
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonitoring tissue-level remodelling during inflammatory arthritis using a three-dimensional synovium-on-a-chip with non-invasive light scattering biosensing†
Mario
Rothbauer‡
 *ab,
Gregor
Höll‡
a,
Christoph
Eilenberger
a,
Sebastian R. A.
Kratz
a,
Bilal
Farooq
a,
Patrick
Schuller
a,
Isabel
Olmos Calvo
c,
Ruth A.
Byrne
b,
Brigitte
Meyer
b,
Birgit
Niederreiter
b,
Seta
Küpcü
d,
Florian
Sevelda
e,
Johannes
Holinka
e,
Oliver
Hayden
f,
Sandro F.
Tedde
g,
Hans P.
Kiener
c and
Peter
Ertl
*a
*ab,
Gregor
Höll‡
a,
Christoph
Eilenberger
a,
Sebastian R. A.
Kratz
a,
Bilal
Farooq
a,
Patrick
Schuller
a,
Isabel
Olmos Calvo
c,
Ruth A.
Byrne
b,
Brigitte
Meyer
b,
Birgit
Niederreiter
b,
Seta
Küpcü
d,
Florian
Sevelda
e,
Johannes
Holinka
e,
Oliver
Hayden
f,
Sandro F.
Tedde
g,
Hans P.
Kiener
c and
Peter
Ertl
*a
aFaculty of Technical Chemistry, Vienna University of Technology, Getreidemarkt 9, 1060 Vienna, Austria. E-mail: mario.rothbauer@tuwien.ac.at; peter.ertl@tuwien.ac.at
bDepartment of Orthopedics and Trauma Surgery, Karl Chiari Lab for Orthopaedic Biology, Medical University of Vienna, 1090 Vienna, Austria
cDivision of Rheumatology, Department of Medicine III, Medical University Vienna, 1090 Vienna, Austria
dInstitute of Synthetic Bioarchitectures, Department of Nanobiotechnology, University of Natural Resources and Life Sciences, 1190 Vienna, Austria
eDepartment of Orthopedics and Trauma Surgery, Medical University of Vienna, 1090 Vienna, Austria
fHeinz-Nixdorf-Chair of Biomedical Electronics, Department of Electrical and Computer Engineering, TranslaTUM, Campus Klinikum rechts der Isar, Technical University of Munich, 81675 Munich, Germany
gSiemens Healthcare GmbH, Technology Center, TI TC BMT, 91058 Erlangen, Germany
First published on 23rd March 2020
Abstract
Rheumatoid arthritis is a chronic, systemic joint disease in which an autoimmune response translates into an inflammatory attack resulting in joint damage, disability and decreased quality of life. Despite recent introduction of therapeutic agents such as anti-TNFα, even the best current therapies fail to achieve disease remission in most arthritis patients. Therefore, research into the mechanisms governing the destructive inflammatory process in rheumatoid arthritis is of great importance and may reveal novel strategies for the therapeutic interventions. To gain deeper insight into its pathogensis, we have developed for the first time a three-dimensional synovium-on-a-chip system in order to monitor the onset and progression of inflammatory synovial tissue responses. In our study, patient-derived primary synovial organoids are cultivated on a single chip platform containing embedded organic-photodetector arrays for over a week in the absence and presence of tumor-necrosis-factor. Using a label-free and non-invasive optical light-scatter biosensing strategy inflammation-induced 3D tissue-level architectural changes were already detected after two days. We demonstrate that the integration of complex human synovial organ cultures in a lab-on-a-chip provides reproducible and reliable information on how systemic stress factors affect synovial tissue architectures.
Introduction
Inflammatory rheumatoid arthritis (RA) is a disease affecting about 1% of the human adult population1 and represents the most common chronic inflammatory joint disease. In arthritis, inflammatory reaction primarily affects the inner layer of the joint capsule and the sublining layer of the synovial membrane. The dominant resident cell type, fibroblast-like synoviocytes, have been recognized in various animal studies and in vitro assays as the key tissue component supporting persistent arthritis. Existing in vitro models describing inflammatory arthritis include analysis of (a) acellular synovial fluids,2,3 (b) different 3D culture models in microtiter plates using hydrogels as extracellular matrix components4–9 and more recently in (c) microfluidic cell culture systems based on two-dimensional (2D) co-cultures of fibroblast-like synoviocytes.10 To date, in vivo animal models are still considered the gold standard in the study of the pathogenesis of arthritis. Here, arthritic diseases in rodents are commonly induced by injection of collagen or collagen antibody-induced arthritis (CIA/CAIA).11,12 Although animal models exhibit many aspects of human arthritic diseases such as RA, they are still unfit for large drug screening due to their inherent low reproducibility and interspecies variations. Additionally, manual injection of the inflammatory stimuli within the joint using needles requires experienced and highly skilled workers to limit animal suffering and pain. In order to reduce the need for large magnitude of costly animal models, it is widely accepted that the next generation in vitro diagnostic systems aside from ‘tissue-slices-on-a-chip’13–15 will be based on the creation of microphysiological in vitro system, so-called organs-on-a-chips based on primary patient cells.16 The premise of any organ-on-a-chip is to replicate the architectural features and/or function of real human tissue as well as pathophysiological cellular responses using three-dimensional (3D) cultures of organ specific cell types.17 These miniaturized systems allow the control of direct as well as indirect cell–cell communications as well as biomechanical cues at the microscale using various strategies such as gradient generation, controllable and defined integration of engineered tissue-like constructs, as well as more recently also integrated actuation and biosensing systems.16,18–21 Despite recent advances in in vitro models for the articular joint, no organ-on-a-chip system emulating functional 3D tissue architecture of the synovium has been reported so far.22From an anatomical viewpoint, the healthy synovium is a highly organized structure that resides between the joint cavity and the fibrous joint capsule and consist of two separate layers: a lining layer formed by condensed cells, up to four cell layers thick, and a loosely organized sublining layer composed of extracellular matrix, scattered fibroblasts, macrophages, mast cells, nerves, blood vessels, and lymphatics.23 In other words, the sublining layer as loose connective tissue forms the microanatomic base of the synovial lining where fibroblast-like synoviocytes form a continuous network of compacted cells. These fibroblast-like synoviocytes express markers common to mesenchymal cells including collagen and they distinctly express cadherin-11, CD55, and uridine diposphoglucose dehydrogenase (UDPDG), thus underlining the importance of cell-to-cell as well as cell–matrix interaction and matrix production and remodelling.24–26 In addition to producing hyaluronan, the lining synoviocytes also synthesize the glycoprotein lubricin, both of which are essential for the lubricating ability of synovial fluid thus maintaining proper joint function.27 It is important to highlight that fibroblast-like synoviocytes are major participants in the active process of synovial extracellular matrix (ECM) production and remodeling.28 Among others fibroblast-like synoviocytes synthesize collagens, fibronectin, and a variety of proteoglycans and distribute these ECM components in distinct regions of the synovium.29 This also means that fibroblast-like synoviocytes constantly produce degradative enzymes required for matrix remodeling and cellular movement,30 thus actively contributing to joint homeostasis. Joint homeostasis is therefore achieved by the maintenance of synovial lining flexibility and integrity, synthesis of lubricating substances, and cartilage nutrition.
