
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA self-calibrating and multiplexed electrochemical lab-on-a-chip for cell culture analysis and high-resolution imaging†
Pablo
Giménez-Gómez
a,
Rosalía
Rodríguez-Rodríguez
b,
Juan Manuel
Ríos
a,
Marta
Pérez-Montero
b,
Estrella
González
a,
Manuel
Gutiérrez-Capitán
 a,
Jose Antonio
Plaza
a,
Xavier
Muñoz-Berbel
*a and
Cecilia
Jiménez-Jorquera
a
a,
Jose Antonio
Plaza
a,
Xavier
Muñoz-Berbel
*a and
Cecilia
Jiménez-Jorquera
a
aInstituto de Microelectrónica de Barcelona (IMB-CNM, CSIC), Campus UAB, 08193 Bellaterra, Barcelona, Spain. E-mail: xavier.munoz@imb-cnm.csic.es; Tel: +34935947700
bBasic Sciences Department, Faculty of Medicine and Health Sciences, Universitat Internacional de Catalunya, E-08195 Sant Cugat del Vallés, Barcelona, Spain
First published on 2nd January 2020
Abstract
In vitro analysis requires cell proliferation in conditions close to physiological ones. Lab-on-a-chip (LoC) devices simplify, miniaturize and automate traditional protocols, with the advantages of being less expensive and faster due to their shorter diffusion distances. The main limitation of current LoCs is still the control of the culture conditions. Most LoCs employ off-chip equipment to determine cell culture activity, which confers limited monitoring capacity. The few systems integrating transducers on-chip present important functional problems mostly associated with the attachment of biomolecules to the transducer surface (i.e., biofouling) and the impossibility of re-calibrating the sensors during cell culturing. This limitation is addressed in the present LoC containing a network of micro-channels and micro-chambers, which allows (i) cell seeding and cultivation, avoiding biofouling risk, (ii) multiplexed analysis of cell culture, reactivation and recalibration of the (bio)sensors without compromising cell viability, (iii) cell imaging and (iv) reference electrode compartmentalization to guarantee stability. The activity of the culture is monitored with four independent electrochemical micro-electrodes for glucose, hydrogen peroxide, conductivity and oxidation reduction potential. Electrochemical analysis is complemented with high-resolution confocal microscopy analysis. This paper demonstrates the suitability of the current configuration for cell culture monitoring and future applications in drug screening or organ-on-a-chip development.
Introduction
Due to important ethical issues arising from animal experimentation,1 cell cultures in relevant environments are positioned as the best alternative for studying the causes of diseases or drug screening.2 Conventional in vitro tests3 are now competing with simpler, miniaturized and automated lab-on-a-chip (LoC) devices,4–7 which minimize human errors and sample contamination while decreasing the cost and duration of the assays.8,9The main limitation of current LoCs for cell culture is cell culture monitoring. Cells proliferating on the chip should be continuously analysed to guarantee that they are growing under optimal physiological conditions. Only then, the changes reported in the culture may be associated with the process or treatment under study. Unfortunately, most LoCs do not integrate transduction systems but require external benchtop equipment (i.e., a microscope or an imaging system10–12) to record cell activity.13 Among the few systems integrating transducers on-chip, three have been already commercialized, demonstrating the potential market of these technologies. These are DOX-96, commercialized by Dalkin Industries Ltd,14 which monitors cell respiration; BIONAS 2500 from the Bionas GmbH company,15 which monitors extracellular pH, cell adhesion and cellular respiratory activity; and the xCELLigence® Real-Time Cell Analyser from ACEA Biosciences,16 which evaluates cell adhesion and cytotoxicity through impedance spectroscopy. It is worth mentioning that of these three technologies, only the last one is still commercially available. The reason for this may be partially associated with the transduction mechanism. xCELLigence uses impedance spectroscopy to report on cell attachment and correlates this measurement with cell migration and viability. Because cell attachment is a particular cause of biofouling, this system is not affected by this common drawback. In contrast, DOX-96 and BIONAS 2500 integrate amperometric sensors, which are very sensitive to biofouling. Biofouling decreases the active area of the electrode, altering amperometric recordings. To obtain reliable results, amperometric sensors should be reactivated and recalibrated between measurements. Regarding the latter, it is becoming increasingly accepted that the integration of electrochemical transducers on-chip should consider important design constraints to guarantee cell integrity in physiological conditions and measurement reliability.17
These constraints have not been completely addressed by current cell culture systems. A number of examples integrate CO2,18 glucose and oxygen19 sensors on poly(dimethylsiloxane) (PDMS) structures. These systems benefit from the low cost, fabrication simplicity and high gas diffusion capacity of PDMS; however, they do not solve the biofouling problem of electrochemical transducers. Additionally, there is some controversy regarding the chemical stability of PDMS because some authors claim that PDMS releases un-polymerized monomers to the medium, which may affect cell activity.20,21
Silicon and glass are more chemically stable but also more expensive and difficult to bond. For this reason, the number of publications using these materials is small. In 2003, a silicon chip with pH-ISFETs and amperometric electrodes was proposed for cell proliferation and analysis.22 Unfortunately, in the design, the fluidic inlets and outlets were on top of the silicon chip platform, which did not allow simultaneous imaging of cells. This limitation was solved in 2014 when Urban et al. developed a new LoC device integrating electrochemical sensors with a transparent tape that enabled imaging.23 However, the microfluidic configuration of the chip, with a single micro-channel connecting the fluidic inlet and the outlet, did not allow sensor reactivation and recalibration between measurements, compromising the reliability of the analysis.
As a step forward, we present a silicon-glass electrochemical LoC with compartmentalisation and smart micro-fluidic interconnections enabling (i) cell seeding and proliferation in an independent micro-chamber, minimizing biofouling; (ii) regular reactivation and recalibration of the amperometric electrodes without compromising cell viability; (iii) compartmentalisation of the Ag/AgCl pseudo-reference electrode to enhance stability; (iv) culture medium and reagent transport, with small dead volumes; and (v) high-resolution imaging of cells through a micro-patterned glass layer. The integration of four electrodes allows multiplexed analysis of key analytes, namely glucose, hydrogen peroxide, conductivity and oxidation reduction potential (ORP). The micro-electrodes have been tested individually and together to evaluate their performance. Human lung fibroblast cells (MRC-5) have been cultured in the multiplexed LoC (ME-LoC), and oxidative stress induction by hydrogen peroxide has been evaluated with the four integrated micro-electrodes and compared to optical analysis by confocal microscopy.
