
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly sensitive mutation quantification by high-dynamic-range capillary-array electrophoresis (HiDy CE)†
Takashi
Anazawa
 *a,
Hiroko
Matsunaga
ac,
Shuhei
Yamamoto
b and
Ryoji
Inaba
b
*a,
Hiroko
Matsunaga
ac,
Shuhei
Yamamoto
b and
Ryoji
Inaba
b
aResearch & Development Group, Hitachi, Ltd., 1-280 Higashi-koigakubo, Kokubunji, Tokyo 185-8601, Japan. E-mail: takashi.anazawa.rc@hitachi.com
bScience & Medical Systems Business Group, Hitachi High-Technologies Corporation, 882 Ichige, Hitachinaka, Ibaraki 312-8504, Japan
cPresent address: Research Organization for Nano and Life Innovation, Waseda University, 513 Waseda-tsurumaki-cho, Shinjuku, Tokyo 162-0041, Japan
First published on 28th February 2020
Abstract
A simple and robust ultra-small four-color-fluorescence detection system was developed by integrating its components, namely, a four-capillary array, an injection-molded-plastic four-lens array, a four-dichroic-mirror array, and a CMOS sensor, as one device. The developed system was applied to a high-dynamic-range capillary-array electrophoresis (HiDy CE) to quantify a rare EGFR mutant (MT) of exon 19 deletion in a large excess of EGFR wild type (WT). Samples with serially diluted MT and constant-concentration WT were co-amplified by competitive PCR and subjected to HiDy CE. The MT peak in each electropherogram was then compared to the WT peak. As a result, MT was quantified with high-sensitivity (LOD of 0.004% MT/WT) and four-orders-of-magnitude dynamic range (0.01–100% MT/WT) by HiDy CE. Moreover, compared with existing methods, HiDy CE achieves higher speed, higher sample throughput, and lower consumable cost per sample. It has therefore great potential to be used in clinical practice.
Introduction
A liquid biopsy for cancer diagnosis, treatment, and prognosis is gaining a great deal of attention because it is non-invasive, and its blood specimen includes circulating tumor DNA (ctDNA) continuously released from tumor cells throughout the body.1–4 Accordingly, it is valuable in regard to not only cancer patients but also healthy people. More importantly, it can be frequently performed to monitor the dynamic change of the highly heterogeneous genetic profile of tumor cells. For example, such a longitudinal liquid biopsy is remarkably useful for monitoring response and resistance to treatment (e.g., drug).5,6 However, ctDNA often represents an extremely small ratio (<1%) in relation to normal circulating cell-free DNA (cfDNA) in blood.2 Therefore, a highly sensitive assay is required to quantify rare mutated DNA (MT) in a large excess of wild-type DNA (WT), that is, the assay with the limit-of-detection (LOD) for mutant-to-wild-type ratio (MT/WT) of <1%.1–4,7 It is thus important to not only detect MT at low concentration but also quantify MT/WT. Moreover, in clinical use, the assay should be cost-effective, high-speed, and high-throughput, especially when the liquid biopsy is repeated. As for a liquid biopsy, various methods with high analytical sensitivity have been developed.Next-generation sequencing (NGS) is the most-used of these methods. Although NGS detects all possible mutations, including unknown mutations, systematic sequencing errors limit its sensitivity to LOD of >1% MT/WT.2,4 To overcome that limitation, several NGS techniques combining barcoding (unique identification of each DNA-template molecule), targeting (previous selection of known mutations), computational-error suppression, and/or deep sequencing have been developed.8–10 As a result, sensitivity can be drastically improved to LOD of <0.01% MT/WT. However, NGS is too expensive and time consuming for clinical use. Moreover, expertise in bioinformatics is required for data analysis and interpretation.2,4 NGS is therefore more suited to discovery and validation of mutations applicable to clinical use rather than clinical use itself.
Droplet digital PCR (ddPCR) is an alternative to NGS.11–14 In principle, sensitivity and dynamic range of ddPCR increases with number of detected droplets for each specimen when false-positive droplets are negligible. For example, LOD is <0.01% MT/WT when >4 × 105 droplets are detected.2,11 However, because multiplexity and sensitivity share a trade-off relationship, one of them could be sacrificed for the other.15–17 In addition, because plural specimens, with each divided into millions of droplets, are sequentially detected in non-reusable microfluidic devices, throughput and cost-effectiveness are still insufficient for clinical use.
Although capillary electrophoresis (CE) is a traditional analog method in contrast to NGS and ddPCR, it is more straightforward and robust. Besides, when combined with laser-induced multicolor-fluorescence detection and parallel processing of CEs, that is, capillary-array electrophoresis,18–24 it obtains high-multiplexity and high-throughput. More importantly, its cost per sample is low because the capillary-array device is reusable and only a small amount of reagent is consumed in each analysis. Furthermore, serial analyses of different specimens with the same capillary-array device is already automated in commercial capillary-array DNA sequencers (e.g., Applied Biosystems® 3500/3500xL Genetic Analyzers). However, sensitivity in the case of rare-mutation detection in a quantitative manner is low. Sanger sequencing by CE has sensitivity with LOD of 5–20% MT/WT.2,22,25,26 This low sensitivity is mainly because adjacent peaks in an electropherogram overlap, and one of them forms high background for the other. Even in the case of fragment analysis by CE, where adjacent peaks are sufficiently separated in an electropherogram, LOD is limited to no less than 1–5% MT/WT.27–30 This limited sensitivity is due to a conventional detection system having dynamic range of less than three orders of magnitude.
In the meantime, an ultra-small (three-orders-of-magnitude smaller than a conventional detection system) multicolor-fluorescence detection system for capillary-array electrophoresis has been recently developed.31 Moreover, as an unexpected but invaluable by-product of that development, dynamic range of more than four orders of magnitude was obtained. In this study, therefore, the system was applied to high-dynamic-range capillary-array electrophoresis, termed “HiDy CE,” for highly sensitive quantification of mutations. An epidermal growth factor receptor (EGFR) exon 19 deletion, which accounts for 45% of all EGFR mutations carried by non-small-cell lung-cancer (NSCLC) patients, was selected as a target mutation.32,33 Then, WT and MT around the locus of the exon 19 deletion were synthesized. After that, model samples containing WT and MT at various concentration ratios were co-amplified by competitive PCR. Finally, highly sensitive mutation quantification with LOD of 0.01% MT/WT and four-orders-of-magnitude dynamic range was demonstrated by using HiDy CE.