In the case of rheumatoid arthritis, this highly organized anatomy of the synovium undergoes striking changes in cellular organization and behavior. For instance, an increase in cell number can be observed in the synovial lining that expands to a thickness of up to 15 cell layers where fibroblast-like synoviocytes comprise a significant protion of this hyperplastic lining.31 The inflamed fibroblast-like synoviocytes display features that are consistent with a metabolically active, secretory cellular state including expansion, thickening and dilatation of the endoplasmic reticulum, lysosomes and Golgi apparatus. As a hallmark of inflammatory synovitis during RA, tissue remodeling results in the formation of a continuous mass of cells spreading out over and invading into the articular cartilage and subchondral bone. If unchecked, uncontrolled inflammation leads to cartilage destruction primarily in the cartilage–pannus junction where both fibroblast-like and macrophage-like synovial cell types release proteinases capable of digesting the cartilage matrix components.32 Additionally, research into the mechanisms influencing fibroblast-like synoviocytes activity in inflammatory arthritis indicated that soluble inflammatory mediators, especially cytokines secreted by infiltrating leukocytes play a key role. For instance, the secretion of IL-1 and tumor necrosis factor strongly stimulate fibroblast-like synoviocytes proliferation in vitro, which promotes synovial hyperplasia. Both proinflammatory cytokines, IL-1 and TNF-α, are known to induce the expression of other cytokines and degradative enzymes in fibroblast-like synoviocytes, thus contributing to the destruction of cartilage in vivo. Moreover, cytokine stimulation of fibroblast-like synoviocytes propagates the additional secretion of cytokines and chemokines. This cytokine-stimulated production of inflammatory mediators by fibroblast-like synoviocytes provides the means by which the synovial inflammatory response is amplified resulting in an autoimmune disease. Especially soluble biochemical cues of three-dimensional organ models including nutrients, cytokines, growth factors can be easily and precisely controlled using microfluidic organs-on-a-chip.18,20,33 To better understand the onset and progression of inflammatory arthritis in response to cytokine-stimulation, a chip-based synovial 3D model containing embedded optical sensor arrays has been developed and characterized in this study. Fig. 1 shows a schematic drawing of the 3D synovium-on-a-chip system containing embedded organic photodiode arrays and the light-scatter detection principle. Since structural reorganisation of the synovium as response to inflammation takes place over the entire tissue architecture – at the surface lining layer and in the deeper sublining layer – continues and non-invasive monitoring of dynamic tissue-level cellular responses such as migration, proliferation and aggregation is needed. While such structural changes of patient-derived human synovial organoids based on isolated fibroblast-like synoviocytes are monitored using inline light-scatter technology, time-resolved cytokine release is additionally detected off-chip using ELISA. Immunohistochemistry as endpoint detection method is conducted to finally link specific tissue reorganization events to 3D light scatter signals. It is important to note that synovial tissue inflammation is predominantly associated with architectural changes in the tissue as a result of increased cell proliferation rates, morphological changes of the synoviocyte network and matrix condensation.34 In the current study we therefore exclusively focus on fibroblast-like synoviocytes as primary cell model instead of a complex co-culture with other involved cell types such as resident macrophages (also known as type A cells or macrophage-like synoviocytes) or endothelial cells. FLS are the most abundant cell population, isolation is easier and they can be expanded very rapidly within a few weeks without loss of function.35 Also, FLS are known to be the key player in inflammatory rheumatoid arthritis, especially during lining layer formation, disease-related hyperplasia (pannus formation) and proinflammatory processes.36–38 For that reason, only purified primary fibroblast-like synoviocytes derived from rheumatoid arthritis patients depleted for at least five passages from immune cells (e.g. T-cells, B-cells and macrophage-like synoviocytes) were used inside microfluidic devices (e.g. for more information on the individual patient's age and treatment please see ESI† Table 1). As artificial biochemical stimulus tumor necrosis factor alpha (TNF-α) was chosen because this potent inflammatory cytokine secreted by monocytes, macrophages, B- and T-cells that has a dominant role in RA triggering enhanced proliferation and cytokine production.39 In our study we demonstrate that on-chip synovial organoids exhibit striking similarities to the synovial membrane of the joint in vivo including the formation of a compact outermost synovial lining layer and a highly organized sublining layer comprising of an interconnected synoviocyte network. Performance evaluation of the light scatter technology involves the assessment of cytokine-induced structural tissue changes that take place over a culture period of 7 days, while verification of disease progression and 3D light scatter signals is performed using ELISA and immunohistochemistry. Overall, only an accurate prediction of dynamic cellular responses to drugs and environmental factors will ultimately open the way for personalized diagnostics and therapy optimization.
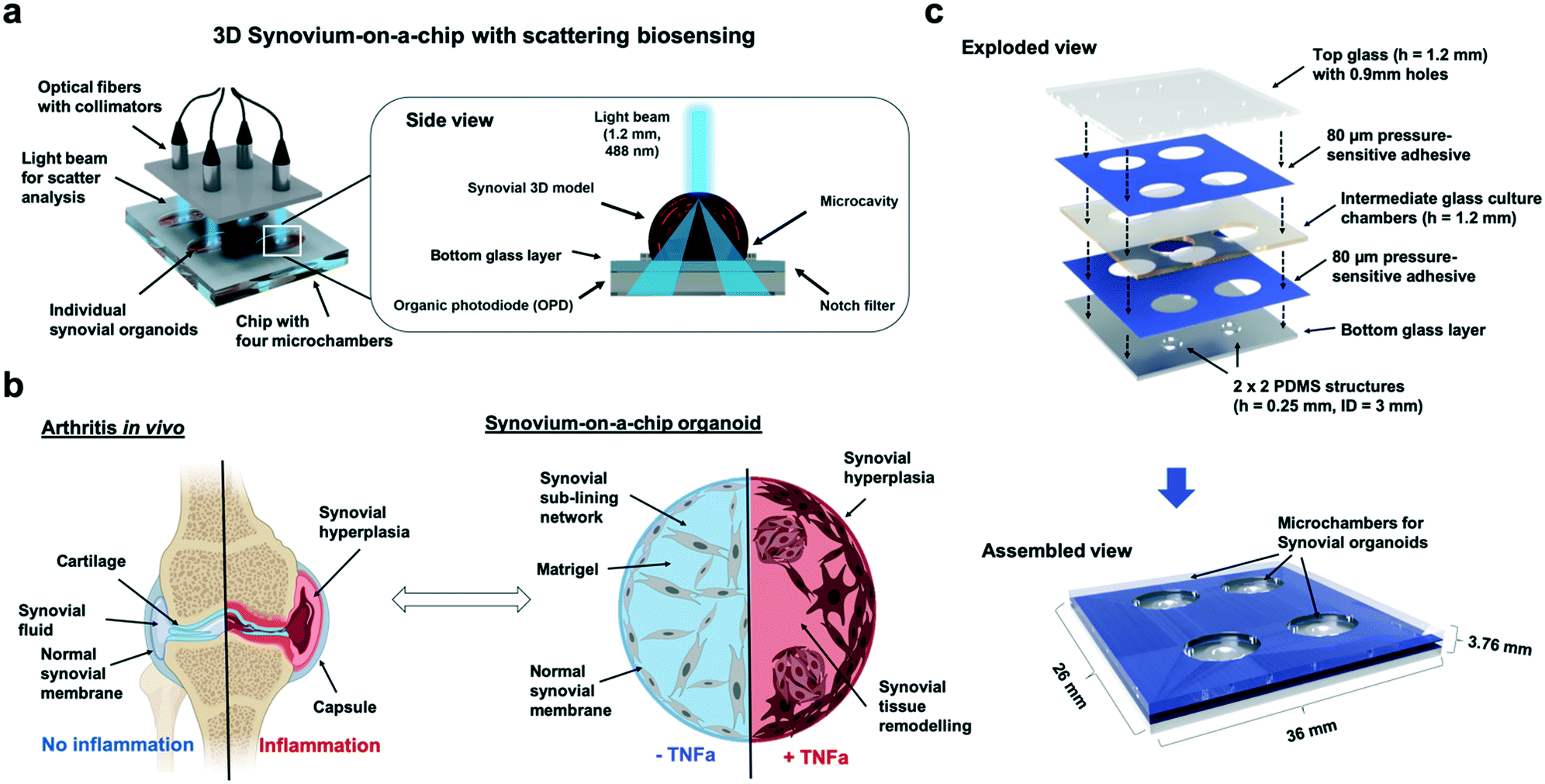 | ||
| Fig. 1 Overview of the synovium-on-a-chip system with integrated time-resolved light scatter biosensing for analysis of tissue-level remodelling during onset of inflammatory arthritis. a) Schematic overview of the synovium-on-a-chip system comprising of four individual microchambers harnessing three-dimensional human synovium organoids. The biosensing principle is based on light scatter measurements through the individual synovial organoids, which is in turn recorded using organic photodiodes underneath the synovium-on-a-chip. b) Comparison of tissue remodelling processes for arthritis in vivo in the human joint and tumour necrosis factor alpha (TNFa) cytokine-induced three-dimensional synovial remodelling on chip. c) Overview of the dimensions and build-up of the synovium-on-a-chip comprising of a combination of layers of structures glass, double sided pressure sensitive biomedical adhesive tape and ring-shaped PDMS waveguide microstructures with 3 mm inner diameter. | ||
Materials and methods
Light scatter platform design
The light scattering station was comprised of a multi-plexed 488 nm sapphire laser (LDP.1116054.091969, Coherent), split by beam splitters (BS010, Thorlabs), fibre couplers (FC488,50B-FC, Thorlabs) and collimators (F240SMA-A, Thorlabs) towards the biochip array measurement platform. In the sample region, a 1.2 mm diameter beam interacts with the biochip samples before the light is being scattered by an angle greater than 20° to the incident light beam (notch filter of 03FIN-series, Melles Griot). This scattered light can pass the notch filter and is detected by an organic photodiode (OPD; Siemens Healthcare GmbH, Germany) generating an electrical signal as potential difference which was in turn recorded using a LabView GUI. To increase dynamic range laser output power can be adjusted between 7 μW and 85 μW. A portable powermeter (LaserCheck, Coherent) was used log the output laser power. A heated water bath (Julabo) connected to the base of the scattering platform allowed temperature control of the scatter biochip array around 37 °C. For each data spot, single measurements were performed for about 10 seconds and data points acquired every 0.1 s. The output laser power of all channels was interpolated in such a way that the laser power on chip was the same for every channel. A mean of the data was calculated and plotted in Microsoft Excel. Limit of detection was calculated as three times the noise amplitude of the random blank signal.Cell culture