Experimental
Design and fabrication of the ME-LoC
The ME-LoC is composed of two layers permanently attached by anodic bonding. The bottom silicon layer contains the electrochemical transducers, connections and pads. Microfluidic elements, i.e. micro-channels and micro-chambers, are defined in the second layer, a glass cover, which seals the system. A schematic of the ME-LoC is shown in Fig. 1A.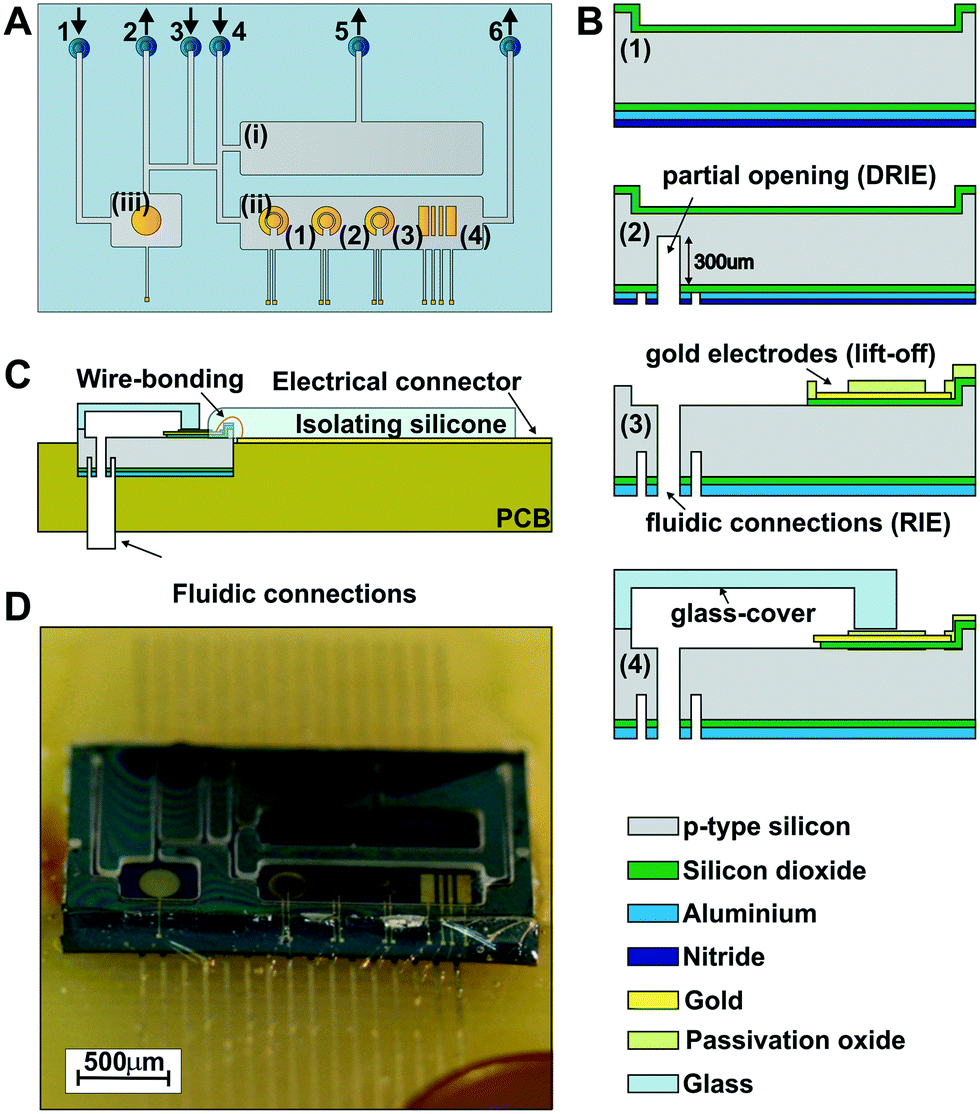 | ||
| Fig. 1 Design and fabrication of the ME-LoC. (A) Schematic of the ME-LOC detailing (i) the cell-culture chamber, (ii) the measurement chamber, and (ii) the pseudo-RE chamber. The sensor chamber includes four thin-film gold electrodes, three of which are in a two electrode configuration (1, 2 and 3) and a 4 bar sensor functioning as a conductimetric (4). The rear microfluidic connections are numbered from 1 to 6, with arrows indicating the direction of the flow (inlet or outlet). (B) Main steps of the silicon chip fabrication and image of the ME-LOC before the dicing step: (1) silicon wafer (grey) protected with a silicon oxide layer (green) on the top side and with silicon oxide (green), aluminium (light blue) and nitride oxide (dark blue) layers on the rear side; (2) silicon wafer after partial opening of the fluidic connections by DRIE and RIE; (3) electrical pads, connections and gold electrodes (yellow) defined by the standard lift-off process. Light green represents the passivation oxide layer protecting the electrical components and enabling the next bonding step. The fluidic connections are totally defined by the RIE process. (4) Glass-cover (light blue structure on the top of the image) and silicon wafer bonding. (C) Wirebonding and electrical connections between the ME-LOC and the PCB. (D) Image of the final ME-LOC. | ||
The ME-LoC contains three independent, but fluidically connected, micro-chambers: (i) a 15 μL cell culture chamber for cell seeding and proliferation; (ii) a 15 μL measurement chamber integrating four electrode systems for amperometric analysis of cells; and (iii) a 6 μL pseudo-reference chamber containing the reference electrode for amperometric measurements. A network of fluidic micro-channels is designed with multiple inlets and outlets for precise microfluidic control while minimizing dead volumes. With this network, it is possible to seed cells without affecting amperometric measurements, to reactivate/recalibrate the electrodes in situ without altering cell proliferation and to set the reference electrode conditions without influencing the measurement or cells.
Five thin-film gold electrode systems are defined in the silicon chip: one in the pseudo-reference chamber and four in the measurement chamber. The pseudo-reference chamber contains a circular gold electrode with a diameter of 3.1 mm2 to operate as a pseudo-reference electrode in electrochemical and ORP measurements (after electrodeposition of the Ag/AgCl layers). In the measurement chamber, systems (1)–(3) in Fig. 1A consist of two circular gold electrodes with diameters of 0.6 mm2 and 1.2 mm2 to be used as working and counter electrodes, respectively, in amperometric measurements. The electrochemical system (4) contains 4 bar gold electrodes for conductivity measurements.
Both the silicon chip and glass cover were fabricated using standard silicon technology, as summarized in Fig. 1B. First (step 1), the silicon wafer was etched in the regions of the pads, electrodes and connections and subsequently passivated by deposition of a thermal silicon oxide layer for protection. Additional silicon nitride and aluminium layers were deposited on the rear side of the wafer as passivation for the subsequent etching steps. Next (step 2), the rear fluidic connections were developed in a multi-step process involving two photolithographic and three etching steps (two deep reactive ion etching, or DRIE, steps and one RIE step). Before completing the opening of the rear fluidic connection (step 3), the gold electrodes, electric connection and pads were defined through a lift-off process.24 The rear fluidic connections were finally defined by RIE. The final step (step 4) consisted of anodic bonding between the silicon chip and the glass cover, which was previously patterned by etching. The anodic bonding was performed at 1000 V and 400 °C for 30 min at wafer level with the SB6 Gen2 SUSS MicroTec bonder (SUSS MicroTec Group, Garching, Germany) (Fig. S1A, in the ESI†). The thickness of the glass cover (about 180 μm) was chosen to enable high-resolution imaging.
Afterwards, the glass-silicon wafer was diced into individual chips (Fig. S1B in the ESI†) and fluidic tubes were inserted into the rear fluidic connections of the chip with epoxy resist (Fig. S2A in the ESI†). Then, the chips were bonded to a printed circuit board (PCB) electrically connected by wire bonding (Fig. 1C) and protected with a thin isolating silicone layer to preserve the electrical connections (Fig. S2C and D in the ESI†). The rear fluidic connections enabled fluid management and electrochemical measurements without interfering with the imaging. The final assembled ME-LoC system is illustrated in Fig. 1D.
