
Liver microphysiological systems development guidelines for safety risk assessment in the pharmaceutical industry

Abstract
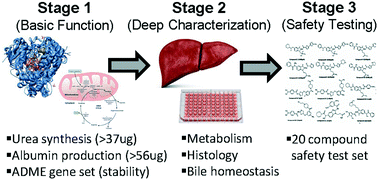
The liver is critical to consider during drug development because of its central role in the handling of xenobiotics, a process which often leads to localized and/or downstream tissue injury. Our ability to predict human clinical safety outcomes with animal testing is limited due to species differences in drug metabolism and disposition, while traditional human in vitro liver models often lack the necessary in vivo physiological fidelity. To address this, increasing numbers of liver microphysiological systems (MPS) are being developed, however the inconsistency in their optimization and characterization often leads to models that do not possess critical levels of baseline performance that is required for many pharmaceutical industry applications. Herein we provide a guidance on best approaches to benchmark liver MPS based on 3 stages of characterization that includes key performance metrics and a 20 compound safety test set. Additionally, we give an overview of frequently used liver injury safety assays, describe the ideal MPS model, and provide a perspective on currently best suited MPS contexts of use. This pharmaceutical industry guidance has been written to help MPS developers and end users identify what could be the most valuable models for safety risk assessment.
- This article is part of the themed collections: Microphysiological systems in pharmaceutical safety and ADME applications and organ-on-a-chip systems: translating concept into practice
 Please wait while we load your content...
Please wait while we load your content...