Human synovial tissues from patients fulfilling the ACR/EULAR classification criteria for RA40 were obtained as discarded post-surgical specimens following synovectomy with approval of the local ethics committee (Medical University of Vienna AKH) and informed consent by the patients. Pseudonymized clinical data for patient FLS cell lines can be viewed in ESI† Table 1. Human fibroblast-like synoviocytes (FLS) were isolated and cultured as previously described.8 Fibroblast-like synoviocytes were used between passages 4 to 8 and maintained in Dulbecco's modified Eagle's medium (DMEM high glucose, D5796-500ML, Sigma-Aldrich) supplemented with 10% HyClone fetal calf serum (SH30070.03, GE Healthcare Life Sciences), 1% antibiotic antimycotic solution (A5955-100ML, Sigma-Aldrich) and 1% MEM non-essential amino acids (11140-050, Gibco). For subcultivation, cells were washed twice with Dulbecco's phosphate buffered saline (DPBS, D8537-500ML, Sigma-Aldrich) and pelleted at 1200 rpm for 5 minutes after enzymatic detachment with 0.4% trypsin–EDTA (T3924-500ML, Sigma-Aldrich). Cells were split in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 and culture medium was exchanged weekly. For biochemical stimulation of arthritic phenotype synoviocytes were incubated with tumour necrosis factor alpha (TNF-α) (Cat# 210-TA-100, R&D Systems) at a concentration of 10 ng ml−1 in fully supplemented culture medium. For all on- and off-chip synoviocyte experiments aside from cell maintenance, culture medium was further supplemented with 1% insulin–transferrin–selenium (ITS; 41400-045, Gibco), 0.0316 mg ml−1 vitamin C (L-ascorbic acid; A5960, Sigma-Aldrich) and 2% 4-(2hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (J848-100ML, Amresco).
:3 and culture medium was exchanged weekly. For biochemical stimulation of arthritic phenotype synoviocytes were incubated with tumour necrosis factor alpha (TNF-α) (Cat# 210-TA-100, R&D Systems) at a concentration of 10 ng ml−1 in fully supplemented culture medium. For all on- and off-chip synoviocyte experiments aside from cell maintenance, culture medium was further supplemented with 1% insulin–transferrin–selenium (ITS; 41400-045, Gibco), 0.0316 mg ml−1 vitamin C (L-ascorbic acid; A5960, Sigma-Aldrich) and 2% 4-(2hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer (J848-100ML, Amresco).
Biochip array microfabrication & operation
Biochips with four individual cultivation chambers consisted of three layers of microscope glass slides (631-1550, VWR) bonded together using 80 μm thick biocompatible pressure-sensitive adhesive tape (ARcare 90445/90445Q, Adhesive Research) as shown in Fig. 1b. For retention and placement of three-dimensional synoviocyte organoids a circular hydrophobic PDMS structure (inner diameter of 3 mm and an outer diameter of 5 mm) with a height of 250 μm (MVQ Silicones) was bonded with oxygen plasma (PDC-002-CE, Harrick Plasma) on the bottom glass layer to prevent movement or floating of the synovial model. Glass chambers were structured by powderblasting with 120 μm aluminium oxide microparticles (Logiblast) using adhesive masking (Sandblast Super, Gemba) that was structured via xurography with a vinyl cutter (Camm-1 GS-24, Roland). Chip inlets and outlets were drilled in the top glass layer using a 0.9 mm diamond drill (805314009, Komet Dental). Before use, synovium-on-a-chip devices were assembled layer-by-layer partially and chips were disinfected with 70% ethanol for 30 minutes and UV irradiation. For three-dimensional synoviocyte cultures, the complete biochip surface was coated with a self-assembled antifouling protein coating as previously described to prevent cell outgrowth and adhesion.41,42 In brief, for S-layer re-assembly on the surface SbpA protein solution (Lysinibacillus sphaericus CCM 2177) was diluted with a recrystallization buffer (0.5 mM Tris-(hydroxymethyl)aminomethane, 10 mM CaCl2, pH 9) to 200 μg ml−1 incubated overnight in sealed and sterile petri dishes at 4 °C. Right before loading of the synovial organoid cultures, the S-layer protein was aspirated, and biochips were rinsed once with deionized water. For three-dimensional synoviocyte on-chip cultures, Matrigel itself was thawed at 4 °C for 30 minutes before cells were mixed with Matrigel in a ratio of 1:4 and stored on ice. A final volume of 15 μl Matrigel-cell-medium mixture was pipetted in the center of the circular PDMS barrier. Biochips finally assembled by applying the top layer, were sealed with PCR Seal (95.1993, Sarstedt) and incubated at 37 °C for 35 minutes for gelation. After gelation the PCR seal was removed, the chips were filled with complete culture medium and again sealed for on-chip cultivation of the three-dimensional synovium organoids. For two-dimensional on-chip cultures, synoviocyte cells were pelleted, resuspended in complete culture medium and loaded at the desired seeding density into the inlet port of the microchambers. To avoid contaminations in the inlet and outlet ports the surface was closed off by the means of adhesive PCR seal.
Preparation of cell- and particle-laden hydrogels and particle suspensions
Polystyrene nano- and microparticles at a range of 46 nm (GK809W, Kisker Biotech), 794 nm (GK893W, Kisker Biotech), 5 μm (GK1810801-42, Kisker Biotech) and 10 μm (GK2891301-08, Kisker Biotech) were used. Particle concentrations included 125 μg ml−1, 250 μg ml−1, 500 μg ml−1 and 1000 μg ml−1 and particle numbers per biochip included 1.25 × 105, 2.5 × 105, 5.0 × 105 particles. Particles were either diluted in DPBS (L 1820, Biochrom) or suspended in Matrigel, PEG–dextran or fibrin hydrogels and subsequently loaded into a biochip with a total chamber volume of 18 μl.For all experiments, Matrigel (Corning, 354234) was used in a ratio of 1:3, whereas a 1:4 ratio was used for fibroblast-like synoviocytes (Corning, 354230). PEG–dextran (Cellendes, G90-1) at a final volume of 18 μl was prepared according to the manufacturer's web-based calculator by mixing conjugate buffer (1.5 μl, pH 7.2), water (0.9 μl, included in kit), maleimide-dextran (3 μl, 30 mM), thioglycerol (6 μl, 20 mM), culture medium containing cells (3 μl) and PEG-linker (3.6 μl, 20 mM) as final addition to the mix swiftly initiating the polymerization process. Tisseel fibrin hydrogels (Baxter, 1503794) were prepared by mixing first thrombin (0.9 μl, 50 U ml−1) with CaCl2 (8.1 μl, 40 mM), secondly culture medium with fibrinogen (5.4 μl, 100 mg ml−1), and finally combining the two components to start polymerization. Preparation of collagen type I gels was performed by mixing collagen type I solution from rat tail (60 μl, C3867-1VL, Sigma-Aldrich) with culture medium (62.5 μl) and NaOH (2.8 μl, 0.5 M solution), yielding a final collagen concentration of 5 mg ml−1. For experiments on uncontrolled crosslinking NaOH was substituted with culture medium. For experiments with no crosslinking only collagen stock solution at a concentration of 10 mg ml−1 was used.
Viability and metabolic activity assays
For evaluation of cell viability, cells were incubated with a fluorescence Live-Dead staining at 37 °C for 20 minutes using culture medium containing ethidiumhomodimer (4 μM, L3224, Invitrogen) and Calcein (2 μM, L3224, Invitrogen) prior fluorescence microscopy read-out. Complementary, metabolic activity was determined using PrestoBlue™ (A13261, ThermoFisher). After culture medium was removed, 1:10 PrestoBlue mix in culture medium (400 μl) was added to the biochip cultures. After two hours of incubation at 37 °C the culture medium was transferred to 96 well plates (655180, Greiner Bio-One) at 100 μl per well and absorbance measurements were performed using a plate reader (Enspire 2300, PerkinElmer) at a wavelength of 535 nm.
