
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An adaptable soft-mold embossing process for fabricating optically-accessible, microfeature-based culture systems and application toward liver stage antimalarial compound testing†
Steven P.
Maher
 *ab,
Amy J.
Conway
*a,
Alison
Roth
a,
Swamy R.
Adapa
a,
Phillip
Cualing
a,
Chiara
Andolina
c,
James
Hsiao
d,
Jessica
Turgeon
a,
Victor
Chaumeau
c,
Myles
Johnson
a,
Chris
Palmiotti
d,
Naresh
Singh
a,
Samantha J.
Barnes
a,
Raahil
Patel
a,
Virginia
Van Grod
d,
Robert
Carter
a,
H.-C. Steve
Sun
d,
Jetsumon
Sattabongkot
e,
Brice
Campo
f,
François
Nosten
c,
Wajeeh M.
Saadi
d,
John H.
Adams
a,
Rays H. Y.
Jiang
a and
Dennis E.
Kyle
*ab
*ab,
Amy J.
Conway
*a,
Alison
Roth
a,
Swamy R.
Adapa
a,
Phillip
Cualing
a,
Chiara
Andolina
c,
James
Hsiao
d,
Jessica
Turgeon
a,
Victor
Chaumeau
c,
Myles
Johnson
a,
Chris
Palmiotti
d,
Naresh
Singh
a,
Samantha J.
Barnes
a,
Raahil
Patel
a,
Virginia
Van Grod
d,
Robert
Carter
a,
H.-C. Steve
Sun
d,
Jetsumon
Sattabongkot
e,
Brice
Campo
f,
François
Nosten
c,
Wajeeh M.
Saadi
d,
John H.
Adams
a,
Rays H. Y.
Jiang
a and
Dennis E.
Kyle
*ab
aCenter for Global Health and Infectious Diseases Research, Department of Global Health, College of Public Health, University of South Florida, Tampa, Florida, USA. E-mail: dennis.kyle@uga.edu
bCenter for Tropical and Emerging Global Diseases, University of Georgia, Athens, GA, USA
cShoklo Malaria Research Unit, Mahidol Oxford Research Unit, Faculty of Tropical Medicine, Mahidol University, Mae Sot, Thailand & Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, UK
dCharles Stark Draper Laboratory, Cambridge, MA, USA
eMahidol Vivax Research Unit, Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand
fMedicines for Malaria Venture, Geneva, Switzerland
First published on 14th February 2020
Abstract
Advanced cell culture methods for modeling organ-level structure have been demonstrated to replicate in vivo conditions more accurately than traditional in vitro cell culture. Given that the liver is particularly important to human health, several advanced culture methods have been developed to experiment with liver disease states, including infection with Plasmodium parasites, the causative agent of malaria. These models have demonstrated that intrahepatic parasites require functionally stable hepatocytes to thrive and robust characterization of the parasite populations' response to investigational therapies is dependent on high-content and high-resolution imaging (HC/RI). We previously reported abiotic confinement extends the functional longevity of primary hepatocytes in a microfluidic platform and set out to instill confinement in a microtiter plate platform while maintaining optical accessibility for HC/RI; with an end-goal of producing an improved P. vivax liver stage culture model. We developed a novel fabrication process in which a PDMS soft mold embosses hepatocyte-confining microfeatures into polystyrene, resulting in microfeature-based hepatocyte confinement (μHEP) slides and plates. Our process was optimized to form both microfeatures and culture wells in a single embossing step, resulting in a 100 μm-thick bottom ideal for HC/RI, and was found inexpensively amendable to microfeature design changes. Microfeatures improved intrahepatic parasite infection rates and μHEP systems were used to reconfirm the activity of reference antimalarials in phenotypic dose–response assays. RNAseq of hepatocytes in μHEP systems demonstrated microfeatures sustain hepatic differentiation and function, suggesting broader utility for preclinical hepatic assays; while our tailorable embossing process could be repurposed for developing additional organ models.
Introduction
In vitro organ models are gaining popularity as tools for drug discovery, due to their potential to serve as more predictive models of human diseases.1 However, medium- and high-throughput drug discovery approaches utilize standard robotic liquid handling and automated high-content and high-resolution imaging (HC/RI) equipment for screening of compound libraries, requiring biological constituents to conform to the basic shape and sizes of standard microtiter plates, the design specifications of which have been set by ANSI SLAS industry standards. Yet many in vitro organ models require microenvironments that are more complex than the simple, two dimensional-bottom architecture of microtiter plates. As such, efforts have focused on engineering platforms that conform to the standards of commercial microtiter plates while maintaining critical features necessary for sophisticated organ-mimetic in vitro models.2,3 Achieving broad applicability of such in vitro model platforms requires fabrication strategies that are compatible with a variety of designs, each having specific features relevant to the organ of interest, and some degree of scalability for use in multiple research settings.4The human liver is the largest internal organ, performs over 500 physiological functions, including metabolism of most xenobiotics, and is subject to a variety of disease states, including those caused by cytotoxic side-effects of xenobiotics and infectious diseases.5,6 Historically, due to rapid de-differentiation ex vivo, primary hepatocytes are used only in short-term experiments (2–3 days) to optimize metabolic liability and hepatic safety of developmental drugs.7 More robust, long-term liver models were not available until the introduction of advanced hepatocyte culture methods, including co-cultures, extracellular matrix overlays, and perfused microfluidic devices, resulting in many two-dimensional and three-dimensional tissue-engineered systems.8 Furthermore, co-cultures have been applied to microtiter plate formats, including 96- and 384-well, permitting an expanded set of preclinical hepatic tests including the effect of repeated drug dosing and culture of infectious diseases such as hepatitis and malaria.9–11 These and other long-term primary hepatocyte culture methods demonstrate the key to long-term culture is to maintain hepatocyte membrane polarization and cuboidal morphology characteristically found in vivo, thereby preventing dedifferentiation and stabilizing function in vitro.12–14 However, intrinsic to advanced culture methods are intricate protocol steps, such as incorporation of a second cell type, addition of a matrix overlay, or setup of media perfusion equipment, which can be difficult or expensive to multiplex.15–17 Conversely, a relatively simple and abiotic method to maintain primary hepatocyte morphology and function is to culture on a surface designed to mechanically confine the spread of the hepatocyte monolayer, including microchannels and microwells.18–20 While many microfeature-based systems have been described, few have been produced in a standard microtiter plate format to gain throughput and amenability to standard laboratory equipment.21 We hypothesized we could engineer microfeatures onto the culture area of a microtiter plate, maintain optical accessibility for HC/RI, and fabricate the plate in a one-step process, thereby making microfeature-based hepatocyte culture more congruent with existing laboratory equipment and in a throughput amendable to many applications, including liver stage antimalarial drug development.
We first performed proof-of-concept testing by fabricating a 4-well microfeature slide from polydimethylsiloxane (PDMS) and glass before transitioning to fabrication from polystyrene (PS), a standard cell culture material. During concept testing we determined the design, materials, and fabrication process were key considerations for meeting utility requirements as we transitioned to fabricating systems from PS. Previously described microfeature systems were designed to minimize hepatocyte attachment to produce a spheroid within microwells,22–24 or ‘float’ hepatocytes on air bubbles to further discourage attachment to a hard surface,21 or encourage hepatocytes to form a monolayer on a culture surface.21 For our specific application, we determined the hepatocytes should be cultured in an adhered monolayer format, producing a relatively two-dimensional cell monolayer in each microfeature, to avoid spheroid formation as multi-cellular structures can be difficult to image and characterize in a single focal plane during high-content imaging.25,26 Regarding material selection, previously described microwell systems have been fabricated from PEG hydrogel,27 heparin hydrogel,28 agarose,22 PDMS,29–31 or PS.21,32 To maintain optical accessibility and take advantage of the bulk-material properties ideal for small molecule testing,33 we fabricated systems from stock PS, and tested several surface treatments and coatings to encourage formation of a hepatic monolayer and promote stable, long-term culture. As we transitioned from conceptual PDMS systems to PS systems, we sought a dynamic fabrication process that could reliably produce an optically accessible (<100 μm) thin bottom; while simultaneously being amendable to arrayed fabrication of macrowells in a microtiter plate footprint. Furthermore, we sought to test the suitability of various microfeature designs for hepatocyte culture and pursued a fabrication method amendable to rapid and inexpensive redesign and prototyping. Injection molding was determined impractical as even commercial prototyping resources involve production of expensive molds, which would require costly tooling for each design iteration. The most recent frontiers of three-dimensional printing technology are capable of forming microfeatures with biocompatible materials,34 but are not yet capable of producing the optically-accessible, smooth, and thin surfaces needed for our HC/RI endpoint. Computer numerical control machining was also found not ideal, as it would likely be very difficult to achieve the surface finish required for high resolution imaging of the wells, was cost prohibitive, and was not easily scalable. Conventional hot embossing was found to not always allow for the molding of very small microfeatures and thin walls due to the differences in thermal expansion coefficients of the molds and plastic. Alternatively, a PDMS soft mold has been used to hot emboss micro-scale sized features into thermoplastics.35,36 As PDMS replica molding allows for nanometer scale resolution, is relatively inexpensive to use as a mold, and allows for ease of design changes with minimal turnaround times, we speculated hot embossing PS with a PDMS mold could be an ideal process to develop our microfeature culture system. Following development of our soft-mold embossing process we generated custom-designed microfeature culture systems (μHEP) in 48-well slide (an eighth of a 384-well plate per slide) and full 384-well microtiter plate formats. To demonstrate the utility of our process and culture model, PS-μHEP systems were used for long term culture of primary human hepatocytes (PHHs) and modeling of Plasmodium liver stage (LS) parasite infection. By exploiting the flexibility of our soft-mold process, our PS-μHEP system was optimized to sustain robust quantities of P. vivax LS parasites in long-term culture such that we could observe reactivation of dormant parasites (hypnozoites). To demonstrate optical accessibility and amenability to HC/RI equipment, μHEP systems were used to perform phenotypic assays for small molecule testing against LS parasites. RNAseq analysis of PHHs cultured in both μHEP and traditional microtiter plate formats suggests the global maintenance of differentiated primary hepatic functional pathways is responsible for PHH stability in μHEP system culture.
Materials and methods
Polydimethylsiloxane–glass μHEP slide fabrication
The PDMS-μHEP device consisted of three layers: a glass coverslip, a microfeatured PDMS layer and a macroscale PDMS layer forming the individual wells and holding culture media (Fig. 1A). To fabricate the microfeatured PDMS layer, a 4-inch Si wafer was patterned with SU-8 (MicroChem, SU-8 3050, Westborough, MA) using standard photolithographic techniques to create a mold with the dimensions shown in Fig. 1A. After patterning SU-8, tridecafluoro-1,1,2,2-tetrahydrooctyl trichlorosilane (Gelest Inc, Morrisville, PA) was vapor deposited on the patterned wafers for ease of PDMS removal. The PDMS (Sylgard 184, Dow Corning, Corning, NY) pre-polymer mixture was spun on top of the wafers at a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 base to curing agent weight ratio at 650 RPM for 1 minute and cured at 65 °C for 1 hour. The spun PDMS layer was then peeled off and bonded to a No.1 glass coverslip (Dow Corning) via oxygen plasma (PE-100, Plasma Etch, Carson City, NV) at 100 mTorr for 10 seconds at 100 Watts. The macroscale PDMS layer was fabricated by pouring PDMS (10:1 base to curing agent weight ratio) into a 150 mm Petri dish (Dow Corning) to a height of 5 mm and curing at 65 °C for 1 hour. The cured PDMS was peeled from the dish, 5 mm media reservoir holes were punched using a biopsy punch, the individual wells were aligned over the microfeatured PDMS layer, and the two PDMS layers were bonded together using oxygen plasma. Completed slides were sterilized using ethylene oxide (Anderson Products, Haw City, NY).
:1 base to curing agent weight ratio at 650 RPM for 1 minute and cured at 65 °C for 1 hour. The spun PDMS layer was then peeled off and bonded to a No.1 glass coverslip (Dow Corning) via oxygen plasma (PE-100, Plasma Etch, Carson City, NV) at 100 mTorr for 10 seconds at 100 Watts. The macroscale PDMS layer was fabricated by pouring PDMS (10:1 base to curing agent weight ratio) into a 150 mm Petri dish (Dow Corning) to a height of 5 mm and curing at 65 °C for 1 hour. The cured PDMS was peeled from the dish, 5 mm media reservoir holes were punched using a biopsy punch, the individual wells were aligned over the microfeatured PDMS layer, and the two PDMS layers were bonded together using oxygen plasma. Completed slides were sterilized using ethylene oxide (Anderson Products, Haw City, NY).