Experimental
Lens array
A four-lens array was fabricated by Nalux CO., LTD., or Enplas Corporation, through injection molding of ZEONEX®, a cycloolefin polymer, with refractive index of 1.528 at 600 nm. It was designed so that four lenses were arrayed at intervals of 1 mm in the center of a 15 × 10 mm plate, and each lens was a plano-convex aspheric lens with diameter of ϕ1 mm, height of 0.68 mm, and focal length of 1.4 mm, i.e., NA of 0.36. The aspheric surface profile z (sag) as a function of r (radial distance from the optical axis) is given bywhere R = 0.7393587 mm, κ = −0.6143667, α4 = 0.024171492, α6 = 0.004130857, α8 = 0.006881871, and α10 = −0.020907492.

Sample preparation and assay design
15 bp in-frame deletion in exon 19 (delE746-A750) (accounting for 68% of EGFR exon 19 deletion) was targeted. As shown in Fig. S-1 in the ESI,† WT and MT were synthesized by Sigma-Aldrich so that they were respectively 107-base and 92-base single-stranded genomic DNAs. Both include the 15-base deletion locus, but only MT includes the 15-base deletion. Samples with 100%, 10%, 1%, 0.1%, 0.01%, and 0% MT/WT were prepared so that molarities of MT were 10 nM, 1 nM, 0.1 nM, 0.01 nM, 0.001 nM, and 0 nM, respectively, and all the samples contained 10 nM WT, 10 mM Tris-HCl (pH 8.0), and 0.1% Tween 20. A forward primer (FP) and a reverse primer (RP) were also synthesized by Sigma-Aldrich so that WT and MT were co-amplified by competitive PCR to give PCR products of 87-base-pair and 72-base-pair DNA fragments, respectively. RP was labeled with 6-FAM™ at its 5′-terminus.For each sample, first, 25 μl of PCR mixture consisting of 1× KAPA HiFi HotStart Ready mix (KAPA Biosystems), 0.3 μM of FP and RP, and 2 μl of the sample was prepared. Next, PCR was performed by initial denaturation at 95 °C for 3 min followed by 25 cycles of denaturation at 98 °C for 20 s, annealing at 62 °C for 15 s, extension at 72 °C for 15 s, and a final extension at 72 °C for 1 min. Then, excess primers and other potential non-specific by-products were removed by using NucleoSpin® Gel and PCR Clean-up kit (MACHEREY-NAGEL) according to the manufacturer's protocol. The PCR products were eluted by 30 μl of Elution Buffer NE (5 mM Tris/HCl, pH 8.5). After that, 10 μl of solution consisting of 1 μl of the 50-times-diluted eluted solution, 1 μl of 50-times-diluted GeneScan™ 500 ROX™ dye Size Standard (Thermo Fisher Scientific), and 8 μl of Hi-Di™ Formamide (Thermo Fisher Scientific) was prepared. Finally, the solution was denatured at 94 °C for 2 min and cooled on ice for 5 min, and then subjected to CE.
Capillary-array electrophoresis
Total lengths of four capillaries with outer and inner diameters of 0.36 mm and 0.05 mm, respectively, were 47 cm. Detection points of the four capillaries were 36 cm from the cathode ends (effective lengths were 36 cm). The four detection points are defined as four positions in the interiors of the capillaries at which a focused laser beam is irradiated and from which fluorescence is emitted. Polyimide coatings of the capillaries around the detection points were removed in advance. The temperature of 32 cm regions of the capillaries (between positions 2 cm and 34 cm from the cathode ends) was kept at 60 °C by placing the regions in a tube in which 60 °C water was continuously flowing. The other regions were left at room temperature. A POP-7™ polymer solution (Thermo Fisher Scientific) were injected into the capillaries from the anode ends by applying a pressure of 3 MPa for 5 min to the polymer solution. Then, for pre-electrophoresis, with the cathode ends immersed in 1× Genetic Analyzer Buffer with EDTA (Thermo Fisher Scientific), 15 kV was applied across both ends for 3 min. Next, for sample injection, with the cathode ends respectively immersed in sample solutions, 1.2 kV was applied for 24 s. After that, for electrophoresis, with the cathode ends immersed in 1× Genetic Analyzer Buffer with EDTA, 8.5 kV was applied for 15 min.Fluorescence image sensing
A CMOS image sensor (C11440-52U, Hamamatsu Photonics K.K.) was used for fluorescence image sensing. The sensor area is 13.312 × 13.312 mm, composed of 2048 × 2048 pixels, each with a size of 6.5 × 6.5 μm. Part of the sensor area with 1320 × 660 pixels (8.58 × 4.29 mm) on which four-color (green, yellow, orange, and red) fluorescence images of the four capillaries (total of sixteen fluorescence images) were formed was used in 2 × 2 binning mode. For each electrophoretic analysis, 1200 frames of sensor images were acquired for 10 min (from 5 to 15 min of electrophoretic migration), with 16 bit A/D conversion, exposure time of 400 ms, and interval of 500 ms.Data analysis
Within four-color fluorescence images of four capillaries in each sensor image, only green and red fluorescence images (total of eight fluorescence images) were used. First, regions of interest (ROIs) with 100 × 100 pixels (0.65 × 0.65 mm) and 100 × 140 pixels (0.65 × 0.91 mm) were defined at positions of green and red fluorescence images of four capillaries, respectively. Second, baseline-subtracted and averaged fluorescence intensities within the ROIs in each sensor image were calculated to give time courses of green and red fluorescence intensities of the four capillaries (Fig. S-2 in the ESI†). Third, green and red fluorescence intensities were deconvoluted to remove spectral overlap between FAM and ROX fluorescence in each sensor image and give time courses of FAM and ROX fluorescence intensities (Fig. S-3†). Fourth, FAM and ROX fluorescence intensities as a function of migration time were converted to FAM and ROX fluorescence intensities as a function of base length (FAM and ROX electropherograms) by using a least-squares quadratic curve fitted to four data points of base lengths of 35-, 75-, 100-, and 139-base fragments of the Size Standard and their migration times. Here, the data point for the 50-base fragment was not used because of its anomalous migration speed. Finally, FAM fluorescence intensity at each base length in each FAM electropherogram was normalized (multiplied by a constant factor) so that FAM fluorescence intensity of WT peak was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 counts (Fig. S-4†). In the same way, ROX fluorescence intensity at each base length in each ROX electropherogram was normalized so that ROX fluorescence intensity of 100-base-fragment peak of the size standard was 10000 counts (Fig. S-4†). In this study, “counts” is used as an arbitrary unit of fluorescence intensity for mutual comparison.