Reagents
All reagents were of analytical grade or equivalent and were purchased from Sigma Aldrich unless otherwise stated. All solutions were prepared using de-ionized water.Equipment
For the electrodeposition of the polypyrrole (PPy) layers used to entrap the enzyme and mediators, an Autolab electrochemical workstation (PGSTAT-100 potentiostat–galvanostat, Ecochemie, Uthecht, The Netherlands) controlled with NOVA software was used. The electrodeposition required an external reference (Orion 92-02-00, Thermo Fisher Scientific Inc., Berverly USA) and counter electrode (platinum layer, Radiometer, Lyon, France). To ensure electrical contact between the on-chip and off-chip electrodes, all the electrodes were immersed in the same 0.1 M KCl solution during electro-synthesis.Multiplexed electrochemical analysis on-chip was performed with a portable homemade multi-parametric system fabricated at the IMB-CNM.25 Briefly, the system generated and received signals from four micro-sensors: (i) the resistance temperature detector (RTD) Pt-100 (Pico Technology, St Neots, UK); (ii) the four-wire conductivity sensor, where an alternating current at 5 kHz is applied to the two external electrodes and the current is recorded between the two internal electrodes; (iii) the ORP sensor, which is directly connected to the acquisition module and provides potential values by comparison with the reference electrode; and (iv) the amperometric sensor, a three-electrode microsensor system where the electrical voltage is fixed between the reference and the working electrodes while recording the current flowing between the working and the counter electrodes. The device incorporated two amperometric terminals that could be used simultaneously for glucose and hydrogen peroxide determination. The digital interface was permitted to establish the communication between the user and the analogue electronic part. The visualization of the results and the configuration of the measurement parameters were carried out employing a virtual instrument programmed with LabView 2013 (National Instruments, Austin, USA).
Fluorescence microscopy images were acquired with a Nikon Eclipse Ti-E inverted microscope (Plan Apo 60× Oil DIC objective; Nikon Instruments, Tokyo, Japan) using the same imaging settings in either non-treated or H2O2-treated conditions. Dihydroethidium (DHE) fluorescence was detected with a 604/40 nm band-pass filter. For quantification, the integrated optical density was calculated in each cell (79 ± 16 cells per experimental condition) using NIS Elements AR 4.13 software (Nikon Instruments, Tokyo, Japan).
Ag/AgCl pseudo-reference electrode
The electro-generation of the Ag/AgCl pseudo-reference electrode is based on the protocol described in ref. 26. First, the thickness of the Ag and AgCl layers was estimated by the combined Faraday law:| th = (I × t × Mw)/(F × ρ × A) |
Based on theoretical data, a Ag layer was galvanostatically electrodeposited on the working electrode. For a 4 μm layer, Ag was electrodeposited for 3760 s at −31 μA in a 10 mM HNO3 solution containing 15 mM tartaric acid and 0.1 M silver nitrate (AgNO3). The Ag layer was then partially chlorinated to obtain a 1.3 μm-thick layer in a 0.1 M HCl solution containing 0.1 M potassium chloride (KCl). The chlorination conditions were +12 μA for 1250 s. The thickness of the layers was confirmed by profilometry (Fig. S3, in the ESI†).
Electrochemical measurements
Four parameters were independently determined through electrochemical transduction, namely hydrogen peroxide, glucose, conductivity and ORP. Each (bio)sensor was characterized first individually and later in combination with the others. Sensor characterization and optimization was conducted on-chip using PB (pH 5.5) as the optimal conditions for glucose oxidase activity. Cell culture analysis was performed in culture medium (pH 7). Before analysing the cell proliferation, the response of the glucose biosensor in the culture medium was determined to evaluate the matrix effects and the influence of the pH on the enzymatic activity. In the characterization, all samples were supplemented with suitable concentrations of the analyte. The sensitivity, limit of detection and linear range were determined. Each parameter was measured in triplicate.Glucose biosensor
The glucose enzymatic biosensor was produced in the electrode system (2) (Fig. 1A) by selective electrodeposition of PPy27,28 layers on the working electrode surface. Two PPy layers were consecutively electrodeposited: the first layer contained the mediator (ferricyanide) and the second layer was the enzyme (glucose oxidase, GOX; GOX activity is described in Fig. S4A in the ESI†), which was deposited on the previous layer. This configuration improved the electron transfer between the transducer and mediator (Fig. S4B in the ESI†). The enzymatically mediated production of ferrocyanide was proportional to the glucose concentration and was determined amperometrically at +0.15 V (vs. Ag/AgCl).The electrodeposition solution contained 0.4 M pyrrole (monomer), 0.1 M KCl (counter-ion) in phosphate buffer (PB = 0.05 M KH2PO4, pH 7) and either 100 mM of potassium ferricyanide (mediator) for the mediator layer or 5 mg mL−1 GOX for the enzymatic layer. The PPy layers were electrodeposited at +0.7 V (vs. Ag/AgCl pseudo-reference) until an accumulation charge of 250 mC cm−2 was reached (estimated time of 25 s; see Fig. S5A in the ESI†) for the mediator or 500 mC cm−2 (approx. 50 s, Fig. S5B in the ESI†) for the enzymatic layer. Electrodeposition was performed using the electrodes integrated on the chip. The glucose biosensor was finally rinsed with buffer and stored in PB solution at 4 °C until use.
Linear sweep potentiometry from 0 to 0.8 V (vs. Ag/AgCl pseudo-reference) at a scan rate of 25 mV s−1 was used to determine the glucose concentration through oxidation of the enzymatically reduced ferrocyanide. Glucose concentrations between 0 and 50 mM in PB were analysed. Matrix effects were determined by comparison of previous PB records with those obtained from culture medium samples spiked with glucose (between 0 and 1 mM glucose).
Cell culture
Human fibroblast cell line MRC-5 from the ATCC Line Bank (Virginia, USA) was used to validate the ME-LoC. MRC-5 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 25 mM glucose concentration and supplemented with 10% foetal bovine serum and 1% antibiotic solution (10 kU mL−1 penicillin and 10 mg mL−1 streptomycin) in a humidified incubator with 5% CO2 at 37 °C. When confluent, the MRC-5 cells were passaged with 0.25% trypsin-EDTA (Gibco, Fisher Scientific, Spain), and an aliquot was stained with trypan blue for cell counting. Then, the cells were centrifuged and re-suspended in complete DMEM at a concentration of 106 cell per mL and seeded either in the ME-LoC or in conventional plastic plates for comparison. Proliferation and oxidative stress induction experiments were conducted with DMEM supplemented with 2 mM glucose. The cells used in the experiments were between the 6th and 8th passages and under 50% to 60% confluence.Oxidative stress induction and detection
Oxidative stress in MRC-5 cultured cells was induced by addition of H2O2 according to previous protocols.30,31 Cells proliferating in conventional 12 mm glass culture coverslips were exposed to H2O2 for 30 min in a humidified cell incubator (H2O2 concentrations: 10, 15, 25 and 50 μM). Intracellular reactive oxygen species (ROS) levels, including superoxide anion, were determined using the oxidation-sensitive fluorescent probe dye DHE (Ex/Em = 535/610 nm). The detection protocol was as follows. After oxidative stress induction, cells were washed with phosphate buffered saline (PBS) and incubated in 2 μM DHE at 37 °C for 30 min.31,32Oxidative stress induction data represent the means of at least two independent experiments (means ± SEM). The data were analysed using GraphPad Prism 5 software (San Diego, CA, USA). Student's t-test or one-way analysis of variance with post hoc analysis using Bonferroni's multiple comparison test was used for parametric data. P < 0.05 was considered to indicate a statistically significant difference.
