
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe elemental analysis and multi-nuclear NMR study of an alkali molten salt used to digest reference and commercial SWCNT powders†
Filipa R. F.
Simoes
 *a,
Edy
Abou-Hamad
b,
Jan
Kamenik
c,
Jan
Kučera
c and
Pedro M. F. J.
Costa
*a
*a,
Edy
Abou-Hamad
b,
Jan
Kamenik
c,
Jan
Kučera
c and
Pedro M. F. J.
Costa
*a
aKing Abdullah University of Science and Technology (KAUST), Physical Science and Engineering Division, Thuwal 23955-6900, Saudi Arabia. E-mail: filipa.fernandessimoes@kaust.edu.sa; pedro.dacosta@kaust.edu.sa
bKing Abdullah University of Science and Technology (KAUST), Core Labs, Thuwal 23955-6900, Saudi Arabia
cNuclear Physics Institute of the Czech Academy of Sciences, 250-68 Husinec-Rez, Czech Republic
First published on 9th October 2020
Abstract
For quite some time, alkaline oxidation (or fusion) has been used to solubilize refractory materials and mineral ores. Recently, its application scope was extended to facilitate batch-scale elemental analysis of nanomaterials such as carbon nanotubes. Here, a sodium tetraborate salt was used to digest four different types of single-walled carbon nanotubes. These samples were produced employing Co–Mo or Fe catalyst systems. Their graphitic matrix was exposed to different melt temperatures for a short period of time, following which the concentration of six transition metals was measured. Recoveries in excess of 80% were obtained, with the melt temperature affecting more the elemental extraction in Fe-catalyzed nanotubes. Together with previous results, the work described allows drawing pertinent conclusions on the advantages and limitations of alkaline oxidation as an alternative sample digestion approach for the routine chemical analysis of nanocarbons.
1. Introduction
Carbon nanotubes (CNTs) have been extensively studied since 1991.1 Today, these nanofibers are part of the industrial landscape and have reached a higher stage of technological development, explaining their year-on-year production output increase.2Classically, there are three main approaches to produce CNTs, namely arc discharge, laser ablation and chemical vapor deposition (CVD).3 Not surprisingly, given the decades-old know-how of growing micron-sized carbon fibers, catalytic CVD has become the preferred industrial method to supply nanotubes.4 For the particular case of single-walled carbon nanotubes (SWCNTs), a variety of transition metals are commonly used in the CVD process as their different combinations enable characteristics such as a tailored length and chirality.5 Key growth catalysts are Fe, Co and Ni,6 but other elements (e.g. Mg, Al, Cr, Mn, Cu and Mo) may be added to promote the feedstock's decomposition, generally a hydrocarbon gas such as methane or ethylene.7–11 Post-growth, eliminating these non-carbon elements is not entirely trivial.12 Furthermore, their presence can disrupt the performance of SWCNTs such as in electrochemical13 and biomedical14 applications.
In view of the above, the quantification of non-carbon growth by-products is a critical step in validating the properties and assessing the responses of nanotubes used in the research, industrial and consumer arenas. Moreover, overlooking proper quantitative analysis of SWCNTs will not only interfere with the lab-scale study conclusions for next-generation technologies but may also result in unforeseen side effects on the environment and public health when they get translated to mass-scale commercial products. In fact, much of the toxicity attributed to carbon nanotubes in the literature could well be related to metallic impurities due to poor protocols of elemental quantification of the as-received powders.15–17 For many years, analytical tools such as inductively coupled plasma optical emission spectroscopy (ICP-OES) have been applied in this context.18–25 Surprisingly, few teams have validated these studies against chemically certified SWCNT standard samples that have been commercialized for almost a decade.26–28 Also, and despite the numerous analytical studies, the pre-treatment step continues to be the main hurdle in routine elemental analysis of graphitic nanocarbons. This problem was recently highlighted in a comprehensive review by Dyjak et al.29 To disintegrate the sp2-C matrix, stringent conditions are needed: if not combustion of the nanotubes (a.k.a. ashing),30 then prolonged high-temperature exposure to concentrated acids and/or oxidizers (e.g. peroxides) is employed.31
In 2016, we proposed alkaline oxidation (a.k.a. fusion) as a viable approach to digest different types of carbons, hence offering a pre-treatment alternative for the elemental analysis of SWCNTs by ICP-OES.12 More recently, we validated fusion for SWCNTs32 and tested it on battery-relevant carbon materials with different types of nanotextures (point, radial and planar).13 On the whole, acceptable recoveries (usually, 80–90%) have been achieved for a number of transition metals. Amongst the recovery optimization steps taken, the most successful was the replacement of a receiving plate (where the melt solidifies as a glass-like bead) by a container with diluted acid (where the melt quenches and solubilizes). Whilst these demonstrations are encouraging, we acknowledge that there is room for improvement as the recovery levels remain lower than those provided by the more mature methods of ashing and wet-digestion.
In fusion, a critical variable is the selection of the alkaline salt (or flux). For our previous work, lithium compounds were used (predominantly). Here, four different SWCNTs were subjected to alkaline oxidation using sodium tetraborate as the flux. The nanotubes were divided in two categories: reference materials and commercial samples. The effect of the reaction temperature on the recovery of several transition metals was examined along with possible interactions (structural and chemical) of the sodium salt with the carbon species.
2. Experimental section
2.1 Materials, reagents and solutions
Two lots of commercial SWCNTs procured from Sigma-Aldrich (named here: SWCNT-7867V, with ≥95% carbon content, lot MKBW7867V, and SWCNT-5848V, with >70% carbon content, lot MKBW5848V) and two SWCNT reference materials (SWCNT-1, from the National Research Council Canada and SRM2483, from the National Institute of Standards and Technology) were analyzed. For the flux, sodium tetraborate (hereafter, NaT) was used (Na2B4O7, CAS 1330-43-4, Sigma-Aldrich). As reaction vessels, Pt/Au crucibles (95/5 wt%, Malvern PANanalytical) were employed during the fusion procedure. For the ICP-OES measurements, the melts were poured into a 10% nitric acid (HNO3) solution, prepared from 70% HNO3 (CAS-7697-37-2, lot 1118092, Aristar Plus, BDH, Canada) and deionized water (resistivity = 18 MΩ cm, produced with a Milli-Q system from Millipore, UK).Standard stock solutions of single elements (PerkinElmer, US) were used to calibrate the ICP-OES instrument, namely 2% (v/v) solutions of: Cr (CAS 7440-47-3, lot 15–65Cr), Mn (CAS 7439-96-5, lot 15–91Mn), Fe (CAS 7439-89-6, lot 15–185Fe), Co (CAS 7440-48-4, lot 16–76Co) and Ni (CAS 7440-02-0, lot 15–121Ni). In addition, a Mo standard on a matrix of H2O/tr NH4OH (lot D2-MO02036, Inorganic Ventures) was employed.
2.2 Alkaline oxidation
The crucibles were thoroughly washed with a diluted acid solution and then cleaned with 70% (v/v) ethanol. The fusion sample was prepared by adding 25 mg of the SWCNT powder to 250 mg of the NaT flux (i.e., in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio) and carefully mixing them, inside a Pt crucible, with the assistance of a vortex. Then, the vessel was placed in a furnace (Claisse LeNeo, Malvern PANalytical), where the melting process was performed at a target temperature (850 °C or 1050 °C) and with a dwell period of four minutes. Afterwards, for the chemical analysis, the crucible was tilted and the melt was dropped into a polypropylene beaker filled with a 10% HNO3 solution. Stirring took place until the total dissolution of the melt (less than five minutes). Note that, for each SWCNT powder, three different aliquots/melts were prepared (N = 3). Differently, for the structural analysis, the melts were allowed to solidify in the crucible and collected as glass-like beads. They were then carefully broken and ground, with an agate pestle and a mortar, until a fine powder was obtained. The fusion blank (control) was prepared by weighing 250 mg of the NaT flux that were subjected to the same melting/collection procedures.
