef PubMed.
- P. Verstraelen, I. Pintelon, R. Nuydens, F. Cornelissen, T. Meert and J. P. Timmermans, Cell. Mol. Neurobiol., 2014, 34, 757–776 CrossRef PubMed.
- J. Kuijlaars, T. Oyelami, A. Diels, J. Rohrbacher, S. Versweyveld, G. Meneghello, M. Tuefferd, P. Verstraelen, J. R. Detrez, M. Verschuuren, W. H. De Vos, T. Meert, P. J. Peeters, M. Cik, R. Nuydens, B. Brone and A. Verheyen, Sci. Rep., 2016, 6, 36529 CrossRef CAS PubMed.
- W. Ma, W. Fitzgerald, Q. Y. Liu, T. J. O'Shaughnessy, D. Maric, H. J. Lin, D. L. Alkon and J. L. Barker, Exp. Neurol., 2004, 190, 276–288 CrossRef CAS PubMed.
- S. M. O'Connor, D. A. Stenger, K. M. Shaffer and W. Ma, Neurosci. Lett., 2001, 304, 189–193 CrossRef.
- S. H. Kim, S.-K. Im, S.-J. Oh, S. Jeong, E.-S. Yoon, C. J. Lee, N. Choi and E.-M. Hur, Nat. Commun., 2017, 8, 14346 CrossRef CAS PubMed.
- Y. Song, N. Lin, C. Liu, H. Jiang, G. Xing and X. Cai, Biosens. Bioelectron., 2012, 38, 416–420 CrossRef CAS PubMed.
- E. Fernández, B. Greger, P. A. House, I. Aranda, C. Botella, J. Albisua, C. Soto-Sánchez, A. Alfaro and R. A. Normann, Front. Neuroeng., 2014, 7, 24–24 Search PubMed.
- A. Lecomte, E. Descamps and C. Bergaud, J. Neural Eng., 2018, 15, 031001 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc01148j |
| This journal is © The Royal Society of Chemistry 2020 |