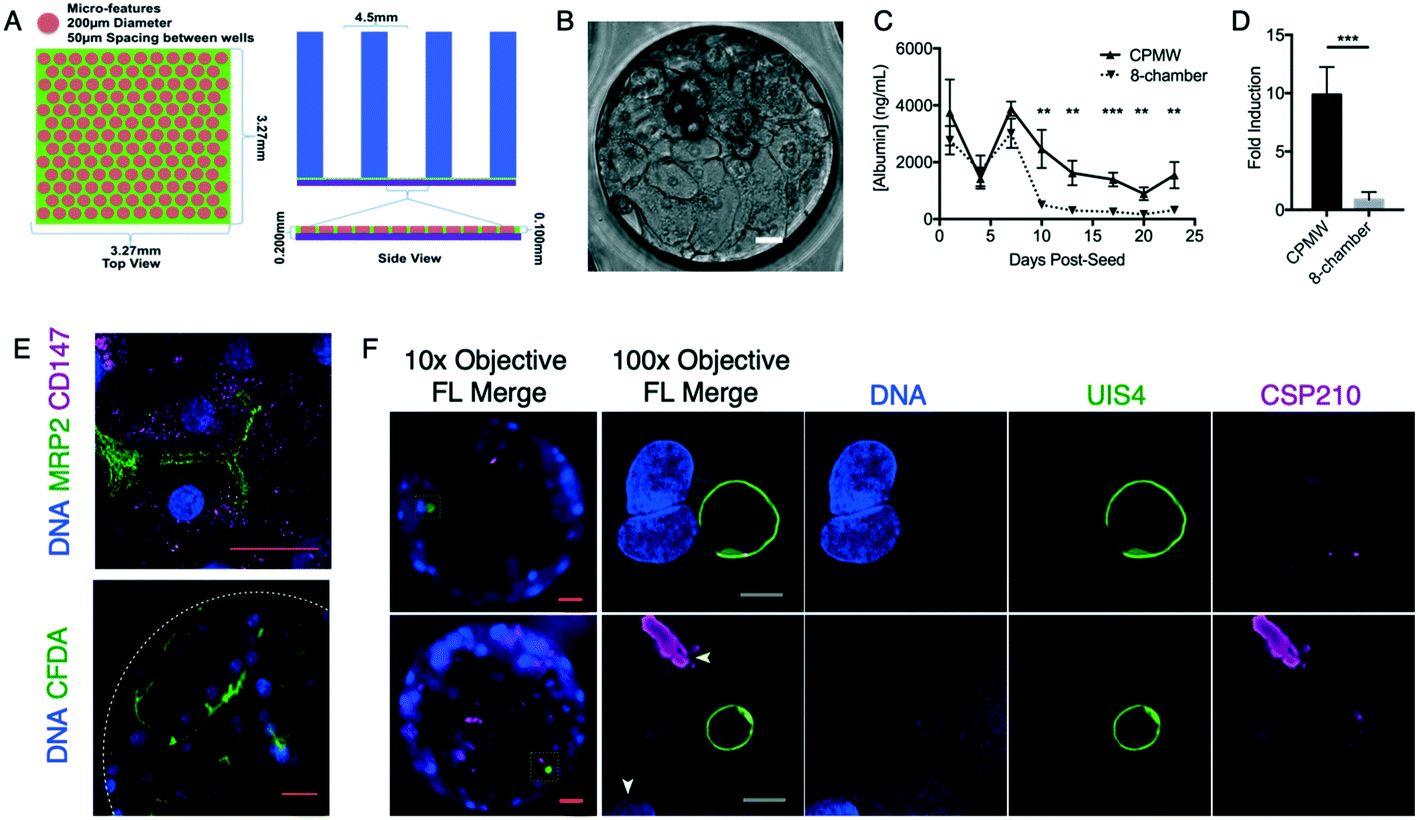 | ||
| Fig. 1 Initial experimentation with primary human hepatocytes (PHHs) cultured in PDMS-μHEP slides. (A) A top and cross-sectional view of the original microwell design fabricated with PDMS. Blue sections were punched from block PDMS, green sections were formed by pressing PDMS in a silicon mold. The PDMS layers were plasma bonded together and then the PDMS assembly plasma bonded to a #1 glass coverslip (purple). (B) Transmitted phase contrast images of cuboidal PHHs within a single 200 μm-diameter close-packed microwell (CPMW) at three weeks post-seed. (C) Albumin production by PHHs cultured in either a PDMS-μHEP slide or 8-chamber glass slide over three weeks. (D) Cytochrome P450 3A4 induction in response to rifampin treatment following three weeks of PHH culture in either PDMS-μHEP slide or 8 chamber glass slide. Error bars indicate S.D. (n = 4), error bars are omitted when shorter than the datapoint marker. Significance determined using multiple unpaired t tests and the Holm–Šídák post-test, with alpha = 0.05. **P < 0.001, ***P < 0.0001. (E) Top: morphological staining of PHH at 11 days post-seed; multidrug resistance protein 2 (MRP2, green) is located in the bile canaliculi of the apical belt between neighboring hepatocytes and CD147 (magenta) is located on the basolateral surface. Bottom: live staining of hepatic transport using fluorescent CFDA (green), a white dotted line indicates the CMPW edge. (F) LS parasites stained and imaged at 3 days post-infection into PHHs cultured in a PMDS-μHEP slide with 200 μm-diameter CPMWs. Slides were imaged with a 10× air and then 100× oil objective. Sporozoites form a parasitophorous vacuole membrane containing UIS4 (green) following invasion of the hepatocyte. Circumsporozoite protein (CSP, subtype VK210, magenta) is strongly expressed in the sporozoite inner membrane complex (yellow arrow), but not in developing parasites (green). Yellow box indicates the area imaged at higher resolution, white arrow marks the host cell nucleus. Red scale bar represents 25 μm, grey bar represents 5 μm. | ||
Polystyrene μHEP system fabrication
Development of the soft mold embossing process required fabrication of a soft PDMS mold used to emboss a stock PS sheet. Using standard photolithography techniques, a 6-inch Si wafer was patterned with SU-8 photo resist (MicroChem SU-8 3050, Westborough, MA), serving as a mold for forming sets of close-packed microwell arrays (384 arrays, 150 microwells/array, with each microwell 200 μm in diameter, 100 μm deep, and 50 μm edge-to-edge spacing) or other geometries in Fig. 2B. Frames containing a 384- or 48-well super structure were machined from acrylic. The patterned Si wafer was then bonded to the corresponding acrylic frame using an epoxy (Hapco R-36, Hapco, Hanover, MA) to serve as the casting frame for the soft mold. PDMS (Sylgard 184, Dow Corning) was cast at a 10:1 base to curing agent ratio into the casting frame and cured at 65 °C for 12 hours. Once cured, the molded PDMS was peeled from the casting frame and baked at 150 °C for 1 hour. The PDMS mold was loaded into a thermal press (DT-3, Sonitek, Milford, CT) by placing a 2 mm thick sheet of stock PS (Goodfellow Corporation, Coraopolis, PA) cut to a standard microtiter plate or microscope slide footprint into an aluminum fixture and the PDMS mold centered on top of the PS. Using pressure (100 PSI) and heat (210 °C), the PDMS mold was embossed into the then-melting PS sheet. The aluminum fixture controlled the flow of the PS such that all PS displaced by the PDMS mold was used to form the microfeatures and macrostructure walls of the molded part. The aluminum fixture also controlled the bottom thickness of the molded PS part as it prevented the PDMS mold from embossing completely through the PS by acting as stopping mechanism. The PS part was fully molded after 20 minutes and the mold and plastic were allowed to cool off-press. Once cooled to below the glass transition temperature of PS (80 °C), the PDMS mold was easily peeled off the embossed PS without damage to the microfeatures (Fig. 2A). Embossed μHEP slides and plates were shipped to commercial sources for surface treatment (PVA Tepla America, Corona, CA or Dow Corning) and then electron beam sterilized (Steris-Synergy Health, San Diego, CA). Sterilized μHEP slides and plates were sealed in plastic packaging and stored at 4 °C to avoid interactions between humidity and surface treatments (Table S1†).
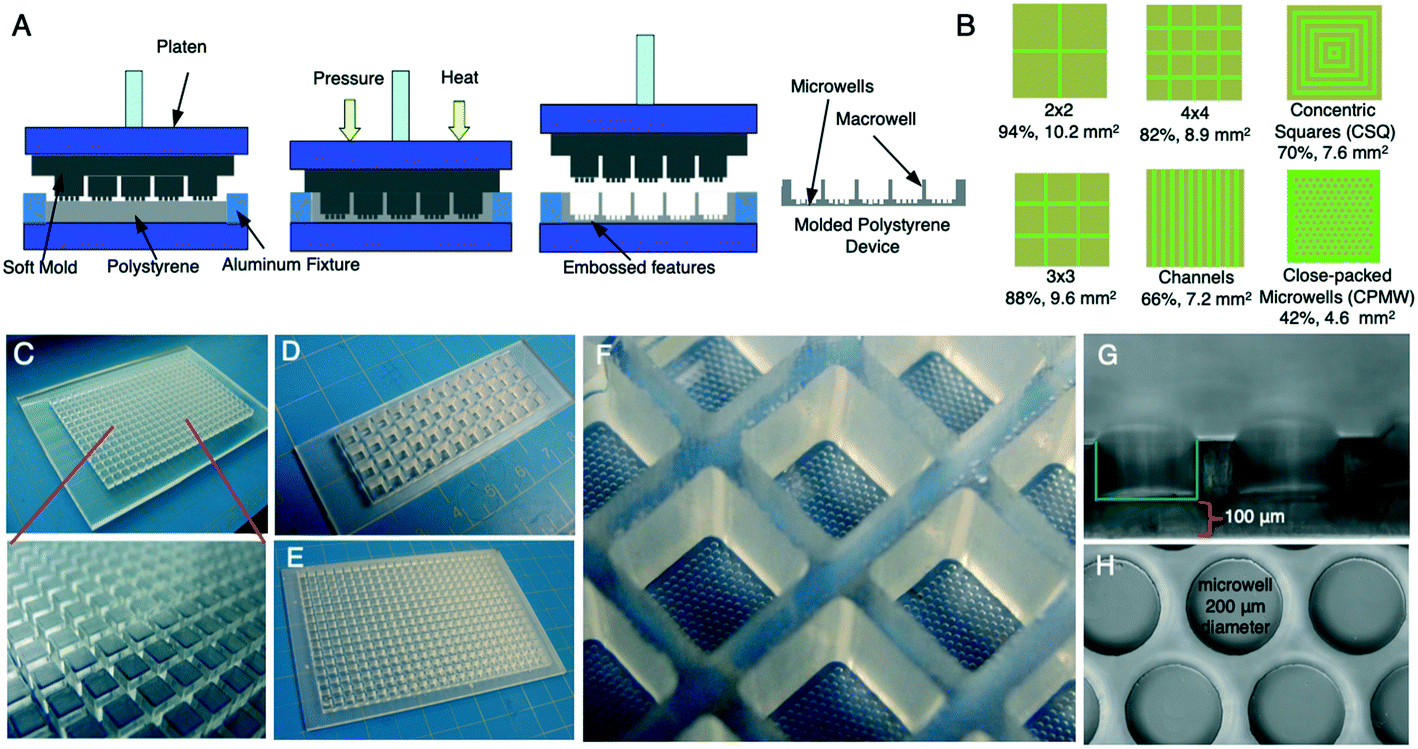 | ||
| Fig. 2 Polystyrene μHEP slide and plate fabrication. (A) Graphical summary of the soft mold embossing fabrication process. The soft mold and polystyrene are loaded in between two platens of a thermal press; as platens apply heat and pressure, the soft mold embosses into polystyrene. The material properties of a soft mold allow for easy mold removal following pressing into polystyrene. (B) Alternative microfeature patterns (green) leave different sized areas for cell culture (brown) within the macrowell. Percentages are proportion of total bottom area for cell culture resulting in the indicated square millimeters of cell culture area per macrowell. (C) A soft mold made out of PDMS used for embossing the microfeatures and macrowells of a 384-well plate into polystyrene. (D and E) Polystyrene prototypes developed. (D) A 48-well microscope slide footprint with wells fabricated to standard 384-well pitch and size. (E) A full 384-well platform for high throughput experiments that meets the standards of commercial 384-well plates for well pitch, length, and width. (F) Embossed microfeatures inside each macrowell. (G) Brightfield cross sectional and (H) top views of 200 μm embossed microwells with a 100 μm-thick bottom. | ||
Hepatocyte culture
Sterilized μHEP slides or plates, or commercially available culture devices such as 8-chamber slides (BD, Franklin Lakes, NJ), were unpackaged in a sterile field and placed in a Petri dish to serve as a lid and control evaporation. The day prior to PHH seeding, wells were collagen coated with 40 μL of 15 μg μL−1 rat tail collagen I (BD) in sterile filtered 0.02 N acetic acid (Thermo Fisher Scientific, Waltham, MA), then left overnight at 37 °C. Immediately prior to seeding the collagen solution was washed out thrice with sterile PBS and then filled with 20 μL In Vitro GRO® CP plate media (BioIVT, Westbury, NY) supplemented with 1× Pen–Strep–Neo solution (Thermo Fisher Scientific) and 20 μM gentamicin (Thermo Fisher Scientific). Vials of cryopreserved PHHs (BioIVT or Yecuris, Tualatin, OR) were thawed by immersion in a 37 °C water bath for 2 minutes, sterilized by 70% ethanol in a sterile field, and contents added directly to 4 mL plate media. Live and dead cells were quantified by trypan blue exclusion on a Neubauer improved hemocytometer, the PHH density was set to 1 × 103 live cells per μL, and 18 μL cell suspension was added to each well. Media was exchanged thrice weekly with CP media with antibiotics.Hepatic phenotype assays
Hepatocyte viability was assessed with the Live/Dead™ kit (Thermo Fisher Scientific). Media was washed out of wells and replaced with 2 mM Calcein AM, 4 mM ethidium homodimer, and 10 μg mL−1 Hoechst 33342 (Millipore Sigma, St. Louis, MO) in serum-free RPMI (Thermo Fisher Scientific) for 30 minutes at 37 °C. Wells were then washed with PBS and live-cell imaging was performed on a Deltavision Elite with an environmental chamber, an upgraded automatic slide and plate scanning stage, and softWoRx® well scanning software (GE Healthcare Life Science, Marlborough, MA). Each well consisted of 16–25 images which were paneled and quantified for PHH nuclei by stain intensity scaling with a 2D polygon filter in softWoRx®. Albumin and factor IX production were measured by collecting media at pre-determined timepoints and freezing samples at −80 °C. Media samples were quantified by human albumin or factor IX ELISA per manufacturer's protocols (AssayPro, St Charles, MO); in summary, samples were thawed together, diluted 100× (for albumin) or 5× (for factor IX) in kit-supplied dilution buffer, and quantified by an Infinite 200 plate reader (Tecan, Männedorf, Switzerland). The CYP3A4 P450 Glo™ luciferin-IPA kit was used to measure CYP3A4 induction (Promega, Madison, WI). Wells were induced for 72 hours with 25 μM rifampicin (Thermo Fisher Scientific) in plate media, or an equivalent volume of DMSO in uninduced controls, and luciferin signal measured by plate reader following the manufacturer's lytic protocol version. PHH nuclei counts were obtained prior to running the lytic assay by Hoechst staining and automated live cell imaging as described above. PHH functional transport of salt to bile canaliculi was assessed by first staining hepatic nuclei with Hoechst (as above) and then adding 2 μg mL−1 carboxy-DCFDA (Thermo Fisher Scientific) in serum free media for 10 minutes at 37 °C, followed by replacement of stain with media, and live-cell imaging as described above.37,38 Immunofluorescent imaging of PHH polarity was performed by fixing wells with 4% paraformaldehyde, washing with PBS, and staining with 1:500 rabbit anti-MRP2 monoclonal IgG (Abcam, Cambridge, UK) and 1:100 mouse anti-CD147 monoclonal IgG (Abcam) in a dilution buffer (1% w/v bovine serum albumin and 0.3% v/v Triton X 100 in PBS) overnight at 4 °C. Wells were then washed thrice with PBS and stained with 2 μg mL−1 Alexafluor 488®-labeled goat anti-rabbit IgG (Thermo Fisher Scientific) and 2 μg mL−1 Alexafluor 568®-labeled goat anti-mouse IgG (Thermo Fisher Scientific). Wells were counter-stained with Hoechst and imaged as described above.
Mosquito rearing and infection, sporozoite collection, in vitro culture of liver stage parasites, and high-resolution imaging