:10 ratio) and carefully mixing them, inside a Pt crucible, with the assistance of a vortex. Then, the vessel was placed in a furnace (Claisse LeNeo, Malvern PANalytical), where the melting process was performed at a target temperature (850 °C or 1050 °C) and with a dwell period of four minutes. Afterwards, for the chemical analysis, the crucible was tilted and the melt was dropped into a polypropylene beaker filled with a 10% HNO3 solution. Stirring took place until the total dissolution of the melt (less than five minutes). Note that, for each SWCNT powder, three different aliquots/melts were prepared (N = 3). Differently, for the structural analysis, the melts were allowed to solidify in the crucible and collected as glass-like beads. They were then carefully broken and ground, with an agate pestle and a mortar, until a fine powder was obtained. The fusion blank (control) was prepared by weighing 250 mg of the NaT flux that were subjected to the same melting/collection procedures.
2.3 Elemental analysis
The instrumental neutron activation analysis (NAA) measurements were performed as described previously.33 Briefly, 30 mg portions of the carbon nanotubes were heat-sealed into irradiation capsules made from two polyethylene discs with 25 mm diameter and 0.15 mm thickness. The NAA procedures developed and routinely employed at the Nuclear Physics Institute laboratory were used with minor modifications. Quantification was carried out by the comparator method. Irradiations were performed in the experimental reactor LVR-15 of the Research Centre Rez, the Czech Republic.For the ICP-OES measurements, an Agilent 5110 instrument, bearing a synchronous vertical dual view configuration and a wavelength window between 167 nm and 785 nm, was employed. The instrumental parameters used are listed elsewhere.32 To calibrate the equipment, single-element solutions (with different concentrations) of Cr, Mn, Fe, Co, Ni and Mo were used. They were all derived from the respective 1000 ppm single-element standards. In order to check the instrument performance and ensure that its precision was not degrading over the period of the analysis, a quality control sample (1 ppm) and solutions for continuing calibration verification (5 ppm) were prepared. All recovery levels are given in percentages relative to the elemental concentration values measured from NAA.
2.4 Structural characterization
For the 1D 13C magic angle spinning (MAS) nuclear magnetic resonance (NMR) analysis, a known amount of sample was placed inside zirconia rotors. The spectrum was acquired on a Bruker AVANCE III, operating at a resonance frequency of 100 MHz and using a conventional double resonance 4 mm cross polarization magic angle spinning (CPMAS) probe. The spinning frequency was set to 10–15 kHz. NMR chemical shifts are reported with respect to an external reference, tetramethylsilane (TMS). The spectra were recorded via a spin echo pulse sequence (pulse length 3.4 μs) with four-phase alternation synchronized with the spinning rate for the MAS experiments (to delete all background signals from the probe). The inter-scan delay was set to 15 s, to allow complete relaxation, and 5000–30000 scans were performed. An apodization function (exponential) corresponding to a line broadening of 80 Hz was applied prior to the Fourier transformation. The 1D 11B (I = 3/2) MAS NMR experiments were performed on a 21.1 T Bruker AVANCE III spectrometer (vL(11B) = 288.826 MHz) with a double resonance 3.2 mm MAS probe and a MAS frequency of 20 kHz with a short pulse (π/12 with a pulse width of 1 μs and repetition time of 2 s). 11B multiple quantum MAS (MQMAS) experiments were carried out using the three pulse MQMAS sequence with a z-filter and a spinning frequency of 20 kHz. The excitation and conversion pulse lengths were 5.7 μs and 1.9 μs, respectively, and the read pulse length was 20 μs. The t1 increment was 50 μs to ensure rotor synchronized data acquisition. Chemical shifts are referenced to an external sample of 1 M boric acid solution. The 1D 23Na (I = 3/2) MAS NMR experiments were performed on a 21.1 T Bruker AVANCE III spectrometer (vL(23Na) = 230.869 MHz) with a double resonance 3.2 mm MAS probe and a MAS frequency of 18 or 20 kHz. 23Na MQMAS experiments were carried out using the three pulse sequence with a z-filter and a spinning frequency of 20 kHz. The excitation and conversion pulse lengths were 5.7 μs and 1.9 μs, respectively, and the read pulse length was 20 μs. The t1 increment was 50 μs to ensure rotor synchronized data acquisition.
Raman spectroscopy was carried out to assess the diameter distribution and structure of the four as-received nanotube samples. A WITeC 300RA system fitted with a 532 nm laser and a UHT300 spectrometer was used.
To complement the NMR and Raman structural analyses, localized imaging of the SWCNTs was performed with a FEI TECNAI G2 Spirit TWIN transmission electron microscope (TEM), operated at 120 kV. To prepare the sample for TEM, a few drops of nanotubes suspended in ethanol were poured onto Au (or Cu) holey carbon grids and dried, in a vacuum oven, at 70 °C.
3. Results and discussion
Subsequent to the fusion and dissolution steps, the elemental analysis of the two sets of SWCNTs (standards and commercial samples) was performed by ICP-OES. Under normal pressure and temperature conditions, NaT melts at 741 °C. Accordingly, to shed light on the effect of the alkaline oxidation temperature on elemental recovery, two reaction temperatures were considered: 850 °C and 1050 °C.3.1 Reference materials
One standard reference (SRM 2483) and one certified reference (SWCNT-1) SWCNTs were used. Of these materials, SWCNT-1 was previously employed to validate the fusion method with a lithium borate flux.32 This Canadian standard differs significantly from SRM2483 (NIST), not just in composition and certified elements but also in the structure and allotrope homogeneity of the carbon powder.34 In fact, the latter is labelled “soot”.As for SWCNT-1, we previously analyzed it employing microwave-assisted acid digestion as the pre-treatment step.26 The recoveries and respective standard deviations were quite satisfactory for the six elements mentioned, further verifying the efficiency of wet-digestion for elemental analysis by ICP-OES. In the present work, the SWCNT-1 beads exhibited a dark blue color after the fusion process. The same hue was seen when a lithium flux was used as the reaction medium and attributed to the presence of Co2+ species.32 Upon pouring the melt into the beaker containing the diluted acid, a colorless and transparent solution was obtained. The bar charts in Fig. 1a and b summarize the recoveries of the six transition metal elements studied. Generally, there were only minor differences in the average values obtained at 850 °C and 1050 °C. The exception was Cr. However, given the large standard deviation in its 1050 °C readings, the variation in the recovery value is questionable. In fact, the higher temperature process seems to increase the uncertainty of measurements for SWCNT-1. It is possible that the additional 200 °C altered the evaporation rate of the elements and, by doing so, led to competing physicochemical processes that weakened the retention of transition metals in the flux. It is interesting to compare these results to those obtained using a lithium salt flux.32 In that study, a melting temperature of 1075 °C was used, close to the current 1050 °C, but the exposure time of SWCNT-1 was four times longer (17 min). Still, the average recovery values obtained for Co, Ni and Mo were also within the interval of 80–90%. Overall, it appears that short exposure times can be used without compromising the recovery levels in nanotube powders.