000 counts (Fig. S-4†). In the same way, ROX fluorescence intensity at each base length in each ROX electropherogram was normalized so that ROX fluorescence intensity of 100-base-fragment peak of the size standard was 10000 counts (Fig. S-4†). In this study, “counts” is used as an arbitrary unit of fluorescence intensity for mutual comparison.
Results and discussion
Ultra-small multicolor fluorescence detection system
As shown in Fig. 1a and b, the plastic four-lens array was injection-molded in accordance with the design. Each optical axis of the four lenses deviated from its designed position by less than 1 μm. The diameter of each lens was ϕ0.997 mm. The form error of the aspherical surface of each lens was also less than 1 μm PV (peak-to-valley value). As a result it became possible to mass produce the four-lens array with a high degree of accuracy and at low cost. | ||
| Fig. 1 Photographs of key components of an ultra-small, four-capillary, four-color fluorescence detection system. a and b: Four-lens array (LA, each lens with f 1.4 mm) fabricated by plastic-injection molding; c and d: four-capillary array (CA, each capillary with outer diameters of 0.36 and inner diameters (ID) of 0.05 mm) around detection points with apertures of ϕ0.25 mm pinholes; e: LA mounted on CA; f: four-dichroic-mirror array (DA); g: DA mounted on an adapter (A). | ||
Meanwhile, as shown in Fig. 1c and d, a four-capillary array was formed by a capillary-array holder. The four capillaries around the detection points were arrayed on the same plane at intervals of 1 mm. Four pinholes with diameters of ϕ0.25 mm were positioned close to (at distances of 0.23 mm from) the four detection points, respectively.
Then, as shown in Fig. 1e, the four-lens array was mounted on the capillary-array holder with screws so that each of the four detection points was on the optical axis of the corresponding lens. They were designed so that distance between each capillary and the corresponding lens was automatically the optimal value (1.54 mm).
The four-dichroic-mirror array shown in Fig. 1f, which is the same as that used in the previous system,31 was mounted on an adapter as shown in Fig. 1g. As shown in Fig. 2a, the adapter with the four-dichroic-mirror array was then fixed on the image-sensor board (the adapter with the four-dichroic-mirror array in Fig. 1g was flipped upside down). After that, as also shown in Fig. 2a, the four-capillary array with the four-lens array was mounted in place on the adapter using positioning pins and magnets (the four-capillary array with the four-lens array in Fig. 1e was also flipped upside down). Perspective and cross-sectional schematics of the system perpendicular and parallel to the axes of the capillaries are shown in Fig. 2b and c, respectively.
 | ||
| Fig. 2 Configuration of integrated ultra-small, four-capillary, four-color fluorescence detection system. A with DA (Fig. 1g) was fixed on the image-sensor board (SB), and then CA with LA (Fig. 1e) was attached on A. a: Photograph of the system. A laser beam (LB) is schematically superimposed. b and c: Perspective and cross-sectional schematics of the system perpendicular (b) and parallel (c) to the axes of the capillaries. Only schematics of CA in b and LB in c are not cross-sectional for ease of comprehension. CA and the image-sensor surface (SS) were parallel and separated by a distance of 11.8 mm. | ||
A 505 nm laser beam with a power of 15 mW was irradiated from the side direction of the four-capillary array. The laser beam was confined in the four-capillary array by multiple-laser-beam-focusing technique.34 As a result, the four detection points were simultaneously irradiated. Because the laser beam intensity gradually decreases by laser-light-reflection loss at each capillary (7% per capillary) as number of capillaries through which the laser beam passes increases, the laser beam intensity of the fourth capillary was as much as 80% (93%3) of that of the first capillary. Therefore, relatively uniform laser-beam-intensity distribution in the four-capillary array can be obtained in this study.
Fluorescence from each detection point was collimated by each respective lens, split into four color fluxes (green: 520–555 nm, yellow: 555–585 nm, orange: 585–620 nm, and red: 620–675 nm) by the four dichroic mirrors, and then directly input into the image sensor. As shown in Fig. 3a, a total of sixteen four-color images, that is, green, yellow, orange, and red fluorescence images of the four detection points, were simultaneously, efficiently, and independently obtained by the image sensor.