Anopheles dirus mosquitoes were reared at the Mahidol Vivax Research Unit, Mahidol University (Bangkok, Thailand). Anopheles dirus sensu stricto and An. crascens were reared at the Shoklo Malaria Research Unit, part of the Mahidol-Oxford Research Unit (Mae Sot, Thailand), following published protocols.39 Following informed consent, patients seeking treatment for vivax malaria at malaria clinics along the Thai-Myanmar border donated up to 10 mL of blood for membrane feeding to Anopheles mosquitoes in secure insectaries. The human subjects protocol for this study was approved by the Ethical Review Committee of Faculty of Tropical Medicine, Mahidol University (MUTM-2011-040-01) and Oxford Tropical Medicine Ethical Committee, Oxford University, England (TMEC 11-008 and OxTREC 17-11; TMEC 14-016 and OxTREC 40-14). Studies were performed at MVRU and SMRU research facilities, or infected mosquitoes were shipped (still pre-infectious) to the University of South Florida via private courier following procurement of approval from Mahidol University, the US Center for Disease Control, the US Department of Agriculture, and the state of Florida Department of Agriculture. Following 14 days of mosquito-stage development, sporozoite-laden salivary glands were aseptically dissected and collected into 100 μL cold Schneiders insect media (Millipore Sigma) without any additives, as described.40,41 Sporozoite density was counted on a Neubauer improved hemocytometer, adjusted to 200 sporozoites per μL with plate media, and then a specific volume of sporozoites was added to wells depending on the experimental condition. Freshly inoculated μHEP slides or plates were then spun at 200 RCF for 5 minutes and media exchanged the next day and thrice weekly thereafter.Subcellular analysis of LS parasite morphology within μHEP cultures was performed by fixation with 4% paraformaldehyde followed by two PBS washes and primary antibody staining overnight at 4 °C. Primary antibodies were diluted into dilution buffer (formulation above) as follows: 1:1000 rabbit anti-upregulated in infectious sporozoites-4 (UIS4) polyclonal antibody purified from immune serum42 or 1:25000 purified mouse anti-UIS4 recombinant IgG,43 and 1:1000 mouse anti-glucose related protein, binding protein (GRP-BiP) monoclonal IgG.44 Primary antibody stained cultures were washed thrice with PBS and secondary stained with 2 μg mL−1 Alexafluor 488®-labeled goat anti-rabbit IgG and 2 μg mL−1 Alexafluor 568®-labeled goat anti-mouse IgG as described above, and simultaneously counterstained with Hoechst as described above. High-resolution Z-stack image series of stained μHEP-cultured LS parasites were acquired with a 100× oil objective (Olympus, Tokyo, Japan) on a Deltavision Elite and deconvoluted with softWoRx®; maximum projections are shown.
Dose response assays and high-content imaging
Following inoculation with sporozoites, μHEP cultures were treated with drug-containing plate media refreshed daily for day 1–4 post-infection (prophylactic mode) or day 5–8 post-infection (radical cure mode) prior to fixation on day 6 post-infection (prophylactic mode) or day 8 post-infection (radical cure mode) as previously described.45 Stock drug powders were sent as a courtesy from Medicines for Malaria Venture. Cultures were fixed with 4% paraformaldehyde and stained with rabbit anti-UIS4 polyclonal antibody and Hoechst as described above. Automated imaging was performed by programming a Deltavision Elite with an upgraded plate stage and well screening module. The footprint of our μHEP slide and plate were programmed as plate types in the Deltavision software, then the imager was programmed to use intensity-based autofocus to focus on the middle of each well before capturing all fields of view in every well at 16× (10× objective and 1.6× optovar). Alternatively, μHEP slides and plates were imaged with a 20× objective on an Operetta (PerkinElmer, Waltham, PA) by programming the slide and plate footprint into the labware type editor. Laser-based autofocus was used to capture all fields of view in all wells, however, occasional autofocus directly on microfeature areas necessitated imaging of all wells at 2 planes, which were combined into a single image using maximal projections prior to image analysis.Statistics
All parasite population metrics were calculated in Graphpad Prism® (Graphpad Software, San Diego, CA). An EC50 for each dose response assay was calculated using nonlinear curve fitting in Graphpad Prism®. The statistical tests used and summary of technical replicates in experiments is included in figure legends. A summary of experimental replication is presented in Table S2.†RNAseq characterization
Total RNA samples for RNA sequencing were extracted from PHHs cultured in the μHEP slides and a variety of commercial microtiter plates (Corning® Biocoat™ collagen-coated 384-well plate, Cat No: Cat No: 356664, Corning; Falcon® 24-well Clear Flat Bottom Tissue Culture-treated Multiwell Cell Culture Plate, Cat No: 353047, Corning; and a 12 Well glass bottom plate with high performance #1.5 cover glass, Cat No: P12-1.5H-N, In Vitro Scientific, CA, USA). The PHHs were seeded at a cell density of 1 × 103 live PHHs per μl for each sample in a particular device based on the surface area (1.5 × 105 cells per cm2). Trizol was added to each sample, incubated for 5 minutes and stored at −80 °C until further isolation. Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific) following the manufacturer's instructions using chloroform, isopropanol, and 75% ethanol. The resulting RNA pellet was resuspended in DEPC-treated water. RNA quantification was performed by Qubit (Thermo Fisher Scientific), RIN values were determined using a TapeStation 2200 (Agilent Technologies, Santa Clara, CA) and A260/280 and A260/230 ratios were assessed using the NanoDrop® Spectrophotometer ND-1000 (Thermo Fisher Scientific). For each sample, 0.5–1.0 μg total RNA with a RIN value greater than 7.0, A260/280 ratio greater than 1.8, and A260/230 ratio greater than 2.0 was used for library preparation. All of the libraries were prepared by using TruSeq Stranded mRNA kit (Illumina, San Diego, CA). The library quantification was conducted by qPCR and measurement of TapeStation (Agilent Technologies). RNAseq reads from each sample were aligned to the human reference genome (GRCh38.p10). A maximum of one mismatch per read was allowed. The mapped reads from TopHat46 were used to assemble known transcripts from the reference and their abundances were estimated using Cufflinks.47 The expression level of each gene was normalized as fragments per kilobase of exon per million mapped reads (FPKM). To identify significantly differentially expressed genes, the Wilcoxon signed-rank test was used to compare the expression FPKM between RNAseq samples from PHHs cultured in μHEP slides and commercial microtiter plates.48 The FDR p-value adjustment was implemented and the genes with adjusted p-value less than 0.05 were identified as significantly differentially regulated genes.49 The Barnard test was used to test the enrichment of specific GO term in each gene groups against the genomic background. To evaluate the PHH metabolic properties, we constructed a master map of important pathways (human KEGG pathway maps) in PHH with genes encoding enzymes that catalyzes the rate-limiting step of each pathway. We then compared the averaged FPKM of these genes following culture in μHEP slides and commercial microtiter plates; and LOG2 fold change was calculated to indicate the changes. Significance of differential expression was computed as described in the RNAseq analysis. All RNA sequencing raw reads have been deposited into NCBI’s Gene Expression Omnibus (GEO) which are accessible through GEO Series accession number GSE144599.Results and discussion
As our first attempt to establish a Plasmodium LS culture platform, we previously developed a microfluidic bilayer model with a porous membrane designed to accurately recreate the liver sinusoid structure and allow for adjustable media perfusion rates and inclusion of several cell types, including hepatocytes, liver endothelial, stellate, and Küpffer cells.19 These studies confirmed the most fundamental element capable of maintaining PHH properties was the abiotic physical confinement offered by the width of the device channel (200 μm), a well-described mechanism also associated with microfluidic culture.18,50 Furthermore, we recently reported commercially-available, Greiner 384-well microtiter plates are capable of maintaining PHHs in long-term culture and robust LS parasite formation, suggesting the dimensions of a 384-well microtiter plate well alone provide superior confinement in comparison to larger culture systems such as 96-well plates and 8-chamber slides.45 To continue development of a miniaturized and efficient LS parasite culture platform we hypothesized microfeatured cultureware would better sustain hepatic differentiation and therefore enhance LS parasite infection rates.Described over 30 years ago, LS parasite culture was first accomplished in an nonpatterned PHH and stromal cell co-culture,51 and then a patterned co-culture,10,11 demonstrating how advanced culture techniques can enhance LS parasite infection rate. Quantity of LS parasites from well-to-well, and the ability to quantify those parasites, is perhaps the first hurdle for LS antimalarial testing.25,45 In turn, the overall efficiency of LS parasite formation from an inoculum of infectious parasites (sporozoites) is driven by the optimization of several factors, including selection of supportive host hepatocytes, quantity of sporozoites available, and ability of the culture system and method to sustain the health of both hepatocytes and LS parasites for 8–30 days of culture.10,45 Therefore, advanced culture methods leading to improved hepatocyte or LS parasite viability or infection rate could drastically increase the throughput of what is currently a resource-limited assay primarily due to the limited availability of P. vivax sporozoites. To fully optimize our μHEP system for LS parasite formation we hypothesized LS parasite formation rate could be dependent on the specific microfeature design, and the identification of an ideal design could be predicted by how each design supports hepatic differentiation, measured by individual hepatic phenotypes such as production of albumin and factor IX, or induction of CYP3A4.