Results and discussion
Characterization of electrochemical (bio)sensors, culturing and imaging in the LoC
The position of the pseudo-reference electrode in an independent chamber and at a distance from the working and counter electrodes may negatively influence amperometric measurements. To evaluate this, the response of the integrated electrodes (working, counter and reference electrodes in the ME-LoC) to ferricyanide was compared with an external double-junction reference electrode (working and counter electrodes of the ME-LoC). The study was complemented with a third experiment with external electrodes operating as working, counter and reference electrodes. Although there was a small shift in the potential peak, the three situations provided comparable results (Fig. S7 in ESI†) with similar current values. No potential drop associated with hysteresis was observed in the case of the integrated electrodes, probably due to the good electrical conductivity through the fluidic micro-channels.
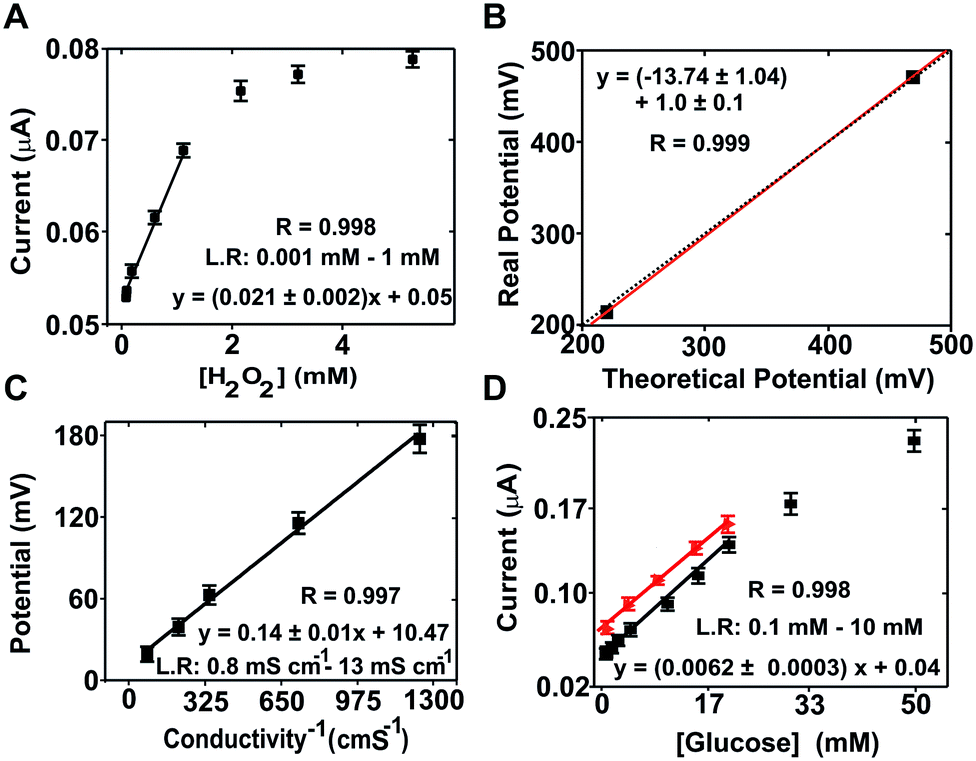 | ||
| Fig. 2 Calibration of the electrochemical (bio)sensors. (A) Calibration curve for the hydrogen peroxide sensor using the mean current value of the last 30 s of the recorded signal for concentrations of hydrogen peroxide of 0, 0.001, 0.01, 0.05, 0.1, 0.5, 1, 2, 3 and 5 mM; (B) least squares analysis of ORP for the standard solutions of 220 mV and 468 mV. The dotted line corresponds to the ideal correlation between the real (measured) and theoretical values; (C) calibration plot for the 4 bar sensor using conductivity PB solutions of 0.80, 1.52, 2.82, 6.87 and 13.07 mS cm−1; (D) calibration curve of the PPY/ferricyanide/GOX glucose biosensor in PB solution (black line) and in the cell culture medium (red line). Each point represents the current value recorded from the sweep linear potentials obtained from glucose concentrations of 0.1, 0.3, 0.6, 1, 2, 3 and 5 mM. Error bars represent the corresponding standard deviation from two chips measured in triplicate (n = 3). | ||
Hydrogen peroxide concentration (Fig. 2A) was determined by chronoamperometry at +0.7 V (vs. the Ag/AgCl pseudo-reference). The average current density increased with the hydrogen peroxide concentration in the range from 0.001 to 1 mM with good sensitivity (slope = 1.8 ± 0.2 μA mM−1) and linearity (r = 0.998). The calculated limit of detection (LOD) was 5 × 10−4 mM according to the 3σ IUPAC criterion.31
Calibration of the ORP sensor was performed by measuring two calibration solutions with known potentials, i.e. 220 and 468 mV according to the supplier. Following conventional calibration protocols for ORP, the theoretical values were compared to the values obtained experimentally (214.3 ± 0.5 mV and 470.4 ± 1.0), which presented a variation coefficient below 3% in each case. A paired representation of the theoretical versus the experimental data (Fig. 2B) resulted in a straight line with a slope of 1 (1.0 ± 0.1), confirming the good performance of the ORP sensor.
The conductivity sensor was characterized using KCl solutions as calibration solutions. The sensor response was compared with the results from a commercial conductimeter. The ME-LoC presented good sensitivity (0.14 ± 0.01 mV Ω−1 cm−1) and linearity (r2 = 0.997) in the range between 0.80 and 13.07 mS cm−1 (Fig. 2C), providing results comparable to those of a commercial conductimeter.
For the glucose biosensor, the current magnitude at +0.15 V (vs. Ag/AgCl pseudo-reference) corresponding to oxidation of the enzymatically produced ferrocyanide was used as an analytical signal. The linear range of the biosensor comprised three orders of magnitude, from 0.1 to 25 mM, with good sensitivity (0.0060 ± 0.0003 μA mM−1) and linearity (r = 0.998) (Fig. 2D). Matrix effects were investigated with the glucose biosensor because it is one of the most sensitive to biofouling and pH. The results are presented in Fig. 2D(red dots/line). As shown, similar responses were obtained in both buffer and culture medium solutions, providing comparable sensitivities (0.052 ± 0.003 and 0.041 ± 0.001 μA mM−1 for PB solutions and cell culture medium, respectively) and linear ranges. However, a small displacement of the calibration curve to higher current magnitudes was observed with the culture medium due to matrix effects. These effects were considered in subsequent measurements.