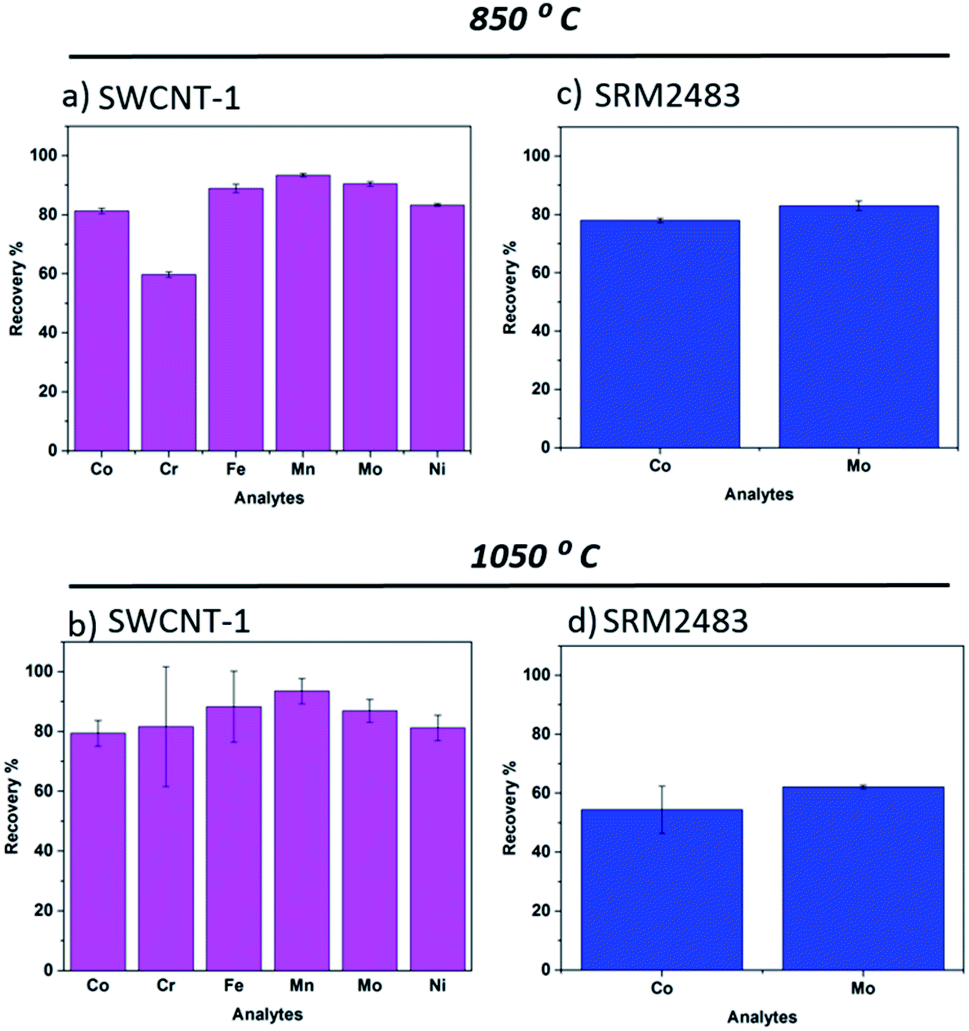 | ||
| Fig. 1 Elemental recovery of catalytically active transition metals in the two SWCNT reference materials, with the averages and standard deviations obtained with N = 3. (a, b) Calculated percentages for SWCNT-1, fused at 850 °C and 1050 °C, respectively; (c, d) SRM2483, fused at 850 °C and 1050 °C, respectively. | ||
Regarding SRM2483, we previously quantified it using wet-digestion with the recovery levels for Co and Mo being well above 80%.26 After exposure to the NaT melts, its glass beads also showed a dark blue color. However, and in contrast to SWCNT-1, there was a pronounced temperature effect on the average recovery values. As shown in Fig. 1c and d, increasing the flux temperature resulted in a 20% decline of elemental recovery. We attribute this to faster evaporation rates and/or elemental heterogeneity. In fact, the certificate of analysis of SRM2483 indicates that there is some degree of heterogeneity in the powdered soot (for the two elements considered, Co and Mo).35
From the above, it seems that a higher fusion temperature (i.e. several hundred degrees above the melting point of the flux) leads to a recovery decrease. As mentioned, one possible reason is faster evaporation rates. In the molten salt, the transition metals are most likely ionized, forming metal oxides upon condensation. Logically, this charged state would considerably decrease the element's vapor pressure when compared to the metallic atomic state. Still, given the limited number of aliquots (and heterogeneity of SRM2483), additional experiments will be required to consolidate this hypothesis.
In Fig. 2a, the 11B 1D MAS spectra of sodium tetraborate are shown for the as-received (in red) and post-fusion states, at 850 °C (in grey) and 1050 °C (in black). Initially, two resonances are present in the NaT salt. They are related to the tetrahedral units of BO4 (or B4) and the trigonal units of BO3 (or B3).36 The corresponding 11B MQMAS (Fig. 2b) shows that the B4 resonance has a double symmetric site with two different orientations. As for B3, it is more diffuse. Its resonance points to a diversity of chemical environments originating from the anisotropic shift contribution. Accordingly, four components may be attributed: two to the symmetric trigonal sites (containing either three bridging or non-bridging oxygen atoms) and two to the asymmetric trigonal sites (with one or two bridging oxygen atoms).13 After the fusion procedure, the salt's resonances in the 11B 1D MAS high temperature spectra became broader (Fig. 2a), which is understandable given the glassy nature of the samples. In Fig. 2c and d, the 11B MQMAS spectra of the 850 °C and the 1050 °C experiments, respectively, showed some variations between them. For the B3 sites, besides the anisotropic shift in some of the four resonance components (by 1 to 2 ppm), the isotropic distribution was changed. They are due to the rearrangement of boron coordination and the existence of multiple B3 sites. As for the B4 resonance, the post-fusion flux contained a single symmetric site.
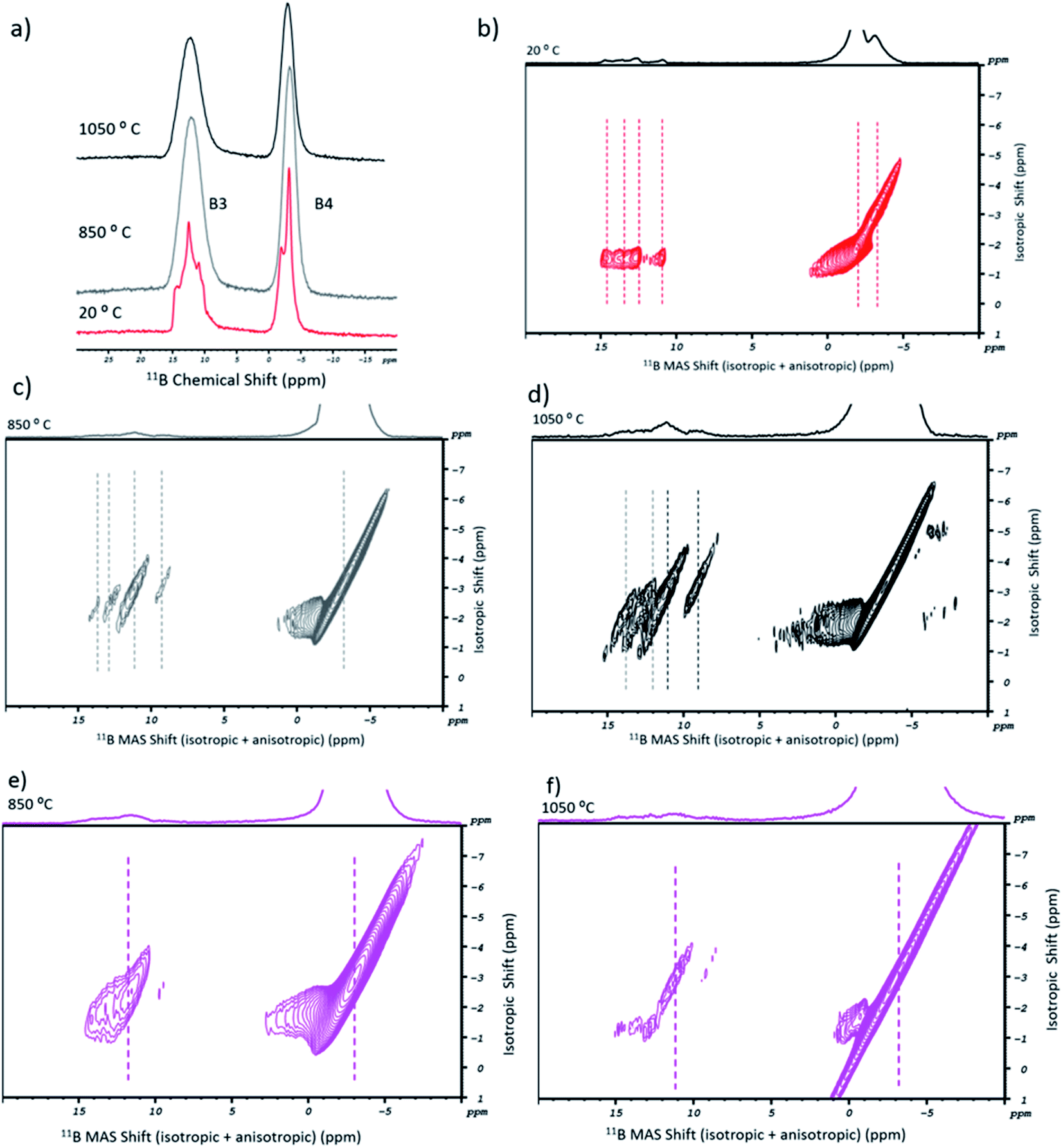 | ||
| Fig. 2 11B solid state NMR of the sodium flux and SWCNT-1. (a) 1D MAS spectra of NaT in the as-received state (red), and after melting at 850 °C (grey) and 1050 °C (black); (b) 2D MQMAS spectrum of the as-received NaT; (c, d) 2D MQMAS of NaT after melting at 850 °C and 1050 °C, respectively; (e, f) 2D MQMAS of the fused mix containing the SWCNT-1 sample, at 850 °C and 1050 °C, respectively. | ||
Concerning SWCNT-1, its post-fusion 11B MQMAS spectra are given in Fig. 2e (850 °C) and Fig. 2f (1050 °C). For the first, when compared to the analogous spectrum of the blank flux (Fig. 2c), there was an isotropic shift and just a single site for both the B3 and B4 regions. Additionally, broadening of the B4 resonance took place. At the highest fusion temperature, both resonances became more diffuse. The 2D profiles indicate that the chemical environment for the B3 units is dominated by the symmetric coordination. The B4 lobes, on the other hand, are indicative of lower ordering of these units in relation to the blank flux.