Our intended application effectively set design requirements early in the development process, including the need for optical accessibility for high-resolution imaging, a standard footprint to make the system adaptable to automated high-content imaging systems, and the need to rapidly and efficiently modify the microfeature designs (geometries). We fabricated systems with either a standard microscope slide (48 wells at 384-well microtiter plate size and pitch) or microtiter plate (a full 384-well microtiter plate) footprint. The smaller slide footprint was found useful for relatively small developmental experiments incorporating single-channel pipetting and was more amendable to visualization on microscopes with stages incompatible with full microtiter plates. The full 384-well footprint was considered a final design goal, was compatible with 16-channel pipetting, and is the standard footprint for high-content imaging systems.
Proof of concept experimentation with PDMS-μHep slides
We initially fabricated a proof-of-concept μHEP slide in which the culture well structure (macrowells) were derived from hole-punching 4–8 wells into PDMS blocks and microfeatures were derived from a thin PDMS layer pressed from a mold developed using photolithography (PDMS-μHEP). The PDMS macrowell and microfeature layers were plasma bonded together and then plasma bonded to a glass coverslip bottom, resulting in a configuration in which hepatocytes would attach to glass and be confined by PDMS microfeatures (Fig. 1A). The PDMS-μHEP slides were ethylene oxide sterilized and were not surface treated, as collagen-coated glass has excellent cell-attachment properties,52 thus the slides were only collagen coated prior to PHH seeding. Our PHH seeding protocol was essentially the same as the protocol used to seed commercial 384-well plates45 and resulted in a stable PHH monolayer within microfeatures (Fig. 1B). We found PHHs cultured in PDMS-μHEP slides better maintained long-term (three-week) production of albumin and rifampin induction of cytochrome P450 3A4 in comparison to PHHs cultured in a commercially-available 8-chamber glass slide, chosen for the comparison as PHHs would then adhere to the same material (collagen-coated glass) in both systems and the 8-chamber glass slide is commonly used for short term PHH culture (Fig. 1C and D). We also further characterized long-term culture of PHHs in PDMS-μHEP slides by localizing polarity markers MRP2 and CD147 (ref. 53) to the apical and basolateral surfaces, respectively, and demonstrating evidence of bile transport by using carboxy-DCFDA38 (Fig. 1E).Following this concept test, we performed the first of several rounds of ‘design screening’ experiments in which various culture conditions were tested simultaneously and ranked by how each supports a desired hepatic phenotype, with the goal of using superior culture conditions to further develop our μHEP systems. Due to the risk of quickly becoming impractically numerous, the total number of test wells was minimized in most design screening experiments by measuring the same singleton wells over an experimental timecourse. The first such design screen included a microfeature geometry of close-packed microwells (CPMWs) of four different diameters (100, 125, 150, 200 μm) as these diameters covered the relative range of diameters produced for our microfluidic platform19 and other microwell culture systems.50 Scored by viability staining and albumin production over three weeks of culture, we found all CPMW diameter tested supported long-term albumin production and none was found superior to the others (Fig. S1,† repeated measure ANOVA, NON: F (1.694, 10.16) = 0.9482, P = 0.4042, NLX: F (1.903, 11.42) = 1.065, P = 0.3733). These results suggested simple phenotypes, such as of albumin production, would not suffice to differentiate the relative improvement of additional μHEP design iterations. Our observation that stabilization of albumin production is relatively simple to achieve with traditional culture methods and not informative of superior or inferior advanced culture conditions reaffirms our previously published conclusions from culturing PHHs and LS parasites.45 In these studies we found stable albumin production by PHHs can be achieved in 384-well plates (but not in larger culture systems the size of 12-well plates and 8-chamber slides), is largely driven by the quality of PHH isolation and cryopreservation by the production company, and is necessary for, but not predictive of, culture conditions ideal for LS parasite formation.45 Thus, we determined early in μHEP system development that hepatic phenotype testing could help elucidate which culture conditions allowed for long-term culture, but lacking a biomarker for ideal conditions for LS parasite formation, LS parasite formation itself would be the only metric useful for identifying designs ideal for a LS parasite culture platform. To confirm the feasibility of this endpoint, PHHs in PDMS-μHEP slides were infected with P. vivax sporozoites harvested from infected mosquitoes shipped from the Mahidol Vivax Research Unit in Bangkok, Thailand to the University of South Florida. The resulting LS parasite formation, as noted by high-resolution imaging (Fig. 1F), demonstrated we had the workflow (including supportive hepatocytes, immunofluorescent staining reagents, and protocols) necessary to perform the complex experiment of infecting μHEP systems.
Fabrication of PS-μHEP systems by a soft PDMS mold embossing process
While the PDMS-μHEP slides were relatively simple and inexpensive to develop and fabricate, we opted to continue μHEP system development using PS as our ultimate goal was to use this platform for antimalarial drug screening; PS has much better bulk properties compared to PDMS, is more amenable to rapid downstream production methods, and is broadly used for cell culture applications.33 To develop our fabrication protocol, we first designed a casting frame that would be used to fabricate the PDMS mold for embossing PS. Using traditional photolithography techniques, the desired microfeature designs were patterned using SU-8 on a 6-inch silicon wafer. A machined acrylic superstructure containing an array of macrowells was aligned with the patterned SU-8 microfeatures and bonded to the wafer. PDMS was poured into the casting frame to produce a mold that would contain arrays of micropillars on top of macropillars that were used to simultaneously emboss macrowells containing arrays of microwells in PS sheets. After curing of the PDMS, the PDMS mold was peeled from the casting frame (Fig. 2C) and centered on top of a PS sheet housed in an aluminum fixture on the thermal press. The PDMS mold then embossed into the PS sheet using pressure and heat (Fig. 2A). The aluminum fixture housing the PS sheet acted a stopping block for the PDMS mold and thus prevented the PDMS mold from embossing all the way through the PS; thereby producing the desired 100 μm bottom thickness. Once the embossed μHEP slide or plate cooled below 80 °C (the glass transition temperature of PS), the PDMS mold could be easily peeled from the embossed part. We attributed the ease of removal of the mold from the embossed PS to differential thermal expansion coefficients between the two materials.36 These thermal expansion material properties, in combination with use of a flexible mold, allowed for mold and embossed PS separation without damage to the multitude of fine microfeature elements of the mold, nor damage to the thin 100 μm PS bottom, at nearly a 100% success rate, which would not be possible through traditional hot embossing methods. We used a PDMS mold to emboss more than 100 μHEP systems before the mold needed replacement. The embossing process itself was carefully optimized by several rounds of trial-and-error and troubleshooting. Since we were developing an embossing process using a soft mold we quickly realized that an aspect ratio of at least 1:2 for the macropillars was required to successfully emboss macrowells without the macropillars buckling or misaligning during application of pressure during embossing. Because the PDMS micropillars (forming the individual microwells in PS) were successfully embossed at a 2:1 micro-scale aspect ratio, we did not further test micropillar aspect ratio limits. The thermal press parameters were also found critical as early iterations resulted in extensive air bubble formation within the PS. To overcome this, experiments were performed to test a combination of different pressures, embossing times, platen heights, and temperatures. We determined that gradually increasing the pressure to 100 PSI and temperature to 210 °C over the course of 20 minutes effectively allowed the melted PS to flow yet remain viscous and prevent bubbles from forming within the plastic. We also found that gradually increasing the platen pressure to the soft mold aided in macrowells being molded with uniform pitch.
Establishment of long-term PHH PS-μHEP culture in various microfeature geometries
Following development of our PS fabrication process, CPMW-μHEP slides were fabricated to serve as a baseline for establishing long-term culture of PHHs on PS. Because stock PS is too hydrophobic for hepatocyte adherence,54 we evaluated multiple commercially-available tissue culture surface treatments from two companies to promote optimal attachment critical for PHH morphology and function.55 We found surface treatment was essential for obtaining a uniform monolayer within microfeatures (Fig. S2A†). Surface treatments are known to not withstand harsh sterilization processes such as autoclaving and ethylene oxide gas, thus we opted to use a commercially-available electron-beam sterilization service to sterilize and package surface-treated μHEP systems. Collagen coating was performed identically to collagen-coating of either commercial microtiter plates or our PDMS-μHEP slide, using commercially-available collagen and the supplied manufacturer's protocol. Thus, our PS-μHEP fabrication process was supplemented to include a surface treatment, sterilization, packaging, and collagen coating, all required for successful seeding and culture of both PHHs and LS parasites using the same standard protocols described for the PDMS-μHEP system or commercial microtiter plates.A second round of design screening was then performed to better understand which surface treatments and collagen coatings were ideal for long-term PHH culture. In this round two different collagen coatings (1×: 5 g cm−2 and 10×: 50 μg cm−2) were applied onto four different surface treatments in an effort to find ideal conditions by scoring cell number and albumin production over three weeks of culture. Each surface treatment was tested with PHH from two different cryopreserved PHH donor lots, NON and NLX. Similar to the inconclusive result noted in our first round of design screening with PDMS-μHEP slides, PHHs in all combinations of treatment and collagen coating reached stable levels of albumin production (Fig. S2B,† repeated measure ANOVA, NON with 1× collagen: F (1.336, 4.007) = 0.1.230, P = 0.3536, NLX with 1× collagen: F (1.428, 4.285) = 2.994, P = 0.1548, NON with 10× collagen: F (1.090, 3.270) = 2.469, P = 0.2092, NLX with 10× collagen: F (1.120, 3.360) = 4.613, P = 0.1111). Nonetheless, it appeared as though a 10× collagen coating resulted in de-differentiation and replication of PHHs, indicating over-attachment, thus all following studies were performed with a 5 μg cm−2 collagen coating. Also, while PHH albumin levels stabilized with the amine treatment, we noted poor cell attachment immediately after seeding and down-selected to using only the active, hydroxy, and carboxy PVA Tepla treatments for following studies.
