
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative imaging of carbon in heterogeneous refining catalysts
L.
Jolivet
ab,
V.
Motto-Ros
 *b,
L.
Sorbier
a,
T.
Sozinho
a and
C.-P.
Lienemann
a
*b,
L.
Sorbier
a,
T.
Sozinho
a and
C.-P.
Lienemann
a
aIFP Energies nouvelles, Rond-point de l'échangeur de Solaize, BP 3, 69360 Solaize, France
bInstitut Lumière Matière UMR 5306, Université Lyon 1 – CNRS, Université de Lyon, 69622 Villeurbanne, France. E-mail: vincent.motto-ros@univ-lyon1.fr
First published on 21st March 2020
Abstract
Heterogeneous catalyst materials are the core of crude oil purification process performance. Catalyst deactivation, which is a result of crude impurity deposits into catalyst pores during this process, is therefore a major concern during refining. However, deactivation mechanisms are not easily investigated, as they involve carbon transport phenomena over a large range of concentrations, typically from hundreds of ppm to tens of mass%. Spatially resolved analysis of carbon conducted at the entire grain scale (∼mm2) with a micrometric resolution is therefore of primary importance to better predict, control, and further improve the purification processes. However, usual spatially resolved approaches are not easily suitable for mm2-scale analysis and/or not adequate in terms of carbon detection limits. In this work, we propose a quantitative methodology dedicated to carbon spatially resolved analysis and the evaluation of its analytical performance. This approach, based on laser-induced breakdown spectroscopy (LIBS), allows for the first time 2D imaging of carbon at different process times as well as quantitative profiling of carbon diffusion over more than two orders of magnitude in concentration.
1. Introduction
Refining industries suffer from several challenges during the purification of petroleum products. Natural sources contain a high amount of unwanted impurities, and the world demand for clean oil products is increasing over time. There is thus an important need for improvements in the conversion and purification processes of crude oil. Most refining processes involve heterogeneous catalysts. These materials are composed of metallic active sites supported by a silica or alumina porous matrix.1 During purification processes, catalysts are poisoned by coke (self-assembling polyaromatic macromolecules formed by polymerization/dehydrogenation reactions)2 and by metallic impurities (Ni, V), which form deposits into their pores. As porosity is occluded, access to active sites is hindered, and catalytic functions are inhibited, resulting in a strong decrease in process performance. Therefore, knowledge of the distributions of carbon and impurities in the catalysts as a function of process time is required to better understand catalytic deactivation mechanisms and kinetics, with the idea of progressing toward a more realistic modeling of such systems and finally improving processes performance.3Several works have focused on refining catalyst poisoning by coke to improve the understanding of deactivation mechanisms, proposing various analytical methods.4,5 In most cases, techniques used for carbon analysis are UV-Visible (UV-Vis)6 or infrared (IR) spectroscopy, as well as thermal analysis such as thermogravimetric analysis (TGA)7 and temperature programmed oxidation (TPO).8 These approaches are essentially bulk analysis techniques and require the whole sample or a significant amount of the sample to be burned. When spatially resolved analysis is required, several techniques can be used, such as Raman spectroscopy,9 X-ray absorption spectroscopy (XANES),10 electron spin resonance (ESR) and nuclear magnetic resonance (NMR) spectroscopies.11 These techniques provide molecular or functional group characterization. However, they are not well adapted for a carbon-poisoning study, as no elemental information can be obtained. In two previous works, 1H NMR imaging of coke was performed, first on ethanol dehydration catalysts12 and later on zeolite-based catalysts.13 However, proton NMR provides indirect detection of carbon, and the results are not quantitative. The technique considered a reference for studying the elemental distribution in catalyst materials is electron probe microanalysis (EPMA).14 Pachulski et al. proposed a quantitative study of carbon repartition from 1.5 wt% to 4.0 wt% over the surface of a spent selective hydrogenation catalyst.15 EPMA is generally relevant for elemental analysis but is not well adapted for the quantitative study of carbon and other light elements (Z < 11) because of carbon contamination and soft X-ray absorption issues.16 Lower detection limits can be obtained for carbon with only a few techniques: time-of-flight secondary ion mass spectrometry (TOF-SIMS),17 scanning transmission X-ray microscopy (STXM),18 or scanning transmission electron microscopy in either energy dispersive X-ray spectroscopy mode (STEM-EDX) or electron energy-loss spectroscopy mode (STEM-EELS). Each of these techniques is performed for micro- or nanosized samples but is not adapted for the characterization of a whole catalyst grain. In addition, their limits of detection are still not adequate for studying coke-poisoned catalysts, which implies a gradient concentration from typically hundreds of ppm to tens of mass%.
Laser-induced breakdown spectroscopy (LIBS) appears to be the proper candidate to address the challenge of carbon impurities in catalysts due both to its high sensitivity for carbon (up to the ppm scale) and its microscale analysis capabilities. In recent years, LIBS-based imaging has been the core of important advances both in terms of performance and applications.19 Industrial concerns are focused on a growing number of works.20 The application of LIBS to catalysts was first reported in TiO2-supported vanadium oxides21 and later in several works by the same group, mostly in the frame of automotive catalytic converters.22–24 More recently, several studies have been conducted on refining catalysts. Trichard et al. demonstrated the benefit of LIBS quantitative imaging on palladium-based porous alumina catalysts.25 Sorbier et al. used LIBS elemental images to evaluate the crust thickness.26 In addition, our group succeeded in detecting Ni and V impurities in hydrodemetallization catalysts at the ppm concentration scale.27 In this work, carbon was also detected but with no possibility to be exploited because of contamination issues occurring during sample preparation. Such preparation, also used for EPMA analysis, consisted of embedding catalyst bodies in an acrylic or epoxy resin, initially liquid, and then curing by increasing the temperature. Unfortunately, this sample preparation is known to diffuse the resin into the pores and cracks of catalyst bodies during the thermal hardening process, inducing carbon contaminations.28
The aim of this work is to propose a LIBS-based quantitative methodology allowing carbon imaging of heterogeneous catalysts. The methodology is based on a specific sample preparation strategy and LIBS data processing and is dedicated to the accurate characterization of C distribution as a function of process time. Such results have, to the best of our knowledge, never been assessed before and could provide a new understanding of diffusion mechanisms in catalysts, improve process modeling and better control refining. More generally, this approach may be profitable to other fields requiring spatially resolved carbon analysis on porous samples and being affected by sample preparation issues.