A similar study was performed for SRM2483 (Fig. S1†). The 11B MQMAS spectra showed patterns that are similar to those of SWCNT-1. The main difference was the B3 resonance component at lower ppm as this showed a more defined peak at both temperatures.
Following the above, an analogous NMR study was carried out for the salt's alkali cation. Ideally, the 23Na spectra of the sodium tetraborate should show two chemical environments for the cation (Na1 and Na2 sites).37Fig. 3a shows the 1D MAS spectra of NaT in its as-received (20 °C, in red) and post-fusion states, at 850 °C (in grey) and 1050 °C (in black). The spectrum of the initial state included a range of sharp peaks with a chemical shift that coincided with a broader and overlapping band. In the corresponding 23Na MQMAS (Fig. 3b), four peaks could be resolved, interpreted as four crystallographically distinct Na+ environments in the asymmetric Na2B407 unit cell. The presence of additional and less-resolved peaks is explained by the highly hygroscopic nature of the salt. Accordingly, as water molecules penetrate through the crystalline network, the chemical environment and mobility of the cation are altered due to its progressive solvation. Post-fusion, the 1D spectra (Fig. 3a) can be read as just one broad peak (tough, at 1050 °C, two small protrusions are observed). The lack of spectral definition is attributed to the glassy state of the flux, a result of the quenching process which impairs the ordered formation of sodium sites. In Fig. 3c and d, the 2D spectra (at 850 °C and 1050 °C, respectively) are shown. Despite being less resolved than the ones from the as-received salt, it is possible to distinguish two peaks, particularly in the higher temperature spectrum. While the anisotropic shift did not change, a noticeable variation occurred in the isotropic part. The two peaks at negative ppm (one of which was the Na1 site, at −12 ppm) either disappeared or lost all definition. On the other hand, the profile shows two peaks on the Na2 site. The shorter isotropic ppm interval of the signals could arise from the glassy-state. In fact, the disappearance of long-range order would compromise the cation mobility in the structure. Besides this, water molecules should have been entirely eliminated. Consequently, the missing Na1 sites (or the resonance at −30 ppm) would originate from sodium surrounded by water.
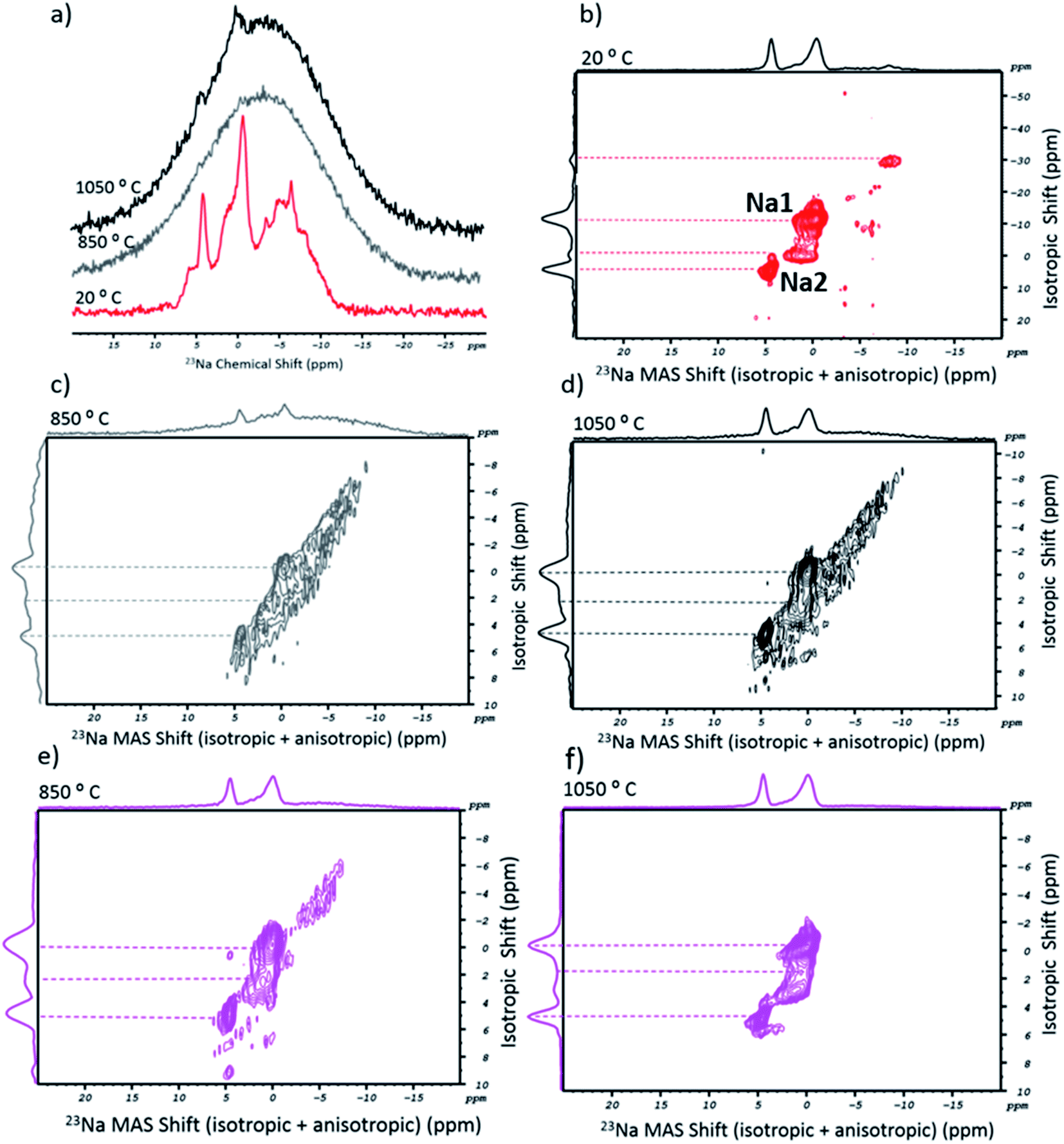 | ||
| Fig. 3 23Na solid state NMR of the sodium flux and SWCNT-1. (a) 1D MAS spectra of NaT in the as-received state (red), and after melting at 850 °C (grey) and 1050 °C (black); (b) 2D MQMAS spectrum of the as-received NaT; (c, d) 2D MQMAS of NaT after melting at 850 °C and 1050 °C, respectively; (e, f) 2D MQMAS of the fused mix containing the SWCNT-1 sample, at 850 °C and 1050 °C, respectively. | ||
With respect to the 23Na MQMAS of SWCNT-1, the post-fusion spectra (Fig. 3e and f) show a set of resolved peaks (more so at the higher temperature), the profiles and chemical shifts of which are similar to those in the 2D spectrum of the flux (at 1050 °C). Essentially, the only difference is the intensity reduction of the baseline-like bump seen in the fused blank 2D spectra (cf.Fig. 3c and d).
For the 23Na MQMAS of the fused SRM2483 sample (Fig. S2†), the 850 °C spectrum is similar to the previous ones. Conversely, the higher temperature spectral profile is entirely stripped of noticeable peaks and only a featureless bump remains. It is possible that the higher fusion temperature and extensive oxidation of the carbon lattice destroyed the short-to-medium range order of the borate units that accommodated the Na2 sites.