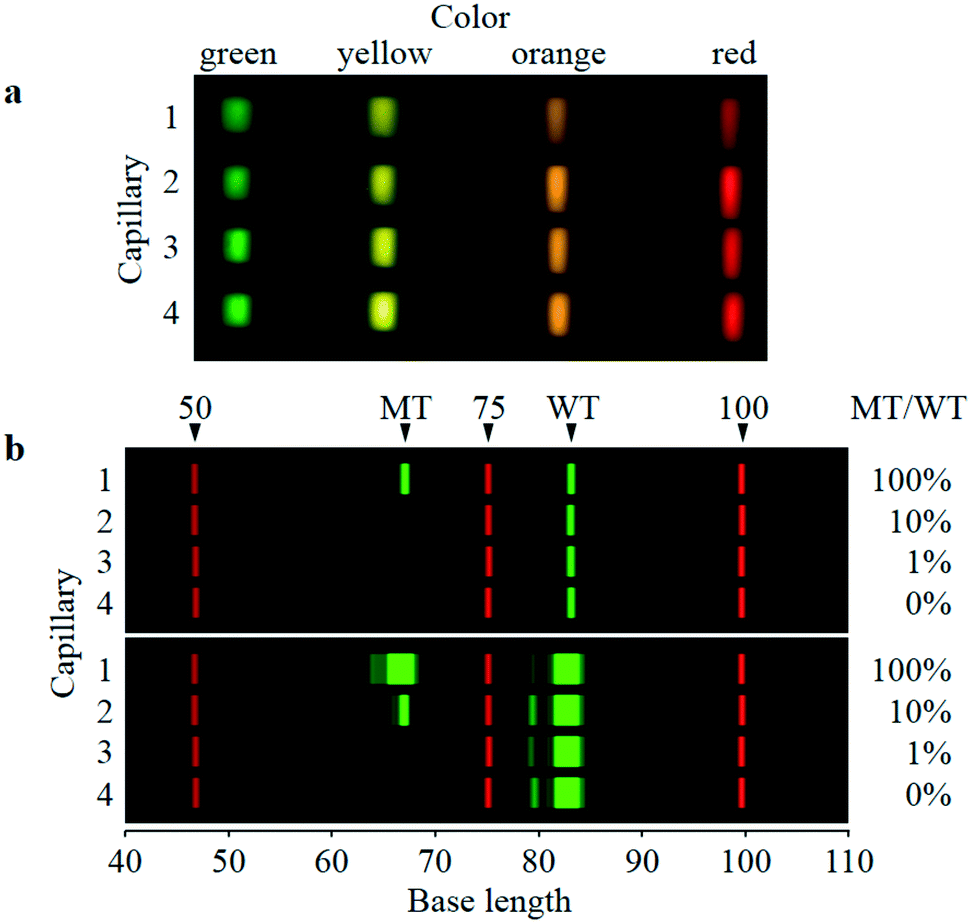 | ||
| Fig. 3 Sixteen four-color-split images and two-color electrophoretic images of the four-capillary array. a: Four-color (green: 520–555 nm, yellow: 555–585 nm, orange: 585–620 nm, and red: 620–675 nm) and four-capillary averaged-fluorescence images taken by the image sensor. Each image is a roughly 12-times-magnified image of the detection point, although the sizes and shapes of the green, yellow, orange, and red images slightly differ because of optical-length difference between each lens and the image-sensor surface. b: Digital-two-color-electrophoretic images (40–110 bases) of the same four-capillary array. Single-stranded DNA fragments of FAM-labeled PCR products of 100% MT/WT (capillary 1), 10% MT/WT (capillary 2), 1% MT/WT (capillary 3), and 0% MT/WT (capillary 4) and ROX-labeled size standard (capillary 1–4) are separated. FAM and ROX fluorescence respectively indicated by green and red were derived from intensities of green and red fluorescence images in a for each capillary. FAM bands of WT and MT and ROX bands of 50, 75, and 100 base fragments of size standard are denoted. Brightness of FAM and ROX fluorescence are both scaled to intensities of 0–10000 counts in upper images, whereas only brightness of FAM fluorescence is changed from the upper images to scale of intensity of 0–1000 counts in lower images. | ||
Advancements from the previous system
This system has made significant advancements over the previous system of ref. 31 in terms of simplicity, reproducibility, and robustness. The previous system was also composed of a four-capillary array, a four-lens array, a four-dichroic-mirror array, and a CMOS image sensor. However, it was necessary to fine-tune the relative positions of these compositional units by using external micromanipulators. In addition, it was difficult to form the four-lens array because it was fabricated by accurately bonding four square-cut glass lenses. To eliminate the need for fine-tuning of the relative positions, therefore, the compositional units were integrated as one device in this system as shown in Fig. 2. Moreover, to fabricate the four-lens array easily, precisely, and at low cost, an injection-molded-plastic four-lens array was developed as shown in Fig. 1a and b. It was therefore made straightforward to fine-tune and maintain the relative positions of the compositional units. As a result, four-color fluorescence detection data for four-capillary array electrophoresis can be acquired in large quantities with high quality and stability.When this system is commercialized in future, a capillary array will be distributed to a user with a lens array fixed in advance as shown in Fig. 1e. The user will only need to mount the capillary array with the lens array on the image sensor with the dichroic-mirror array as shown in Fig. 2 and will not have to fine-tune the relative positions of the compositional units. Reproducibility of the mounting was demonstrated in “Reproducibility of capillary-array mounting.” with Fig. S-5 and S-6 in the ESI.† As with the current commercial capillary-array DNA sequencers (e.g., Applied Biosystems® 3500/3500xL Genetic Analyzer), the same capillary array can be used over 100 times. It is also easy to remove the old capillary array with the lens array after more than 100-times use from the image sensor and mount a new one with high accuracy and high reproducibility.
From the above discussion, this system is simpler, more reproducible, and more robust than the previous system, and can therefore be applied to highly sensitive mutation assay proposed in this study.
Highly sensitive mutation quantification
As shown in Fig. 3b, four electrophoretic images (40–110 bases) of FAM-labeled PCR products of WT and MT (green signals) along with ROX-labeled fragments of Size Standard (red signals) were simultaneously obtained by the same four-capillary array. Samples of 100%, 10%, 1%, and 0% MT/WT are separated in capillaries 1, 2, 3, and 4, respectively. The upper and lower images are the same data, but the brightness of FAM fluorescence in both images is changed. In the upper images, whereas the WT bands are clearly observed in all the capillaries, the MT band is clearly observed in capillary 1, faintly observed in capillary 2, but not observed in capillaries 3 and 4. Contrarily, in the lower images, the MT band is made faintly visible in capillary 3 but is still invisible in capillary 4, although the MT band in capillary 1 and the WT bands in all the capillaries are oversaturated. Electropherograms corresponding to the upper and lower images for 100% MT/WT are shown in Fig. S-4.† These results are satisfactory because MT-band brightness increases with increasing MT/WT, whereas WT-band brightness is constant regardless of MT/WT.Base lengths of the MT and WT bands in Fig. 3b are estimated to be respectively 67 and 83 bases. These values are respectively 5 and 4 bases shorter than actual base lengths of the MT and WT bands (72 and 87 bases). Because the horizontal axis (base length) in Fig. 3b is derived from electrophoretic mobility of ROX-labeled fragments, these differences in base length are caused by dye-induced mobility shift;35,36 that is, FAM-labeled fragments with a certain base length electrophoretically migrate faster than ROX-labeled fragments with the same base length. However, in this study, the dye-induced mobility shift was not corrected because the MT and WT bands were detected at the same base lengths with a high degree of reproducibility, as shown below; therefore, the MT and WT bands can be certainly identified.