ELISA /alpha-ELISA
A human IL-6 uncoated ELISA (88-7066, Invitrogen) was used for quantitative detection of inflammatory stimulation of synoviocyte cultures. Steps indicated in the supplier manual were followed closely, using a purified anti human IL-6 antibody as a capture antibody, which was incubated overnight at 4 °C. After a blocking agent was added and another hour of incubation at room temperature the samples were added at either 1:10, 1:20 or 1:40 dilution depending on the sample volume-to-cell ratio. For detection biotin conjugated anti human IL-6 detection antibody was incubated for one hour at room temperature and followed by avidin–HRP for 30 min and tetramethylbenzidine (TMB) solution for 15 min. Absorbance at 450 nm was analyzed using a plate-reader. For ELISA of two-dimensional cultures, a daily sample aspiration of 10% of the chamber volume with subsequent refilling was necessary. For three-dimensional synoviocyte cultures sample volume with subsequent medium refill amounted for 150 μl at day 1 and 80 μl day 2–4 of a total chamber volume of 400 μl. For Alpha cytokine analysis of IL-6, IL-8, MMP-1 and MMP-3 (AL223C, AL224C, AL284C, AL242C, Perkin Elmer) the supernatant samples were analysed undiluted according to the manufacturer's protocol. The respective standard curve was diluted in fully supplemented FLS culture medium.
Histological analysis
After all endpoint measurements microtissues were fixed in 4% paraformaldehyde, dehydrated and stored in 70% ethanol, embedded in paraffin and sectioned at a thickness of 2.0–3.0 μm. Thin sections were deparaffinized using Neoclear for 7 minutes at room temperature thrice and washed with absolute ethanol, 96% ethanol and distilled water. For hematoxylin and eosin staining following main steps were observed: Meyer's hemalum (1.09249.0500, Merck) diluted 1:5 with distilled water for 10 minutes at room temperature, 1% HCl ethanol rinse, Eosin (300 ml, 318906, Sigma-Aldrich) diluted in distilled water (600 ml) and acetic acid 100% (0.1 ml) for 15 minutes at room temperature and n-butyl-acetate rinse. H/E staining resulted in a colour separation of nuclei (blue), eosinophilic granules (red-orange) and cytoplasm (pink-violet). For Ki-67 immunostaining anti-Ki67 antibody [B126.1] (ab8191, Abcam) was used diluted 1:50 as primary antibody and goat anti-mouse IgG H&L (Alexa Fluor® 647) (ab150115, Abcam) diluted 1:200 as secondary antibody. Additionally, nuclei were stained with DAPI (ThermoFisher) in a 1:200 dilution. 10× Tris-buffered saline (TBS) pH 7.6 (Sigma-Aldrich) with 5% BSA was used as diluent in all cases. BAFF immunostaining was performed as previously described.43 In brief, sections were incubated with primary polyclonal goat anti-human BAFF/BLyS/TNFSF13B antibody (AF124, R&D Systems) at 1:8000 dilution after blocking with rabbit serum. After incubation with a biotinylated rabbit anti-goat antibody (Vector), sections were incubated with Vectastain Elite reagent and visualized using 3,3-diaminobenzidine (Vector). Sections were counterstained with hematoxylin (Merck) for cell nuclei visualisation.
Data acquisition and output
Figures were plotted by Microsoft Excel, GraphPad Prism and Origin. Microscope images were taken with Olympus IX71 and fluorescence images with Olympus IX83. Figures and images were background subtracted using GIMP Image Editor. Confocal micrographs were taken with a Leica SP5 using an Argon and a multiphoton Laser (MaiTai, Spectra Physics, for Second Harmonic Generation imaging of collagen structures).Statistical analysis
Data shown is expressed as the mean in combination with standard deviation. Sample number (referring to distinct samples of the same patient) and patient ID is stated at the individual experiments. If not otherwise stated, statistical significance was determined by a two-sided Student's t-test. P-values below 0.05 were considered as significant (*), below 0.01 as very significant (**) and below 0.001 as highly significant (***).Results and discussion
Characterization of 3D-light scattering biosensing of on-chip synovium organoids cultures and optimization of the chip design
Detection of 3D tissue-level cellular responses is accomplished by illuminating synovial organoids with a collimated laser beam (488 nm wavelength; 1.2 mm diameter; 10 s) and collection of the scattered fraction of the beam using embedded organic photodiodes (see Fig. 1a and ESI† Fig. 1). It is important to note that only light scattered by more than 20° can pass the integrated notch filter indicating the presence and number of macromolecular structures, particles and whole cells as well as cell assemblies in the illuminated volume.44 Initial method characterization investigated sensitivity, detection limit and linear range of the 3D optical bioassay. Results shown in Fig. 2 and ESI† Fig. 2 display sensor response to varying particle sizes and concentrations as well as cells in the presence and absence of hydrogel matrix compositions. In particular, a variety of particle sizes ranging from 50 nm to 10 μm were selected to represent either whole cells and cellular components such as cell nuclei and different intracellular organelles. Light scattering signals (voltage) shown in ESI† Fig. 2a and b revealed a linear voltage increase as a function of particle concentration from 125 μg ml−1 to 1 mg ml−1 and size 46 nm to 10 μm as well as particle numbers ranging from 1.25 to 5.0 × 105 total particle count. Interestingly, particles with 794 nm diameter showed highest fraction of scattered light, indicating a reduction in scatter angle and/or increasing back scattering events in the presence of particles sizes above 1–5 μm.45 Additional experiments using increasing numbers of patient-derived primary FLS synoviocytes embedded inside a Matrigel hydrogel exhibited a linear signal increase over the entire dynamic OPD sensor voltage range (0–10 V) resulting in a detection limit of approximately 500 cells μl−1 with a sensitivity of 1.9 mV (cell μl−1)−1 as shown in Fig. 2a. An additional benefit our optical measurement setup is the ability to readily regulate power output of the solid-state laser, thus providing the means to adjust detection range and sensitivity to any tissue construct (see also ESI† Fig. 3). For instance, simple matrix or hydrogel composition and density may shift light scatter signals depending on their inherent opacity. To estimate the influence of hydrogel type on light scatter changes three frequently used biocompatible hydrogels containing 794 nm diameter polystyrene particles at a total particle count of 5.0 × 105 particles. Results in Fig. 2b show light scatter behaviour of the pure hydrogel and particles