2. Experimental
2.1 Catalyst samples
The catalyst samples used in this study are γ-alumina supports of hydrodemetallization catalysts. They are cylindrical extrudates, 6 to 8 mm long, with a 2 mm diameter and a single-mode pore size distribution centered at 14 nm.29 Their texture characteristics, obtained from porosity characterization performed by nitrogen adsorption and mercury porosimetry techniques (according the American Society for Testing and Materials (ASTM) D3663-03 and D4284-03 methods) are given in Table 1.| Specific surface area, SBET (m2 g−1) | 200 ± 20 |
| Porous volume, VBJH (cm3 g−1) | 0.80 ± 0.04 |
| Mean pore diameter, dm (nm) | 13.7 ± 0.7 |
| Structural density, ρS (g cm−3) | 3.01 ± 0.35 |
| Porosity, ε (%) | 69 ± 3 |
To simulate process conditions and interactions with crude impurities, alumina supports were immersed with varying impregnation times in a 2 wt% asphaltene solution prepared by dissolution in toluene at a target temperature of 250 °C and a high pressure in a hermetic cell.30 Asphaltenes are a heavy cut from crude that contain polyaromatic hydrocarbons that form aggregates over several size ranges and are considered under strong diffusion limitations in heterogeneous hydrodemetallization refining catalysts.31,32 The asphaltenes were previously extracted by n-heptane precipitation from a Safaniya vacuum residue, as described in the French standard NF T60-115 method.33 Six asphaltene-impregnated samples were prepared with impregnation times varying from 30 minutes to 6 weeks. The 30 minute impregnation time means that samples were immersed in the solution during only the increase and decrease in temperature to reach the target temperature.
For each sample, the global carbon concentration in the bulk was estimated by considering the concentration of carbon in Safaniya asphaltenes measured by combustion analysis (equal to 82.3 wt% ± 0.4) and the asphaltene concentrations in supports, estimated by mass balance calculations achieved for each batch.30 The fresh catalyst support (not impregnated with asphaltenes) is known to hold a residual homogeneous carbon content (equal to 0.12 wt%, as measured by combustion analysis). This fresh support will be considered as the reference sample in the following sections.
2.2 LIBS setup
The experimental LIBS setup has been previously described.34,35 It included a Nd:YAG laser source operating at 100 Hz and emitting at 1064 nm (Centurion, Quantel) with an 8 ns pulse duration. A ×15 magnification objective microscope (LMM-15X-P01, Thorlabs) was used to focus the laser beam onto the sample surface, and the sample was moved using a motorized XYZ stage during the scanning sequence. The laser energy was set at 1 mJ in all performed experiments. The step size between each laser shot was fixed to 25 μm. The acquisition was performed at ambient pressure and temperature. An argon flow (1.6 L min−1) blowing through the plasma was used both to prevent surface contamination by ablated material deposition of the previous laser shots and to obtain a better sensitivity due to better plasma confinement. The entire LIBS imaging setup was controlled by homemade software developed in the LabVIEW environment, allowing definition of the scanning sequence as well as automatic spectral acquisition. The plasma emission was collected by a lens-fiber system coupled to a Czerny–Turner spectrometer (Shamrock 500, Andor Technology). This spectrometer was equipped with an intensified charge-coupled device (ICCD) camera (iStar, Andor Technology) and a 600 L mm−1 grating blazed at 300 nm. In this configuration, such a device allows an 80 nm spectral window to be recorded, with a spectral resolution of approximately 0.15 nm. The ICCD camera was synchronized to the Q-switch of the laser, and acquisition was performed with a typical delay of 750 ns and a gate of 4000 ns.2.3 LIBS signal extraction
In our mapping experiment, each recorded spectrum is the result of a single laser shot without any accumulation. Typical single-shot spectra obtained from the different catalyst samples are shown in Fig. 1. We used the spectral range from 220 to 300 nm to detect both the C I line at 247.9 nm and several other lines of interest, particularly for Al (lines doublets at 226, 237, 256 and 265 nm) and Cu (lines at 261, 282 and 296 nm). Note that the carbon line at 193.1 nm was not appropriate because of spectral interference by the Al emission lines. As shown in Fig. 1, several emission lines of Al were detected, as it is the major element constituting the catalyst. We used the Al I 257.5 nm line since it does not interfere with any other elements or is saturated. Several trace impurities from the alumina matrix were also detected, such as Mg and Si, but these emissions will not be considered in the following. The magnified image in Fig. 1 emphasizes the evolution of the carbon line signal for three different samples—the reference sample and two asphaltene-impregnated samples—after 3.5 hours and 5 days. Note that the upper level of the carbon line for a 3.5 hour impregnated sample is 2 times lower than the same line measured on a 5 day impregnated sample. A typical imaging conducted on a catalyst section corresponds to a scanning sequence of approximately 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 pixels (i.e., plasma) and represent an experiment time of approximately 2.5 min.
000 pixels (i.e., plasma) and represent an experiment time of approximately 2.5 min.