FAM electropherograms (60–90 bases) derived from parts of the electrophoretic images in Fig. 3b, along with other FAM electropherograms obtained by different electrophoretic analyses, are overlaid in Fig. 4a to d. The same data set is used and only vertical scales are changed. Red, green, orange, purple, blue, and black lines indicate electropherograms of samples of 100% MT/WT (n = 5), 10% MT/WT (n = 5), 1% MT/WT (n = 5), 0.1% MT/WT (n = 4), 0.01% MT/WT (n = 5), and 0% MT/WT (n = 6), respectively. WT base length was 83.16 ± 0.06 bases (average ± standard deviation for all the electropherograms (n = 30)). Similarly, MT base length was 67.12 ± 0.09 bases (for all the electropherograms except those of 0% MT/WT (n = 24)). It is therefore possible to reproducibly identify WT and MT peaks in any electropherogram with an accuracy of less than 0.1 bases.
 | ||
| Fig. 4 Normalized electropherograms of 0% and 0.01–100% MT/WT samples. a–d: Overlaid FAM electropherograms (60–90 bases) where 100% MT/WT (red lines, n = 5), 10% MT/WT (green lines, n = 5), 1% MT/WT (orange lines, n = 5), 0.1% MT/WT (purple lines, n = 4), 0.01% MT/WT (blue lines, n = 5), and 0% MT/WT (black lines, n = 6) are separated. The same data set is used and only vertical scales are changed in a–d. Fluorescence intensity in each electropherogram is normalized so that WT-peak intensity is 10000 counts. e–h: Averaged FAM electropherograms (63–70 bases) derived from a–d, respectively. Red, green, orange, purple, blue, and black areas indicate 100%, 10%, 1%, 0.1%, 0.01%, and 0% MT/WT, respectively. | ||
When FAM fluorescence intensity in each FAM electropherogram was not normalized, WT-peak intensity varied widely with coefficient of variation (CV) of 26% for all the electropherograms (n = 30). Meanwhile, as shown in Fig. 4a to d, when FAM fluorescence intensity was normalized so that WT-peak intensity was aligned with 10000 counts, MT-peak intensity increased in proportion to MT/WT. Moreover, MT-peak intensities for samples of 100%, 10%, 1%, 0.1%, and 0.01% MT/WT respectively varied narrowly with CV of 2.4% for 100% MT/MT (n = 5), CV of 5.7% for 10% MT/WT (n = 5), CV of 3.0% for 1% MT/WT (n = 5), CV of 1.7% for 0.1% MT/WT (n = 4), and CV of 12.3% for 0.01% MT/WT (n = 5). As a result, as shown in Fig. 4d, even the smallest MT peaks of 0.01% MT/WT (blue lines) were clearly distinguished from backgrounds of 0% MT/WT (black lines) at MT base length.
Averaged FAM electropherograms around MT peaks (63–70 bases) of 100%, 10%, 1%, 0.1%, 0.01%, and 0% MT/WT are derived from Fig. 4a to d and respectively indicated by red, green, orange, purple, blue, and black areas in Fig. 4e to h. Although maximum values on the vertical axes in Fig. 4e to h are the same as those in Fig. 4a to d, respectively, minimum values on the vertical axes are changed from 0 to 3 counts in order to remove noisy backgrounds and highlight the difference between the MT peak of 0.01% MT/WT (blue area) and the background of 0% MT/WT (black area).
MT peaks were quantified as follows. First, in a similar manner as that applied in Fig. 4e to h, fluorescence intensity below 3 counts was cut off from each FAM electropherogram in Fig. 4a to d. Next, the average of the cut-off electropherograms of 0% MT was subtracted from each of the cut-off electropherograms. Finally, for each of the subtracted electropherograms, fluorescence intensity ranging from 65 to 68 bases was integrated at an interval of 0.1 bases to give MT-peak area. As a result, as shown in Table 1 and Fig. 5, a close linear correlation between MT/WT and MT-peak area, with a slope of 1 and r2 (coefficient of determination) of 0.99792, was obtained. Moreover, four-orders-of-magnitude dynamic range covering 0.01% to 100% MT/WT was obtained. That is, precise quantification ranging from MT in a 10000-fold excess of WT to the same amount of MT as that of WT was demonstrated.
| MT/WT | MT normalized peak area (counts) | |
|---|---|---|
| Average | Standard deviation | |
| 100% | 113511 |
2147.36 |
| 10% | 15049 |
730.04 |
| 1% | 1583 | 78.82 |
| 0.1% | 118 | 3.13 |
| 0.01% | 16 | 2.86 |
| 0% | 0 | 2.34 |
 | ||
| Fig. 5 Sensitivity and dynamic range of MT quantification. A close linear correlation between MT/WT and MT normalized peak area (with a slope of 1 and r2 of 0.99792) is shown. Plots and error bars represent average and ± standard deviation shown in Table 1. | ||
Because MT-peak area of 0% MT/WT was 0 ± 2.34 counts and MT-peak area of 0.01% MT/WT was 16 ± 2.86 counts, as shown in Table 1, MT-peak area of 0.01% MT/WT was detected at signal-to-noise ratio (S/N) of 6.8 when noise equaled standard deviation of MT-peak area of 0% MT/WT. Therefore, lower limit of detection (LOD) defined by S/N of 3 was 0.004% MT/WT, and lower limit of quantification (LOQ) defined by S/N of 10 was 0.015% MT/WT. The lower limit is determined by fluorescence-intensity fluctuation of 0%-MT/WT FAM electropherograms indicated by black lines around the MT peak in Fig. 4d. The fluctuation is caused by non-specific by-products in PCR. Consequently, it is possible to further enhance sensitivity of quantification of MT in a large excess of WT by optimizing design and concentration of each primer and thermal-cycling conditions for PCR.