After establishment of PS-μHEP fabrication and long-term culture protocols, we next re-designed the soft mold for fabrication of six different microfeature geometries in a 48-well PS-μHEP slide (8 replicate wells of each geometry per slide) to enable direct comparisons of how each geometry supports long-term culture of PHHs (Fig. S3†). A third round of design screening was then performed in which albumin and factor IX production was measured from PHHs cultured in each geometry on PS-μHEP slides treated with an active, hydroxy, and carboxy surface treatment. Again, we found relatively stable production across all combinations tested, such that a clearly ideal combination was not found (Fig. S4,† albumin two-way repeated measure ANOVA by treatment: F (2, 15) = 1.572, P = 0.2490, by geometry: F (5, 12) = 2.663, P = 0.0763; factor IX two-way repeated measure ANOVA by treatment: F (2, 15) = 1.812, P = 0.1973, by geometry: F (5, 12) = 0.3150, P = 0.8945). However, the carboxy treatment was eliminated from further contention as it was found to lead to the strongest PHH attachment and most variable production of albumin and factor IX over three weeks of culture. These results confirmed further design screening would be dependent on directly assessing LS parasite formation rates in each design combination, which was dependent on being able to infect several μHEP systems in each of many followup experiments. To this end, μHEP slides and plates were fabricated and shipped to Shoklo Malaria Research Unit in Mae Sot, Thailand to perform studies in close proximity to where P. vivax sporozoites are available.
High-content and high-resolution imaging of P. vivax parasites within PS-μHEP systems
In addition to performing LS parasite studies abroad, using LS parasite formation as an endpoint necessitated further development of HC/RI protocols for μHEP systems. To develop these protocols and demonstrate the optical properties of our PS-μHEP systems, we imaged fluorescently-stained PHH organelles and LS parasites in PS-μHEP slides and plates using several manually-operated and automatic inverted microscopes. High-resolution imaging is dependent on culture systems possessing an optically clear, thin bottom to allow focal access within the working distance of 60× and 100× objectives (typically less than 150 μm). The PS-μHEP fabrication process was precisely tailored to press PS microfeatures such that the bottom PS surface of the cell culture area was approximately 100 μm thick throughout the culture area of each microfeature (Fig. 2G). A 100× oil objective on a Deltavision Elite was used to capture high-resolution Z-stacks of LS parasites, revealing fine subcellular details, including the drastic expansion of LS parasite endoplasmic reticulum in developing schizonts, but not dormant hypnozoites (Fig. 3A), as we have previously published for P. vivax LS parasites.45 Quantification of LS parasites (or other biological constituents) by high-content imaging methods is dependent on maintaining focus across fields of view within wells and from well-to-well across a slide or plate. Navigation of stage travel is dependent on the precise location of well A1 in relation to the plate edge (or ‘skirt’), regular X, Y and Z dimensions across all wells, and regular X and Y spacing from well-to-well. While commercial plates and high-content imaging systems are designed for mutual compatibility, especially in terms of maintaining consistent dimensions, the compatibility of our first PS-μHEP systems was unknown, thus we chose to program a Deltavision microscope with a microtiter plate scanning stage to image and quantify initial LS parasite infection as this system was found ideally flexible for mapping new labware types. We programmed the dimensions of both a PS-μHEP slide and plate in the software labware type editor to serve as an imaging map, and then captured a panel of images covering the entire area of each PS-μHEP system well at 16× (10× objective and 1.6× optivar). Focus was maintained by programming the imager to perform an intensity-based autofocus at the center of each well, followed by travel to the top left of each well for acquisition of panels. Panels were digitally stitched together, and then hepatic nuclei and LS parasite forms were defined and quantified using two-dimensional object filters available in the Deltavision software. This semi-automated high-content imaging technique was used to quantify LS parasites in experiments designed to understand which microfeature geometry was best for LS parasites formation rate (Fig. 3B and 4A), to characterize LS parasite populations (Fig. 4B and C), and to capture data during antimalarial activity testing (Fig. 4D). Following successful semi-automated imaging, we seeded PHHs into all 384 wells of PS-μHEP plates, fixed and stained PHH nuclei with Hoechst, and imaged all wells with the 20× objective on an Operetta high-content imager. The Operetta is a turn-key HC/RI imaging system, with a broad range of objective power (2×–100×), fully automated infrared laser-based autofocus, and comprehensive analysis modules to quantify biological data. The Operetta autofocus function utilizes a laser to find the plate bottom and well bottom and then uses a pre-programmed distance offset from the well bottom to look for and image the biological specimen. We found this system was confounded when the autofocus laser fell on a microfeature itself, thus we programmed the imager to capture images at both the culture area and micro-feature planes and then reduced the stack to a single image using a maximal projection. This setup was used to quantify the number of PHH nuclei per well, demonstrating compatibility of both the PS-μHEP plate design and overall fabrication process for HCI (Fig. 3C).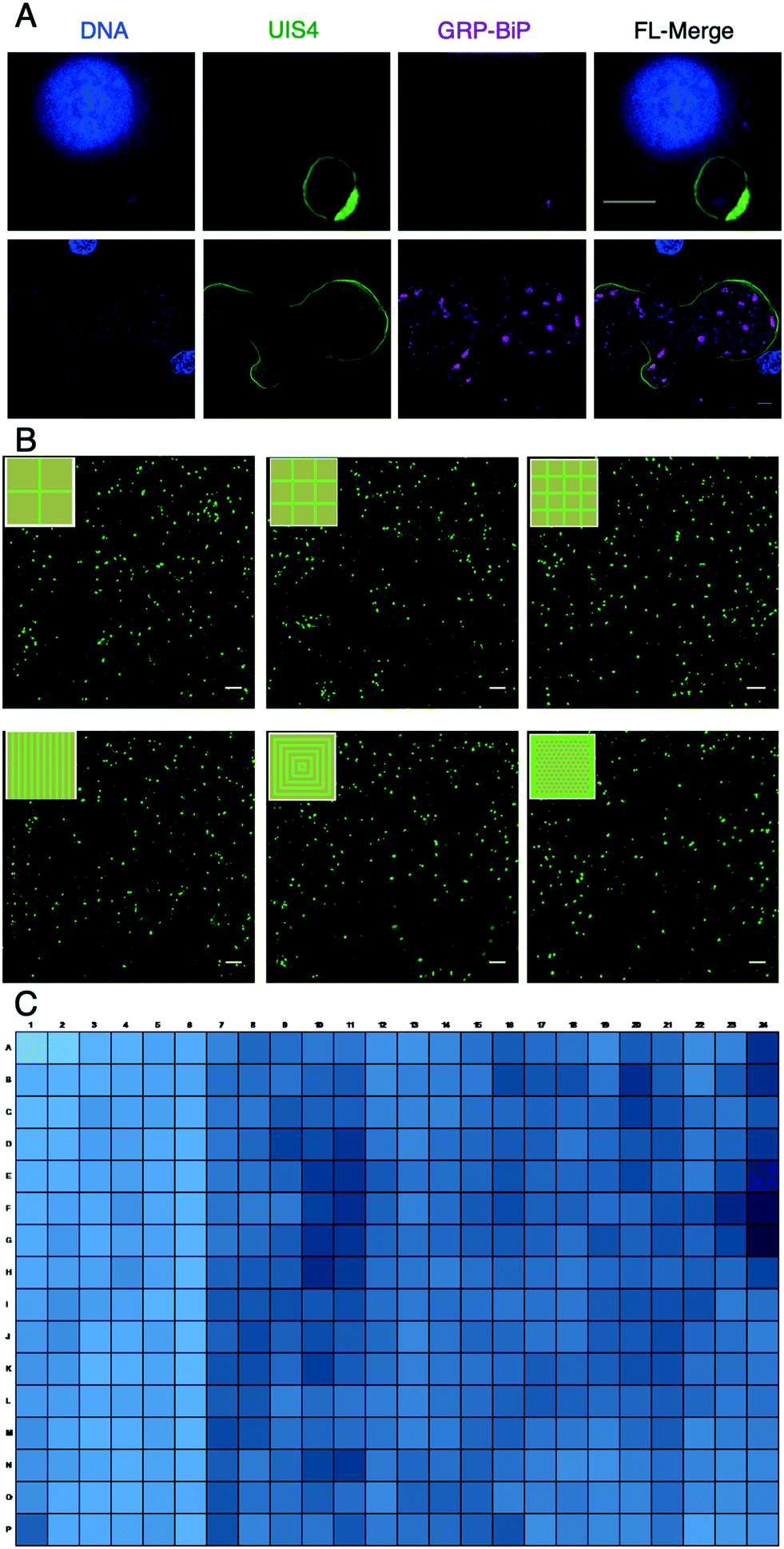 | ||
| Fig. 3 High-resolution and high-content imaging of PS-μHEP slides and plates. (A) A P. vivax hypnozoite (top row) and schizont (bottom row) fixed and stained following 8 days of culture in a PS-μHEP slide. Parasites reside in a parasitophorous vacuole membrane containing UIS4 (green) and developing schizonts harbor an extensive endoplasmic reticulum containing GRP-BiP (magenta). DNA from both the host cell nucleus and parasite nuclei are counterstained with Hoechst. Images were obtained from a deconvoluted Z-stack acquired with a 100× oil objective (NA = 1.4, working distance = 0.13 mm). (B) Automated imaging and analysis of six different PS-μHEP geometries containing PHHs supporting a high-density of developing LS parasites. Developing parasites were imaged and quantified using the slide-scanning function on a Deltavision with a 10× air objective and 1.6× optivar. Shown are stitched images of all fields of view from one of each well with the indicated microfeature geometry (inset), each green dot is a developing P. vivax liver stage parasite. (C) All wells of a μHEP 384-well plate were seeded with PHHs, cultured for 3 days, stained with Hoechst, and imaged on an Operetta CLS with a 20× objective. Each well was covered by 5 × 5 fields of view and nuclei per well quantified, normalized, and represented by a blue scale heat map. The first 6 columns were seeded with fewer hepatocytes to show contrasting quantification of nuclei. Grey bar represents 5 μm, white bar represents 200 μm. | ||
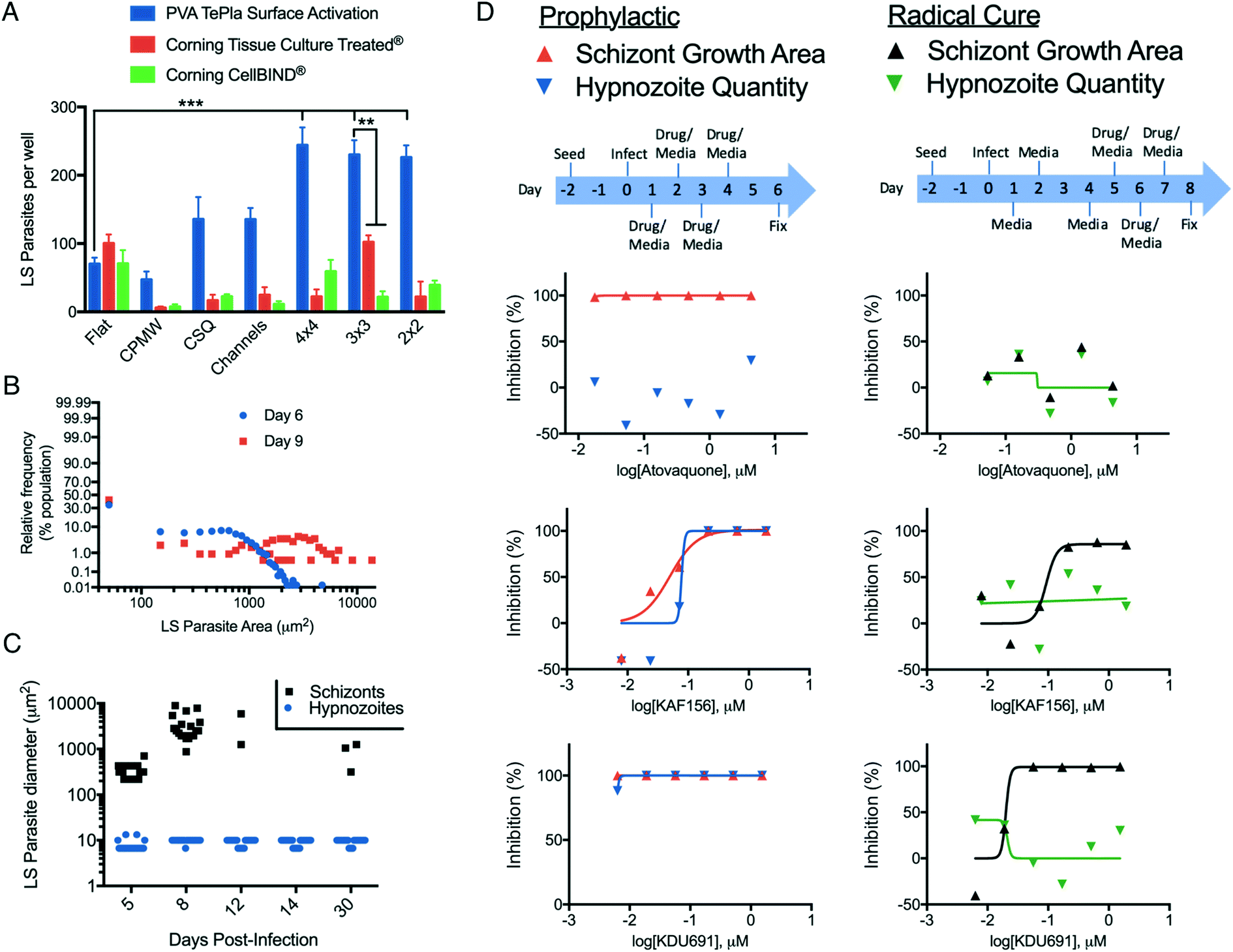 | ||
| Fig. 4 Optimization of μHEP design for P. vivax infection and antimalarial compound testing in a μHEP platform. (A) Quantity of LS parasites per well after six days of culture following infection of PHHs with 5000 P. vivax sporozoites per well. Hepatocytes were seeded into μHEP slides with six different geometrical microfeatures, or a no microfeature (flat) control. Bars represent S.D. (n = 4), significance determined using two-way ANOVA followed by Dunnett's multiple comparisons (to flat control), alpha = 0.05, **P < 0.002, ***P < 0.0001. (B) Frequency histogram of LS parasite populations at 6 and 9 days post-infection. Each datapoint represents a LS parasite, hypnozoites are binned at <100 μm2. (C) Reactivation experiment in which wells with PHHs are infected together and then fixed at time intervals to track development progression of schizonts as well as persistence and growth reactivation of hypnozoites over 30 days post-infection. A small quantity of schizonts are noted at day 30, presumably from reactivated hypnozoites. (D) Schematic of hepatocyte seed, infection, dosing, and endpoint for characterizing early (prophylactic) and late (radical cure) compound activity against LS parasites. | ||
Optimization of PS-μHEP systems for PHH culture and P. vivax infection
