
Degassed micromolding lithography for rapid fabrication of anisotropic hydrogel microparticles with high-resolution and high uniformity†
Hyeon Ung
Kim
 ,
Yong Jun
Lim
,
Hyun Jee
Lee
,
Nak Jun
Lee
and
Ki Wan
Bong
*
,
Yong Jun
Lim
,
Hyun Jee
Lee
,
Nak Jun
Lee
and
Ki Wan
Bong
*
Department of Chemical and Biological Engineering, Korea University, Seoul 02841, Republic of Korea. E-mail: bong98@korea.ac.kr; Tel: +82 02 3290 3294
First published on 8th November 2019
Abstract
Replica molding techniques, which are used to synthesize microparticles inside anisotropic micromolds, have been developed to enable the mass production of hydrogel particles. However, these techniques are limited in their ability to synthesize only a narrow range of particle compositions and shapes because of the difficulty in loading precursors into the micromolds as well as the low particle homogeneity due to the uneven evaporation of the precursors. Herein, we describe a simple yet powerful technique, called degassed micromolding lithography, which can load precursors within 1 min regardless of the wettability. This technique is based on the gas-solubility of a degassed micromold that acts as a suction pump to completely fill the mold by drawing precursor liquids in. The semi-closed system within the micromold prevents the uneven evaporation of the precursor, which is essential for the production of homogeneous particles. Furthermore, controlled uniformity of the hydrogel microparticles (C.V. < 2%) can be achieved by engineering the design of the micromold array.
Introduction
Unlike spherical particles, highly-engineered anisotropic hydrogel microparticles can engage in intricate interactions with the surroundings due to their orientation-specific morphology and their enhanced surface to volume ratio. This characteristic has led to their wide use in various areas, such as multiplex assays,1–3 pharmaceutics,4–6 photonics,7,8 self-assemblies,9,10 and tissue engineering.11 Currently, there is growing interest in the fabrication methods of anisotropic particles, which often necessitate a sophisticated procedure because small particles tend to form a spherical shape that gives minimal surface energy. In order to further expand the applications of anisotropic hydrogel particles, one key concern to be addressed is the mass production of anisotropic hydrogel microparticles with high homogeneity and resolution in a cost-efficient way. The most widely recognized strategy for the synthesis of anisotropic particles is photolithography. Anisotropic particles are synthesized by exposing micropatterned ultraviolet (UV) on a UV-curable precursor, which results in the formation of particles shaped in the 2D-extrusion of the UV pattern.12,13 To extend the anisotropy beyond the 2D-extrusion or the cone shape,14,15 photolithography can be performed inside microfluidic channels that are engineered to allow selective polymerization along the vertical axis.16–19 However, this method yields low productivity since the number of particles that can be simultaneously synthesized is restricted by the size of the microchannel. Furthermore, an automated production cycle is interrupted by the sticking and clogging of the particles on the device, which can result from the repeated UV irradiation on the same spot.20,21 Layer-by-layer techniques, such as multiphoton lithography22 and 3D printing,11,23 offer the highest degree of freedom on the particle shape, but their productivity is critically low and inversely proportional to the resolution.A notable method that allows mass production of anisotropic particles without any specialized equipment is replica molding.24,25 This approach uses an elastomer mold with negative relief patterns that allow a precursor to be filled and polymerized into particles that are shaped according to the patterns of the positive relief. The elastic mold, such as polydimethylsiloxane (PDMS),26,27 perfluoropolyether (PFPE)24,25 and polyurethane,28 which is produced by soft lithography on a micropatterned master template, allows the repeated synthesis of particles in various shapes. Replica molding techniques have been also used commercially for a long time with a rotary drum,29 roll-to-roll and roll-to-plate.30 Yet, despite the powerful approach, this method is barred from wide application due to an inherent technical limitation. Normally, a prepolymer solution is loaded into the mold depending on the capillary action, so the hydrophilic mold is loaded with a hydrophilic liquid31,32 or the hydrophobic mold is loaded with a hydrophobic liquid,33 which makes it difficult to achieve chemical diversity of the particles using one kind of mold. When loading precursors with low wettability, various techniques have been developed, including solvent assisted micromolding,34 plasma treatment,27 scrubbing35 and vacuum casting.36 Meanwhile, as the volume of each pattern is usually in the nanoliter range, the contents of the precursor are prone to rapid evaporation that can proceed in the order of tens of seconds, which would greatly compromise the homogeneity of the resultant particle composition. To minimize the evaporation of the precursor, the synthesis can be conducted in a humidity chamber.35 However, such modification is labor-intensive and requires sizable equipment, which are non-permissive to large-scale particle synthesis. Therefore, there is a need to develop a general technique capable of loading all kinds of precursors in a short time without defects.
Furthermore, due to the oxygen solubility and permeability of PDMS, replica molding in PDMS micromolds, which is the most commonly used material for soft lithography, is presented with additional difficulty. The influx of oxygen molecules from the micromold can inhibit the radical polymerization of the precursor near the micromold surface and greatly limit the production of anisotropic particles that require high resolution, such as fine protrusions and internal holes.
Here, we propose a straight-forward yet powerful replica molding technique called degassed micromolding lithography (DML), which permits the efficient loading of the precursors into the gas-permeable micromold without defects and results in the synthesis of highly homogeneous anisotropic hydrogel microparticles in high resolution. Different from the existing replica molding techniques, the mold template is degassed in a vacuum chamber prior to the particle synthesis. Therefore, as soon as the degassed mold template is removed from the vacuum and brought into contact with the precursor, it compulsorily draws the liquid into the negative relief patterns of the micromold by absorbing the entrapped air bubbles between the micromold and the liquid. In this work, we optimized the degassing duration and demonstrated the versatility of the technique to precursor solutions of various liquid properties. Additionally, we show that degassing of the mold template can enhance the resolution of the synthesized particles through reducing the influx of oxygen into the precursor, thereby alleviating the oxygen inhibition effect37,38 near the walls of the micromolds. We also characterized the oxygen inhibition with respect to the degassing duration and presented the synthesis of encoded microparticles that contain internal holes. Moreover, our method precludes the problem of precursor evaporation since the precursor is enclosed in the mold template with an underlying cover. We verified the high homogeneity within both the intra-batch and inter-batch particle populations synthesized using this method. Lastly, we illustrate the feasibility of particle synthesis using 3D-printed micromolds. We envisage DML to greatly advance the field of replica molding by expanding the selection of precursors and promote its wider application in homogeneous particle synthesis.