as well as particle-embedded hydrogel. As an example, the commercial TISSEEL fibrin hydrogel was deemed incompatible for our 3D biosensing method due to its turbidity, while fibrin hydrogels polymerized in the absence of calcium chloride are compatible with the current optical setup (data not shown). Relevant for our study, however, is the ability to detect any shifts in optical property of the 3D construct that take place during the establishment and cultivation of synovial organoids. Fig. 2c displays light scatter signals over a period of four days of pure Matrigel, 2D synovial fibroblast culture and hydrogel-based synovial organoids were compared and revealed stable light scatter signals in the presence of pure Matrigel, while a small linear signal increase with 2D fibroblast monolayers was detected indicating cell proliferation events and increase in cell number. In turn, cultivation of synovial fibroblasts embedded in a hydrogel under 3D conditions yielded significant signal changes over the four-day cultivation period. Following an initial linear signal increase from 4 to 10 V within the first 2 days, a stable plateau was reached after day 3, indicating the creation of a lining layer and formation of complex 3D cellular networks. Since cell-free hydrogel clots yielded stable signals following polymerization, the ability of the 3D light scattering method to distinguish between complete and compromised cross-linking events was investigated. Early identification of failed and partial hydrogel polymerization has long been recognized as one of the main limitations in reproducible 3D cell cultures systems. Fig. 2d shows light scatter results from synoviocytes embedded into a tunable pH-sensitive collagen I-type hydrogel where controlled and uncontrolled polymerization took place in the presence and absence of sodium peroxide (acidic pH), while the pure collagen stock solution was used as control. Here, light scatter values of controlled polymerization yielded reproducible scatter values between 2.2 to 2.4 V, while uncontrolled polymerization imitated using an inappropriate pH resulted in a significant signal variation ranging from 2 to 4.5 V. Furthermore, in the absence of any crosslinking events minimal light scattering behaviour of the collagen gel was detected, thus demonstrating the ability for initial quality control measures during on-chip cell-laden hydrogel loading and polymerization.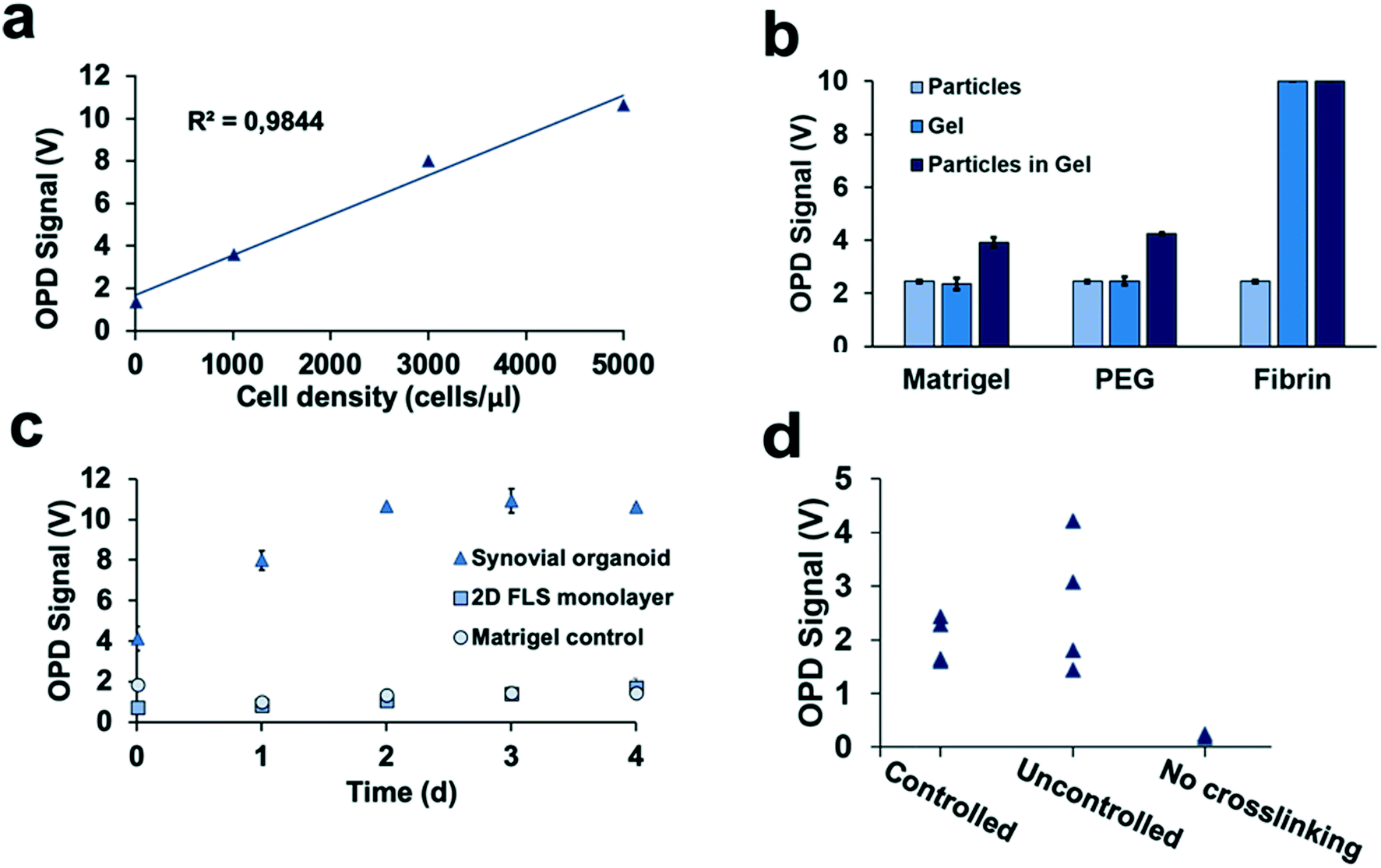 | ||
| Fig. 2 Biophysical characterization of the light scatter bioassay using the synovium-on-a-chip system. a) Sensitivity and limit of detection of light scattering for detection of increasing numbers of primary human synoviocytes (FLS) propagated in Matrigel hydrogel matrix with initial seeding density between 0 and 5000 cells per μl. b) Influence of hydrogel type on light scattering analysis of 28000 microparticles per ml with a diameter of 794 nm suspended in either PBS buffer, plain hydrogels and microparticles suspended in hydrogels on the synovium-on-a-chip system (n = 2–3). c) Time-resolved light scatter analysis of acellular Matrigel, 2D synoviocyte cultures and Matrigel-embedded 3D synovial organoids aver a culture period of four days (n = 2, patient sample ID FLS #68). d) Light scatter analysis for quality control of hydrogel polymerization quality during seeding of synovial organoids at day 0 using collagen I hydrogel as model system for either controlled (+sodium peroxide and correct pH), uncontrolled (−sodium peroxide and acidic pH) and non-existent hydrogel polymerization (pure collagen stock). | ||
Time-resolved monitoring of inflammatory tissue-level remodelling processes using the 3D synovium-on-a-chip screening platform
Prior to time-resolved monitoring of tissue-level remodelling during inflammatory processes, first the reproducibility of light scattering analysis of healthy synovial organoid generation was analysed for the synovium-on-a-chip system. As comparison, well-established synovial organoids of similar volume and cell density were generated in anti-adhesion microtiter plates for four days according to a previously reported protocol.8 At day 4, synovial organoids were placed inside the ring-shaped phaseguide of the synovium-on-a-chip and compared to on-chip healthy synovial organoids. Fig. 3a demonstrates that the on-a-chip approach for synovial organoid formation and positioning is key for reproducible light scatter analysis of tissue-level structures with 5.9 ± 0.25 V signal variation at a RSD of 4.3% at day 4 post-seeding. In contrast, synovial organoids that have been harvested from low-attachment microtiter plates and positioned in the ring-shaped phaseguide yielded a scatter signal of 4.0 ± 1.2 V at a 10-fold higher RSD of 42.2%. These results clearly demonstrate that on-chip organoid growth within the ring-shaped PDMS waveguide structures is key for reproducible light scatter measurements on the synovium-on-a-chip station.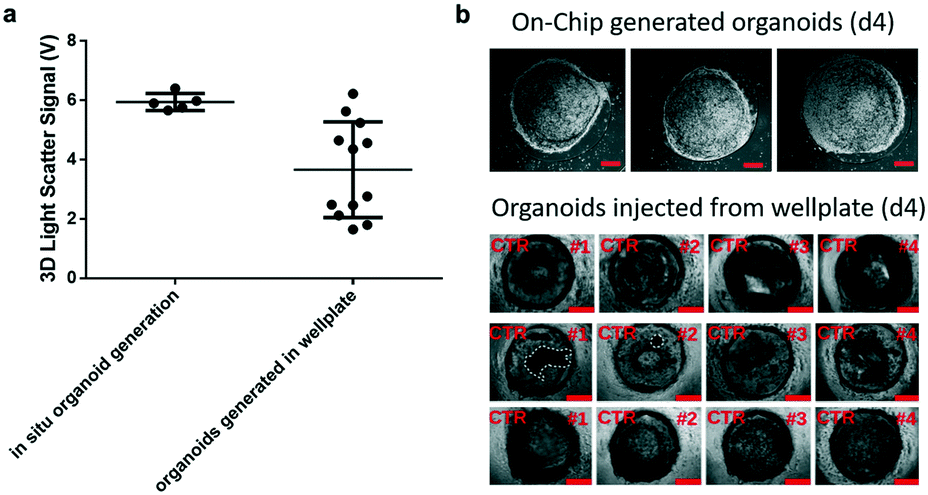 | ||
| Fig. 3 a) Comparison of light scatter data from healthy synovial organoids directly generated within the phase guide structures of the biochips (n = 5) and organoids generated in 12 well plates (n = 12) at day 4 post-seeding. Well-plate generated synovial organoids were injected at day 4 onto the synovium-on-a-chip system and supplied with culture medium b) representative phase contrast images of healthy synovial organoids directly generated within the phase guide structures of the biochips (n = 4) in comparison to off-chip organoids generated in 12 well plates (n = 12) at day 4 post-seeding within the synovium-on-a-chip system with ring-shaped PDMS phaseguide microstructures. Scale bars, 500 μm (a and b: patient sample ID FLS #13, #15, #16 and #68). | ||
Encouraged by the earlier-mentioned time-resolved light scatter results during the formation of synovial organoids on chip (see also Fig. 2c), the ability to identify the onset and progression of tissue remodelling processes following exposure to pro-inflammatory cytokines was investigated in a next set of experiments. To discard the concern of unforeseeable phototoxic events to cyclic exposure of a 488 nm laser beam, life/ dead staining in absence and presence of laser illumination was conducted. As shown in ESI† Fig. 4 at the highest power output no detectable loss of cell viability within the organoids was observable throughout 8 days of continuous operation using 50 msec pulses every 30 s at 37 μW laser power. Even in the presence of TNF-α no significant phototoxic events on organoids were detected, thus demonstrating the suitability of our 3D light-scattering method for biological applications.