In this study, the lowest detected concentration of MT of 0.01% MT/WT is 8 × 10−14 M (=1 pM × 2 μl/25 μl) in PCR. On the contrary, the lowest detectable concentration of ctDNA from liquid biopsy is desired to be less than 2 × 10−17 M (=100 copies/6 × 1023 copies per mol/10 μl) in PCR. Therefore, the desired concentration is more than three-orders-of-magnitude lower than the detected concentration. However, it is expected that such low-concentration ctDNA in a large excess of normal cfDNA can be co-amplified by PCR while maintaining the quantitative ratio of ctDNA to normal cfDNA and detected by HiDy CE. Meanwhile, in this study, only pure model samples are analyzed to evaluate sensitivity of the proposed assay with HiDy CE. In the next stage of research, it is planned to analyze clinical samples including rare mutated DNA in a large excess of wild-type DNA as well as contaminating DNA or other interfering compounds.
Throughput and cost
In this assay with HiDy CE, because time required for PCR, purification, and CE are 30, 15, and 25 min, respectively, turnaround time (TAT) after template preparation is 70 min. Besides, because four different samples are simultaneously assayed by the four-capillary-array electrophoresis, throughput is 3.4 samples per hour. It is easy to enhance throughput to more than 10.3 samples per hour by implementing a twelve-capillary array with a twelve-lens array in the current system. Automated serial analyses of different samples with the same capillary-array device further facilitate high-throughput assay with minimal hands-on time. On the other hand, consumable cost other than PCR and purification reagents, that is, consumable cost of the polymer solution, buffer, and capillary-array device, is only $1–2 per sample, because the same capillary-array device is reusable up to at least 100 times.As for ddPCR by RainDance, up to 107 droplets per sample are generated and read-out after PCR, so LOD is down to 0.0005% MT/WT.11 Since time required for droplet generation, PCR, and droplet read-out are 4, 60, and 30 min, respectively, TAT is 94 min. Eight samples are sequentially processed in droplet generation and droplet read-out, so droplet generation, PCR, and droplet read-out for eight samples take 30, 60, and 240 min, respectively. As a result, throughput is 1.5 samples per hour. Consumable cost for ddPCR other than PCR reagents, that is, consumable cost of microfluidic devices and oil, is as much as $30 per sample, because the microfluidic devices are single-use and not reusable. In summary, although the sensitivity of ddPCR is one-order-of-magnitude higher than that of HiDy CE, its cost is also one-order-of-magnitude higher.
In contrast, as for ddPCR by Bio-Rad, 2 × 104 droplets per sample are generated and read-out after PCR.13 Therefore, when LOD is defined as that in the case of ddPCR by RainDance,11 LOD is 0.2% MT/WT. To decrease LOD to 0.01% MT/WT, more than 4 × 105 droplets per sample must be generated and read-out after PCR. In other words, the sample must be divided into 20 samples, which are sequentially analyzed, and the analysis results are combined to give a result for the original sample. In this case, droplet generation, PCR, and droplet read-out take 5, 90, and 30 min, respectively; throughput is therefore 0.5 samples per hour, and consumable cost is $60 per sample. Therefore, throughput is lower and consumable cost is higher than those for ddPCR by RainDance.
Multiplexing capability
Although only the duplex assay for the single locus was demonstrated in this study, CE with multicolor-fluorescence detection is inherently advantageous in regard to multiplex assays. One of the most-used multiplex assays by CE is human identification by 16–35-plex STR loci typing.18,19 One-color fluorescence is used for internal size standard, and the other is used for STR typing. Fragment sizes of PCR products for STR loci range from 50 to 500 bases. In contrast, in this study, only two colors (green and red) of the four-color images and the limited region of base length (67–83 bases) in the electropherograms were used. It is therefore possible to perform higher multiplex assays for more loci by using all colors (green, yellow, orange, and red) and the entire region of base length (e.g., 50–500 bases) in electropherograms. For example, FAM, VIC, and NED electropherograms for a multiplex assay and a ROX electropherogram for the internal size standard can be simultaneously obtained for each capillary. In each fluorophore electropherogram, ten pairs of WT and MT peaks with 15-base difference in base length can be sufficiently separated by positioning them at intervals of 40 bases. Consequently, up to 60-plex (2-plex × 10 pairs × 3 fluorophores) is feasible with the current system.Meanwhile, as shown in Fig. 4d, even the farthest left end of the WT peak does not overlap the center of the MT peak, because the difference between base lengths of the WT and MT peaks is large (15 bases). As a result, highly sensitive MT quantification was possible. Therefore, when smaller insertion or deletion (i.e., less than 15 bases in base length) or other mutations without insertion or deletion (e.g., point mutation or SNP) is assayed by HiDy CE, sensitivity of MT quantification might be reduced because the WT peak might overlap the MT peak. To overcome this problem, it is necessary to spread two peaks by a different method of chemical reaction from the standard PCR performed in this study. One of the most-promising methods is MLPA™ (multiplex ligation-dependent probe amplification, MRC-Holland).21,37–39 In the assay using MLPA™, for each locus, it is possible to individually and freely design lengths of ligased probes for WT and MT. PCR products of the ligased probes for WT and MT can therefore be sufficiently separated by CE even in case of mutations without insertion or deletion. As for point-mutation or SNP assay, SNaPshot® (Thermo Fisher Scientific) is another promising method.20,22,23 In the assay, for each locus, single-base-extended primers for WT and MT are labeled with different fluorophores according to the extended-base species, but they are the same base length. Nevertheless, they are slightly separated by CE due to the dye-induced mobility shift.35,36 Accordingly, they can be sufficiently separated by increasing the difference in physical or chemical properties of the labeled fluorophores for WT and MT, for example, by changing net electric charge of the labeled fluorophores in the polymer solution.
Multiplexing capability is one of the features of ddPCR systems by RainDance and Bio-Rad.12,40 Up-to-6-plex assays by ddPCR have been demonstrated.17,41 On a two-dimensional scatter plot of all detected droplets, when clusters derived from different target sequences (loci) are sufficiently separated, different target sequences can be assayed simultaneously (i.e., with high multiplexity) and independently (i.e., with high sensitivity). However, multiplexing capability and sensitivity obviously share a trade-off relationship, because the limited space in the two-dimensional scatter plot is occupied by multiple clusters.15–17 Therefore, higher multiplexing (i.e., more clusters) might sacrifice detection and quantification sensitivity in the case of ddPCR.