Materials & methods
Materials
The photoresist (SU-8, 25) was purchased from MicroChem (MA, USA). PDMS (SYLGARD® 184 Silicone Elastomer Kit) was purchased from Dow Corning (MI, USA). The Darocur 1173 (2-hydroxy-2-methylpropiophenone) photoinitiator, rhodamine-B-methacrylate, Tween 20, poly(ethylene glycol) (PEG, MW 200 Da), poly(ethylene glycol)diacrylate (PEG-DA, MW 700 Da), trichloro(1H,1H,2H,2H-perfluorooctyl)silane, and fluorescein isothiocyanate conjugated albumin from bovine serum (FITC–BSA) were purchased from Sigma-Aldrich (Gillingham, UK).Preparation of the precursor solution for the fabrication of various particles
All precursors were composed of 95% (v/v) PEG-DA and 5% (v/v) photoinitiator unless specified otherwise. The shape-encoded particles shown in Fig. 3C were synthesized with 30% (v/v) PEG-DA, 5% (v/v) photoinitiator, 30% (v/v) PEG200 and 35% (v/v) DI water. Rhodamine incorporated particles were synthesized with 94% (v/v) PEG-DA, 5% (v/v) photoinitiator and 1% (v/v) rhodamine-B-methacrylate in PEG200 (1 mg mL−1). Protein encapsulated particles were synthesized with 25% (v/v) PEG-DA, 5% (v/v) photoinitiator, 20% (v/v) PEG200 and 50% FITC–BSA in PBS (10 mg mL−1).DML procedure
A master mold was fabricated by hardening SU-8 (negative photoresist) structures on a 4 in silicon wafer with photolithography. The PDMS micromold and cover were generated by pouring PDMS (mixed in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of base to curing agent) over the master mold in a Petri dish to 3 mm and then baking it for 4 h at 70 °C. The mold template and the PDMS cover placed on a slide glass were degassed in a vacuum chamber at 0.1 atm. After the required degassing time had elapsed, the cover and the micromold were removed from the vacuum chamber. Afterwards, 40 μL of the precursor solution or water was added on the top of the cover and the mold template was pressed down against the precursor solution with the micromolds facing downward. The mold template was removed from the vacuum chamber and the precursor was then added, which took about 20 s. 1 min after the addition of the liquid, the mold template and the cover were compressed at 1 atm for 20 s, and the micromolds were then exposed to UV irradiation. A UV-LED lamp (SOLIS-365C, Thorlabs, Cambridge, UK) was controlled using LabVIEW software (National Instruments, TX, USA) and a DAQ board (Ni-DAQ, national instruments). The UV intensity was measured using a UV light tester (GT510, Giltron, Seoul, Korea) and was controlled using a power driver (DC20, Thorlabs).
:1 ratio of base to curing agent) over the master mold in a Petri dish to 3 mm and then baking it for 4 h at 70 °C. The mold template and the PDMS cover placed on a slide glass were degassed in a vacuum chamber at 0.1 atm. After the required degassing time had elapsed, the cover and the micromold were removed from the vacuum chamber. Afterwards, 40 μL of the precursor solution or water was added on the top of the cover and the mold template was pressed down against the precursor solution with the micromolds facing downward. The mold template was removed from the vacuum chamber and the precursor was then added, which took about 20 s. 1 min after the addition of the liquid, the mold template and the cover were compressed at 1 atm for 20 s, and the micromolds were then exposed to UV irradiation. A UV-LED lamp (SOLIS-365C, Thorlabs, Cambridge, UK) was controlled using LabVIEW software (National Instruments, TX, USA) and a DAQ board (Ni-DAQ, national instruments). The UV intensity was measured using a UV light tester (GT510, Giltron, Seoul, Korea) and was controlled using a power driver (DC20, Thorlabs).
Particle recovery
After UV exposure, the cover was detached from the mold template where synthesized particles were embedded. Then, 100 μL of the recovery solution composed of 70% ethanol, 30% water, and 0.05% Tween 20 was dropped onto the mold template or the mold template was placed in a Petri dish containing the recovery solution. Particles released from the mold template to the recovery solution were transferred to a micro centrifuge tube (1.5 mL) or conical tube (5 mL) using a pipette.Observation of precursor filling in the micromold
The PDMS cover on a glass slide was mounted on a microscope (Axiovert 200, Zeiss), and the entire duration of the filling process was recorded using a microscope equipped with a DSLR camera (EOS 6D, Canon, Ota, Japan). 40 μL of the precursor solution or water was applied and covered with the mold template as previously described.Size measurement of the particles
Synthesized particles were collected by directly pipetting from the mold template and transferring onto a glass slide without any rinsing step. The glass slide was covered with a cover slip and mounted on a microscope. Images of the particles were captured using a DSLR camera equipped to the microscope. The sizes of the particles were measured using Image J software (National Institutes of Health, MD, USA). The start of the shadow peaks was used as the standard for determining the edges of the particle.Micromold fabrication from the 3D printed master mold