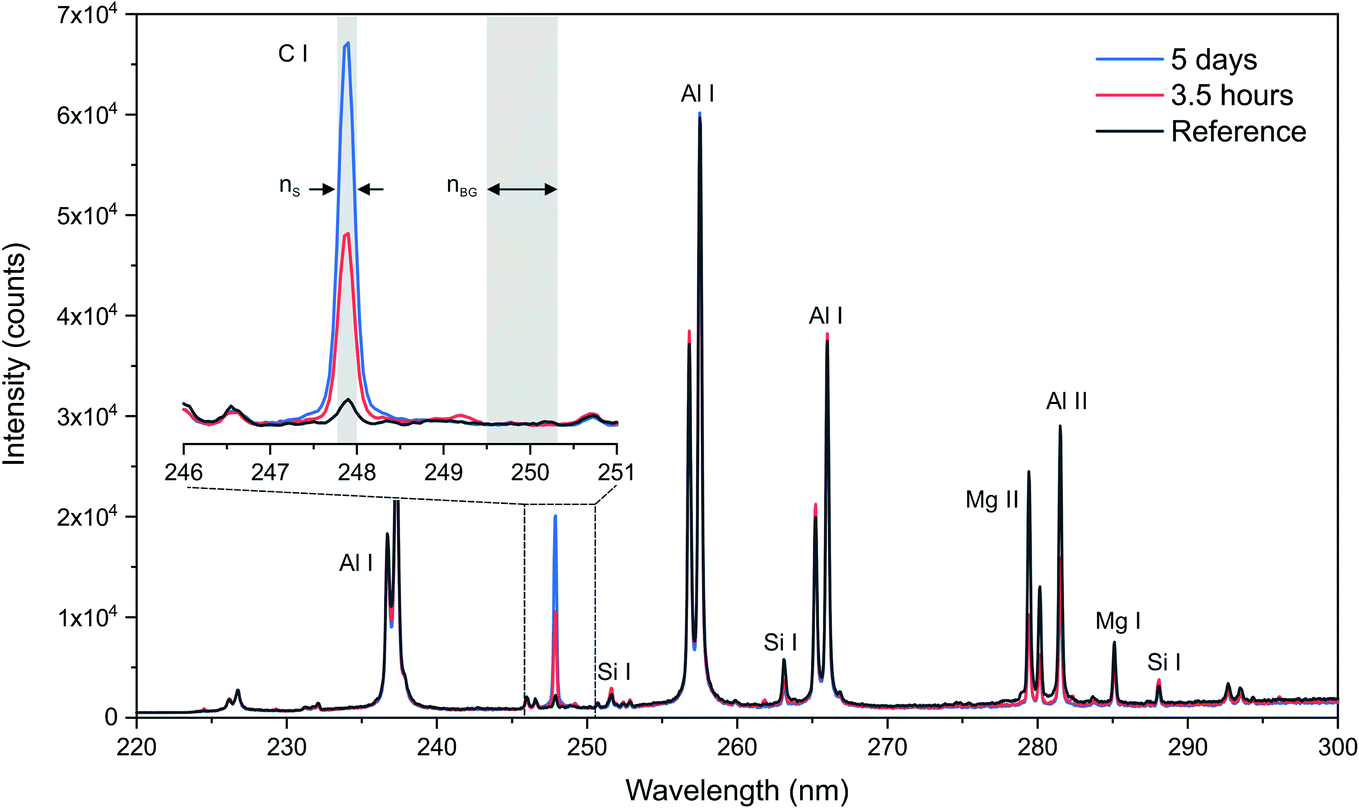 | ||
| Fig. 1 Example of single-shot spectra over the 220–300 nm spectral range for the reference support, a 3.5 hour impregnated support and a 5 day impregnated support and comparison of their respective carbon emission lines at 247.9 nm. | ||
Signal extraction processing was performed according to a previous study.36 The signal of interest IS was extracted by defining two spectral windows: a first window covering the line of interest and a second window covering the surrounding background, containing a number of points equal to nS and nbg, respectively (cf.Fig. 1). We calculate the total intensity measured in the first window by summing the raw signal measured on each of the nS points 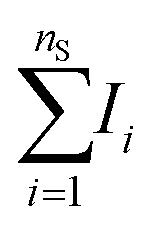 . To this value we subtract the average intensity of the background
. To this value we subtract the average intensity of the background 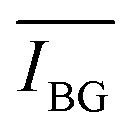 calculated in the second window and multiplied by nS, the number of points considered in the previous sum. Finally, the value for IS was obtained, using the equation:
calculated in the second window and multiplied by nS, the number of points considered in the previous sum. Finally, the value for IS was obtained, using the equation:
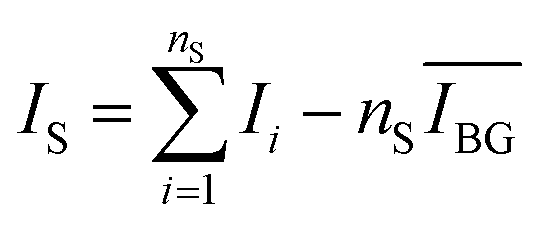 | (1) |
3. Results
3.1 LIBS imaging processing
In the case of heterogeneous catalysts, carbon species penetrate during impregnation in all directions inside the alumina bead with a concentration gradient that can be considered uniform with respect to the catalyst border. It is therefore possible to obtain the average elemental profile by averaging the entire spectra associated with the same distance from the border. The method used to extract the average diffusion profiles is illustrated in Fig. 2. First, the Al image is computed from the Al I 257.5 nm line (Fig. 2a) and a threshold is applied to obtain a binary image with a null pixel under the threshold (Fig. 2b). The resulting binary image represents the catalyst section. The applied threshold was set to 15% of the maximum intensity of the Al image. Then, the image of carbon is obtained by applying the binary mask on the global image of carbon extracted from the C I line at 247.9 nm (Fig. 2c). This step is required to discern the carbon in the catalyst section from the carbon in the resin around the section. Finally, a distance transform algorithm is applied to the binary mask with the aim of obtaining a distance map of the section associated with each pixel a value equal to the distance, in pixel, from the edges of the section. The resulting map (Fig. 2d) associates the same value with each pixel located at the same distance from the catalyst border. From this distance map, it is then possible to average all spectra at the same distance from the border of the catalyst. The average diffusion profiles of carbon in the catalyst are then obtained by extracting the carbon signal of this series of averaged spectra. In addition to providing global information regarding the diffusion process, such a method allows both a significant improvement of the analytical performances due to the averaging and the evaluation of associated statistical errors. Such averaging indeed allows the signal-to-noise ratio (SNR), and thus the limit of detection (LoD), to be improved by a factor of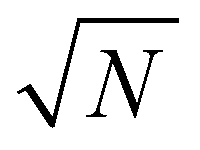 , where N is the number of averaged spectra. Such improvement is, however, not constant throughout the profile since each averaged spectrum is obtained from a different number of measurements.