For the fourth and final round of design screening, PS-μHEP slides were surface treated with the active treatment from PVA Tepla, the Tissue Culture treatment from Corning, or the CellBIND treatment from Corning, in order to better understand how our treatment performed compared to those from a company specializing in cell culture products. Following seeding with PHHs, infection with P. vivax sporozoites, culture for 8 days, and quantification of LS parasites by high-content imaging we found PHHs cultured in iterations of more open geometries (4 × 4, 3 × 3, and 2 × 2) with an active treatment from PVA Tepla were superior for supporting LS parasite formation in comparison to other combinations (Fig. 4A). The surface treatment and geometry significantly contributed to LS parasite infection rate (two-way ANOVA, alpha = 0.05, F (12, 63) = 42.91, P < 0.0001). To better understand this effect, the infection rates per well for μHEP geometries was charted by available cell attachment area, revealing a correlation between infection rate and attachment area (Fig. S5†). From these data and the data from design screening experiments we concluded an ideal surface treatment is required for long-term PHH culture, but the increase in LS parasite infection rate compared to a no microfeature (flat) control is made possible by microfeatures themselves. However, the relative difference between geometries is simply a function of available area for LS parasites to form. Thus, provided microfeatures are present to increase confinement in a well, there is a tradeoff in that an excess of PS forming microfeature walls themselves can diminish a desired phenotype which is dependent on cell number or area, such as LS parasite infection rate. This finding further demonstrates our rapidly and inexpensively amendable fabrication process is ideal for developing advanced culture systems.Based upon these findings, we decided to freeze our μHEP design to consist of the 3 × 3 geometry and active surface treatment for fabrication of subsequent PS-μHEP systems. The 3 × 3 geometry was selected over other open geometries (2 × 2 and 4 × 4) because the large, open area in the center of the 3 × 3 design (the central cell attachment area) provided an ideal location to program the well-to-well autofocus function of high-content imaging systems. Many autofocus functions begin the focusing process within a well by autofocusing on the well center. To this end, we re-designed our soft mold to only emboss the 3 × 3 pattern into all assay wells and had >100 slides and plates embossed, surface treated, and sterilized for followup studies. To further compare our optimized μHEP design to commercial products, we measured albumin production and CYP3A4 induction from PHHs over three weeks of culture in both an active-treated 3 × 3 μHEP slide and Corning 384-well plate, finding sustained phenotypes in both cultures. Albumin production was found slightly higher in the μHEP culture while CYP3A4 induction was found slightly higher in the Corning product (Fig. S6C and D†).
Following hepatocyte invasion, P. vivax sporozoites form into either a developing schizont or dormant hypnozoite, which can resume development and cause a relapse blood infection weeks, months or even years afterwards.56,57 To better understand the LS parasite populations, we assessed the ratios of schizont and hypnozoite formation from multiple experiments with different isolates of P. vivax sporozoites and found approximately 40–50% of the LS population formed hypnozoites, while the remaining forms were schizonts (Fig. 4B), similar to the ratio obtained in a commercial 384-well plate.45 To better understand the LS parasite populations over long-term μHEP culture, we infected P. vivax sporozoites into PHHs in active-treated 3 × 3 μHEP slides and fixed a slide at day 5, 8, 12, 14, and 30 post-infection (Fig. 4C). We noted most of the population of developing schizonts disappear after day 8 post-infection, presumably because they completed development and burst from their host hepatocytes, but hypnozoites persisted throughout the time course. Interestingly, at 30 days post-infection we noted the re-emergence of schizonts in culture. While there is currently no self-reporting P. vivax strain available to enable live-cell imaging and direct observation of a hypnozoite resuming development in vitro (this type of experiment being precluded by our inability to propagate and genetically modify the parasite), because at least 18 days passed between the first wave of schizont development and schizont re-emergence, we presume these schizonts are indeed the result of relapsing hypnozoites, similar to that seen in a P. vivax humanized mouse model.58
Because only the malaria blood stages cause disease, and because Plasmodium parasites complete an asymptomatic and obligatory liver stage prior to infection of the blood, killing LS parasites can prevent disease manifestation in the affected individual and prevent transmission via uptake of blood stage parasites by a mosquito.59 Thus the LS parasite population is widely considered as an underutilized and ideal therapeutic target.60 Imidazolo-piperazines and phosphatidyl-inositol 4 kinase (PI4K) inhibitors represent two novel group of antimalarial compounds with several compounds in development that possess potent inhibitory activity at a nanomolar range against blood and liver stages of the Plasmodium parasite. Recent studies have demonstrated that the lead imidazole-piperazine KAF156 and PI4K inhibitor KDU691 have prophylactic activities against both liver-resident parasite forms.61,62 We replicated prophylactic activity of KAF156 and KDU691 against P. vivax LS parasites in active-treated 3 × 3 μHEP slides. Using HC/RI to quantify the drug effect on LS parasite formation, we found both compounds extremely potent at reducing numbers of both LS populations. However, both compounds were able to kill schizonts but failed to clear hypnozoites in radical cure mode (Fig. 4D). Our findings are consistent with the recent primate model experiment with P. cynomolgi, a zoonotic species closely related to P. vivax, and recently published P. vivax results.45,63 Our in vitro confirmation of these data, in conjunction with high-resolution imaging of LS parasites, demonstrate utility of μHEP systems for small molecule testing. These studies also included primaquine, an 8-aminoquinoline, which is currently the only FDA approved drug class for radical cure treatment against liver hypnozoites.64 However, as has previously been shown for in vitro culture of P. vivax LS parasites,11 primaquine treatment results in malformed and likely nonviable LS parasite forms which persist in culture for weeks following treatment (Fig. S7†), thus a primaquine dose response was found negative over the 8-day course of our activity assay.
RNAseq characterization of PHH in μHEP versus traditional culture systems
Having found the basic viability and hepatic functional phenotypes of albumin production, factor IX production, and CYP3A4 induction of PHHs in long-term μHEP culture were not remarkably different than that found in a commercial 384-well plate (Fig. S6C and D†), we broadened our assessment of μHEP culture effect by performing RNAseq of PHH cultured for two weeks in active-treated 3 × 3 μHEP slides and three types of commercial microtiter culture systems. Two of the systems chosen, a 384-well plate and 24-well plate, were collagen-coated Corning products, in line with the other comparisons to commercial products described above. RNA was harvested on day 2, 8, and 14 post-seed for μHEP slides and on day 3 post-seed from the other systems. We defined the μHEP-cultured PHH properties by examining gene expression patterns for canonical primary hepatic functions of phase I & II metabolism, innate immunity, and principle hepatic differentiation regulators (Fig. 5A). We also generated a master hepatic metabolic map by using genes encoding rate-limiting enzymes catalyzing important hepatic pathways and found enhanced expression of most of these critical metabolic genes in μHEP culture (Fig. 5B). As our immediate goal is to use the μHEP concept for P. vivax biology and small molecule testing, we confirmed that hepatic genes associated with sporozoite invasion such as CD81 (day 14 FPKM > 100) and SCARB1 (day 14 FPKM > 300) are well-expressed.65 As studies of hypnozoites will require that the μHEP plate support long-term expression of PHH phenotypes, we demonstrated that the μHEP preserves most primary hepatocyte properties including the cellular capacity to support long-term LS culture. In particular, by day 8, PHHs in μHEP culture had self-aggregated into multicellular functional units including bile canaliculi between neighboring cells (Fig. 1E). This was confirmed by the expression of bile acid production-related genes. Taken together, these μHEP-cultured PHH RNAseq results are highly consistent with findings from our albumin production assays, cell viability stains, and P. vivax infections (Fig. 5C).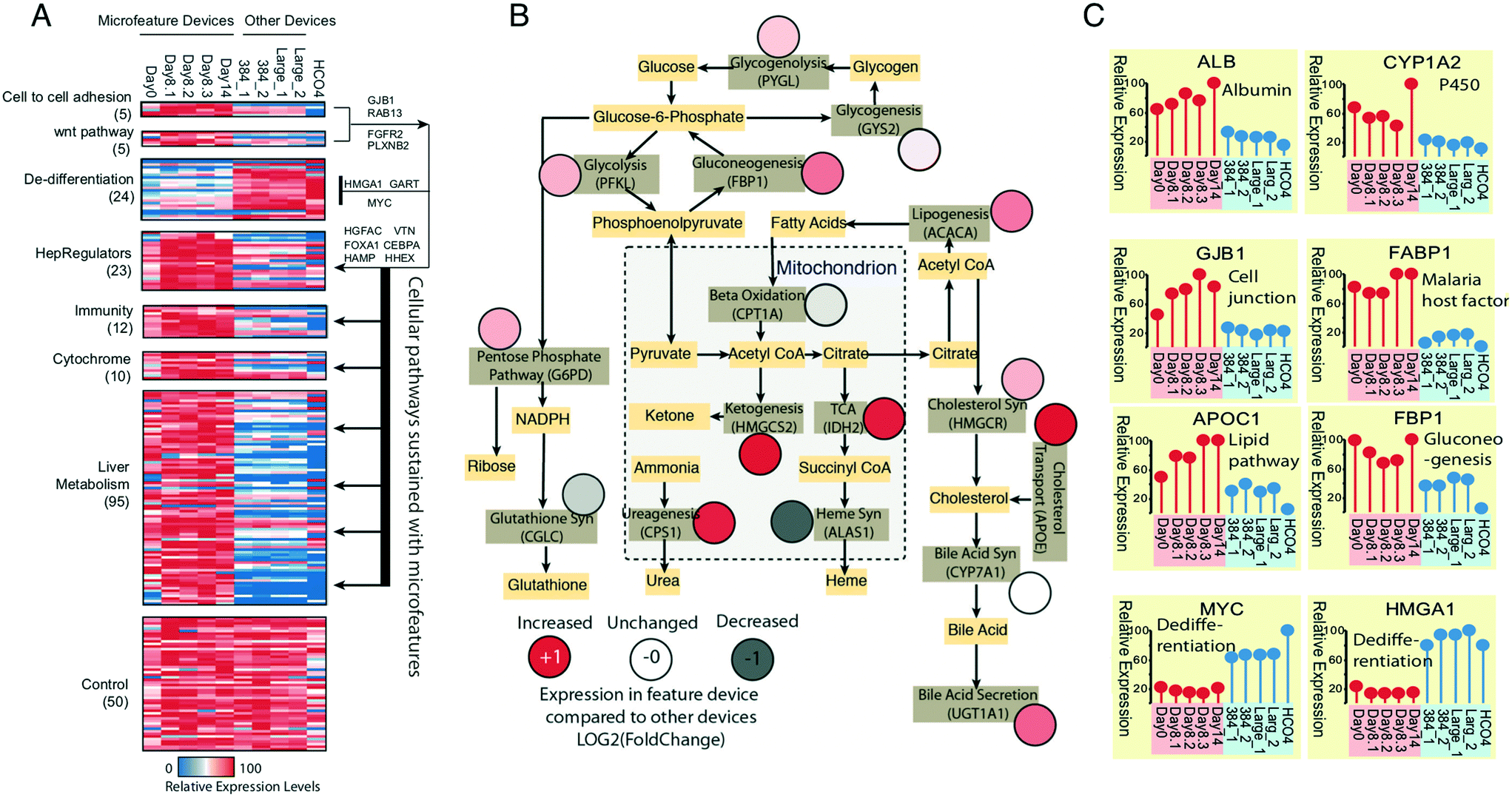 | ||