To complete the structural analysis by NMR, 13C 1D MAS spectra were acquired for the two standards in the as-received and post-fusion states (Fig. 4). Note that an insufficient amount of carbon residues in the flux beads (after the melting procedure) precluded the 13C 2D studies. In the initial state (20 °C), both nanotube samples showed a single resonance with the same chemical shift (125 ppm). The peak can be assigned straightforwardly to the sp2-hybridised carbon atoms that make up the nanotube's lattice.38 In contrast, after the fusion, there was a visible difference in the spectral response of the standards.
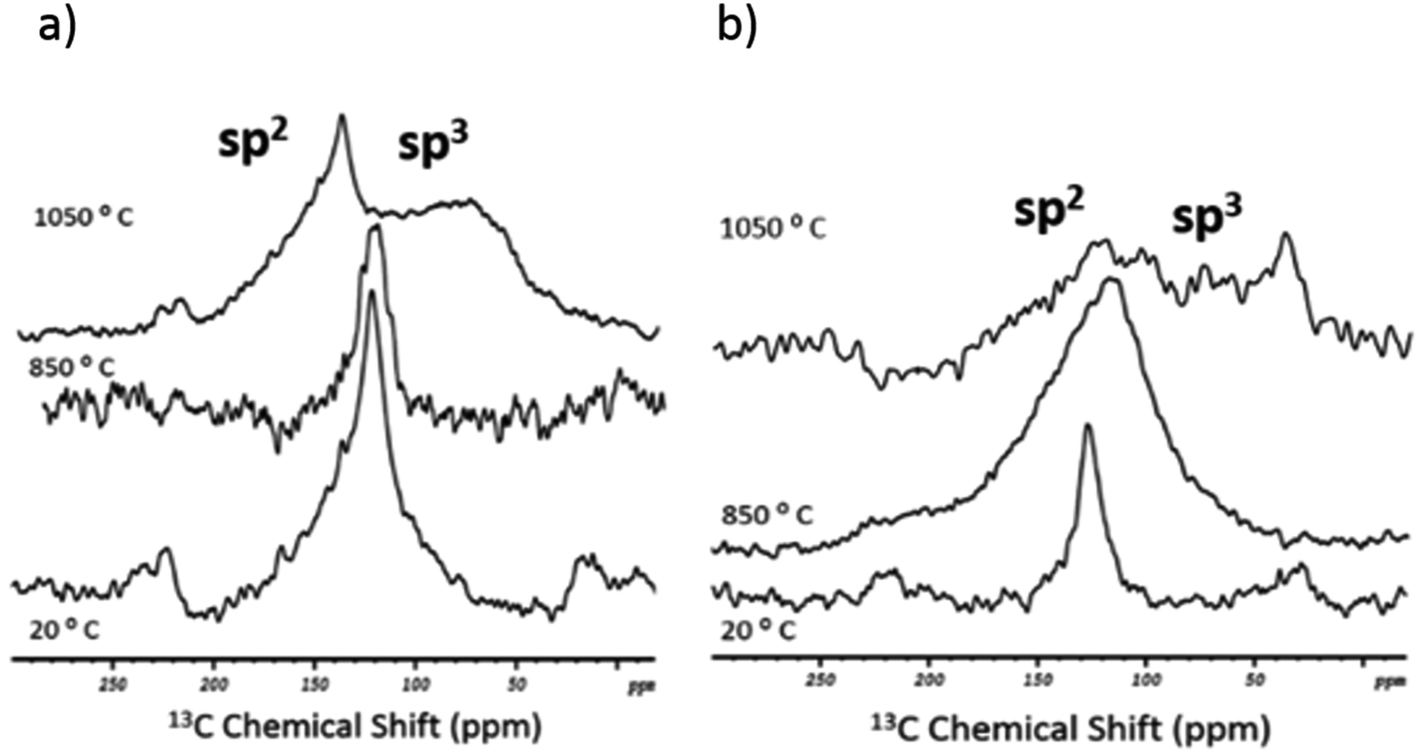 | ||
| Fig. 4 13C solid state 1D MAS NMR of the two standards. (a) SWCNT-1 in the as-received state (bottom spectrum) and after melting (top spectra), at 850 °C and 1050 °C; (b) SRM2483, in the as-received (bottom spectrum) and after melting (top spectra), at 850 °C and 1050 °C. | ||
With respect to SWCNT-1 (Fig. 4a), the chemical shift and shape of the peak were mostly maintained at the lower melting temperature. However, at 1050 °C, two overlapping resonances were observed. The sharp peak at 145 ppm is still within the region where the sp2-C peaks are expected. The 20 ppm shift is reasoned by the presence of graphitized debris and their interaction with electronegative elements (e.g. oxygen from the borate units). At 75 ppm, the less intense and broader peak corresponds to the carbon fragments richer in sp3-C. In our previous study,32 SWCNT-1 was also subjected to alkaline oxidation but a different flux was used. The post-fusion 13C NMR analysis identified a ∼10 ppm upfield shift and broadening of the sp2-C resonance. In comparison, the present changes are more pronounced and imply that the NaT melt results in a higher degree of structural degradation of the nanotubes (despite the shorter exposure time and similar fusion temperature). Further experiments will be required to confirm and understand this observation.
As for SRM2483 (Fig. 4b), the initial sp2-C peak shifted to 110 ppm and became significantly broader at 850 °C. When the melt temperature was raised further, the resulting spectrum showed weaker spectral features. Still, two overlapping resonances could be identified. The first was the sp2-C peak that remained centered at about 110 ppm. The second was a diffuse band that ranged from 90 to 25 ppm and had an intensity spike at 35 ppm. This falls in the known interval of sp3-C resonances. The more dispersed spectral signature of SRM2483 implies that a faster and more extensive degradation took place, when compared to SWCNT-1. This is understandable given the lower structural purity of the NIST standard.39
The above multi-nuclei NMR analysis provided some interesting results. To start with, SRM2483 undergoes oxidative structural degradation of the carbon lattice at lower temperatures and gets more digested than SWCNT-1. Also, the resilience of sp2-C is remarkable and suggests that some sections of the well-structured nanotubes (or, more likely, sections of these nanotubes) resist the alkaline oxidation, in particular when bundled together. This chemical sturdiness justifies the constancy of the 23Na spectra for SWCNT-1 as opposed to SRM2483 which, at 1050 °C, loses spectral definition. From this, the higher concentration of sp3-C would contribute to the disruption of the formation of regular sodium sites in the glassy borate salt. Hence, the carbon lattice degradation induces a more extensive effect on the alkali metal sites than on the borate units in the fused salt. Finally, there was no evidence of the presence of carbonates, carbides or dopants.
3.2 Commercial materials
The two commercial samples, SWCNT-7867V and SWCNT-5848V, were selected after a preliminary screening of several nanotube materials. Besides the degree of carbon content (chemical purity), differences in their elemental composition (transition metals) were taken into consideration.With respect to SWCNT-7867V (the one with the highest carbon purity), its fused beads exhibited a dark blue color, similar to that of the two nanotube standards. Fig. 5a and b show the average elemental recoveries (and corresponding standard deviation bars) obtained at 850 °C and 1050 °C, respectively. Given the lack of benchmark values for Fe and Ni, these elements were not considered. With the exception of Co, minor differences were observed (for Cr, Mn and Mo). The Co recovery at 850 °C was 81 ± 9% whilst, at 1050 °C, it increased to 89 ± 6%. Again, the deviation bars are relatively large meaning that the improvement in Co extraction is indicative. Interestingly, the uncertainties did not differ as widely with temperature as in the SWCNT-1 standard. In the past, we tested SWCNT-7867V under different fusion conditions.13 A lithium borate flux was used and the exposure lasted six minutes, at 1050 °C. The recoveries obtained were higher than those here, namely 97 ± 2% (for Cr), 107 ± 0% (for Mn), 86 ± 3% (for Co) and 93 ± 1% (for Mo). The additional two minutes of melt exposure may have contributed to leaching more of these elements into the lithium salt.