, where N is the number of averaged spectra. Such improvement is, however, not constant throughout the profile since each averaged spectrum is obtained from a different number of measurements.
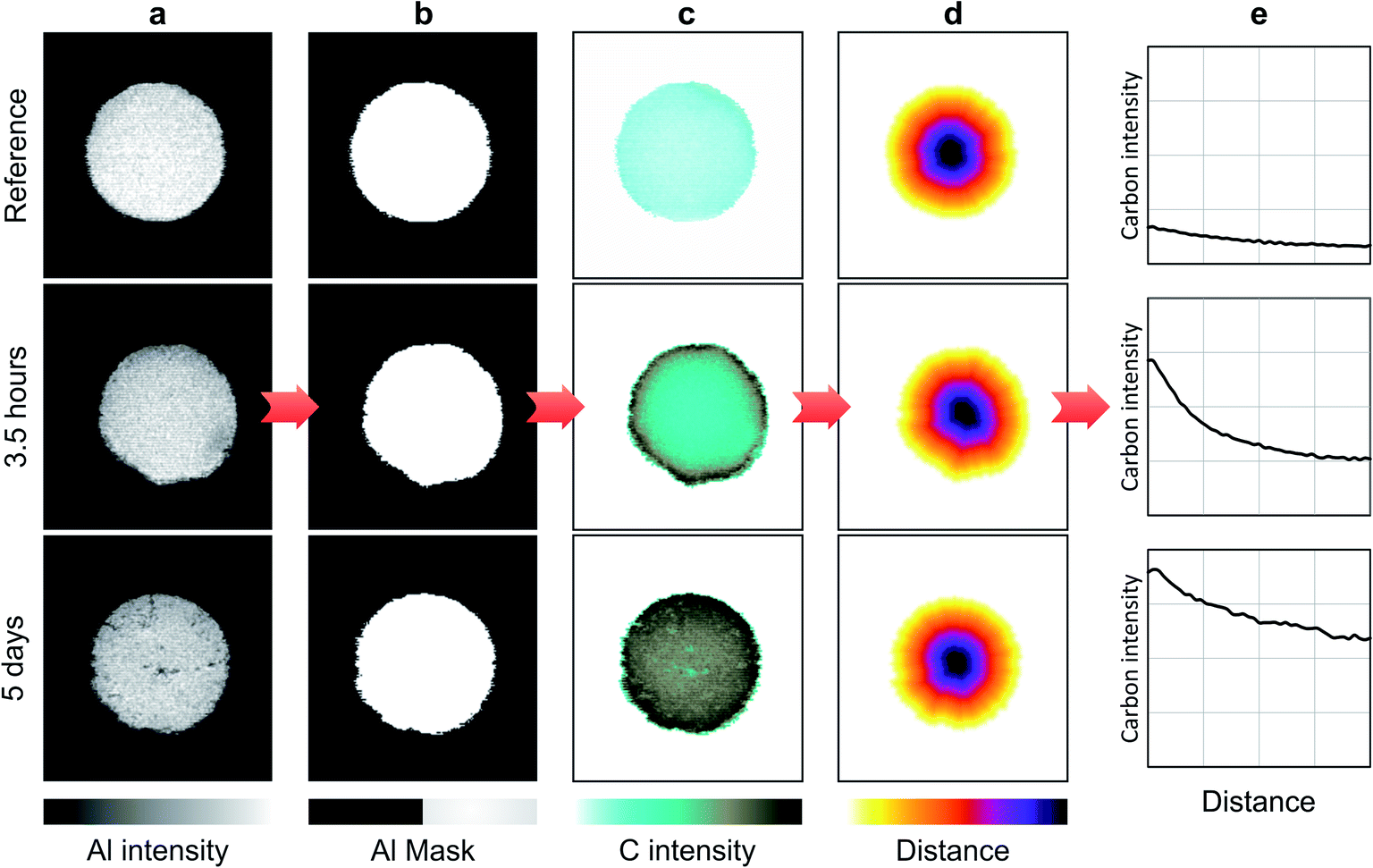 | ||
| Fig. 2 LIBS imaging processing principle, illustrated for low (top) and high (bottom) C contents. (a) Al images representing the Al2O3-matrix. (b) Binary masks obtained from Al images. (c) Masked C images. (d) Distance transformed representation applied to the mask. (e) Intensity profiles illustrating the C repartition from the border to the core of the catalyst section. | ||
3.2 Carbon-free sample preparation
Usual sample preparation for LIBS analysis of catalysts is the same as the sample preparation for microprobe analysis. It consists of embedding catalyst bodies in an acrylic or epoxy resin, initially liquid, which is hardened by increasing the temperature, followed by polishing of catalyst sections with silicon carbide-coated abrasive foils.37 As mentioned above, this procedure is not adapted for carbon quantification due to contamination issues because carbon species from the resin are diffusing into pores and cracks of alumina catalyst bodies.27 To overcome this issue, a specific update of the usual preparation procedure is required. We propose to apply a film on catalyst bodies to act as a barrier to the resin during the embedding process and prevent carbon diffusion from the resin. We first investigated the workability of the analysis, depending on the nature of the film, with the idea to choose the most appropriate one. First, the material used for the coating must not interfere with the emission lines of interest. This means that the major constituent of the film should not have emission lines at approximately 247.9 nm and must be as pure as possible. The behavior of the film through polishing is the second critical point. In particular, catalyst bodies may not be sufficiently attached on the coating and could break away during polishing. This depends on the grip between the film and the catalyst material. Considering these different aspects, the best candidate was the use of a high-purity copper film (GoodFellow, copper foil, 0.25 mm thick, 99.9% pure), avoiding spectral interference and providing good attachment onto alumina. Small strips of 25 × 13 mm2 were cut and then applied by hand all around the catalyst body (Fig. 3b). Each film extremity was properly curled to avoid resin penetration from the top and the bottom of the extrudate. After this Cu coating, the samples were embedded in resin as usual (Fig. 3c). It was possible to embed up to 20 catalysts simultaneously in a 2.5 cm diameter mold. We finally used silicon carbide-coated abrasive foils for surface polishing by following a procedure of up to 5-micron granulometry. An example of the resulting stub is shown in Fig. 3d. The total preparation time for a 10-catalyst stub was approximately 3 h and 30 min, including 10 minutes to apply the film, 3 h of resin hardening, and 20 minutes of polishing. | ||