3D printed master molds were fabricated using S350 (Solidscape, Inc., Merrimack, NH, USA) through 3D MAKERS (Seoul, Korea). The master molds were coated with trichloro(1H,1H,2H,2H-perfluorooctyl)silane by chemical deposition in a 0.1 atm vacuum chamber for 24 h. PDMS (curing agent mixed) was then poured onto the master mold, and the master patterns were replicated by soft lithography. The cured PDMS was again replicated with Norland Optical Adhesive 81 (NOA81) to complete a durable 3D patterned master mold. Afterwards, the NOA81 master mold was replicated with PDMS which was then used as the mold template for the synthesis of complex shaped particles using the methods described above. To allow the polymerization of particles in discrete micromolds, we reduced the UV intensity from 100 mW cm−2 to 10 mW cm−2.Calculation of the oxygen concentration of the PDMS mold template
We applied the finite-element method provided in COMSOL software for calculating the change in oxygen concentration of the PDMS block in a vacuum chamber. The initial oxygen concentration in PDMS is 1.02 mol m−3,39 the boundary condition is 0.102 mol m−3, the diffusivity of oxygen is 3.4 × 10−9 m2 s−3,39 the thickness of the PDMS block is 3 mm, and the bottom surface of the PDMS block is blocked with glass.Results and discussion
Loading liquids into degassed micromolds
When a hydrophilic liquid is in contact with a PDMS micromold that is not degassed, a strong cohesive force between the liquid molecules and their low adhesion to the micromold leads to capillary repulsion. As a result, air becomes entrapped between the liquid and the micromold, and this air pocket hampers the complete filling of the micromold. Recently, PDMS blocks have been used as power-free vacuum pumps for drawing liquids into microfluidic devices owing to their solubility and permeability to gas.39–41 PDMS is initially placed in a vacuum chamber to be degassed of all the air dissolved in the PDMS. When the PDMS block is removed from the chamber and placed under atmospheric pressure, the PDMS block takes up air from the surroundings until the air concentration inside the PDMS reaches an equilibrium with the ambient pressure (Fig. 1A). By using degassed PDMS as the micromold, the air pocket that impeded the complete filling of the micromold is effectively removed, thereby drawing the precursor into the micromold. Subsequently, by compressing the mold template unto the underlying cover, any excess precursor is discarded and each negative relief pattern forms an enclosed compartment in which an individual particle is generated through photoinduced polymerization (Fig. 1B and C).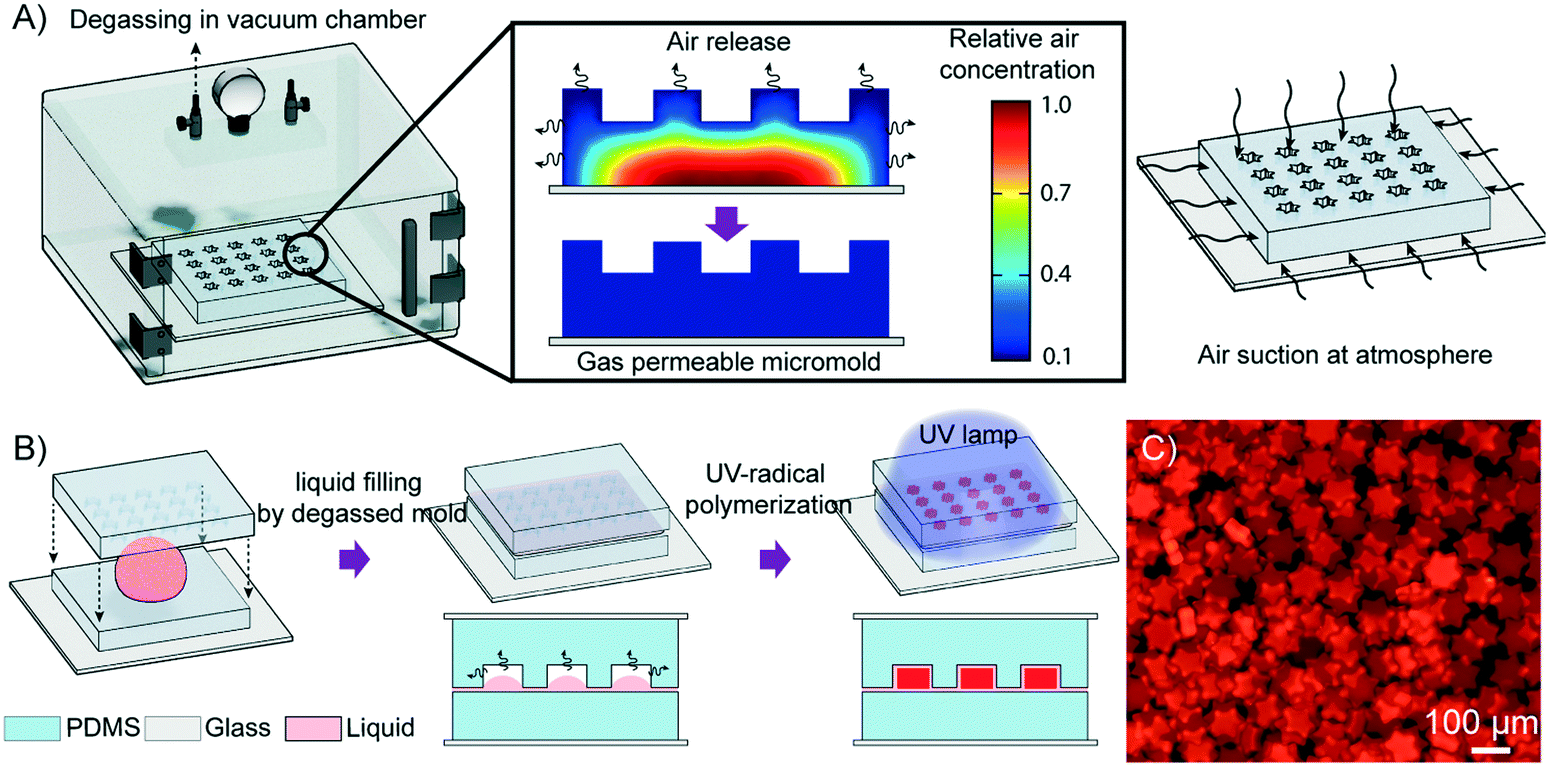 | ||
| Fig. 1 Schematic of degassed micromolding lithography. A) Degassing of a gas-soluble PDMS mold template in a vacuum chamber. The air concentration in the mold template decreases to a new equilibrium concentration after degassing. When taken out from the chamber into the atmosphere, the degassed mold template can act as an air absorbing pump. B) Microparticle fabrication in a degassed micromold. The precursor is sandwiched between the cover and the degassed mold template. The entrapped air bubbles between the precursor and the micromold are removed through gas uptake by the degassed micromolds, which are then filled with the precursor. With the cover compressed onto the mold template, UV irradiation was applied to polymerize the precursor. C) An image of fluorescent particles synthesized by the DML method. | ||
To characterize the profile of the liquid filling kinetics of the degassed micromold, we used a PDMS block (thickness: 3 mm) with square prism-shaped micromolds (100 × 100 × 25 μm3) and two hydrophilic liquids, water and a polyethylene glycol diacrylate (PEG-DA) based precursor. The contact angles of the water and the precursor on PDMS were 113.4° and 87.8°, respectively. Although the loading of the precursor into the non-degassed micromolds occurred spontaneously, it took about three hours to completely fill the micromolds as shown in Fig. 2A. On the other hand, all the degassed micromolds were completely filled within a few minutes, which could be cut down to around 1 min if the degassing time of the PDMS block was longer than 20 min (Movie S1, ESI†). When water was used as the loading liquid, the non-degassed micromolds could not be filled at all due to the low wettability of water on PDMS. However, the loading profile of water into the degassed micromolds was similar to that of the precursor, in which the loading time was also about 1 min when the degassing time exceeded 20 minutes. The relationship of the degassing time and the loading time of the liquids is shown in Fig. 2B. Furthermore, a mathematical model is proposed in order to estimate the approximate loading time according to the geometry of the micromold (see the ESI† for details). In Fig. 2B, the recorded loading times are independent of the wettability of the liquid on the degassed micromolds (see the ESI† for details). Moreover, we also observed that the loading times are not significantly affected by the changes in liquid viscosity from 16 cp to 190 cp (see Fig. S1A, ESI† for details). However, the liquid loading time was found to be dependent on the volume of the micromold. A micromold of volume below 1 nL was filled with the precursor within 1 minute, while the loading time lengthened with the increases in the volume of the micromold (Fig. S1B, ESI†).