Next, the establishment of physiological synovial tissue-level architecture relating to healthy synovium within the 3D organoids was confirmed using histology analysis and fluorescent microscopy. Fig. 4a shows a representative histology picture a of a cross section of the 3D synovial organoid confirming the formation of a thin lining layer at the fluid-hydrogel interface and loosely connected sub-lining layer over a period of 3 to 4 days. Additional confocal laser scanning images (lower panel of Fig. 4b) further verified the presence of a thin, homogenous and tight synovial lining layer at the surface as well as a complex network of loosely interconnected fibroblast-like synoviocytes throughout the sub-lining compartment. Using second harmonics generation microscopy (upper panel in Fig. 4b) further revealed the deposition of collagen fibres (type I and II) by FLS synoviocytes into the Matrigel matrix.8 This deposition of organotypic reticular fibres (purple) at day 4 post-seeding, strongly points at the generation of a functional and healthy 3D synovial organoids within the synovium-on-a-chip platform.
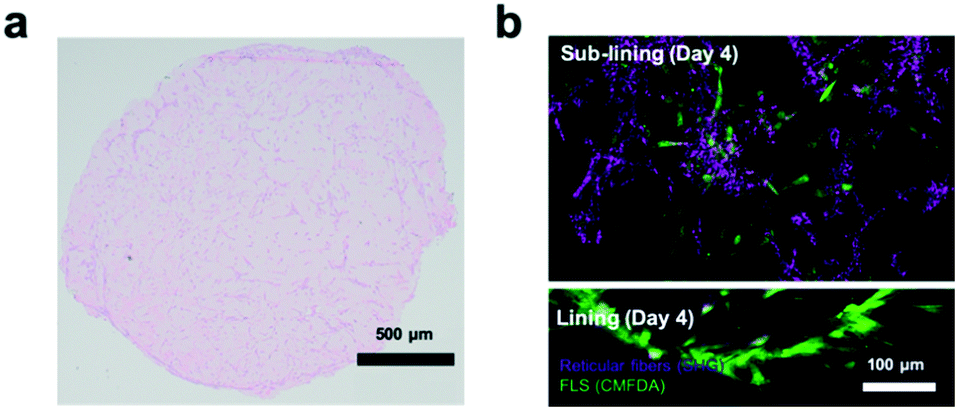 | ||
| Fig. 4 a) Histological and b) confocal laser scanning imaging of healthy synovial organoid-on-a-chip at day 4 post-seeding. For histological evaluation at day 4 post-seeding hematoxylin (blue cytosol and nuclei)/eosin (pink-colored collagen) double staining was used on paraffin embedded thin sections. (patient sample ID FLS #16) For confocal analysis of the organoid prior embedding, synovial organoids were stained with CMFDA staining (green) and while performing second harmonic generation (SHG) analysis of collagenous extracellular matrix structures (magenta) at day 4. Images were rendered using Imaris ©Bitplane software. | ||
To recreate the pathology of inflammatory arthritis in our chip-based synovial organoids, the culture media was supplemented with 10 ng ml−1 TNF-α during cultivation.7 Lining hyperplasia and condensation/aggregation of FLS synoviocytes in the sublining area has been documented both in vitro and in vivo in response to TNF-α8,9 exposure a well-known and potent inflammatory cytokine. Although structural changes of the synovial tissue architecture has been previously described,10 little is known about the dynamics of cellular network degradation, proliferation and cell aggregation that take place during inflammatory arthritis. To assess the ability of our chip-based 3D light scatter biosensing method to detect the onset and progression of tissue-level remodelling processes, 3D synovial organoids were cultivated on chip in the absence and presence of TNF-α. Time-resolved light scattering signals of 3D synovial organoids are shown in Fig. 5a, exhibiting a signal increase of 16% and 21% in the presence of cytokine-stimulation at day 3 and 4 respectively. This significant increase in light scatter behaviour can most likely be attributed to structural changes within the synovial organoid. For instance, corresponding live-cell microscopic analysis (lower panel of Fig. 5a) revealed an increase of secondary structures in the sublining over time, indicating cellular hyperproliferation and network remodelling upon cytokine stimulation. In an attempt to confirm the presence of inflammatory processes, complementary on-chip and off-chip analysis were performed to determine (1) sites of tissue remodelling, (2) the release of inflammatory cytokines, (3) overall cell viability and (4) metabolic activity as well as (5) proliferation events within the 3D organoids. Interestingly, cell viability and metabolic activities were similar in untreated and TNF-α stimulated 3D organoids (see Fig. 5b and c and ESI† Fig. 5) thus pointing at an excellent tissue constitution. An overall cytokine increase was recorded in TNF-α stimulated organoids with respect to the untreated control (see Fig. 5d and ESI† Fig. 6), pointing at a strong inflammatory tissue response. Also, IL-6 production in our chip-based 3D organoids in the time-course of four days were also significantly higher than found in 2D monolayer culture of FLS, thus highlighting the impact of an in vivo-like 3D chip-based environment. In addition to interleukin increase (IL-6 and IL-8) also a significant increase of matrix-metalloprotease (MMP-1 and MMP-3) activity was observed indicating degradative capacity towards tissue matrices (e.g. collagen II in cartilage).46,47
 | ||
| Fig. 5 Screening of synovial tissue remodelling processes in vitro in the presence of inflammatory cytokines using the light scattering analysis of synovial organoids within the synovium-on-a-chip system. a) Non-invasive and continuous light scatter analysis (top) and corresponding bright-field images (bottom) of patient-derived three-dimensional synovial organoids seeded at an initial cell density of 3000 cells μl−1 over four days in the presence (dark blue triangles) and absence (light blue squares) of 10 ng ml−1 TNF-α (n = 3, patient sample ID FLS #16). b) Representative fluorescence images of untreated and TNF-α stimulated synovial organoids at day 4 post-seeding in the light scattering biochip array. Organoids were stained with a Live/Dead cytotoxicity assay using 2 μM calcein AM (green-fluorescent live cells) and 4 μM ethidium-homodimer (red-fluorescent dead cells). White arrows highlight lining layer. Scalebar, 500 μm (patient sample ID FLS #16) c) greyscale intensity of a Live/Dead cytotoxicity assay using 2 μM calcein AM and 4 μM ethidium-homodimer at day 4 was calculated by using FIJI to measure the mean grey scale value of the synovial organoids. A Student t-test showed no significant differences between organoids cultured with and without TNF-α (10 ng ml−1). (p > 0.05, n = 3) d) Comparative secretion of inflammatory cytokines of three-dimensional Matrigel FLS organoids maintained within the synovium-on-a-chip platform at day four post-seeding in the presence and absence of the inflammatory cytokine TNF-α at a concentration of 10 ng ml−1 (control: n = 6, TNF-α: n = 8, patient sample ID FLS #68; data was normalized against acellular Matrigel control experiments). | ||
When analysing overall tissue-level morphology and structure in more detail (see Fig. 5) distinct differences are observable in inflamed organoids. Importantly, a thickened hyperplastic synovial lining layer can only be identified in TNF-α stimulated synovial organoids, thus confirming the presence of an inflamed phenotype of the synovial tissue (see Fig. 5a, upper panel). Similar findings (black arrows) can be seen in animal tests where mice challenged with TNF-α caused hyperproliferation and hyper-survival of synoviocytes resulting in lining layer hyperplasia in inflamed joints.48 Additionally, the presence of multi-layered network protrusions following TNF-α stimulation seen in the centre region of the organoid-on-chip shown in Fig. 6a (black arrowheads in lower panel) point at increased cell migration and changes in cell-to-cell interactions caused by inflammatory overexpression of adhesion molecules such as cadherin-11. The occurrence of inflammatory tissue reactions was further confirmed in subsequent histological evaluations. For instance, Fig. 6b shows the formation of thick and aggregated synovial networks of proliferative KI-69 positive synoviocytes in contrast to more homogenous and thinner networks seen in healthy organoids.49 Another hallmark of an inflammatory synovial tissue response constitutes the presence of cell-free regions, which are highlighted in Fig. 6b (right panel) using dashed white lines in TNF-α stimulated arthritic organoids. In addition to changes of morphology and adhesion molecules, also B cell activating factor BAFF was overexpressed as shown in Fig. 6c by 1.6 ± 0.1-fold for TNF-α stimulated organoids compared to untreated control samples indicating functional changes during RA induction in the synovial models. Overall, the above results demonstrate that dynamic tissue remodelling processes such as cell proliferation, network formation or cell-matrix interactions take place in synovial organoids cultured on chip following stimulation with TNF-α, which accurately mimics the onset and progression of inflammatory arthritis.