| Fig. 3 Carbon-free sample preparation. (a) Catalyst supports. (b) Copper-coated catalysts. (c) Preparation of the catalysts for resin embedding. (d) Final stub after embedding and polishing. | ||
3.3 Evaluation of the carbon-free sample preparation
Two batches of reference catalyst supports samples (i.e., not impregnated with asphaltenes) were prepared. The first batch was prepared according to the carbon-free preparation described above, and the other batch was prepared by the usual preparation process (cf.Fig. 4a). The corresponding images of aluminum and carbon are shown in Fig. 4b and c, respectively. As mentioned above, the resin regions were removed from carbon images by applying the Al mask. As expected, an important carbon contamination was observed on samples prepared via the usual procedure. As shown in the left images of Fig. 4b, this preparation induced a carbon gradient associated with a strong signal in the periphery of the catalyst sections (brown color), which was also observed in the cracks of the catalyst body (the cracks are visible in the Al images in Fig. 4b). In contrast, the results obtained from the carbon-free prepared samples show a homogeneous carbon signal with an intensity reduced by more than half. This carbon signal is due to the grade of the used alumina matrix that contains residual carbon (0.12 wt%) (cf. Section 2.1) and possibly by uniform carbon contamination during polishing. Importantly, no carbon gradient from the border of the catalyst section was observed, which validates the performance of the proposed carbon-free preparation. | ||
| Fig. 4 Evaluation of the carbon-free preparation for LIBS analysis. (a) Optical images. (b) Corresponding LIBS images of Al and Cu. The Cu signal was extracted from the 261.8 nm emission line. (c) Corresponding LIBS images of carbon. | ||
In addition, as observed in Fig. 4, the Al signal is higher for the carbon-free preparation than for the usual preparation. It is finally important to mention that for each prepared stub of a given process time (i.e., impregnated with asphaltenes), we always included one or several reference samples (i.e., not impregnated), as shown in Fig. 3d. In this way, the residual carbon signal (i.e., measured from the reference sample) was subtracted from the impregnated supports with the aim of obtaining only the carbon signal related to asphaltenes.