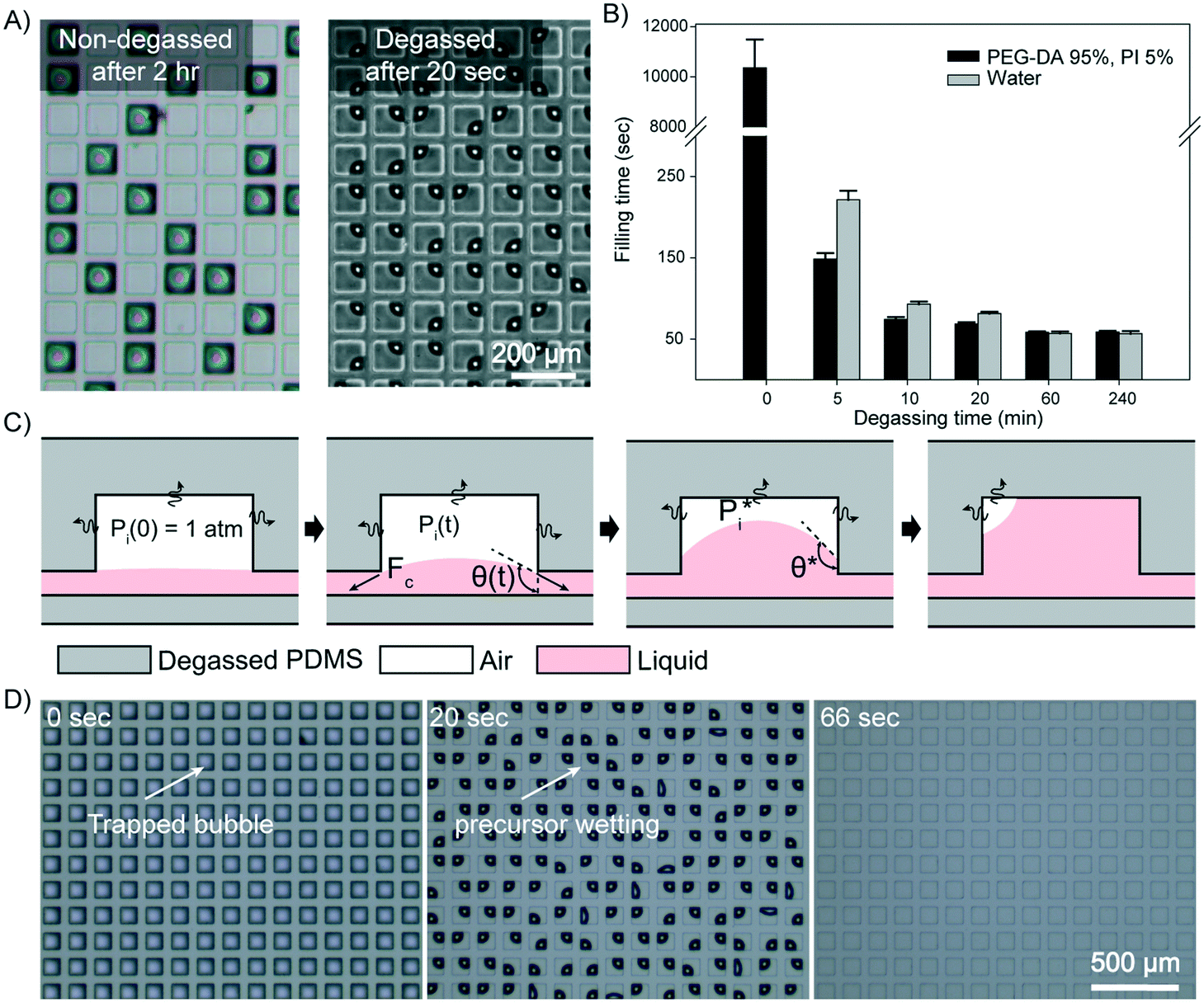 | ||
Fig. 2 Liquid filling in the degassed micromold. A) A comparison between the outcomes of PEG-DA loading into a non-degassed mold block (left) and a degassed mold block (right). B) The liquid filling time as a function of the degassing time. Each bar represents the mean from three independent experiments. Error bars represent the standard deviation. C) Schematic illustrations of the stepwise liquid loading process. The initial air pressure inside the micromold (Pi(0)) is 1 atm, and the pressure drops as the air is taken up by the degassed PDMS. The pressure drop (ΔP) is balanced by the capillary pressure (PC), which is characterized by a curved air–liquid interface. After the contact angle of the air–liquid interface reaches  , PC and ΔP are balanced and the liquid is subsequently filled into the micromold. D) Uniform liquid loading can be achieved in a large-scale synthesis (6000 particles) in the degassed micromold. , PC and ΔP are balanced and the liquid is subsequently filled into the micromold. D) Uniform liquid loading can be achieved in a large-scale synthesis (6000 particles) in the degassed micromold. | ||
A schematic illustration of the stepwise loading process of the precursor is depicted in Fig. 2C. When the precursor is first in contact with the PDMS micromold, the air pressure inside the enclosed compartment (Pi(t)) is at 1 atm, and the pressure starts to drop due to gaseous absorption by the degassed PDMS. The capillary pressure (PC = 2γcosθc) and the pressure difference between the enclosed compartment and the atmosphere (ΔP = P0 − Pi(t)) are gradually balanced by the formation of a curved surface at the air–liquid interface (Fig. S2A, ESI†), in which the contact angle (θc) consequently increases from 0°, where γ is the surface tension of the liquid and P0 denotes the atmospheric pressure. When the curvature is fully developed with the critical contact angle 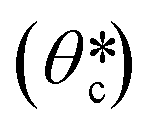 being reached, the liquid begins to be drawn into the micromold at a rate equivalent to the gaseous absorption rate of the degassed micromold, since PC and ΔP are estimated to be balanced at a constant value. The time required to reach this state has been defined as the induction time in previous studies.40,42 After the induction time has elapsed, the precursor is being filled into the micromold. When the liquid reaches the inmost surface of the micromold, the residual air trapped in the compartment now occupies a corner of the micromold and adopts a quarter-of-a-sphere conformation to minimize the surface tension as shown in Fig. 2D. Then the entrapped air is perfectly removed by air uptake of the degassed mold. To support our model of the liquid filling process, we monitored the side-views of the air–liquid interface (Fig. S2B, ESI†). From the mathematical model, the calculated induction time is around 0.22 s (see the ESI† for details). From the simulation for the calculation of the air flux, the liquid loading time according to the diffusivity of the mold is calculated (Table S1, ESI†). The diffusivity of the mold which allows loading of the precursor in minutes is 0.1 to 10 times that of PDMS. As the liquid loading process is independent of the number of micromolds in the PDMS block, it is possible to load the precursor simultaneously into a large array of micromolds, without any labor-intensive step (Fig. S3A, ESI†), which is compared with vacuum casting (Fig. S3B, ESI†).
being reached, the liquid begins to be drawn into the micromold at a rate equivalent to the gaseous absorption rate of the degassed micromold, since PC and ΔP are estimated to be balanced at a constant value. The time required to reach this state has been defined as the induction time in previous studies.40,42 After the induction time has elapsed, the precursor is being filled into the micromold. When the liquid reaches the inmost surface of the micromold, the residual air trapped in the compartment now occupies a corner of the micromold and adopts a quarter-of-a-sphere conformation to minimize the surface tension as shown in Fig. 2D. Then the entrapped air is perfectly removed by air uptake of the degassed mold. To support our model of the liquid filling process, we monitored the side-views of the air–liquid interface (Fig. S2B, ESI†). From the mathematical model, the calculated induction time is around 0.22 s (see the ESI† for details). From the simulation for the calculation of the air flux, the liquid loading time according to the diffusivity of the mold is calculated (Table S1, ESI†). The diffusivity of the mold which allows loading of the precursor in minutes is 0.1 to 10 times that of PDMS. As the liquid loading process is independent of the number of micromolds in the PDMS block, it is possible to load the precursor simultaneously into a large array of micromolds, without any labor-intensive step (Fig. S3A, ESI†), which is compared with vacuum casting (Fig. S3B, ESI†).
Optimization of the degassing time for oxygen-assisted particle synthesis
To evaluate the reproducibility of the resolution of the synthesized particles using the DML method, we prepared a precursor with a high concentration of crosslinker (95% PEG-DA) to minimize the effect of particle swelling, which can complicate the interpretation of particle resolution. A non-patterned PDMS block that functions as an underlying cover to enclose the micromolds is degassed along with the micromolds to ensure a symmetrical oxygen level in the z-axis direction during the synthesis. It is critical to control the oxygen level because particle polymerization is inhibited by the presence of oxygen. When the precursor is exposed to ultraviolet (UV), the photoinitiators are decomposed to free radicals which then initiate the polymerization process. The presence of oxygen in the precursor inhibits the polymerization process by reacting with the radicals or the propagated polymer chain, and thereby terminating the reaction.37,38 Upon UV exposure, generated radicals are continuously depleted by the oxygen present in the precursor. After the oxygen concentration falls below a critical level, the weaker inhibitory effect will be overcome by the chain propagation and particles can then be synthesized. Due to the high oxygen permeability of PDMS,37 there is an uncured precursor layer (oxygen inhibition layer (δ)) on all the surrounding walls of the micromold.During the first 10 s after the precursor comes into contact with the degassed micromold, the oxygen concentration in the precursor rapidly decreases before reaching an equilibrium state (Fig. S4, ESI†). The equilibrium oxygen concentration of the precursor can be controlled by regulating the degassing conditions of the micromold, which can vary the depth of the oxygen inhibition layer (δ) as shown in Fig. 3A. In the non-degassed micromold, continuous diffusion of oxygen from the PDMS micromold prevents polymerization of the precursor near the edge of the mold. As a result, only hexagonal shaped particles could be synthesized in the star-shaped micromolds. On the other hand, reduced oxygen diffusion from the degassed micromold allows the accurate synthesis of star-shaped particles at the same UV dosage applied.