It is worth mentioning that the linear range of the sensor was in agreement with the expected magnitudes of each parameter: between 0 to 2 mM for glucose (considering that the culture medium was supplemented with 2 mM glucose and that this parameter should decrease during the experiment due to cell metabolism); from 100 to 250 mV for ORP considering the composition of the culture medium; around 25 mS cm−1 for conductivity, which may slightly increase or decrease during the experiment; and from 1 to 50 mM in the case of hydrogen peroxide, which is the range conventionally used to induce oxidative stress in mammalian cells.
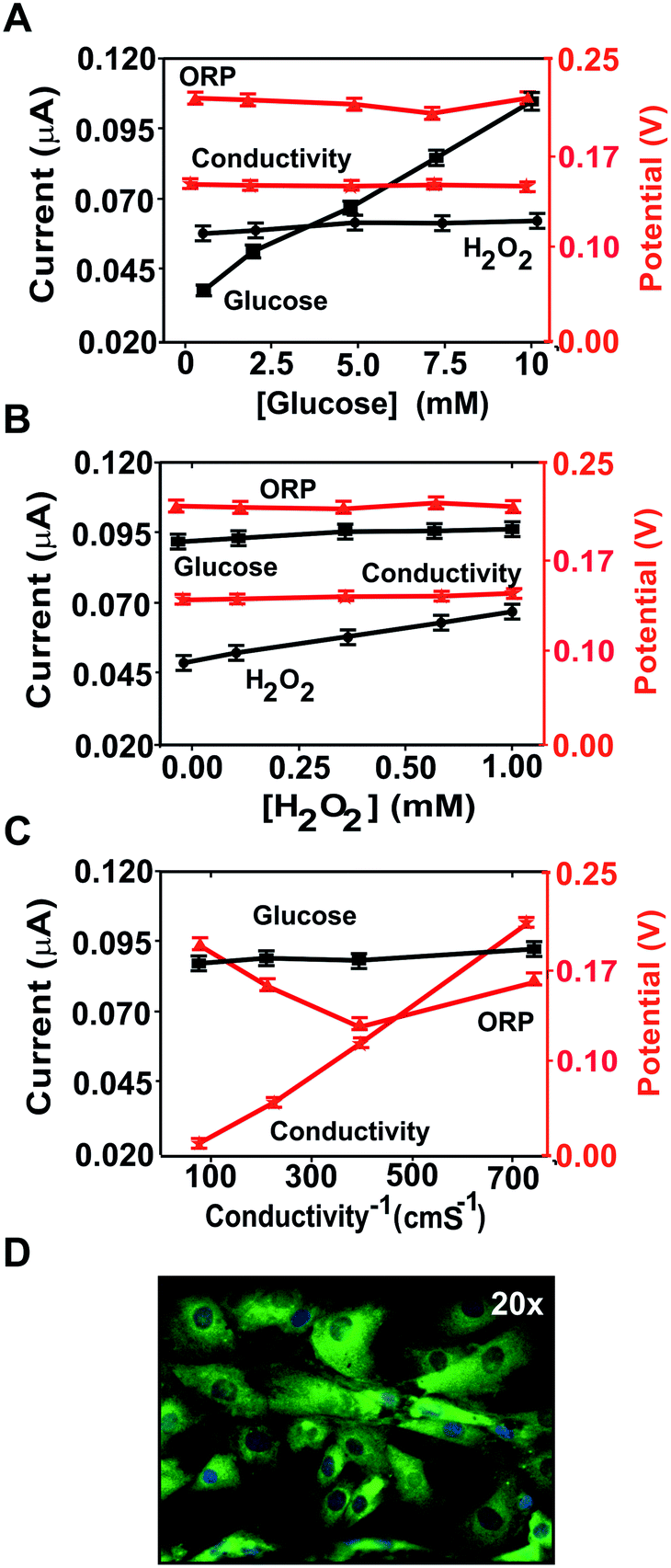 | ||
| Fig. 3 Multiplexed electrochemical analysis and cell culture imaging. Cross-talk results changing the glucose (A) in a concentration range from 0 to 2 mM, the hydrogen peroxidase (B) concentration in a range from 0 to 1 mM and (C) the conductivity in a range from 0.8 to 13.07 mS cm−1. (D) Cell culture microscopy image. Error bars represent the corresponding standard deviation from two chips measured in triplicate (n = 3). | ||
The glucose (Fig. 3A) and hydrogen peroxide (Fig. 3B) concentrations were selectively recorded by the corresponding (bio)sensors and did not interfere with the other measurements. Thus, there was no cross-talk in the determination of these two parameters.
Although changes in medium conductivity have been reported to influence ORP,35 only a minor variation (around 30 mV) was observed in this case (Fig. 3C), as expected from the small influence of conductivity on the Nernst equation. Thus, the conductivity and glucose and hydrogen peroxide concentrations were not expected to interfere with the determination of any of the other analytes of interest in these assays. Additionally, any influence of conductivity was minimized by using culture media with high and constant conductivity.
 | ||
| Fig. 4 Fluidic control of the ME-LoC. (1) Filling of the pseudo-reference electrode with a blue solution; (2) filling of the cell culture chamber with a red solution; (3) initial calibration of the electrochemical (bio)sensors using a purple solution; (4) medium culture measurement by the electrochemical (bio)sensors in the measurement chamber; (5) final calibration of the electrochemical (bio)sensors. | ||
To demonstrate (i), the pseudo-reference micro-chamber was filled with a blue colorant using fluidic connection number 1 as the inlet and number 2 as the outlet (Fig. 4.1). As shown in the magnification of the figure, the blue colorant was confined in the micro-chamber with minimal diffusion to other chambers due to the small diameter of the connecting micro-channels. Based on that, the pseudo-reference electrode micro-chamber could be filled with a 0.1 M KCl solution to ensure the stability of the reference electrode potential without compromising electrochemical measurements or cell viability. In a second step (Fig. 4.2), the culture micro-chamber was filled with a red colorant to demonstrate (ii). Selective filling was achieved by using connection number 4 as the inlet and number 5 as the outlet. The minimal diffusion of the red colorant (see the magnification of the image) guaranteed the confinement of cells in the cell culture micro-chamber, minimizing their flow to other regions of the chip. This minimizes the biofouling risk during cell culture. The demonstration of (iii) required selective filling of the measurement chamber. As shown in Fig. 4.3, the measurement chamber was filled with a purple solution. Fluidic connection number 3 was used as the inlet and number 6 as the outlet. As shown, there was minimal diffusion to the other chambers (see image magnification), which demonstrates that it is possible to reactivate and recalibrate the electrodes before measurement without compromising cell proliferation. Finally, to prove (iv), the culture medium was selectively transferred from the culture micro-chamber to the measurement micro-chamber for measurement (Fig. 4.4). The red culture medium was thus transported from one chamber to the other using fluidic connection number 5 as the inlet and number 6 as the fluidic outlet. As an additional step, the measurement micro-chamber was again filled with the purple solution for reactivation and recalibration of the electrodes before a new measurement was performed (Fig. 4.5). In this case, fluidic connections number 3 and number 6 were used again as the inlet and outlet, respectively. A video recording all these steps in real time is included in the ESI† of this article.