| Fig. 5 Evaluation of long-term PHH properties in μHEP culture with RNAseq. (A) Primary human hepatocytes (PHHs) were seeded into active-treated 3 × 3 μHEP cultures and total RNA collected at the time of seeding (‘Day 0’), from three replicates after 8 days of culture, and from a single replicate after 14 days of culture. Commercially available collagen-coated Corning 384-well plates (two replicates, ‘384_1’ and ‘384_2’), a collagen-coated Corning 24-well plate (‘Large_1’) and a collagen-coated glass bottom 12-well plate (‘Large_2’) were also seeded and RNA collected after 3 days, to serve as a comparison to commercial devices. Genes encoding crucial primary cell functions such as cell-to-cell adhesion, innate immunity, and hepatic metabolic pathways were highly expressed during two weeks of μHEP culture but exhibit lower expression levels in a variety of commercial devices. In contrast, the expression of dedifferentiation marker genes such as HMGA1 and MYC were significantly lower in μHEP culture (P < 0.01, Fisher's exact test and P value adjusted with Benjamini–Hochberg corrections). The pathway on the right of the heatmap indicates distinct molecular events initiated with μHEP devices. (B) A master metabolic map of PHHs is generated with rate-liming enzymes catalyzing important metabolic pathways in hepatocytes. The color-filled circles represent the relative changes in gene expression between μHEP culture and the average value of all other control devices. Mitochondrial metabolic pathways are marked with dotted lines. Most metabolic functions show enhanced expression (pink or red) in μHEP devices as compared to commercial devices. (C) Representatives of gene differential expression patterns in μHEP versus other devices. All expression levels were normalized to the scale of 1 to 100. Fisher's exact test were performed on unscaled FPKM data with P < 0.01. Red indicates gene expression levels in μHEP culture; blue indicates gene expression levels from commercial devices. | ||
Conclusions
In the field of tissue engineering and advanced cell culture models, often enhancement of organ-like function in vitro comes at the cost of methodological and structural complexity which can diminish or completely eliminate important assay elements such as optical accessibility and scale. Herein we describe how a simple and fundamental cell culture concept of cell monolayer confinement can be used to enable long-term culture of PHHs and developed a novel fabrication process to produce a multiplex culture (“μHEP”) slide and plate with microfeatures embossed into the bottom of culture wells. Our novel soft-mold embossing process was used to nimbly form alternative microfeature geometries into wells to systematically identify ideal designs for our specific culture needs: PHHs infected with P. vivax LS parasites. We characterized the growth characteristics of both the host PHHs and LS parasites in long-term culture, culminating in a proof-of-concept antimalarial susceptibility assay using HC/RI to gather endpoint data. RNAseq analysis suggests microfeatures do not promote a particular hepatic phenotype, rather, μHEP culture is uniquely capable of maintaining hepatic differentiation on a whole-cell level. Ongoing μHEP system development aims to incorporate additional “assay-ready” fabrication elements, such as pre-collagen-coated systems, and application of soft-mold embossing toward alternative organ models.Author contributions
Conceptualization, S. P. M., A. J. C., J. H., H. C. S. S., B. C., W. M. S., J. H. A., D. E. K.; data curation, S. P. M., A. J. C., S. R. A., P. C., R. H. Y. J.; formal analysis, S. P. M., S. R. A., P. C., R. H. Y. J., funding acquisition, S. P. M., W. M. S., J. H. A., D. E. K., investigation, S. P. M., A. J. C., A. R., C. A., J. T., M. J., C. P., S. R. A., N. S., P. C., V. C., V. V. G., R. C., R. P.; methodology, S. P. M., A. J. C., J. T., J. H., H. C. S. S., R. H. Y. J.; project administration, S. P. M., C. A., S. J. B., V. C.; resources, S. P. M., A. J. C., C. A., N. S., S. J. B., J. S., V. C., B. C.; supervision, H. C. S. S., W. M. S., J. H. A., D. E. K.; validation, S. P. M., A. J. C., W. M. S.; writing—original draft, S. P. M., A. J. C., S. R. A.; writing—review & editing, A. R., P. C., V. C., W. M. S., J. H. A., D. E. K.Conflicts of interest
B. C. is an employee of Medicines for Malaria Venture. The soft mold embossing process has been disclosed as a lapsed patent application US20150368599A1 (authors S. P. M., W. M. S., A. J. C., H. C. S. S., D. E. K., and J. H. A.). The other authors have no conflicts to declare.Acknowledgements
We thank the Thai P. vivax patients and insectary staff of MVRU and SMRU for their participation in this study. Shoklo Malaria Research Unit is part of the Mahidol Oxford University Research Unit, supported by the Wellcome Trust of Great Britain (F. N.) We thank Dr. Ron Faris at Corning for surface treatment applications. We thank Dr. Joseph Cuiffi at Florida Gulf Coast University for invaluable advice. We thank staff at The Charles Stark Draper Laboratory for assistance with photolithography and fabrication techniques and the Draper Laboratory Fellowship (DLF) program for support (S. P. M.). Funding support was provided by the American Cancer Society (ACS-IRG-14-189-19 to R. H. Y. J.), the Women's Health Collaborative (#310033 to R. H. Y. J.), and the Bill & Malinda Gates Foundation (OPP1023643 to J. H. A. and OPP1023601 to R.H.Y.J. and D. E. K.).Notes and references
- Y. Zhao, R. K. Kankala, S. B. Wang and A. Z. Chen, Molecules, 2019, 24, 675 CrossRef CAS PubMed.
- I. Meyvantsson, J. W. Warrick, S. Hayes, A. Skoien and D. J. Beebe, Lab Chip, 2008, 8, 717–724 RSC.
- P. Joshi, A. Datar, K. N. Yu, S. Y. Kang and M. Y. Lee, Toxicol. In Vitro, 2018, 50, 147–159 CrossRef CAS PubMed.
- B. Zhang, A. Korolj, B. F. L. Lai and M. Radisic, Nat. Rev. Mater., 2018, 3, 257–278 CrossRef.
- R. Gebhardt, Pharmacol. Ther., 1992, 53, 275–354 CrossRef CAS PubMed.
- P. H. Hayashi and E. S. Bjornsson, Curr. Hepatol. Rep., 2018, 17, 292–299 CrossRef PubMed.
- P. Maurel, Adv. Drug Delivery Rev., 1996, 22, 105–132 CrossRef CAS.
- V. M. Lauschke, D. F. Hendriks, C. C. Bell, T. B. Andersson and M. Ingelman-Sundberg, Chem. Res. Toxicol., 2016, 29, 1936–1955 Search PubMed.
- B. Y. Winer, T. S. Huang, E. Pludwinski, B. Heller, F. Wojcik, G. E. Lipkowitz, A. Parekh, C. Cho, A. Shrirao, T. W. Muir, E. Novik and A. Ploss, Nat. Commun., 2017, 8, 125 Search PubMed.
- S. March, S. Ng, S. Velmurugan, A. Galstian, J. Shan, D. J. Logan, A. E. Carpenter, D. Thomas, B. K. Sim, M. M. Mota, S. L. Hoffman and S. N. Bhatia, Cell Host Microbe, 2013, 14, 104–115 CrossRef CAS PubMed.
- N. Gural, L. Mancio-Silva, A. B. Miller, A. Galstian, V. L. Butty, S. S. Levine, R. Patrapuvich, S. P. Desai, S. A. Mikolajczak, S. H. I. Kappe, H. E. Fleming, S. March, J. Sattabongkot and S. N. Bhatia, Cell Host Microbe, 2018, 23, 395–406 CrossRef CAS PubMed , e394.
- S. R. Khetani and S. N. Bhatia, Nat. Biotechnol., 2008, 26, 120–126 CrossRef CAS PubMed.
- P. V. Moghe, F. Berthiaume, R. M. Ezzell, M. Toner, R. G. Tompkins and M. L. Yarmush, Biomaterials, 1996, 17, 373–385 CrossRef CAS PubMed.
- P. Gissen and I. M. Arias, J. Hepatol., 2015, 63, 1023–1037 CrossRef PubMed.
- S. March, V. Ramanan, K. Trehan, S. Ng, A. Galstian, N. Gural, M. A. Scull, A. Shlomai, M. M. Mota, H. E. Fleming, S. R. Khetani, C. M. Rice and S. N. Bhatia, Nat. Protoc., 2015, 10, 2027–2053 CrossRef CAS PubMed.
- K. Domansky, W. Inman, J. Serdy, A. Dash, M. H. Lim and L. G. Griffith, Lab Chip, 2010, 10, 51–58 RSC.
- L. Dembele, J. F. Franetich, A. Lorthiois, A. Gego, A. M. Zeeman, C. H. Kocken, R. Le Grand, N. Dereuddre-Bosquet, G. J. van Gemert, R. Sauerwein, J. C. Vaillant, L. Hannoun, M. J. Fuchter, T. T. Diagana, N. A. Malmquist, A. Scherf, G. Snounou and D. Mazier, Nat. Med., 2014, 20, 307–312 CrossRef CAS PubMed.
- Y. Wang, Y. C. Toh, Q. Li, B. Nugraha, B. Zheng, T. B. Lu, Y. Gao, M. M. Ng and H. Yu, Integr. Biol., 2013, 5, 390–401 CrossRef CAS PubMed.
- S. P. Maher, R. B. Crouse, A. J. Conway, E. C. Bannister, A. K. Achyuta, A. Y. Clark, F. L. Sinatra, J. D. Cuiffi, J. H. Adams, D. E. Kyle and W. M. Saadi, Biomed. Microdevices, 2014, 16, 727–736 CrossRef PubMed.
- V. N. Goral, S. H. Au, R. A. Faris and P. K. Yuen, Biomicrofluidics, 2014, 8, 046502 CrossRef PubMed.
- V. N. Goral, S. H. Au, R. A. Faris and P. K. Yuen, Lab Chip, 2015, 15, 1032–1037 RSC.
- E. Gevaert, L. Dollé, T. Billiet, P. Dubruel, L. van Grunsven, A. van Apeldoorn and R. Cornelissen, PLoS One, 2014, 9, e105171 CrossRef PubMed.
- G. H. Lee, J. S. Lee, W. Y. Joung, S. H. Kim, S. H. Lee, J. Y. Park and D. H. Kim, Biofabrication, 2017, 10, 015001 CrossRef PubMed.
- R. Mori, Y. Sakai and K. Nakazawa, J. Biosci. Bioeng., 2008, 106, 237–242 CrossRef CAS PubMed.
- S. Meister, D. M. Plouffe, K. L. Kuhen, G. M. Bonamy, T. Wu, S. W. Barnes, S. E. Bopp, R. Borboa, A. T. Bright, J. Che, S. Cohen, N. V. Dharia, K. Gagaring, M. Gettayacamin, P. Gordon, T. Groessl, N. Kato, M. C. Lee, C. W. McNamara, D. A. Fidock, A. Nagle, T. G. Nam, W. Richmond, J. Roland, M. Rottmann, B. Zhou, P. Froissard, R. J. Glynne, D. Mazier, J. Sattabongkot, P. G. Schultz, T. Tuntland, J. R. Walker, Y. Zhou, A. Chatterjee, T. T. Diagana and E. A. Winzeler, Science, 2011, 334, 1372–1377 CrossRef CAS PubMed.