The carbon images obtained with the carbon-free preparation were used to evaluate the limit of detection (LoD) in a single-shot configuration. We obtained a LoD value of 305 ppm by using definition:38
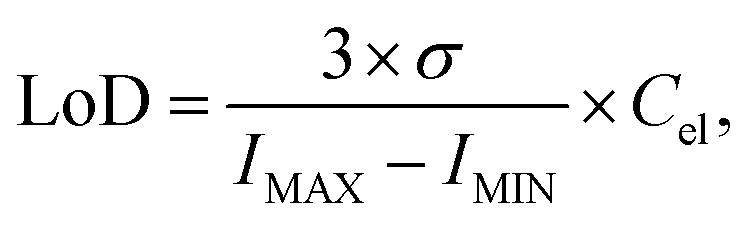 | (2) |
3.4 Imaging
We analyzed the seven samples previously described, including the reference and the six samples impregnated with asphaltenes over a time range from 30 min to 6 weeks (cf.Table 2). For each sample, five catalyst sections were analyzed. The optical and elemental images of two of these 5 sections are shown in Fig. 5. Al images may reveal the sample morphology by evidencing eventual cracks in or deterioration of the matrix (Fig. 5b). The carbon images shown in Fig. 5c demonstrate that carbon is successfully detected over the whole concentration range involved. In addition, carbon gradients are clearly evidenced, attesting the diffusion of carbonaceous molecules contained in the asphaltenes as well as the expected radial symmetry of the diffusion processes. Whereas optical images do not show any change in contrast after only 6.5 hours of impregnation, carbon imaging reveals the accurate poisoning of catalysts as a function of time. These results illustrate the relevance of LIBS imaging for the study of carbon transport into catalyst materials. The stability of the Al intensity as a function of impregnation time allows us to consider that the matrix effects can be neglected over the concentration range involved, despite a significant change in the color and composition between the samples.| Impregnation duration | 30 min | 3.5 h | 6.5 h | 5 days | 3 weeks | 6 weeks |
| Carbon concentration (wt%) | 2.50 ± 0.3 | 7.03 ± 0.3 | 8.51 ± 0.3 | 13.53 ± 0.2 | 16.2 ± 0.2 | 16.99 ± 0.2 |
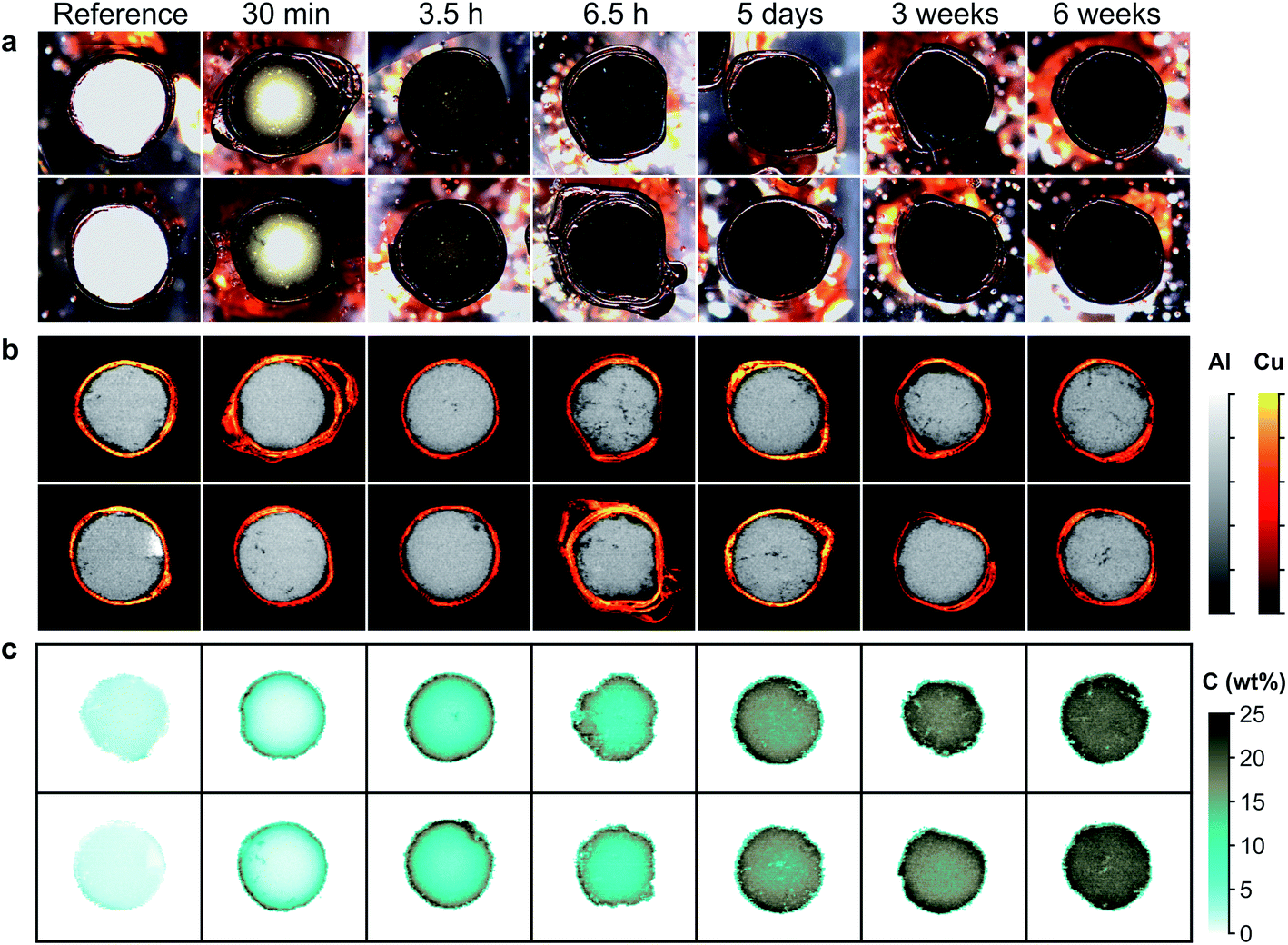 | ||
| Fig. 5 Carbon distribution on alumina refining catalyst supports impregnated with asphaltenes during different times of contact between the supports and the asphaltenes solution. (a) Optical images of the catalyst support sections. (b) LIBS images of the Al matrix and the Cu foil coating. (c) LIBS images of the C distribution. | ||
3.5 Intensity calibration
Homogeneous standards of porous alumina catalysts are difficult to develop mainly because of their heterogeneous texture and porosity. To overcome this issue, we propose to calibrate the LIBS intensities by considering the global amount of carbon contained in the supports for the different impregnation times. As mentioned above, the diffusion of carbon follows a radial distribution. It is therefore possible to hypothesize that the surface analysis conducted by LIBS is representative of the bulk concentrations. The global carbon LIBS intensity can then be obtained by summing all the pixel intensities contained in the alumina matrix (positive pixels in the Al mask). It is important to mention that our aim is to focus only on carbon coming from asphaltenes, so the residual carbon signal measured on reference samples has to be subtracted for each sample of interest. Fig. 6 shows these corrected intensities as a function of the bulk carbon concentration. For each point, the y error bars correspond to the standard deviation of the retrieved intensities for the five analyzed sections. As seen, these data can be modeled accurately by a quadratic regression (i.e., determination coefficient of R2 = 0.995). The use of a quadratic model is justified since the used carbon line appears to be self-absorbed at high concentrations (typically > 10 wt%).39 However, this line of saturation remains moderate and may not affect the quantification performance.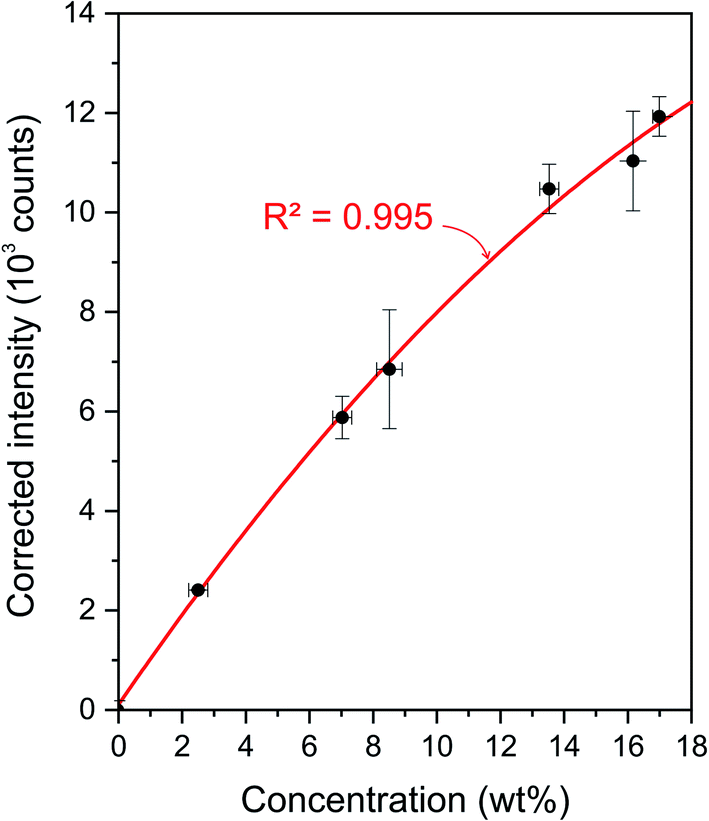 | ||