These results demonstrated the compartmentalization, minimal diffusion and versatility of the intricate network of micro-channels designed in the chip, proving high control of cell culture for the integration of multiple micro-electrode systems in the chip and their self-calibration capacity.
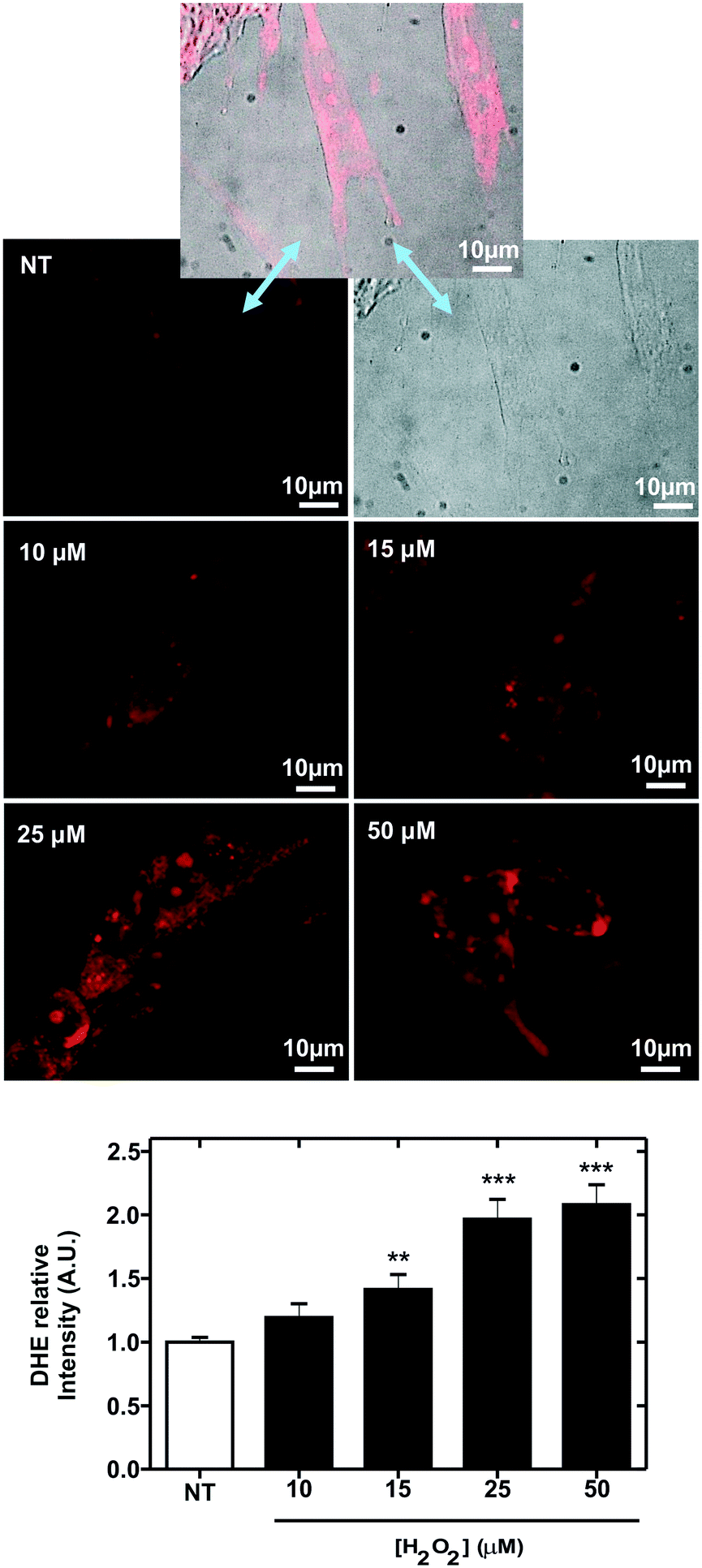 | ||
| Fig. 5 Induction of reactive oxygen species (ROS) in MRC-5 cells. The top images correspond to optical (right), fluorescence (left) and mergence (middle) images of MRC-5 cells (non-treated cells, NT) after staining with the membrane-permeable ROS-sensitive dye dihydroethidium (DHE). Below, fluorescence images of MRC-5 cells stained with DHE after 30 minutes of incubation with the hydrogen peroxide (H2O2) indicated in the image. The plot in the bottom represents the variation of the relative intensity of DHE-dye, from image quantification, with the H2O2 concentration. **P < 0.01; ***P < 0.001 compared to NT cells. Error bars represent the corresponding standard deviation. Three technical (n = 3) and biological (N = 3) replicates were conducted. | ||
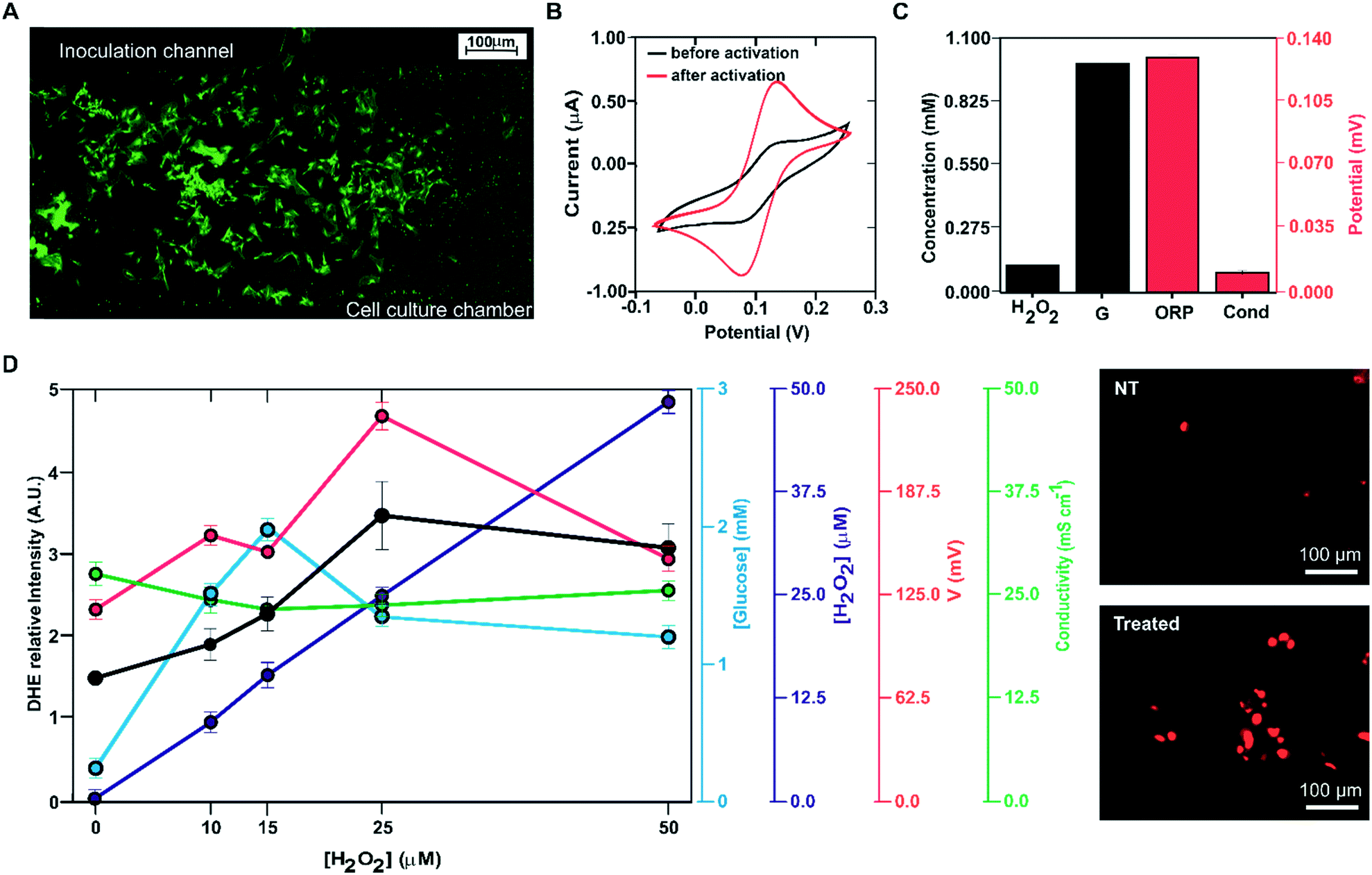 | ||
| Fig. 6 Multiplexed analysis of oxidative stress induction in MRC-5 cell cultures on-chip. (A) Image of MRC-5 cells selectively inoculated in the cell culture chamber; (B) cyclic voltammograms recorded in 0.1 M KCl 1 mM potassium ferricyanide before (black) and after (red) re-activation of the gold microelectrodes; (C) microelectrode calibration using calibration solutions with 0.1 mM H2O2, 1 mM glucose (G), 220 mV of ORP and 2.82 mS cm−1 (Cond); (D) electrochemical signal recorded during the oxidation stress assays on-chip for glucose (blue), hydrogen peroxide (purple), ORP (red) and conductivity (green). The variation of the relative intensity of the DHE-dye with the hydrogen peroxide concentration is represented in black. The images on the right correspond to fluorescence images of MRC-5 cells after staining with the membrane-permeable ROS-sensitive dye dihydroethidium (DHE) when the cells were not treated (NT, top image) and treated with 25 μM of hydrogen peroxide (treated, bottom image). Error bars represent the corresponding standard deviation. Three technical (n = 3) replicates were conducted from five cultures (N = 5). | ||
Oxidative stress was induced by incubation of the cells with hydrogen peroxide. After 30 min of incubation with the reagent, the culture medium was driven to the measurement micro-chamber and analysed using the four electrode systems implemented in the ME-LoC. Glucose, hydrogen peroxide, conductivity and ORP were thus determined.
The study was complemented with confocal microscopy imaging and analysis of oxidative stress induction using the fluorescent indicator DHE. To this end, the DHE solution was inoculated in the cell culturing micro-chamber, incubated for 30 min and imaged by confocal microscopy.
After analysis at one hydrogen peroxide concentration, the ME-LoC was regenerated for future experiments as follows. Cells were detached with 0.025% trypsin (5 min). The micro-chamber was then washed with 70% ethanol (3 times) and PBS (5 min, 3 times), removing any remaining cells present in the chip and leaving it ready for the next assay. The performance of the electrodes was also evaluated at this point; the glucose biosensor was regenerated by electrodeposition with PPy, as described in the Experimental section.
The oxidative stress induction procedure was repeated with the following concentrations of hydrogen peroxide: 10, 15, 25 and 50 μM. Electrochemical analysis of the cell culture provided the following results, illustrated in Fig. 6D. The hydrogen peroxide sensor reported an increase in the response (blue line, blank circles) with increasing concentration of this molecule in the culture medium. Considering calibration, the reported hydrogen peroxide concentration was very close to the inoculated concentration. The glucose biosensor signal (blue line, filled blue circles) increased with the hydrogen peroxide concentration up to 25 μM H2O2, where it