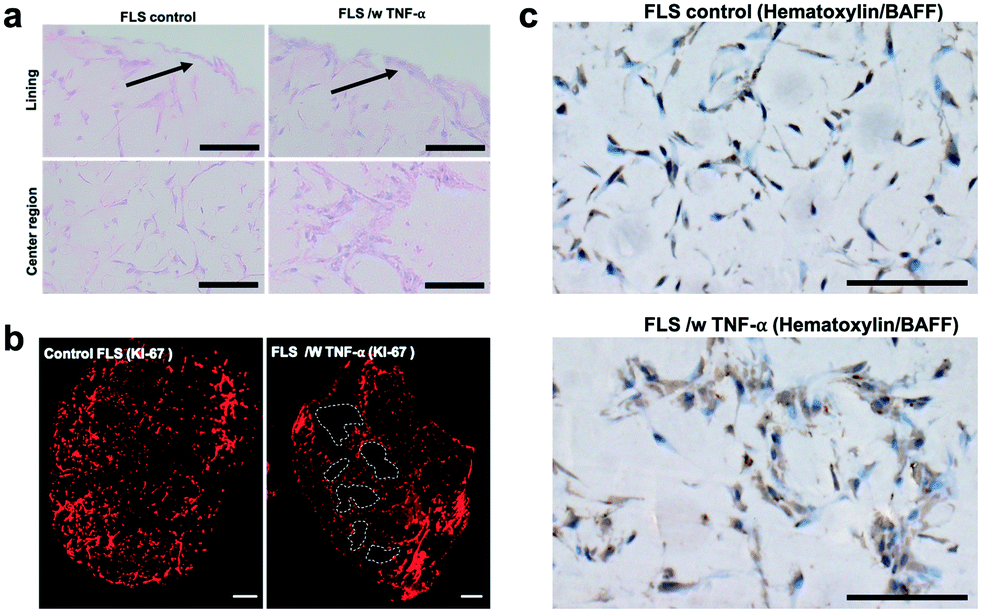 | ||
| Fig. 6 Histological evaluation of morphology of untreated healthy and TNF-α stimulated arthritic synovial organoids at day 4 post-seeding harvested from the synovium-on-a-chip systems. a) Brightfield images of hematoxylin (blue cytosol and nuclei)/eosin (pink-colored collagen) stained synovial on-chip organoids in the presence and absence of TNFa. Black arrows indicate lining layer. Scalebar, 100 μm (patient sample ID FLS #16) b). Histological evaluation of proliferating cells of untreated and TNF-α stimulated synovial microtissues at day 4 post-seeding using anti KI-67 immunostaining (red fluorescing proliferative cells). White dashed lines highlight areas cell-free areas. Scalebar, 100 μm. (patient sample ID FLS #16) c) Brightfield images of hematoxylin (blue cytosol and nuclei)/B cell activating factor BAFF stained brown) of synovial on-chip organoids in the presence and absence of TNFa. Scalebar, 100 μm (patient sample ID FLS #16). | ||
Conclusion
Next generation organ-on-a-chip systems are expected to (a) provide reliable information on the health status of the integrated complex biological system, (b) reproducibly generate functional tissue structures and (c) non-invasively monitor dynamic tissue responses to external stimuli. The more complex the integrated in vitro models get from a biological viewpoint, the rarer biosensors are being integrated for non-invasive and time-resolved online monitoring. To address these mandates and demonstrate that biosensors are a valuable tool for microphysiological systems, we have developed a synovium-on-a-chip system containing an embedded organic photodetector array to study the onset and progression of inflammatory arthritis using light scattering technology. Using patient-derived primary synoviocytes we initially demonstrated the ability to reliably generate 3D synovial organoids on chip. Reproducible generation of 3D synovial organoids with similar size and structure was accomplished by loading cell-laden hydrogels into ring-shaped phase guides located within the cultivation chambers. The integrated ring-shaped phase guides not only allowed the generation of functional synovial tissue structures featuring a tight lining layer at the surface and a loosely populated sub-lining layer, but also served as an anchor to confine and localize synovial organoids. Moreover, this microfluidic organoid localization is key for the implementation of any biosensing strategy,20,50–53 where precise positioning is needed. For instance, we showed that the application of a simple light scatter method is ideally suited to non-invasively detect cell motility, proliferation, invasion and even matrix condensation processes within a 3D tissue constructs without the need of tedious and expensive staining and imaging techniques. As a practical application of our microfluidic 3D light-scattering technology we show that a diseased phenotype can already be distinguished as early as day 2–3 post seeding, which is normally distinguished around 14 to 21 days post-seeding for conventional microtiter-based 3D synovial models with proliferation assays and cytokine assays such as ELISA. We believe that our synovium-on-a-chip system may serve as a rapid screening tool that can be potentially be applied in drug screening as well as future efforts in personalized medicine for inflammatory arthritis or other joint-related disease models.Author contributions
M. R., G. H., C. E., S. R. A. K., S. K., R. B., B. F., P. S., I. O. C., B. N., B. M. performed the experiments and analysed the data. F. S. and J. H. provided primary patient tissue from synovectomy. O. H. and S. F. T. provided OPDs. H. P. K. and P. E. supervised the work and edited the manuscript. All authors co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Vienna Science and Technology Fund (WWTF-LS-013).Notes and references
- G. S. Firestein, Nature, 2003, 423, 356 CrossRef CAS PubMed.
- J. C. Krebs, Y. Alapan, B. A. Dennstedt, G. D. Wera and U. A. Gurkan, Biomed. Microdevices, 2017, 19, 20 CrossRef.
- W.-T. Hsu, W.-H. Hsieh, S.-F. Cheng, C.-P. Jen, C.-C. Wu, C.-H. Li, C.-Y. Lee, W.-Y. Li, L.-K. Chau and C.-Y. Chiang, Anal. Chim. Acta, 2011, 697, 75–82 CrossRef CAS PubMed.
- T. P. Lozito, P. G. Alexander, H. Lin, R. Gottardi, A. W.-M. Cheng and R. S. Tuan, Stem Cell Res. Ther., 2013, 4, S6 CrossRef PubMed.
- Y. Peck, L. T. Leom, P. F. P. Low and D. A. Wang, J. Tissue Eng. Regener. Med., 2018, 12, e237–e249 CrossRef CAS PubMed.
- M. Broeren, C. Waterborg, R. Wiegertjes, R. Thurlings, M. Koenders, P. van Lent, P. van der Kraan and F. van de Loo, ALTEX, 2019, 36(1), 18–28 CrossRef.
- H. P. Kiener, D. M. Lee, S. K. Agarwal and M. B. Brenner, Am. J. Patho.l, 2006, 168(5), 1486–1499 CrossRef CAS PubMed.
- H. P. Kiener, G. F. Watts, Y. Cui, J. Wright, T. S. Thornhill, M. Sköld, S. M. Behar, B. Niederreiter, J. Lu and M. Cernadas, Arthritis Rheum., 2010, 62, 742–752 CrossRef CAS PubMed.
- D. M. Lee, H. P. Kiener, S. K. Agarwal, E. H. Noss, G. F. Watts, O. Chisaka, M. Takeichi and M. B. Brenner, Science, 2007, 315(5814), 1006–1010 CrossRef CAS PubMed.
- H.-P. Ma, X. Deng, D.-Y. Chen, D. Zhu, J.-L. Tong, T. Zhao, J.-H. Ma and Y.-Q. Liu, R. Soc. Open Sci., 2018, 5, 180528 CrossRef PubMed.
- D. Wernicke, C. Schulze-Westhoff, P. Petrow, R. Bräuer, J. Zacher, S. Gay and E. Gromnica-Ihle, Arthritis Rheum., 2002, 46, 64–74 CrossRef CAS PubMed.
- D. L. Asquith, A. Miller, I. B. McInnes and F. Y. Liew, Eur. J. Immunol., 2009, 39, 1991–2058 CrossRef PubMed.