 | ||
| Fig. 3 Microparticle fabrication in DML. A) A comparison of the replication quality of particles synthesized in a non-degassed micromold (left) and a degassed micromold (middle). Images of particles synthesized in a micromold degassed for 0 min (i), 5 min (ii), and 60 min (iii) (right). Scale bars = 50 μm. B) The depth of the inhibition layer as a function of the degassing time and the UV dosage. The duration of the UV irradiation is kept constant (1 s). The change in oxygen concentration in the micromold was calculated using the finite element method in COMSOL software. The dimensions of the PDMS block are 10 × 10 × 3 mm3, and the oxygen diffusivity is 3.4 × 10−9 m2 s−1. Each bar represents the mean from three independent experiments. Error bars represent the standard deviation. C) Encoded particles with internal structures were synthesized in a non-degassed micromold (left) and a degassed micromold (right). D) A microscopy image of a homogeneous population of encoded microparticles. | ||
Based on the data generated from simulation (Fig. 3B), the oxygen concentration of the mold template (3 mm thickness) can reach a near-equilibrium (99%) state after being placed inside the vacuum chamber for 1 h. To determine the optimal degassing time, we evaluated the δ values under various degassing conditions and found that δ gradually decreased with the increase in the degassing time (Fig. S5, ESI†). In agreement with the simulation result, a minimum δ value can be obtained with more than 1 h of degassing. Meanwhile, the degassing time should be increased in order for the near-equilibrium oxygen concentration to be attained in a thicker PDMS mold template.43 The approximate optimal degassing times in relation to the thickness of the mold template are provided (Fig. S6, ESI†).
The PDMS mold template is pressed onto the underlying PDMS cover to seal each micromold with a wall that separates it from the neighboring micromolds. However, complete sealing is impossible because the precursor is flooded over the surface of the PDMS44,45 and therefore a residual precursor layer of about 2.2 μm in height will be inevitably left between the block and the cover (see Fig. S7, ESI† for details). Nevertheless, the diffusion of oxygen into the center of the residual layer is approximately 100 times faster than that into the precursor inside the micromold,37 which almost totally inhibits the polymerization process at the residual layer. Therefore, DML is capable of fabricating isolated hydrogel microparticles on a bulk scale (Fig. 1C). In the case of a short height micromold (height = 5 μm), the residual layer was slightly polymerized with particles (Fig. S8, ESI†). The minimum height of the mold for the individual particle fabrication is estimated (see the ESI† for details).
To demonstrate the feasibility of synthesizing geometrically diverse particles using the DML method, we fabricated a template with two pillars placed inside each 100 × 100 μm2 rectangular micromold. Since the inserted pillars can serve as local channels for oxygen diffusion, the oxygen inhibition layer of these pillar-containing micromolds is expanded compared to that of the simple rectangular micromolds. Using the non-degassed micromold, the precursor near the pillars could not be sufficiently polymerized and therefore the synthesized particles failed to achieve the intended design with low replicable resolution (Fig. 3C). In contrast, the minimum δ under the optimal conditions is below 1 μm in the degassed micromolds, which allows the synthesis of defect-free particles with the intended morphology. As the precursor near the mold is not polymerized by oxygen inhibition, the particles do not stick to the surface of the mold but float on the precursor. Therefore, when the recovery solution is dropped onto the mold, the particles come out of the micromold, which takes about 15 s and the recovery rate is about 99.7% where the coefficient of variability is 0.12% (Fig. S9 and Movie S2, ESI†). Furthermore, due to the oxygen inhibition, the crosslinker in the precursor is not crosslinked with the porous network of the mold.37 Therefore, the mold template can be reused after several times (Fig. S10, ESI†). We conclude that the DML technique allows the reproducible synthesis of microparticles with internal structures, which is applicable to the incorporation of various graphical schemes into the microparticles.46,47
Micromold design for uniform microparticle fabrication
In our experiments, a recurring issue of using micromold arrays to fabricate microparticles was that the particles synthesized in micromolds located at the edge of the mold template were usually smaller than those synthesized in micromolds located at the center of the template (Fig. 4A). The particles were smaller in size when they were synthesized in micromolds lying within 1000 μm from the edge of the mold template, with the smallest particles being synthesized in micromolds closest to the edge. As a result, the particles synthesized in the same block have as much as 6.0% coefficient of variation (C.V.) in the widths. Since the UV intensity is found to be constant over more than 95% of the illumination area (Fig. S11, ESI†), the only factor that can contribute to the observed position-dependent differences in the degree of polymerization is the equilibrium oxygen concentration of the precursor. The oxygen level in the precursor is likely to be higher at the edge of the template than at the center of the template during the polymerization process because atmospheric oxygen can penetrate from the vertical sides of the template, whereas the top and bottom surfaces are in contact with gas-impermeable glass.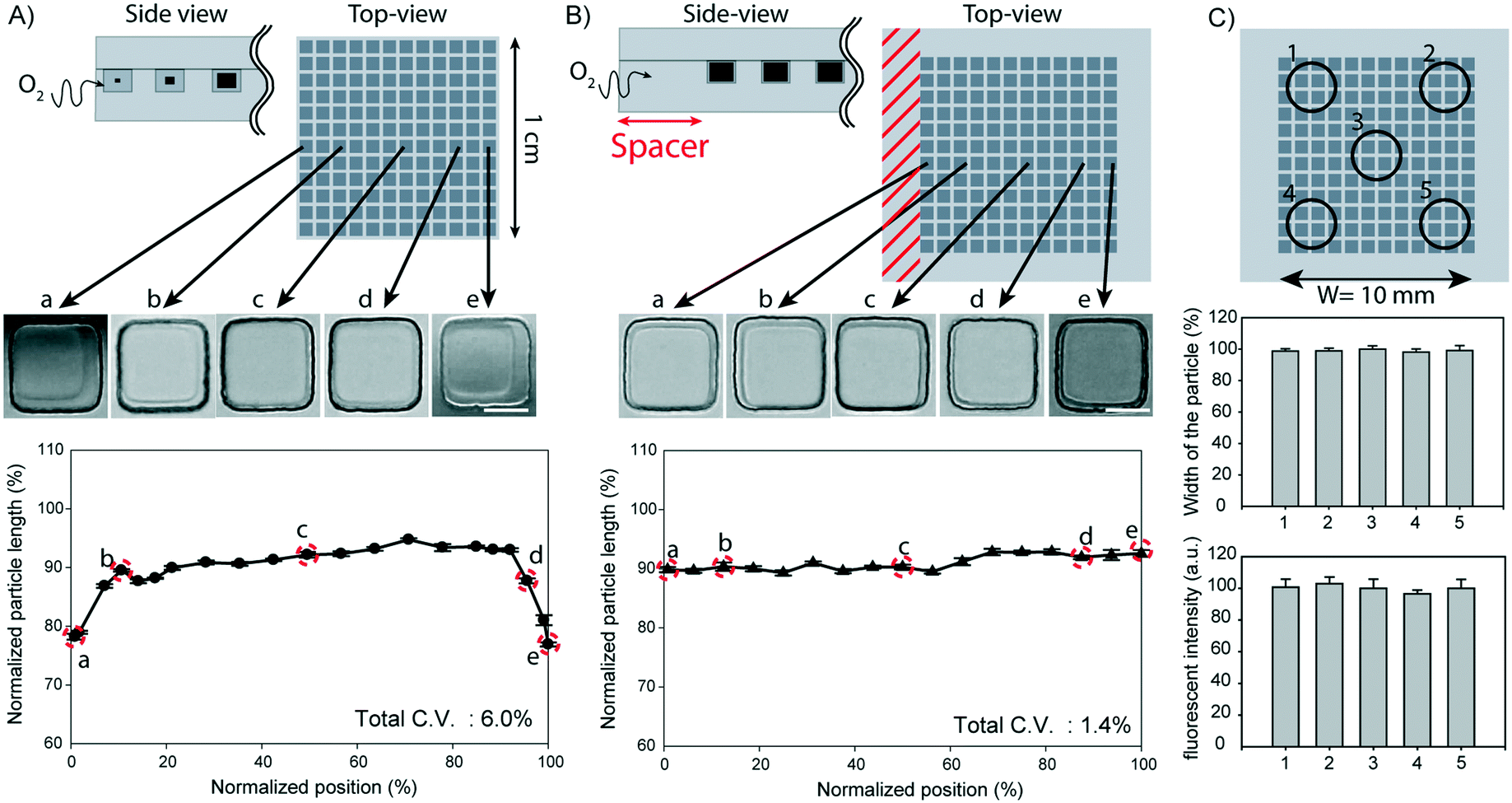 | ||