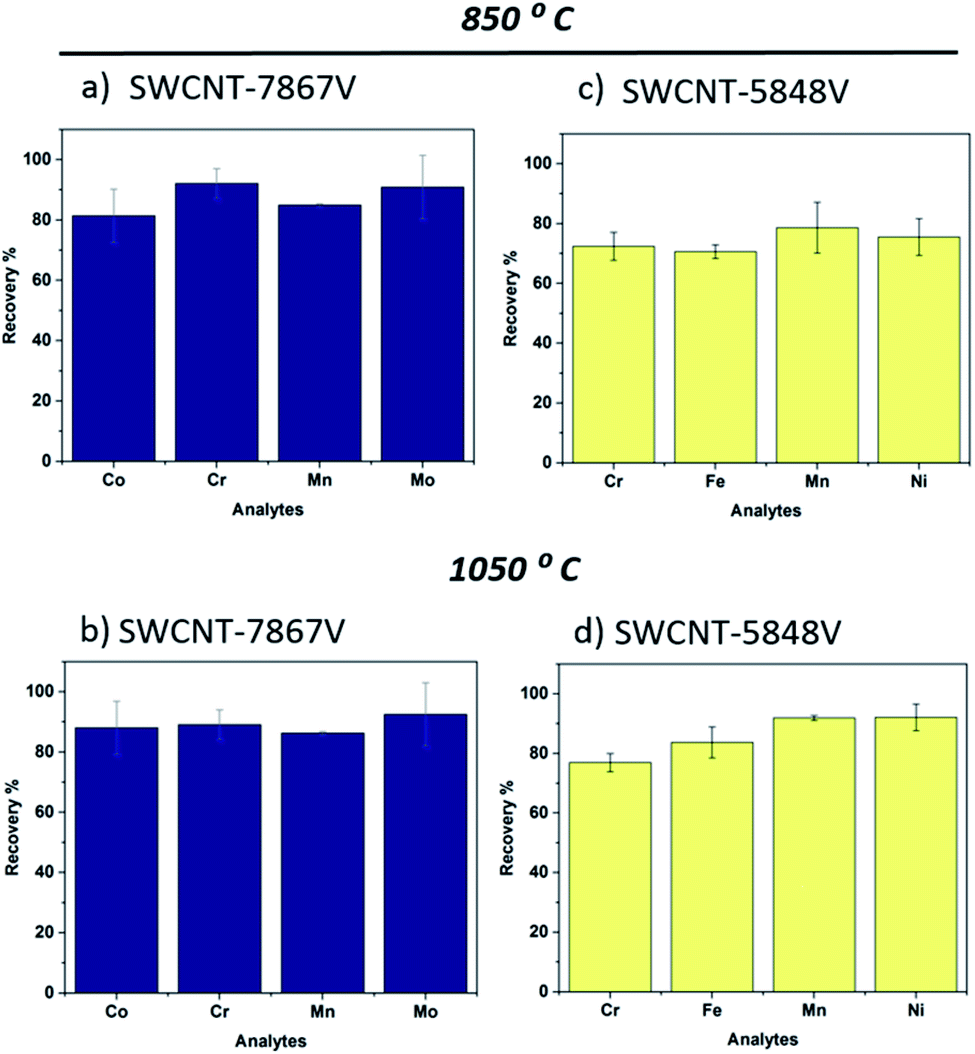 | ||
| Fig. 5 Elemental recovery of selected transition metals present in commercial SWCNT samples (average recoveries and standard deviations based on N = 3). (a, b) Calculated percentages for SWCNT-7867V, at 850 °C and 1050 °C, respectively; (c, d) SWCNT-5848V, at 850 °C and 1050 °C, respectively. | ||
As for the SWCNT-5848V sample, NAA revealed that Cr, Mn, Fe and Ni were the predominant transition metals in the as-received powder (Table S1†). In fact, Fe was several orders of magnitude more abundant than the other three elements. Co and Mo were present in relatively small quantities. Post-fusion, the quenched glassy beads were dark yellow, a color consistent with the oxidation of Fe. From Fig. 5c and d, it is clear that an increase in the recovery levels occurred in the higher temperature experiment. While for Cr and Mn this improvement may be disputed (given the magnitude of the deviation bars at 850 °C), there is no doubt about the other two elements. In the case of Fe, the average value jumped from 70% to 83%. For Ni, the increment was even more pronounced, from 75% to 92%. Remarkably, this response to the fusion temperature contrasts with that of all other nanotube samples.
From the elemental analysis of the commercial samples, it is possible to infer that different catalysts were used to produce the nanotubes. Whereas SWCNT-7867V follows the well-known CoMoCAT CVD approach (as disclosed by the vendor), where Co and Mo are prominently used,39 the growth of SWCNT-5848V was based on an Fe-catalyzed reaction, likely promoted by a free-floating CVD process.42 If so, this would help understand the exceptional increment in recovery levels at 1050 °C. A common by-product of Fe-catalyzed free-floating CVD is metallic nanoparticles enveloped by a thin shell of turbostratic carbon. It is logical that the hotter melt was more efficient in disintegrating them, accelerating the dilution of the trapped Fe (and Ni, if alloyed).
The morphology and structural characteristics of SWCNT-7867V were previously described by us.26 Generally, the as-received nanotubes were well-structured, with an average diameter of 0.96 ± 0.29 nm and arranged in bundles of less than ten SWCNTs. However, by-products such as catalyst particles (alloys of Co and Mo) were also identified. With this information in hand, the 11B and 23Na MQMAS NMR spectra were acquired for the SWCNT-7867V fusion products. At 850 °C (Fig. S3a†), the anisotropic shift contribution in the 11B 2D spectra showed a diversity of chemical environments for the B3 units. They were similar in the number, shift and intensity of the resonances to those present in blank NaT (cf.Fig. 2c). Conversely, the B4 sites showed a resonance profile that was sharper than that of the analogous of the SWCNT-1 and SRM2483 samples. Remarkably, the increase in the melt temperature led to a slight enlargement of the boron resonances (Fig. S3b†). This contrasts with the response of the standards where the opposite trend was observed. In SWCNT-7867V, the higher disorder of the anionic B3 and B4 units could be a consequence of increased leaching of trapped cobalt and the formation of glassy cobalt(II) borate domains (cf.Fig. 5a and b). The analysis of the alkali cation sites by 23Na MQMAS NMR (Fig. S4†) showed a slightly more diffuse spectrum at the higher temperature. While the induced disorder was not as large as that seen for SRM2483 (cf. Fig. S2†), it showed a different response from that of the blank and the SWCNT-1 samples. Cobalt leaching, or the oxidation of the nanotubes and other carbon aggregates in this sample (see TEM analysis in ref. 23),26 could be the reason for this behavior. Finally, the 13C 1D MAS NMR spectra for the as-received nanotubes and the post-fusion samples were acquired (Fig. 6a). The initial spectrum (20 °C) of SWCNT-7867V is similar to those of the standards, with a single resonance centered at 125 ppm. However, the peak is less symmetric possibly due to a greater presence of sp2-C species that are not nanotubes (e.g. carbon aggregates).26 Post-fusion, the 13C spectra are comparable to those of the standards. At 850 °C, the resonance's broadening is quite visible indicating higher anisotropy and less structural order in the C lattice. At 1050 °C, the spectrum follows a profile that somehow falls in between that of SWCNT-1 and SRM2483. On one hand, the sp3-C peak shows up with a comparable chemical shift to that of SRM2483 (30 ppm). On the other hand, the sp2-C resonance is maintained but its sharpness and intensity are akin to those of the SWCNT-1 sample. From this, a considerable degradation of sp2-C in SWCNT-7867V took place but not as extensively as in SRM2483. It is worth recalling that SWCNT-7867V was also subjected to oxidation using a lithium salt.32 Interestingly, at 1050 °C, the spectrum was constituted solely by the sp3-C peak, centered at 37 ppm. The absence of the sp2-C resonance implies a larger disintegration of the nanotubes, which agrees well with the higher recoveries obtained with the lithium salt when compared to the present sodium tetraborate.13 As stated before, the additional two minutes of melt exposure could account for this.
 | ||
| Fig. 6 Structural characterization of commercial nanotube samples. (a) 13C solid state 1D MAS NMR for the SWCNT-7867V sample in the as-received state (bottom spectrum) and after melting (top spectra), at 850 °C and 1050 °C; (b) high magnification TEM micrograph of the as-received SWCNT-5848V; (c) Raman spectrum of the nanotube sample shown in (b). | ||
For SWCNT-5848V, the morphological and structural characterization is shown in Fig. 6b and c. Besides the bundles of SWCNTs (marked with a white arrow in Fig. 6b), the presence of metallic catalyst particles was observed (red arrows). Judging from the elemental analysis, they are composed of Fe. The corresponding Raman spectrum identified the typical bands for SWCNTs, namely the radial breathing mode (RBM), the graphitic peak (G) and the 2D overtone. The defect-related peak (D) was barely noticeable, verifying the structural quality of these nanotubes. The estimated diameter distribution was 1.7 ± 0.5 nm (see Fig. S5b†). Unfortunately, due to the high concentration of magnetic Fe particles in the as-received sample, it was not possible to carry out the structural analysis by solid state NMR.