- A. C. Y. Chua, A. Ananthanarayanan, J. J. Y. Ong, J. Y. Wong, A. Yip, N. H. Singh, Y. Qu, L. Dembele, M. McMillian, R. Ubalee, S. Davidson, A. Tungtaeng, R. Imerbsin, K. Gupta, C. Andolina, F. Lee, K. S.-W. Tan, F. Nosten, B. Russell, A. Lange, T. T. Diagana, L. Rénia, B. K. S. Yeung, H. Yu and P. Bifani, Biomaterials, 2019, 216, 119221 CrossRef CAS PubMed.
- C. M. Williams, G. Mehta, S. R. Peyton, A. S. Zeiger, K. J. Van Vliet and L. G. Griffith, Tissue Eng., Part A, 2011, 17, 1055–1068 CrossRef CAS PubMed.
- J. You, D. S. Shin, D. Patel, Y. Gao and A. Revzin, Adv. Healthcare Mater., 2014, 3, 126–132 CrossRef CAS PubMed.
- S. F. Wong, D. Y. No, Y. Y. Choi, D. S. Kim, B. G. Chung and S. H. Lee, Biomaterials, 2011, 32, 8087–8096 CrossRef CAS PubMed.
- J. Lee, B. Choi, D. Y. No, G. Lee, S. R. Lee, H. Oh and S. H. Lee, Integr. Biol., 2016, 8, 302–308 CrossRef CAS PubMed.
- G. Lee, J. Lee, H. Oh and S. Lee, PLoS One, 2016, 11, e0161026 CrossRef PubMed.
- J. Fukuda and K. Nakazawa, Tissue Eng., 2005, 11, 1254–1262 CrossRef CAS PubMed.
- M. W. Toepke and D. J. Beebe, Lab Chip, 2006, 6, 1484–1486 RSC.
- T. H. Lücking, F. Sambale, B. Schnaars, D. Bulnes-Abundis, S. Beutel and T. Scheper, Eng. Life Sci., 2015, 15, 57–64 CrossRef.
- V. N. Goral, Y. C. Hsieh, O. N. Petzold, J. S. Clark, P. K. Yuen and R. A. Faris, Lab Chip, 2010, 10, 3380–3386 RSC.
- J. Narasimhan and I. Papautsky, J. Micromech. Microeng., 2003, 14, 96–103 CrossRef.
- M. J. Zamek-Gliszczynski, H. Xiong, N. J. Patel, R. Z. Turncliff, G. M. Pollack and K. L. R. Brouwer, J. Pharmacol. Exp. Ther., 2003, 304, 801 CrossRef CAS PubMed.
- S. R. Khetani and S. N. Bhatia, Nat. Biotechnol., 2007, 26, 120 CrossRef PubMed.
- C. Andolina, J. Landier, V. Carrara, C. S. Chu, J. F. Franetich, A. Roth, L. Renia, C. Roucher, N. J. White, G. Snounou and F. Nosten, Malar. J., 2015, 14, 312 CrossRef PubMed.
- E. J. Lupton, A. Roth, R. Patrapuvich, S. P. Maher, N. Singh, J. Sattabongkot and J. H. Adams, Parasitol. Int., 2015, 64, 211–218 CrossRef PubMed.
- A. Roth, S. R. Adapa, M. Zhang, X. Liao, V. Saxena, R. Goffe, S. Li, R. Ubalee, G. S. Saggu, Z. R. Pala, S. Garg, S. Davidson, R. H. Y. Jiang and J. H. Adams, Sci. Rep., 2018, 8, 12183 CrossRef PubMed.
- S. A. Mikolajczak, A. M. Vaughan, N. Kangwanrangsan, W. Roobsoong, M. Fishbaugher, N. Yimamnuaychok, N. Rezakhani, V. Lakshmanan, N. Singh, A. Kaushansky, N. Camargo, M. Baldwin, S. E. Lindner, J. H. Adams, J. Sattabongkot and S. H. Kappe, Cell Host Microbe, 2015, 17, 526–535 CrossRef CAS PubMed.
- C. Schafer, N. Dambrauskas, R. W. Steel, S. Carbonetti, V. Chuenchob, E. L. Flannery, V. Vigdorovich, B. G. Oliver, W. Roobsoong, S. P. Maher, D. Kyle, J. Sattabongkot, S. H. I. Kappe, S. A. Mikolajczak and D. N. Sather, Malar. J., 2018, 17, 370 CrossRef CAS PubMed.
- W. Roobsoong, S. P. Maher, N. Rachaphaew, S. J. Barnes, K. C. Williamson, J. Sattabongkot and J. H. Adams, Malar. J., 2014, 13, 55 CrossRef PubMed.
- A. Roth, S. P. Maher, A. J. Conway, R. Ubalee, V. Chaumeau, C. Andolina, S. A. Kaba, A. Vantaux, M. A. Bakowski, R. T. Luque, S. R. Adapa, N. Singh, S. J. Barnes, C. A. Cooper, M. Rouillier, C. W. McNamara, S. A. Mikolajczak, N. Sather, B. Witkowski, B. Campo, S. H. I. Kappe, D. E. Lanar, F. Nosten, S. Davidson, R. H. Y. Jiang, D. E. Kyle and J. H. Adams, Nat. Commun., 2018, 9, 1837 CrossRef PubMed.
- D. Kim, G. Pertea, C. Trapnell, H. Pimentel, R. Kelley and S. L. Salzberg, Genome Biol., 2013, 14, R36 CrossRef PubMed.
- C. Trapnell, B. A. Williams, G. Pertea, A. Mortazavi, G. Kwan, M. J. van Baren, S. L. Salzberg, B. J. Wold and L. Pachter, Nat. Biotechnol., 2010, 28, 511–515 CrossRef CAS PubMed.
- F. Wilcoxon, J. Econ. Entomol., 1946, 39, 269 CrossRef CAS PubMed.
- Y. Benjamini, D. Drai, G. Elmer, N. Kafkafi and I. Golani, Behav. Brain Res., 2001, 125, 279–284 CrossRef CAS PubMed.
- K. Nakamura, R. Mizutani, A. Sanbe, S. Enosawa, M. Kasahara, A. Nakagawa, Y. Ejiri, N. Murayama, Y. Miyamoto, T. Torii, S. Kusakawa, J. Yamauchi, M. Fukuda, H. Yamazaki and A. Tanoue, J. Biosci. Bioeng., 2011, 111, 78–84 CrossRef CAS PubMed.
- D. Mazier, I. Landau, P. Druilhe, F. Miltgen, C. Guguen-Guillouzo, D. Baccam, J. Baxter, J. P. Chigot and M. Gentilini, Nature, 1984, 307, 367–369 CrossRef CAS PubMed.
- D. M. Bissell, S. C. Stamatoglou, M. V. Nermut and R. C. Hughes, Eur. J. Cell Biol., 1986, 40, 72–78 CAS.
- S. Zhang, L. Xia, C. H. Kang, G. Xiao, S. M. Ong, Y. C. Toh, H. L. Leo, D. van Noort, S. H. Kan, H. H. Tang and H. Yu, Biomaterials, 2008, 29, 3993–4002 CrossRef CAS PubMed.
- A. S. Zeiger, B. Hinton and K. J. Van Vliet, Acta Biomater., 2013, 9, 7354–7361 CrossRef CAS PubMed.
- C. J. Flaim, S. Chien and S. N. Bhatia, Nat. Methods, 2005, 2, 119–125 CrossRef CAS PubMed.
- W. A. Krotoski, Trans. R. Soc. Trop. Med. Hyg., 1985, 79, 1–11 CrossRef CAS PubMed.
- W. A. Krotoski, W. E. Collins, R. S. Bray, P. C. Garnham, F. B. Cogswell, R. W. Gwadz, R. Killick-Kendrick, R. Wolf, R. Sinden, L. C. Koontz and P. S. Stanfill, Am. J. Trop. Med. Hyg., 1982, 31, 1291–1293 CrossRef CAS PubMed.
- S. A. Mikolajczak, A. M. Vaughan, N. Kangwanrangsan, W. Roobsoong, M. Fishbaugher, N. Yimamnuaychok, N. Rezakhani, V. Lakshmanan, N. Singh, A. Kaushansky, N. Camargo, M. Baldwin, S. E. Lindner, J. H. Adams, J. Sattabongkot, J. Prachumsri and S. H. Kappe, Cell Host Microbe, 2015, 17, 526–535 CrossRef CAS PubMed.
- T. N. Wells, J. N. Burrows and J. K. Baird, Trends Parasitol., 2010, 26, 145–151 CrossRef PubMed.
- S. E. Lindner, J. L. Miller and S. H. Kappe, Cell. Microbiol., 2012, 14, 316–324 CrossRef CAS PubMed.
- C. W. McNamara, M. C. Lee, C. S. Lim, S. H. Lim, J. Roland, O. Simon, B. K. Yeung, A. K. Chatterjee, S. L. McCormack, M. J. Manary, A. M. Zeeman, K. J. Dechering, T. S. Kumar, P. P. Henrich, K. Gagaring, M. Ibanez, N. Kato, K. L. Kuhen, C. Fischli, A. Nagle, M. Rottmann, D. M. Plouffe, B. Bursulaya, S. Meister, L. Rameh, J. Trappe, D. Haasen, M. Timmerman, R. W. Sauerwein, R. Suwanarusk, B. Russell, L. Renia, F. Nosten, D. C. Tully, C. H. Kocken, R. J. Glynne, C. Bodenreider, D. A. Fidock, T. T. Diagana and E. A. Winzeler, Nature, 2013, 504, 248–253 CrossRef CAS PubMed.
- K. L. Kuhen, A. K. Chatterjee, M. Rottmann, K. Gagaring, R. Borboa, J. Buenviaje, Z. Chen, C. Francek, T. Wu, A. Nagle, S. W. Barnes, D. Plouffe, M. C. Lee, D. A. Fidock, W. Graumans, M. van de Vegte-Bolmer, G. J. van Gemert, G. Wirjanata, B. Sebayang, J. Marfurt, B. Russell, R. Suwanarusk, R. N. Price, F. Nosten, A. Tungtaeng, M. Gettayacamin, J. Sattabongkot, J. Taylor, J. R. Walker, D. Tully, K. P. Patra, E. L. Flannery, J. M. Vinetz, L. Renia, R. W. Sauerwein, E. A. Winzeler, R. J. Glynne and T. T. Diagana, Antimicrob. Agents Chemother., 2014, 58, 5060–5067 CrossRef PubMed.
- A. M. Zeeman, S. B. Lakshminarayana, N. van der Werff, E. J. Klooster, A. Voorberg-van der Wel, R. R. Kondreddi, C. Bodenreider, O. Simon, R. Sauerwein, B. K. Yeung, T. T. Diagana and C. H. Kocken, Antimicrob. Agents Chemother., 2016, 60, 2858–2863 CrossRef CAS PubMed.
- A. Llanos-Cuentas, M. V. G. Lacerda, T. T. Hien, I. D. Vélez, C. Namaik-Larp, C. S. Chu, M. F. Villegas, F. Val, W. M. Monteiro, M. A. M. Brito, M. R. F. Costa, R. Chuquiyauri, M. Casapía, C. H. Nguyen, S. Aruachan, R. Papwijitsil, F. H. Nosten, G. Bancone, B. Angus, S. Duparc, G. Craig, V. M. Rousell, S. W. Jones, E. Hardaker, D. D. Clover, L. Kendall, K. Mohamed, G. C. K. W. Koh, V. M. Wilches, J. J. Breton and J. A. Green, N. Engl. J. Med., 2019, 380, 229–241 CrossRef CAS PubMed.
- G. Manzoni, C. Marinach, S. Topcu, S. Briquet, M. Grand, M. Tolle, M. Gransagne, J. Lescar, C. Andolina, J. F. Franetich, M. B. Zeisel, T. Huby, E. Rubinstein, G. Snounou, D. Mazier, F. Nosten, T. F. Baumert and O. Silvie, eLife, 2017, 6, e25903 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00921c |
| This journal is © The Royal Society of Chemistry 2020 |