| Fig. 4 Micromold design for uniform microparticle synthesis. A) and B) Comparisons of the variations in the sizes of the particles synthesized at positions a, b, c, d, and e in (A) a mold template without spacers and (B) a mold template with spacers (B). Scale bars = 50 μm. Each data point represents the mean value from 10 particles. Error bars represent the standard deviation. C) The sizes and fluorescence intensities were uniform among particles synthesized at positions 1, 2, 3, 4, and 5. | ||
In order to eliminate the effect of uneven distribution of oxygen concentration among the micromolds, we added a spacer, a layer of PDMS margin measuring 3 mm thick, around all the vertical sides of the micromold array. The introduction of the spacer effectively insulates the array from atmospheric oxygen, as the oxygen will take a much longer diffusion path to reach the micromolds at the edge of the array. As shown in Fig. 4B, the particle population synthesized in a spacered-mold template displayed a more homogeneous size distribution with a C.V. value of less than 2%. We further tested the particle synthesis in templates with varying thickness of the spacer, and we found that a minimum of 2 mm-spacer was required to eliminate the edge effect (Fig. S12, ESI†).
To further verify a uniform degree of polymerization in all micromolds, regardless of their position in the array, we used a rhodamine-B-methacrylate-containing precursor to synthesize the particles. Rhodamine-B-methacrylate is a fluorescent dye that is conjugated to the polymer network during polymerization, therefore, quantitative assessment of the polymerization process can be achieved by comparing the fluorescence intensities of the particles. As shown in Fig. 4C, we confirmed that there are no differences in both the fluorescence intensities and particle sizes among the synthesized particles, irrespective of their positions in the mold array. In our experimental setup, we were able to synthesize 40000 particles, with a cross-sectional area of 25 × 25 μm2 each, in a 1 cm2 array area upon a single dose of UV exposure. Therefore, 4 million particles can be theoretically synthesized with a single dose of UV exposure if the area exposed to the irradiation can be increased to 10 × 10 cm2. Extrapolating from this, the DML technique can ideally feature a productivity of 790 million particles per h with a 60 s loading time, a 1 s exposure time, and a 120 s processing time for any other steps combined. If DML is continuously operated by the roll-to-roll process, the productivity can increase by more than 100 times (Table S2, ESI†).
Synthesis of protein encapsulated particles and 3D-shaped particles
An advantage of the DML method is that there is minimal evaporation of the liquid used to fill the micromolds. Therefore, protein-containing precursors, in which dispersion is determined by the water content, can be used to fill a degassed micromold without causing concomitant protein aggregation in the particles. This property is essential for synthesizing particles used for enzyme immobilization,48 immunoassays,3 controlled protein delivery,49,50etc. To demonstrate the potential of using the DML method for the synthesis of protein-encapsulated particles, we synthesized particles using a precursor containing well-dispersed fluorescein isothiocyanate (FITC) labeled bovine serum albumin (BSA). We then loaded the precursor into non-degassed micromolds in order to make a comparison between the DML method and a previously described replica molding technique. Non-degassed micromolds were filled with the precursor after manual removal of the air bubbles by scrubbing with a pipette. Due to the time required to completely fill the micromolds by scrubbing, the precursor had to be exposed to the ambient pressure for up to 2 minutes during this procedure. Consequently, the water content in the precursor evaporated during this step due to its high volatility compared to any other components in the precursor, and caused the proteins to aggregate as shown by the uneven particle surface (Fig. 5A). The 3D surface plots of the particles are shown in Fig. S13A and B, ESI.† In contrast, the particles synthesized in degassed micromolds displayed a smooth surface and a uniform fluorescence signal (Fig. 5A and S13C, ESI†).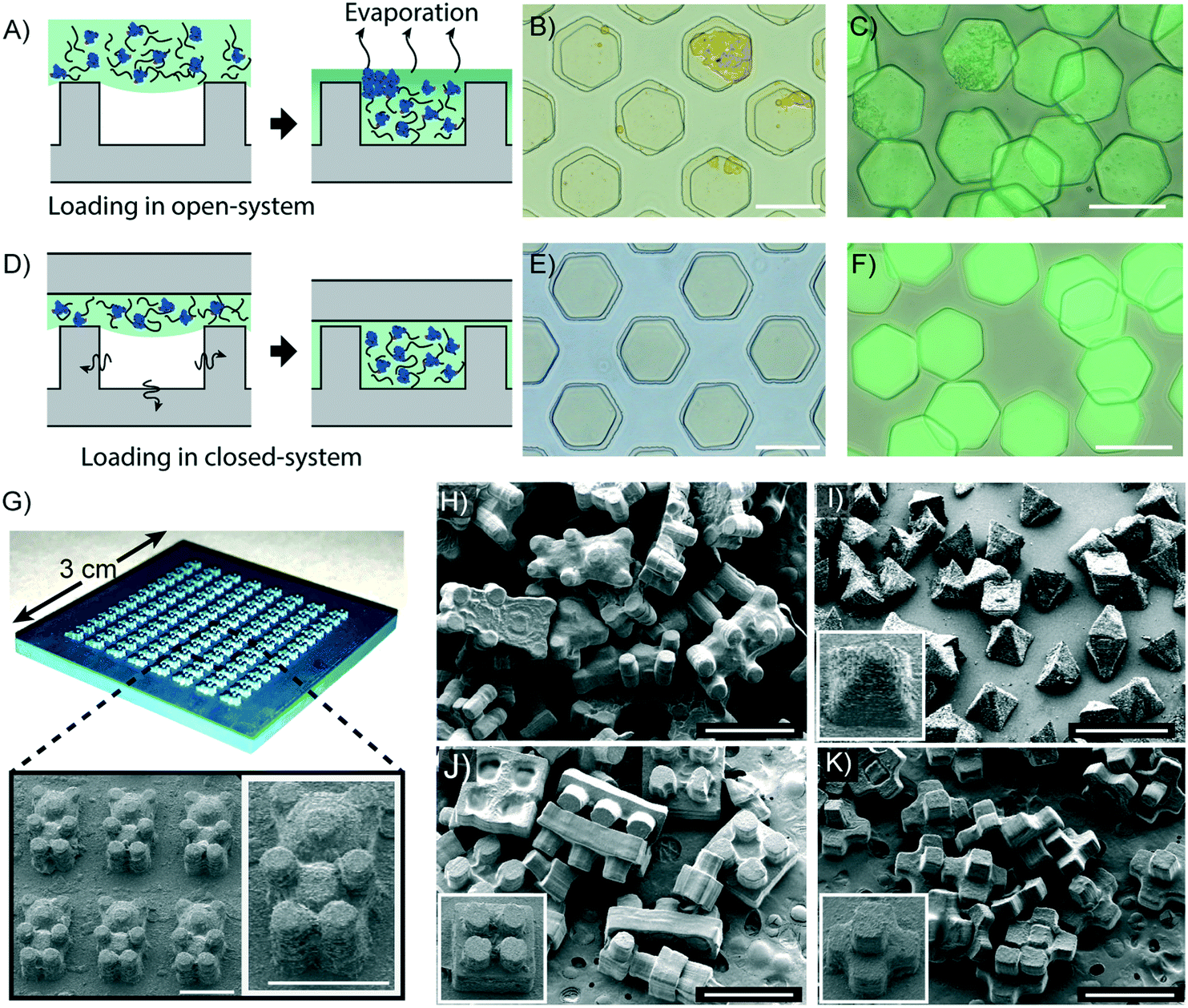 | ||
| Fig. 5 Synthesis of protein-loaded microparticles and 3D particles. A–C) The precursor is exposed to the ambient pressure for 2 min, during which the water in the precursor solution evaporates and causes the proteins in the precursor solution to aggregate. D–F) Homogeneous protein encapsulation can be achieved in particles synthesized by DML. B and E) Bright field images of protein encapsulated particles in the micromolds. C and F) Overlaid fluorescence and bright field images of protein encapsulated particles. Scale bars = 60 μm. G) A photograph of a 3D-printed master mold and its SEM image. Scale bar = 1 mm. H–K) SEM images of particles adopting complex shapes: H) bear-shaped, I) pyramid-shaped, J) toy brick, and K) polyhedral. Insets of I–K) show the SEM images of the corresponding 3D micromold. Scale bars = 1 mm. | ||