3.3 Final discussion
It is common knowledge that the synthesis of SWCNTs, may that be via CVD or other methods, relies on the use of transition metals. They act as catalysts that promote the cracking of carbon feedstocks (e.g. a hydrocarbon gas or vaporized graphite powders) as well as the subsequent integration of atomic carbon species in a tubular graphitic lattice. Not surprisingly, catalyst nanoparticles are the most common by-product of these processes. In general, the metal remainders are found nested inside the nanotubes or encapsulated in onion-like carbon particles. These graphitic shells are quite sturdy and act as a formidable barrier to liquids capable of solubilizing transition metals. Hence, destroying the carbon matrix is a necessary step to ensure appropriate recovery of transition metals when analyzing SWCNT samples by ICP-OES.Throughout this and past reports, we have explored different flux systems, carbon materials and molten salt reaction conditions to evaluate how feasible is alkaline oxidation as an ICP-OES pre-treatment step for the analysis of transition metals in nanocarbons. Given the availability of chemically certified SWCNTs, a special emphasis was placed on this class of allotropes. With the results of the present study, an interesting perspective arises on the points described hereafter.
4. Conclusions
Four different types of SWCNTs were subjected to alkaline oxidation using a sodium tetraborate flux. For each sample, two different temperatures were considered. The solubilized products of the fusion processes were used to perform elemental analysis by ICP-OES. With reference to benchmark concentrations, the recoveries obtained for six transition metals were commonly in excess of 80%. Depending on the catalyst system employed to synthesize the nanotubes, the fusion temperature induced a different effect. For those samples using a Co–Mo catalyst, a lower temperature was sufficient. For the one sample produced via an Fe catalyst, the best recoveries were obtained at 1050 °C. When put into context with previous literature, the present results show that the structural purity of the nanotubes and the characteristics of the catalyst remainders are the key drivers for the selection of the reaction conditions. A balance between the extent of graphitic matrix digestion and evaporation of the leached target elements needs to be attained. Remarkably, the fusion processes can be quite fast, with reaction times (melt exposure) as short as four minutes. Overall, alkaline oxidation is a relatively safe and simple alternative to digest carbon materials, one that may be particularly useful in settings where batches are handled at a high throughput.Conflicts of interest
I have no conflict of interest to declare.Acknowledgements
KAUST is acknowledged for funding (URF/1/2980-01-01 and BAS/1/1346-01-01). We are grateful for the continuous assistance and availability of the facilities at the Core Labs, KAUST.References
- H.-C. Wu, X. Chang, L. Liu, F. Zhao and Y. Zhao, Chemistry of carbon nanotubes in biomedical applications, J. Mater. Chem., 2010, 20(6), 1036–1052 RSC.
- N. Hamzah, M. F. M. Yasin, M. Z. M. Yusop, A. Saat and N. A. M. Subha, Rapid production of carbon nanotubes: a review on advancement in growth control and morphology manipulations of flame synthesis, J. Mater. Chem. A, 2017, 5(48), 25144–25170 RSC.
- A. Eatemadi, H. Daraee, H. Karimkhanloo, M. Kouhi, N. Zarghami, A. Akbarzadeh, M. Abasi, Y. Hanifehpour and S. W. Joo, Carbon nanotubes: properties, synthesis, purification, and medical applications, Nanoscale Res. Lett., 2014, 9(1), 393 CrossRef.
- A. Magrez, J. W. Seo, R. Smajda, M. Mionić and L. Forró, Catalytic CVD Synthesis of Carbon Nanotubes: Towards High Yield and Low Temperature Growth, Materials, 2010, 3(11), 4871–4891 CrossRef CAS.
- G. Allaedini, S. M. Tasirin, P. Aminayi, Z. Yaakob and M. Z. Meor Talib, Carbon nanotubes via different catalysts and the important factors that affect their production: a review on catalyst preferences, Int. J. Nano Dimens., 2016, 7(3), 186–200 CAS.
- V. Jourdain and C. Bichara, Current understanding of the growth of carbon nanotubes in catalytic chemical vapour deposition, Carbon, 2013, 58, 2–39 CrossRef CAS.
- K. Donaldson, R. Aitken, L. Tran, V. Stone, R. Duffin, G. Forrest and A. Alexander, Carbon Nanotubes: A Review of Their Properties in Relation to Pulmonary Toxicology and Workplace Safety, Toxicol. Sci., 2006, 92(1), 5–22 CrossRef CAS.
- J. Prasek, J. Drbohlavova, J. Chomoucka, J. Hubalek, O. Jasek, V. Adam and R. Kizek, Methods for carbon nanotubes synthesis—review, J. Mater. Chem., 2011, 21(40), 15872–15884 RSC.
- D. Yuan, L. Ding, H. Chu, Y. Feng, T. P. McNicholas and J. Liu, Horizontally Aligned Single-Walled Carbon Nanotube on Quartz from a Large Variety of Metal Catalysts, Nano Lett., 2008, 8(8), 2576–2579 CrossRef CAS.
- M. A. Kazakova, V. L. Kuznetsov, S. N. Bokova-Sirosh, D. V. Krasnikov, G. V. Golubtsov, A. I. Romanenko, I. P. Prosvirin, A. V. Ishchenko, A. S. Orekhov, A. L. Chuvilin and E. D. Obraztsova, Fe–Mo and Co–Mo Catalysts with Varying Composition for Multi-Walled Carbon Nanotube Growth, Phys. Status Solidi B, 2018, 255(1), 1700260 CrossRef.
- Y. Yan, J. Miao, Z. Yang, F.-X. Xiao, H. B. Yang, B. Liu and Y. Yang, Carbon nanotube catalysts: recent advances in synthesis, characterization and applications, Chem. Soc. Rev., 2015, 44(10), 3295–3346 RSC.
- S. Patole, F. Simões, T. F. Yapici, B. H. Warsama, D. H. Anjum and P. M. F. J. Costa, An evaluation of microwave-assisted fusion and microwave-assisted acid digestion methods for determining elemental impurities in carbon nanostructures using inductively coupled plasma optical emission spectrometry, Talanta, 2016, 148, 94–100 CrossRef CAS.
- F. R. F. Simoes, E. Abou-Hamad, J. Smajic, N. M. Batra and P. M. F. J. Costa, Chemical and Structural Analysis of Carbon Materials Subjected to Alkaline Oxidation, ACS Omega, 2019, 4(20), 18725–18733 CrossRef CAS.
- R. Alshehri, A. M. Ilyas, A. Hasan, A. Arnaout, F. Ahmed and A. Memic, Carbon Nanotubes in Biomedical Applications: Factors, Mechanisms, and Remedies of Toxicity, J. Med. Chem., 2016, 59(18), 8149–8167 CrossRef CAS.
- Y. Liu, Y. Zhao, B. Sun and C. Chen, Understanding the Toxicity of Carbon Nanotubes, Acc. Chem. Res., 2013, 46(3), 702–713 CrossRef CAS.
- V. Amenta and K. Aschberger, Carbon nanotubes: potential medical applications and safety concerns, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7(3), 371–386 CAS.
- Z. Gao, J. A. Varela, L. Groc, B. Lounis and L. Cognet, Toward the suppression of cellular toxicity from single-walled carbon nanotubes, Biomater. Sci., 2016, 4(2), 230–244 RSC.
- E. Zolfonoun and S. R. Yousefi, Simultaneous Determination of Rare Earth Elements by ICP OES After On-Line Enrichment Using Multi-Walled Carbon Nanotubes Coated Cellulose Acetate Membrane, J. Braz. Chem. Soc., 2016, 27, 2348–2353 CAS.