| Fig. 6 Calibration of carbon LIBS intensities. | ||
3.6 Diffusion profiles
The diffusion profiles obtained from the method described in Section 3.1 and by applying the calibration model (cf.Fig. 6) is shown in Fig. 7 for the 6 process times. A logarithm scale is used for the concentration axis to highlight the dynamic range of measurements. These profiles represent an average for each impregnation time of the results obtained for the five catalyst sections. These results show that carbon impurities transport from the border to the core of the catalyst sections. Two classes of carbon molecules are observed. First, there is an increasing carbon content reaching the core of the section as a function of impregnation time, and second, there is a continuously increasing carbon content at the catalyst support border, showing nonhomogeneous carbon profiles even at the equilibrium state. Such representation is perfectly adapted to compare with modeling of the diffusion process. It is important to emphasize that they represent the global diffusion (all the pixels of the images are considered) and allow the relative statistical errors to be evaluated (not shown in Fig. 7 for better clarity). Such errors depend on the number of averaged spectra and typically range from 0.2% at the section periphery to 3% at the center. As an example, in EPMA, elemental profiles are obtained from one unique catalyst section and contain information from only one line that crosses a section. A deviation in the results was observed between the 3.5 and 6.5 hour samples. The carbon content at the border should indeed be higher for the 6.5 hour condition. This may be explained by sample alteration. As can be observed in Fig. 5, the 6.5 hour sections show irregular shapes, cracks and important alterations in the external regions. However, we did not exclude this batch from the study because reliable information is obtained for the core of the sample.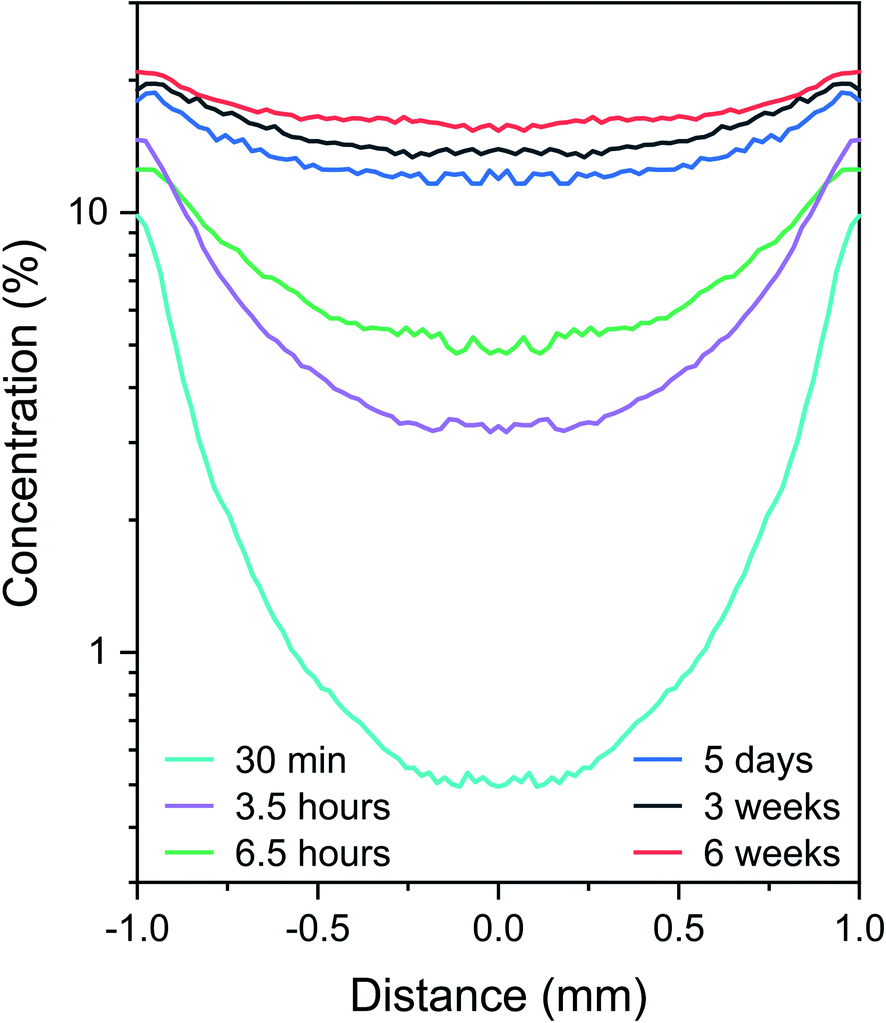 | ||
| Fig. 7 Averaged carbon diffusion profiles for different asphaltene impregnation times. | ||
The two classes of observed carbon molecule diffusion behaviors can be correlated with the aggregation tendency and size polydispersity of asphaltene molecules,40 with the smallest molecules diffusing at the core earlier in the impregnation process and the larger aggregates accumulating slowly at the near border. A similar behavior was proposed for Ni and V species in our previous work, and several hypothesis have been proposed to explain the asphaltene transport phenomena into catalyst bodies.40 The simple diffusion models generally considered suppose reversible adsorption and imply a flat profile at equilibrium. In light of these final results, such models are apparently not adequate to describe the transport of carbon impurities into catalysts. This is an unprecedented conclusion that could be further complemented with an appropriate diffusion model able to reproduce the observed profiles.
4. Conclusions