Molding techniques allow particles to be synthesized beyond 2D-extruded shapes. We took advantage of this capacity and synthesized particles with complex morphologies using a master mold fabricated from a commercial 3D printer (Fig. 5B). The master mold was then replicated with PDMS for the fabrication of a 3D micromold. Due to the limited resolution of the 3D printer (6.3 μm), the master mold was produced with a rough surface that differed by a maximum of 14.7 μm between the lowest point of a depression and the highest point of an elevation (Fig. S14, ESI†).
In order to replicate the master mold with elastic materials, the design of the mold is restricted to a hemi-3D shape. Nonetheless, it still gives more morphological diversity than designs with only a 2D-extruded shape. Since air tends to be entrapped at the edge of the mold,51 defects can arise during precursor loading into a complex micromold with many uneven shapes. Excitingly, we found that the precursor loaded into the degassed micromold was free from any defect when the DML method was applied. With the degassed 3D micromold, the precursor could be completely loaded into the 3D micromold with a loading time of about 23 minutes (Fig. S15, ESI†). Furthermore, we also demonstrated the successful synthesis of bear-shaped, pyramid-shaped, toy brick, and polyhedral particles using the 3D printed master mold (Fig. 5D). In conclusion, we present the use of a commercial 3D printer in replica molding for the production of particles that adopt complex morphologies that are beyond traditional 2D-extruded shapes.
Conclusions
DML greatly expands the freedom of precursor choice by eliminating the effects of two major obstacles in particle synthesis: interfacial tension and evaporation. The most critical advantage of DML is that the precursors can be filled into the elastomeric micromolds without being obstructed by the interfacial tension because the air suction force generated by degassed PDMS can effectively pull the liquid into the micromold. As the precursor can be loaded regardless of the area of the mold template, the productivity of the particles can be proportionally increased by using a mold template with a large area. Another advantage of DML is that the technique allows the precursor to be filled into the micromolds free of evaporation. As the liquid is sandwiched between the micromold and the cover, the particles are fabricated in a semi-closed environment, thus preventing the evaporation of the liquid. As an example, protein aggregation could be prevented during the synthesis of protein-encapsulated particles using the DML method. Furthermore, DML also expands the freedom of controlling the morphology, in which we showed that a high resolution of replication can be realized in DML through reducing the oxygen inhibition of the photoinduced polymerization process. The oxygen inhibition layer can be reduced to 1 μm, which is permissible for the production of anisotropic particles with internal structures. Also, we showed the benefit of modifying the mold template with the addition of a spacer, so that particles synthesized in all positions of the mold array are ensured to have a uniform size and the same extent of polymerization. We also demonstrated that DML can expand the geometrical diversity of the synthesis from 2D to 3D-shapes using a 3D-printed master mold. Through the use of DML, molds with complex shapes can be efficiently loaded with the precursor and particles can be fabricated in isolation. With its ability to synthesize anisotropic particles in mass and with uniformity, DML proves itself as a powerful particle synthesis method for various applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering Research Center of Excellence Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2016R1A5A1010148) and the Next-Generation Biogreen 21 Program funded by the Rural Development Administration of Republic of Korea (No. PJ013158). This research was also supported by the Basic Science Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2018R1D1A1B07046577).References
- D. C. Pregibon, M. Toner and P. S. Doyle, Science, 2007, 315, 1393–1396 CrossRef CAS.
- H. Lee, J. Kim, H. Kim, J. Kim and S. Kwon, Nat. Mater., 2010, 9, 745–749 CrossRef CAS PubMed.
- H. J. Lee, Y. H. Roh, H. U. Kim, S. M. Kim and K. W. Bong, Lab Chip, 2019, 19, 111–119 RSC.
- S. W. Song, S. D. Kim, D. Y. Oh, Y. Lee, A. C. Lee, Y. Jeong, H. J. Bae, D. Lee, S. Lee, J. Kim and S. Kwon, Adv. Sci., 2018, 6, 1801380 CrossRef PubMed.
- Y. He and K. Park, Mol. Pharmaceutics, 2016, 13, 2164–2171 CrossRef CAS.
- K. J. McHugh, T. D. Nguyen, A. T. Linehan, D. Yang, A. M. Behrens, S. Rose, Z. L. Tochka, S. Y. Tzeng, J. J. Norman, A. C. Anselmo, X. Xu, S. Tomasic, M. A. Taylor, J. Lu, R. Guarecuco and R. Langer, Science, 2017, 357, 1138–1142 CrossRef CAS.
- S. S. Lee, J. B. Kim, Y. H. Kim and S.-H. Kim, Sci. Adv., 2018, 4, eaat8276 CrossRef PubMed.
- J. Y. Sim, J. H. Choi, J. M. Lim, S. Cho, S. H. Kim and S. M. Yang, Small, 2014, 10, 3979–3985 CrossRef CAS.
- S. C. Glotzer and M. Solomon, Nat. Mater., 2007, 6, 557–562 CrossRef PubMed.