- M. T. Martnez, M. A. Callejas, A. M. Benito, M. Cochet, T. Seeger, A. Ansn, J. Schreiber, C. Gordon, C. Marhic, O. Chauvet and W. K. Maser, Modifications of single-wall carbon nanotubes upon oxidative purification treatments, Nanotechnology, 2003, 14(7), 691–695 CrossRef.
- R. Andrews, D. Jacques, D. Qian and E. C. Dickey, Purification and structural annealing of multiwalled carbon nanotubes at graphitization temperatures, Carbon, 2001, 39(11), 1681–1687 CrossRef CAS.
- J.-H. Lim, V. G. Bairi and A. Fong, Quantification of impurities in carbon nanotubes: development of ICP-MS sample preparation methods, Mater. Chem. Phys., 2017, 198, 324–330 CrossRef CAS.
- M.-L. Avramescu, P. E. Rasmussen and M. Chénier, Determination of Metal Impurities in Carbon Nanotubes Sampled Using Surface Wipes, J. Anal. Methods Chem., 2016, 2016, 3834292 Search PubMed.
- C. Ge, F. Lao, W. Li, Y. Li, C. Chen, Y. Qiu, X. Mao, B. Li, Z. Chai and Y. Zhao, Quantitative analysis of metal impurities in carbon nanotubes: efficacy of different pretreatment protocols for ICPMS spectroscopy, Anal. Chem., 2008, 80(24), 9426–9434 CrossRef CAS.
- R. S. Mortari, R. C. Cocco, R. F. Bartz, L. V. Dresssler and M. E. Flores de Morais, Fast Digestion Procedure for Determination of Catalyst Residues in La- and Ni-Based Carbon Nanotubes, Anal. Chem., 2010, 82(10), 4298–4303 CrossRef.
- L. Ayouni-Derouiche, M. Méjean, P. Gay, M.-L. Milliand, P. Lantéri, L. Gauthier and E. Flahaut, Development of efficient digestion procedures for quantitative determination of cobalt and molybdenum catalyst residues in carbon nanotubes, Carbon, 2014, 80, 59–67 CrossRef CAS.
- F. R. Simoes, N. K. Batra, B. H. Warsama, C. G. Canlas, S. P. Patole, T. F. Yapici and P. M. Costa, Elemental Quantification and Residues Characterisation of Wet Digested Certified and Commercial Carbon Materials, Anal. Chem., 2016, 88(23), 11783–11790 CrossRef CAS.
- P. Grinberg, R. E. Sturgeon, L. d. O. Diehl, C. A. Bizzi and E. M. M. Flores, Comparison of sample digestion techniques for the determination of trace and residual catalyst metal content in single-wall carbon nanotubes by inductively coupled plasma mass spectrometry, Spectrochim. Acta B Atom Spectrosc., 2015, 105, 89–94 CrossRef CAS.
- A. Zhang, H. Wang, P. Zha, M. Wang, H. Wang, B. Fan, D. Ren, Y. Han and S. Gao, Microwave-induced combustion of graphene for further determination of elemental impurities using ICP-OES and TXRF, J. Anal. At. Spectrom., 2018, 33(11), 1910–1916 RSC.
- W. Kiciński and S. Dyjak, Transition Metal Impurities in Carbon-Based Materials: pitfalls, Artifacts and Deleterious Effects, Carbon, 2020 Search PubMed.
- J. S. F. Pereira, F. G. Antes, L. O. Diehl, C. L. Knorr, S. R. Mortari, V. L. Dressler and E. M. M. Flores, Microwave-induced combustion of carbon nanotubes for further halogen determination, J. Anal. At. Spectrom., 2010, 25(8), 1268 RSC.
- K. MacKenzie, O. Dunens and A. T. Harris, A review of carbon nanotube purification by microwave assisted acid digestion, Sep. Purif. Technol., 2009, 66(2), 209–222 CrossRef CAS.
- F. R. F. Simoes, N. M. Batra, A.-H. Emwas and P. M. F. J. Costa, Validation of alkaline oxidation as a pre-treatment method for elemental quantification in single-walled carbon nanotubes, Anal. Methods, 2019, 11(14), 1884–1890 RSC.
- J. Kameník, F. R. F. Simões, P. M. F. J. Costa, J. Kučera and V. Havránek, INAA and ion-beam analysis of elemental admixtures in carbon-based nanomaterials for battery electrodes, J. Radioanal. Nucl. Chem., 2018, 318(3), 2463–2472 CrossRef.
- Z. Mester, Certificate of Analysis, SWCNT-1 Single-Wall Carbon Nanotubes Certified Reference Material, National Research Council (NRC), Ottawa, Canada, 2013 Search PubMed.
- K. Eric, C. Lin, R. L. Watters and J. Chief, Certificate of Analysis Standard Reference Material 2483 Single-Wall Carbon Nanotubes (Raw Soot), National Institute of Standards & Technology, Department of Commerce, USA, 2011 Search PubMed.
- M. Murakami, T. Shimizu, M. Tansho, Y. Takano, S. Ishii, E. A. Ekimov, V. A. Sidorov and K. Takegoshi, 10B/11B 1D/2D solid-state high-resolution NMR studies on boron-doped diamond, Diamond Relat. Mater., 2009, 18(10), 1267–1273 CrossRef CAS.
- H. Al-Johani, E. Abou-Hamad, A. Jedidi, C. M. Widdifield, J. Viger-Gravel, S. S. Sangaru, D. Gajan, D. H. Anjum, S. Ould-Chikh, M. N. Hedhili, A. Gurinov, M. J. Kelly, M. El Eter, L. Cavallo, L. Emsley and J.-M. Basset, The structure and binding mode of citrate in the stabilization of gold nanoparticles, Nat. Chem., 2017, 9(9), 890–895 CrossRef CAS.
- C. Goze Bac, P. Bernier, S. Latil, V. Jourdain, A. Rubio, S. H. Jhang, S. W. Lee, Y. W. Park, M. Holzinger and A. Hirsch, 13C NMR investigation of carbon nanotubes and derivatives, Curr. Appl. Phys., 2001, 1(2), 149–155 CrossRef.
- D. Resasco, R. Silvy and S. Nanotechnologies, Structure and Applications of Single-Walled Carbon Nanotubes (SWCNTs) Synthesized Using the CoMoCAT® Method, Norman, OK, Sigma-Aldrich, 2011 Search PubMed.
- R. R. Greenberg, P. Bode and E. A. De Nadai Fernandes, Neutron activation analysis: a primary method of measurement, Spectrochim. Acta B Atom Spectrosc., 2011, 66(3), 193–241 CrossRef CAS.
- J. Kučera, P. Bode and V. Štepánek, Uncertainty evaluation in instrumental and radio-chemical neutron activation analysis, Quantifying uncertainty in nuclear analytical measurements, IAEA-TECDOC-1401, IAEA Vienna, 2004, pp. 77–102 Search PubMed.
- A. Hussain, Y. Liao, Q. Zhang, E.-X. Ding, P. Laiho, S. Ahmad, N. Wei, Y. Tian, H. Jiang and E. I. Kauppinen, Floating catalyst CVD synthesis of single walled carbon nanotubes from ethylene for high performance transparent electrodes, Nanoscale, 2018, 10(20), 9752–9759 RSC.
- J. Kučera, J. W. Bennett, R. Oflaz, R. L. Paul, E. A. De Nadai Fernandes, M. Kubešová, M. A. Bacchi, A. J. Stopic, R. E. Sturgeon and P. Grinberg, Elemental Characterization of Single-Wall Carbon Nanotube Certified Reference Material by Neutron and Prompt γ Activation Analysis, Anal. Chem., 2015, 87(7), 3699–3705 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization of some of the nanotube materials and fusion products, namely, the NAA reference values and structural analysis (by TEM and Raman) of the as-received SWCNT-5848V, the 11B MQMAS NMR spectra for SRM2483 and SWCNT-7867V, and the 23Na MQMAS NMR spectra for SRM2483 and SWCNT-7867V. See DOI: 10.1039/d0ja00325e |
| This journal is © The Royal Society of Chemistry 2020 |