Conclusion
Mutation quantification by the proposed assay with HiDy CE offers high sensitivity (LOD of 0.004% MT/WT), four-orders-of-magnitude dynamic range (0.01–100% MT/WT), and high throughput (3.4 samples per hour) at the lowest analysis cost ($1–2 per sample). Throughput can be easily enhanced by the twelve-capillary array with the twelve-lens array and automated serial analyses. Moreover, multiplexity can be greatly enhanced by maximal use of HiDy CE and combination with other conventional methods. Therefore, it has great potential for clinical applications, in particular, frequent quantification of rare ctDNA in liquid biopsies to monitor dynamic change of highly heterogeneous genetic profiles of tumor cells.Author contributions
T. A. developed the system, conducted experiments, analyzed data, and wrote the manuscript. H. M. designed the assay and prepared samples. S. Y. and R. I. discussed the results and gave inputs on the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors thank Chihiro Uematsu and Yoshinobu Kohara of Hitachi, Ltd., Taro Nakazawa and Motohiro Yamazaki of Hitachi High-Technologies Corporation for their valuable discussions on the manuscript, and Tomoyuki Sakai of Hitachi, Ltd. for creating a favorable environment for this research.References
- E. Crowley, F. Di Nicolantonio, F. Loupakis and A. Bardelli, Nat. Rev. Clin. Oncol., 2013, 10, 472–484 CrossRef CAS PubMed.
- L. A. Diaz, Jr. and A. Bardelli, J. Clin. Oncol., 2014, 32, 579–586 CrossRef PubMed.
- G. Siravegna, S. Marsoni, S. Siena and A. Bardelli, Nat. Rev. Clin. Oncol., 2017, 14, 531–548 CrossRef CAS PubMed.
- M. Elazezy and S. A. Joosse, Comput. Struct. Biotechnol. J, 2018, 16, 370–378 CrossRef CAS PubMed.
- M. Murtaza, S. J. Dawson, D. W. Tsui, D. Gale, T. Forshew, A. M. Piskorz, C. Parkinson, S. F. Chin, Z. Kingsbury, A. S. Wong, F. Marass, S. Humphray, J. Hadfield, D. Bentley, T. M. Chin, J. D. Brenton, C. Caldas and N. Rosenfeld, Nature, 2013, 497, 108–112 CrossRef CAS PubMed.
- G. Siravegna, B. Mussolin, M. Buscarino, G. Corti, A. Cassingena, G. Crisafulli, A. Ponzetti, C. Cremolini, A. Amatu, C. Lauricella, S. Lamba, S. Hobor, A. Avallone, E. Valtorta, G. Rospo, E. Medico, V. Motta, C. Antoniotti, F. Tatangelo, B. Bellosillo, S. Veronese, A. Budillon, C. Montagut, P. Racca, S. Marsoni, A. Falcone, R. B. Corcoran, F. Di Nicolantonio, F. Loupakis, S. Siena, A. Sartore-Bianchi and A. Bardelli, Nat. Med., 2015, 21, 795–801 CrossRef CAS PubMed.
- V. Marx, Nat. Methods, 2016, 13, 295–299 CrossRef PubMed.
- M. W. Schmitt, S. R. Kennedy, J. J. Salk, E. J. Fox, J. B. Hiatt and L. A. Loeb, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14508–14513 CrossRef CAS PubMed.
- A. M. Newman, S. V. Bratman, J. To, J. F. Wynne, N. C. Eclov, L. A. Modlin, C. L. Liu, J. W. Neal, H. A. Wakelee, R. E. Merritt, J. B. Shrager, B. W. Loo, Jr., A. A. Alizadeh and M. Diehn, Nat. Med., 2014, 20, 548–554 CrossRef CAS PubMed.
- A. M. Newman, A. F. Lovejoy, D. M. Klass, D. M. Kurtz, J. J. Chabon, F. Scherer, H. Stehr, C. L. Liu, S. V. Bratman, C. Say, L. Zhou, J. N. Carter, R. B. West, G. W. Sledge, J. B. Shrager, B. W. Loo, Jr., J. W. Neal, H. A. Wakelee, M. Diehn and A. A. Alizadeh, Nat. Biotechnol., 2016, 34, 547–555 CrossRef CAS PubMed.
- D. Pekin, Y. Skhiri, J. C. Baret, D. Le Corre, L. Mazutis, C. B. Salem, F. Millot, A. El Harrak, J. B. Hutchison, J. W. Larson, D. R. Link, P. Laurent-Puig, A. D. Griffiths and V. Taly, Lab Chip, 2011, 11, 2156–2166 RSC.
- Q. Zhong, S. Bhattacharya, S. Kotsopoulos, J. Olson, V. Taly, A. D. Griffiths, D. R. Link and J. W. Larson, Lab Chip, 2011, 11, 2167–2174 RSC.
- B. J. Hindson, K. D. Ness, D. A. Masquelier, P. Belgrader, N. J. Heredia, A. J. Makarewicz, I. J. Bright, M. Y. Lucero, A. L. Hiddessen, T. C. Legler, T. K. Kitano, M. R. Hodel, J. F. Petersen, P. W. Wyatt, E. R. Steenblock, P. H. Shah, L. J. Bousse, C. B. Troup, J. C. Mellen, D. K. Wittmann, N. G. Erndt, T. H. Cauley, R. T. Koehler, A. P. So, S. Dube, K. A. Rose, L. Montesclaros, S. Wang, D. P. Stumbo, S. P. Hodges, S. Romine, F. P. Milanovich, H. E. White, J. F. Regan, G. A. Karlin-Neumann, C. M. Hindson, S. Saxonov and B. W. Colston, Anal. Chem., 2011, 83, 8604–8610 CrossRef CAS PubMed.
- L. B. Pinheiro, V. A. Coleman, C. M. Hindson, J. Herrmann, B. J. Hindson, S. Bhat and K. R. Emslie, Anal. Chem., 2012, 84, 1003–1011 CrossRef CAS PubMed.
- V. Taly, D. Pekin, L. Benhaim, S. K. Kotsopoulos, D. Le Corre, X. Li, I. Atochin, D. R. Link, A. D. Griffiths, K. Pallier, H. Blons, O. Bouche, B. Landi, J. B. Hutchison and P. Laurent-Puig, Clin. Chem., 2013, 59, 1722–1731 CrossRef CAS PubMed.