- K. Han, C. W. Shields, N. M. Diwakar, B. Bharti, G. P. López and O. D. Velev, Sci. Adv., 2017, 3, e1701108 CrossRef.
- M. Neufurth, X. Wang, S. Wang, R. Steffen, M. Ackermann, N. D. Haep, H. C. Schroder and E. G. Muller, Acta Biomater., 2017, 64, 377–388 CrossRef CAS.
- B. Li, M. H. He, L. Ramirez, J. George and J. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 4158–4162 CrossRef CAS.
- H. U. Kim, D. G. Choi, H. J. Lee, M. S. Shim and K. W. Bong, Lab Chip, 2018, 18, 754–764 RSC.
- J. H. Kim, K. Je, T. S. Shim and S. H. Kim, Small, 2017, 13, 1603516 CrossRef.
- K. Choi, M. Salehizadeh, R. B. Silva, N. Hakimi, E. Diller and D. K. Hwang, Soft Matter, 2017, 13, 7255–7263 RSC.
- S. Habasaki, W. C. Lee, S. Yoshida and S. Takeuchi, Small, 2015, 11, 6391–6396 CrossRef CAS.
- K. S. Paulsen, Y. Deng and A. J. Chung, Adv. Sci., 2018, 5, 1800252 CrossRef.
- R. Yuan, M. B. Nagarajan, J. Lee, J. Voldman, P. S. Doyle and Y. Fink, Small, 2018, 14, 1803585 CrossRef.
- K. W. Bong, D. C. Pregibon and P. S. Doyle, Lab Chip, 2009, 9, 863–866 RSC.
- D. C. Appleyard, S. C. Chapin, R. L. Srinivas and P. S. Doyle, Nat. Protoc., 2011, 6, 1761 CrossRef CAS.
- D. Baah and T. Floyd-Smith, Microfluid. Nanofluid., 2014, 17, 431–455 CrossRef CAS.
- M. B. Dickerson, P. B. Dennis, V. P. Tondiglia, L. J. Nadeau, K. M. Singh, L. F. Drummy, B. P. Partlow, D. P. Brown, F. G. Omenetto, D. L. Kaplan and R. R. Naik, ACS Biomater. Sci. Eng., 2017, 3, 2064–2075 CrossRef CAS.
- H. Yin, Y. Ding, Y. Zhai, W. Tan and X. Yin, Nat. Commun., 2018, 9, 4096 CrossRef.
- J. P. Rolland, B. W. Maynor, L. E. Euliss, A. E. Exner, G. M. Denison and J. M. DeSimone, J. Am. Chem. Soc., 2005, 127, 10096–10100 CrossRef CAS.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS.
- E. Kang, S. Jung, J. H. Abel, A. Pine and H. Yi, Langmuir, 2016, 32, 5394–5402 CrossRef CAS.
- K. Parratt, J. Jeong, P. Qiu and K. Roy, Lab Chip, 2017, 17, 2861–2872 RSC.
- S. Yu, H. Cho, J. P. Hong, H. Park, J. C. Jolly, H. S. Kang, J. H. Lee, J. Kim, S. H. Lee, A. S. Lee, S. M. Hong, C. Park, S. Yang and C. M. Koo, Nat. Commun., 2017, 8, 721 CrossRef.
- K. Park, S. M. Skidmore, L. Fultz, Y. Yun and B. K. Lee, U.S. Pat. 9855218, 2018 Search PubMed.
- J. G. Ok, Y. J. Shin, H. J. Park and L. J. Guo, Appl. Phys. A: Mater. Sci. Process., 2015, 121, 343–356 CrossRef CAS.
- M. D. Francesco, R. Primavera, D. Romanelli, R. Palomba, R. C. Pereira, T. Catelani, C. Celia, L. D. Marzio, M. Fresta, D. D. Mascolo and P. Decuzzi, ACS Appl. Mater. Interfaces, 2018, 10, 9280–9289 CrossRef.
- G. Acharya, C. S. Shin, M. McDermott, H. Mishra, H. Park, I. C. Kwon and K. Park, J. Controlled Release, 2010, 141, 314–319 CrossRef CAS.
- R. J. Jackman, D. C. Duffy, E. Ostuni, N. D. Willmore and G. M. Whitesides, Anal. Chem., 1998, 70, 2280–2287 CrossRef CAS.
- J. Guan, N. Ferrell, L. J. Lee and D. Hansford, Biomaterials, 2006, 27, 4034–4041 CrossRef CAS PubMed.
- C. L. Lewis, C. H. Choi, Y. Lin, C. S. Lee and H. Yi, Anal. Chem., 2010, 82, 5851–5858 CrossRef CAS PubMed.
- P. Laulia, M. B. Chan-Park, Y. Yan, S. Q. Wang, C. M. Li and Y. C. Lam, Small, 2008, 4, 69–76 CrossRef CAS.
- D. Dendukuri, P. Panda, R. Haghgooie, J. M. Kim, T. A. Hatton and P. S. Doyle, Macromolecules, 2008, 41, 8547–8556 CrossRef CAS.
- C. Decker and A. D. Jenkins, Macromolecules, 1985, 18, 1241–1244 CrossRef CAS.
- L. F. Xu, H. Lee, D. Jetta and K. W. Oh, Lab Chip, 2015, 15, 3962–3979 RSC.
- G. Li, Y. H. Luo, Q. Chen, L. Y. Liao and J. L. Zhao, Biomicrofluidics, 2012, 6, 14118–1411816 CrossRef.
- N. Tanaka, H. Moriguchi, A. Sato, T. Kawai, K. Shimba, Y. Jimbo and Y. Tanaka, RSC Adv., 2016, 6, 54754–54762 RSC.
- Z. Han, X. Tang and B. Zheng, J. Micromech. Microeng., 2007, 17, 1828–1834 CrossRef CAS.
- H. Z. An, H. B. Eral, L. Chen, M. B. Chen and P. S. Doyle, Soft Matter, 2014, 10, 7595–7605 RSC.
- H. Schmitt, L. Frey, H. Ryssel, M. Rommel and C. Lehrer, J. Vac. Sci. Technol., B, 2007, 25, 785–790 CrossRef CAS.
- J. Lee, J. Y. Kim, J. H. Choi, J. G. Ok and M. K. Kwak, ACS Omega, 2017, 2, 1097–1103 CrossRef CAS PubMed.
- S. Birtwell and H. Morgan, Integr. Biol., 2009, 1, 345–362 CrossRef CAS PubMed.
- Y. H. Roh, H. J. Lee, H. J. Moon, S. M. Kim and K. W. Bong, Anal. Chim. Acta, 2019, 1076, 110–117 CrossRef CAS PubMed.
- S. I. Matsuura, M. Chiba, T. Tsunoda and A. Yamaguchi, J. Nanosci. Nanotechnol., 2018, 18, 104–109 CrossRef CAS PubMed.
- U. Bilati, E. Allémann and E. Doelker, Eur. J. Pharm. Biopharm., 2005, 59, 375–388 CrossRef CAS PubMed.
- H. U. Kim, D. G. Choi, Y. H. Roh, M. S. Shim and K. W. Bong, Small, 2016, 12, 3463–3470 CrossRef CAS PubMed.
- J. Wang, M. Li, J. Qiu and Y. Zhou, J. Micromech. Microeng., 2018, 28, 035011 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9lc00828d |
| This journal is © The Royal Society of Chemistry 2020 |