stabilized. This increase in glucose concentration in the medium can be attributed to decreased glucose consumption due to oxidative stress and cell death. At 15 μM, the magnitude of the recorded current corresponded to a glucose concentration of 2 mM; this suggests that at this hydrogen peroxide concentration, oxidative stress induction killed almost all the cells in the ME-LoC. The signal corresponding to the conductivity sensor (red line, blank circles) was almost constant over time, providing a constant potential of 24 ± 2 mS cm−1. This magnitude coincided with the medium conductivity; thus, conductivity changes associated with breaking of the cell membrane and the massive release of ions could not be recorded with the current configuration of the ME-LoC. On the other hand, the ORP signal (red line, filled red circles) increased with increasing hydrogen peroxide concentration up to 25 mM. This increase in the ORP magnitude may be due to decreased metabolite production, as suggested in the literature,39 or increased hydrogen peroxide concentration.
The electrochemical results were complemented with high-resolution confocal imaging of the oxidative stress indicator DHE (black line, filled black circles; image of stained cells in Fig. 6E). An increase of oxidative stress was recorded up to 25 μM hydrogen peroxide, coinciding with the electrochemical data. This result confirmed the good performance of the ME-LoC in the analysis of oxidative stress induced by hydrogen peroxide as an example of a process with high impact in pathology development.
Conclusions
The results presented in this article demonstrate that precise control of cell culture is possible with a self-calibrating, reusable and multiplexed LoC. The ME-LoC presents a complex network of micro-channels and micro-chambers that allow compartmentalization of the reference electrode; cell seeding and proliferation without biofouling; electrode reactivation and recalibration; and multiple analyte detection, namely glucose and hydrogen peroxide concentrations, conductivity and ORP, as a way to monitor cell metabolism. Electrochemical analysis is completed with high-resolution imaging by confocal microscopy after labelling with fluorescent dyes. The evaluation of the device with MRC-5 cells by induction of oxidative stress with hydrogen peroxide demonstrates a dose-dependent reduction of glucose consumption and a proportional increase of DHE emission. ORP is also affected by the decrease in cell metabolism; however, changes in conductivity due to cell lysis are not observed. These results confirmed the good performance of the system in the determination of oxidative stress, a process observed in the initial stages of pathologies such as cancer or cardiovascular diseases. For simplicity, integration, automation, compartmentalisation and micro-fluidic control, we envisage the current technology as a promising alternative for in vitro testing and organ-on-a-chip development in the near future.Authors contribution
P. G.-G. and J. M. R. carried out the characterization and optimization of the integrated biosensors as well as the final measurements with cell cultures proliferating in the ME-LoC device. E. G., J. A. P. and C. J.-J. designed and manufactured the device under cleanroom conditions. M. G.-C. contributed to optimizing the reference electrode. R. R.-R. and M. P.-M. conducted the assays with cells and oxidative stress induction. P. G.-G. R. R.-R. and X. M.-B. were responsible for managing and coordinating the execution of the research activity. P. G.-G., C. J.-J., R. R.-R. and X. M-B. were involved in the preparation of the draft and revision and edition of the final manuscript, in collaboration with the other authors. C. J.-J. and X. M.-B. obtained financial support to carry out the project associated with this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish R & D National Program (MEC Project TEC2014-54449-C3-1-R and RTI2018-100773-B-C31).Notes and references
- L. J. Bishop and A. L. Nolen, Kennedy Inst. Ethics J., 2001, 11, 91–112 CrossRef CAS PubMed.