- A. S. Whale, J. F. Huggett and S. Tzonev, Biomol. Detect. Quantif., 2016, 10, 15–23 CrossRef CAS PubMed.
- M. Watanabe, T. Kawaguchi, S. I. Isa, M. Ando, A. Tamiya, A. Kubo, H. Saka, S. Takeo, H. Adachi, T. Tagawa, O. Kawashima, M. Yamashita, K. Kataoka, Y. Ichinose, Y. Takeuchi, K. Watanabe, A. Matsumura and Y. Koh, EBioMedicine, 2017, 21, 86–93 CrossRef PubMed.
- J. M. Butler, E. Buel, F. Crivellente and B. R. McCord, Electrophoresis, 2004, 25, 1397–1412 CrossRef CAS PubMed.
- A. A. Westen, T. Kraaijenbrink, E. A. Robles de Medina, J. Harteveld, P. Willemse, S. B. Zuniga, K. J. van der Gaag, N. E. Weiler, J. Warnaar, M. Kayser, T. Sijen and P. de Knijff, Forensic Sci. Int.: Genet., 2014, 10, 55–63 CrossRef CAS PubMed.
- A. Farina Sarasqueta, E. Moerland, H. de Bruyne, H. de Graaf, T. Vrancken, G. van Lijnschoten and A. J. van den Brule, J. Mol. Diagn., 2011, 13, 199–205 CrossRef PubMed.
- M. Gausachs, P. Mur, J. Corral, M. Pineda, S. Gonzalez, L. Benito, M. Menendez, J. A. Espinas, J. Brunet, M. D. Iniesta, S. B. Gruber, C. Lazaro, I. Blanco and G. Capella, Eur. J. Hum. Genet., 2012, 20, 762–768 CAS.
- M. Perizzolo, B. Winkfein, S. Hui, W. Krulicki, J. A. Chan and D. J. Demetrick, Brain Pathol., 2012, 22, 619–624 CrossRef CAS PubMed.
- M. Fondevila, C. Phillips, C. Santos, A. Freire Aradas, P. M. Vallone, J. M. Butler, M. V. Lareu and A. Carracedo, Forensic Sci. Int.: Genet., 2013, 7, 63–74 CrossRef CAS PubMed.
- J. Lin, Y. Goto, H. Murata, K. Sakaizawa, A. Uchiyama, T. Saida and M. Takata, Br. J. Cancer, 2011, 104, 464–468 CrossRef CAS PubMed.
- A. C. Tsiatis, A. Norris-Kirby, R. G. Rich, M. J. Hafez, C. D. Gocke, J. R. Eshleman and K. M. Murphy, J. Mol. Diagn., 2010, 12, 425–432 CrossRef CAS PubMed.
- C. J. Davidson, E. Zeringer, K. J. Champion, M. P. Gauthier, F. Wang, J. Boonyaratanakornkit, J. R. Jones and E. Schreiber, BioTechniques, 2012, 53, 182–188 CrossRef CAS PubMed.
- Q. Pan, W. Pao and M. Ladanyi, J. Mol. Diagn., 2005, 7, 396–403 CrossRef CAS PubMed.
- Z. Su, D. Dias-Santagata, M. Duke, K. Hutchinson, Y. L. Lin, D. R. Borger, C. H. Chung, P. P. Massion, C. L. Vnencak-Jones, A. J. Iafrate and W. Pao, J. Mol. Diagn., 2011, 13, 74–84 CrossRef CAS PubMed.
- L. V. Furtado, H. C. Weigelin, K. S. Elenitoba-Johnson and B. L. Betz, J. Mol. Diagn., 2013, 15, 592–599 CrossRef CAS PubMed.
- Z. Yang, C. Steentoft, C. Hauge, L. Hansen, A. L. Thomsen, F. Niola, M. B. Vester-Christensen, M. Frodin, H. Clausen, H. H. Wandall and E. P. Bennett, Nucleic Acids Res., 2015, 43, e59 CrossRef PubMed.
- T. Anazawa and M. Yamazaki, Lab Chip, 2017, 17, 4231–4242 RSC.
- S. V. Sharma, D. W. Bell, J. Settleman and D. A. Haber, Nat. Rev. Cancer, 2007, 7, 169–181 CrossRef CAS PubMed.
- Y. Kobayashi and T. Mitsudomi, Cancer Sci., 2016, 107, 1179–1186 CrossRef CAS PubMed.
- T. Anazawa, S. Takahashi and H. Kambara, Anal. Chem., 1996, 68, 2699–2704 CrossRef CAS PubMed.
- O. Tu, T. Knott, M. Marsh, K. Bechtol, D. Harris, D. Barker and J. Bashkin, Nucleic Acids Res., 1998, 26, 2797–2802 CrossRef CAS PubMed.
- J. T. Sutton, B. C. Robertson and I. G. Jamieson, Mol. Ecol. Resour., 2011, 11, 514–520 CrossRef CAS PubMed.
- D. A. Koolen, W. M. Nillesen, M. H. Versteeg, G. F. Merkx, N. V. Knoers, M. Kets, S. Vermeer, C. M. van Ravenswaaij, C. G. de Kovel, H. G. Brunner, D. Smeets, B. B. de Vries and E. A. Sistermans, J. Med. Genet., 2004, 41, 892–899 CrossRef CAS PubMed.
- J. P. Schouten, C. J. McElgunn, R. Waaijer, D. Zwijnenburg, F. Diepvens and G. Pals, Nucleic Acids Res., 2002, 30, e57 CrossRef PubMed.
- D. J. Bunyan, D. M. Eccles, J. Sillibourne, E. Wilkins, N. S. Thomas, J. Shea-Simonds, P. J. Duncan, C. E. Curtis, D. O. Robinson, J. F. Harvey and N. C. Cross, Br. J. Cancer, 2004, 91, 1155–1159 CrossRef CAS PubMed.
- D. Pretto, D. Maar, C. M. Yrigollen, J. Regan and F. Tassone, Clin. Chem., 2015, 61, 182–190 CrossRef CAS PubMed.
- D. Dobnik, D. Stebih, A. Blejec, D. Morisset and J. Zel, Sci. Rep., 2016, 6, 35451 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00853e |
| This journal is © The Royal Society of Chemistry 2020 |