- M. A. Catterton, A. F. Dunn and R. R. Pompano, Lab Chip, 2018, 18, 2003–2012 RSC.
- S. Shim, M. C. Belanger, A. R. Harris, J. M. Munson and R. R. Pompano, Lab Chip, 2019, 19, 1013–1026 RSC.
- I. C. McLean, L. A. Schwerdtfeger, S. A. Tobet and C. S. Henry, Lab Chip, 2018, 18, 1399–1410 RSC.
- J. M. Rosser and P. Ertl, Curr. Trends Biomedical Eng. & Biosci., 2017, 9(2), 555756 Search PubMed.
- K. H. Benam, S. Dauth, B. Hassell, A. Herland, A. Jain, K.-J. Jang, K. Karalis, H. J. Kim, L. MacQueen and R. Mahmoodian, Annu. Rev. Phytopathol., 2015, 10, 195–262 CrossRef CAS PubMed.
- M. Rothbauer, V. Charwat, B. Bachmann, D. Sticker, R. Novak, H. Wanzenböck, R. A. Mathies and P. Ertl, Lab Chip, 2019, 19, 1916–1921 RSC.
- M. Rothbauer, H. Zirath and P. Ertl, Lab Chip, 2018, 18, 249–270 RSC.
- H. Zirath, M. Rothbauer, S. Spitz, B. Bachmann, C. Jordan, B. Müller, J. Ehgartner, E. Priglinger, S. Mühleder, H. Redl, W. Holnthoner, M. Harasek, T. Mayr and P. Ertl, Front Physiol., 2018, 9, 815 CrossRef PubMed.
- S. R. A. Kratz, G. Höll, P. Schuller, P. Ertl and M. Rothbauer, Biosensors, 2019, 9(3), 110 CrossRef PubMed.
- S. Piluso, Y. Li, F. Abinzano, R. Levato, L. M. Teixeira, M. Karperien, J. Leijten, R. van Weeren and J. Malda, Trends Biotechnol., 2019, 37(10), 1063–1077 CrossRef CAS PubMed.
- D. Hamerman and P. Barland, Bull. Rheum. Dis., 1966, 16, 396–399 CAS.
- X. Valencia, J. M. Higgins, H. P. Kiener, D. M. Lee, T. A. Podrebarac, C. C. Dascher, G. F. Watts, E. Mizoguchi, B. Simmons, D. D. Patel, A. K. Bhan and M. B. Brenner, J. Exp. Med., 2004, 200, 1673–1679 CrossRef CAS PubMed.
- J. Hamann, J. O. Wishaupt, R. A. van Lier, T. J. Smeets, F. C. Breedveld and P. P. Tak, Arthritis Rheum., 1999, 42, 650–658 CrossRef CAS PubMed.
- A. A. Pitsillides, L. S. Wilkinson, S. Mehdizadeh, M. T. Bayliss and J. C. Edwards, Int. J. Exp. Pathol., 1993, 74, 27–34 CAS.
- D. K. Rhee, J. Marcelino, M. Baker, Y. Gong, P. Smits, V. Lefebvre, G. D. Jay, M. Stewart, H. Wang, M. L. Warman and J. D. Carpten, J. Clin. Invest., 2005, 115, 622–631 CrossRef CAS PubMed.
- P. A. Revell, N. al-Saffar, S. Fish and D. Osei, Ann. Rheum. Dis., 1995, 54, 404–407 CrossRef CAS PubMed.
- E. H. Danen, P. Sonneveld, C. Brakebusch, R. Fassler and A. Sonnenberg, J. Cell Biol., 2002, 159, 1071–1086 CrossRef CAS PubMed.
- G. Cunnane, O. FitzGerald, K. M. Hummel, R. E. Gay, S. Gay and B. Bresnihan, Rheumatology, 1999, 38, 34–42 CrossRef CAS PubMed.
- B. Henderson and E. R. Pettipher, Semin. Arthritis Rheum., 1985, 15, 1–32 CrossRef CAS.
- M. Otero and M. B. Goldring, Arthritis Res. Ther., 2007, 9, 220 CrossRef PubMed.
- J. Rosser, B. Bachmann, C. Jordan, I. Ribitsch, E. Haltmayer, S. Gueltekin, S. Junttila, B. Galik, A. Gyenesei, B. Haddadi, M. Harasek, M. Egerbacher, P. Ertl and F. Jenner, Mater. Today Bio, 2019, 4, 100023 CrossRef CAS PubMed.
- V. Charwat, M. Purtscher, S. F. Tedde, O. Hayden and P. Ertl, Lab Chip, 2013, 13(5), 785–797 RSC.
- J. Tu, W. Hong, P. Zhang, X. Wang, H. Körner and W. Wei, Front. Immunol., 2018, 9, 1467 CrossRef PubMed.
- B. Bartok and G. S. Firestein, Immunol. Rev., 2010, 233, 233–255 CrossRef CAS PubMed.
- E. H. Noss and M. B. Brenner, Immunol. Rev., 2008, 223, 252–270 CrossRef CAS PubMed.
- A. Mor, S. B. Abramson and M. H. Pillinger, Clin. Immunol., 2005, 115, 118–128 CrossRef CAS PubMed.
- P. Vasanthi, G. Nalini and G. Rajasekhar, Int. J. Rheum. Dis., 2007, 10, 270–274 Search PubMed.
- D. Aletaha, T. Neogi, A. J. Silman, J. Funovits, D. T. Felson, C. O. Bingham III, N. S. Birnbaum, G. R. Burmester, V. P. Bykerk and M. D. Cohen, Arthritis Rheum., 2010, 62, 2569–2581 CrossRef PubMed.
- M. Rothbauer, S. Küpcü, D. Sticker, U. B. Sleytr and P. Ertl, ACS Nano, 2013, 7(9), 8020–8030 CrossRef CAS PubMed.
- S. Moreno-Flores and S. Küpcü, Soft Matter, 2015, 11(7), 1259–1264 RSC.
- T. Karonitsch, R. K. Kandasamy, F. Kartnig, B. Herdy, K. Dalwigk, B. Niederreiter, J. Holinka, F. Sevelda, R. Windhager, M. Bilban, T. Weichhart, M. Saemann, T. Pap, G. Steiner, J. S. Smolen, H. P. Kiener and G. Superti-Furga, Cell Rep., 2018, 23, 2157–2167 CrossRef CAS.
- V. Charwat, M. Rothbauer, S. F. Tedde, O. Hayden, J. J. Bosch, P. Muellner, R. Hainberger and P. Ertl, Anal. Chem., 2013, 85, 11471–11478 CrossRef CAS.
- J. R. Mourant, J. P. Freyer, A. H. Hielscher, A. A. Eick, D. Shen and T. M. Johnson, Appl. Opt., 1998, 37(16), 3586–3593 CrossRef CAS PubMed.
- T. C. A. Tolboom, E. Pieterman, W. H. van der Laan, A. L. Huidekoper, R. Nelissen, F. C. Breedveld and T. W. J. Huizinga, Arthritis Res., 2001, 3, P046 CrossRef.
- A. Fragoulis, J. Laufs, S. Müller, U. Soppa, S. Siegl, L. K. Reiss, M. Tohidnezhad, C. Rosen, K. Tenbrock, D. Varoga, S. Lippross, T. Pufe and C. J. Wruck, Arthritis Res. Ther., 2012, 14, R220 CrossRef CAS PubMed.
- J. Tu, W. Hong, P. Zhang, X. Wang and W. Wei, Front Immunol., 2018, 9, 1467 CrossRef PubMed.
- H. P. Kiener and M. B. Brenner, Arthritis Res. Ther., 2005, 7(2), 49–54 CrossRef CAS PubMed.
- J. Ehgartner, M. Strobl, J. M. Bolivar, D. Rabl, M. Rothbauer, P. Ertl, S. M. Borisov and T. Mayr, Anal. Chem., 2016, 88(19), 9796–9804 CrossRef CAS PubMed.
- L. Richter, V. Charwat, C. Jungreuthmayer, F. Bellutti, H. Brueckl and P. Ertl, Lab Chip, 2011, 11(15), 2551–2560 RSC.
- M. Rothbauer, D. Wartmann, V. Charwat and P. Ertl, Biotechnol. Adv., 2015, 33, 948–961 CrossRef PubMed.
- A. Susloparova, D. Koppenhöfer, J. Law, X. Vu and S. Ingebrandt, Lab Chip, 2015, 15(3), 668–679 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc01097a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |