
Microphysiological systems for ADME-related applications: current status and recommendations for system development and characterization

Abstract
Over the last decade, progress has been made on the development of microphysiological systems (MPS) for absorption, distribution, metabolism, and excretion (ADME) applications. Central to this progress has been proof of concept data generated by academic and industrial institutions followed by broader characterization studies, which provide evidence for scalability and applicability to drug discovery and development. In this review, we describe some of the advances made for specific tissue MPS and outline the desired functionality for such systems, which are likely to make them applicable for practical use in the pharmaceutical industry. Single organ MPS platforms will be valuable for modelling tissue-specific functions. However, dynamic organ crosstalk, especially in the context of disease or toxicity, can only be obtained with the use of inter-linked MPS models which will enable scientists to address questions at the intersection of pharmacokinetics (PK) and efficacy, or PK and toxicity. In the future, successful application of MPS platforms that closely mimic human physiology may ultimately reduce the need for animal models to predict ADME outcomes and decrease the overall risk and cost associated with drug development.
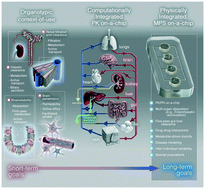
- This article is part of the themed collections: Microphysiological systems in pharmaceutical safety and ADME applications and organ-on-a-chip systems: translating concept into practice
 Please wait while we load your content...
Please wait while we load your content...