- R. M. Pearson, Hum. Reprod., 1986, 1, 559–560 CrossRef CAS PubMed.
- D. Sun, L. X. Yu, M. A. Hussain, D. A. Wall, R. L. Smith and G. L. Amidon, Curr. Opin. Drug Discovery Dev., 2004, 7, 75–85 CAS.
- P. S. Dittrich and A. Manz, Nat. Rev. Drug Discovery, 2006, 5, 210–218 CrossRef CAS PubMed.
- L. Kang, B. G. Chung, R. Langer and A. Khademhosseini, Drug Discovery Today, 2008, 13, 1–13 CrossRef CAS PubMed.
- B. H. Weigl, R. L. Bardell and C. R. Cabrera, Adv. Drug Delivery Rev., 2003, 55, 349–377 CrossRef CAS PubMed.
- X. Muñoz-Berbel, R. Rodríguez-Rodríguez, N. Vigués, S. Demming, J. Mas, S. Büttgenbach, E. Verpoorte, P. Ortiz and A. Llobera, Lab Chip, 2013, 13, 4239 RSC.
- M.-H. Wu, S.-B. Huang and G.-B. Lee, Lab Chip, 2010, 10, 939 RSC.
- N. Khalid, I. Kobayashi and M. Nakajima, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2017, 9, e1381 Search PubMed.
- H. J. Kim, D. Huh, G. Hamilton and D. E. Ingber, Lab Chip, 2012, 12, 2165 RSC.
- D. Huh, B. D. Matthews, A. Mammoto, M. Montoya-Zavala, H. Y. Hsin and D. E. Ingber, Science, 2010, 328, 1662–1668 CrossRef CAS PubMed.
- R. Rodriguez-Rodriguez, X. Muñoz-Berbel, S. Demming, S. Büttgenbach, M. D. Herrera and A. Llobera, Biomed. Microdevices, 2012, 14, 1129–1140 CrossRef CAS PubMed.
- P. Neužil, S. Giselbrecht, K. Länge, T. J. Huang and A. Manz, Nat. Rev. Drug Discovery, 2012, 11, 620–632 CrossRef PubMed.
- J. Karasinski, L. White, Y. Zhang, E. Wang, S. Andreescu, O. A. Sadik, B. K. Lavine and M. Vora, Biosens. Bioelectron., 2007, 22, 2643–2649 CrossRef CAS PubMed.
- Bionas-discovery, http://www.bionas-discovery.com/prodservices/instruments/system2500/index.html, (accessed 7 June 2018).
- https://www.aceabio.com/, Application Protocol: Using the xCELLigence RTCA DP Instrument to Perform Cell Invasion and Migration Assays The xCELLigence ® RTCA DP Instrument Application Protocol: Using the xCELLigence RTCA DP Instrument to Perform Cell Invasion and Migration Assays, https://www.aceabio.com/wp-content/uploads/CIM-Protocol.pdf, (accessed 7 June 2018).
- P.-A. Auroux, D. Iossifidis, D. R. Reyes and A. Manz, Anal. Chem., 2002, 74, 2637–2652 CrossRef CAS PubMed.
- S. P. Forry and L. E. Locascio, Lab Chip, 2011, 11, 4041 RSC.
- N. Pereira Rodrigues, Y. Sakai and T. Fujii, Sens. Actuators, B, 2008, 132, 608–613 CrossRef CAS.
- E. Berthier, E. W. K. Young and D. Beebe, Lab Chip, 2012, 12, 1224 RSC.
- K. J. Regehr, M. Domenech, J. T. Koepsel, K. C. Carver, S. J. Ellison-Zelski, W. L. Murphy, L. A. Schuler, E. T. Alarid and D. J. Beebe, Lab Chip, 2009, 9, 2132 RSC.
- M. Brischwein, E. R. Motrescu, E. Cabala, A. M. Otto, H. Grothe and B. Wolf, Lab Chip, 2003, 3, 234 RSC.
- A. Weltin, K. Slotwinski, J. Kieninger, I. Moser, G. Jobst, M. Wego, R. Ehret and G. A. Urban, Lab Chip, 2014, 14, 138–146 RSC.
- J. Orozco, G. Suárez, C. Fernández-Sánchez, C. McNeil and C. Jiménez-Jorquera, Electrochim. Acta, 2007, 53, 729–736 CrossRef CAS.
- P. Gimenez-Gomez, R. Escude-Pujol, C. Jimenez-Jorquera and M. Gutierrez-Capitan, IEEE Sens. J., 2015, 15, 6517–6523 CAS.
- Water Network Sensors for Widespread Use|Projects| FP7-SME | CORDIS | European Commission, https://cordis.europa.eu/project/rcn/109491_en.html, (accessed 7 June 2018).
- P. Giménez-Gómez, M. Gutiérrez-Capitán, F. Capdevila, A. Puig-Pujol, C. Fernández-Sánchez and C. Jiménez-Jorquera, Anal. Chim. Acta, 2016, 905, 126–133 CrossRef PubMed.
- P. Giménez-Gómez, M. Gutiérrez-Capitán, F. Capdevila, A. Puig-Pujol, C. Fernández-Sánchez and C. Jiménez-Jorquera, Anal. Chim. Acta, 2017, 954, 105–113 CrossRef PubMed.
- L. Kjaergaard, in Advances in Biochemical Engineering, Springer-Verlag, Berlin/Heidelberg, 1977, vol. 7, pp. 131–150 Search PubMed.
- M. Farnoodian, J. B. Kinter, S. Yadranji Aghdam, I. Zaitoun, C. M. Sorenson and N. Sheibani, Physiol. Rep., 2015, 3, e12266 CrossRef PubMed.
- A. Wojtala, M. Bonora, D. Malinska, P. Pinton, J. Duszynski and M. R. Wieckowski, Methods Enzymol., 2014, 542, 243–262 CAS.
- H. Cai, S. Dikalov, K. K. Griendling and D. G. Harrison, Methods Mol. Med., 2007, 139, 293–311 CAS.
- J. Zhou, K. Ren, Y. Zheng, J. Su, Y. Zhao, D. Ryan and H. Wu, Electrophoresis, 2010, 31, 3083–3089 CrossRef CAS PubMed.
- J. Wang, in Analytical Electrochemistry, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2006, pp. 29–66 Search PubMed.
- J. M. Steininger and C. Pareja, NSPI Symposium Series, 1996, vol. I Search PubMed.
- A. G. Georgakilas, Cancer Lett., 2012, 327, 3–4 CrossRef CAS PubMed.
- D. A. Butterfield, J. Drake, C. Pocernich and A. Castegna, Trends Mol. Med., 2001, 7, 548–554 CrossRef CAS PubMed.
- I. M. Fearon and S. P. Faux, J. Mol. Cell. Cardiol., 2009, 47, 372–381 CrossRef CAS PubMed.
- C. Hwang and A. J. Sinskey, in Production of Biologicals from Animal Cells in Culture, Elsevier, 1991, pp. 548–568 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc01051c |
| This journal is © The Royal Society of Chemistry 2020 |