In this work, we propose to address the challenge of carbon analysis with an innovative LIBS-based analytical procedure. The procedure includes a carbon-free sample preparation as well as a specific data processing procedure. Such a procedure is able to provide, for the first time, quantitative imaging and profiling of the diffusion of carbon impurities into alumina catalysts. A large range of carbon concentrations, covering more than two orders of magnitude from 0.12 wt% to 17 wt%, has been successfully analyzed in a very small amount of time. The single-shot LoD of carbon is estimated at 305 ppm, which is small enough to quantify carbon on a large variety of catalyst samples. The final quantitative profiling provides new and meaningful data that can be directly compared to the data obtained from carbon diffusion models. These models will allow a better understanding of the hydrocarbon transport mechanisms and kinetics.LIBS-based imaging offers many advantages, such as multielemental capabilities, high acquisition speed extended to the kHz range and operation at atmospheric pressure. LIBS images contain elemental information with ppm-scale sensitivity and μm-range resolution. Light elements can be detected with very interesting performance, whereas they are not detected with the commonly used EPMA at the same speed. We believe that LIBS imaging applied to the field of catalysis has the potential to characterize the elemental repartition on various catalytic systems, including light elements such as H, Li, C and S.
As an extension, the proposed methodology can be applied to the investigations of coke deposition in catalysts or to the study of the carbonaceous additives employed during the catalyst preparation step. Such a methodology may also benefit the carbon analysis of porous samples from various other fields. We can cite natural materials such as reservoir rocks, soil samples and wood (corks) or body organs such as bones. Synthetic materials can also be considered, such as concretes containing many carbonate phases, anatomical prostheses and innovative materials such as open-cell foams (polymer foams with introduction of ceramic or metal) considered as in-growth materials in the aerospace or industrial fields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Pulsalys (#L0978-L1294) and the French region Rhônes Alpes Auvergne (Optolyse, CPER2016). In addition, we gratefully acknowledge Frédéric Pelascini from the Cetim Grand Est and Florian Trichard from Ablatom S. A. S. for fruitful discussions.References
- H. Topsøe, B. S. Clausen and F. E. Massoth, Hydrotreating Catalysis, Springer Verlag, Berlin, 1996 Search PubMed.
- P. Dufresne, Appl. Catal., A, 2007, 322, 67–75 CrossRef CAS.
- E. Rodríguez, G. Félix, J. Ancheyta and F. Trejo, Fuel, 2018, 225, 118–133 CrossRef.
- C. H. Bartholomew, Appl. Catal., A, 2001, 212, 17–60 CrossRef CAS.
- M. D. Argyle and C. H. Bartholomew, Catalysts, 2015, 5, 145–269 CrossRef CAS.
- F. L. Bleken, K. Barbera, F. Bonino, U. Olsbye, K. P. Lillerud, S. Bordiga, P. Beato, T. V. W. Janssens and S. Svelle, J. Catal., 2013, 307, 62–73 CrossRef CAS.
- A. Sadeq Al-Fatesh, S. Olajide Kasim, A. Aidid Ibrahim, A. Hamza Fakeeha, A. Elhag Abasaeed, R. Alrasheed, R. Ashamari and A. Bagabas, Catalysts, 2019, 9, 2073–4344 CrossRef.
- R. F. Howe, E. K. Gibson, C. R. A. Catlow, A. Hameed, J. McGregor, P. Collier, S. F. Parker and D. Lennon, Faraday Discuss., 2017, 197, 447–471 RSC.
- J. J. H. B. Sattler, A. M. Mens and B. M. Weckhuysen, ChemCatChem, 2014, 6, 3139–3145 CrossRef CAS.
- H. Shimada, M. Imamura, N. Matsubyashi, T. Saito, T. Tanaka, T. Hayakawa and S. Kure, Top. Catal., 2000, 10, 265–271 CrossRef CAS.
- S. R. Bare, F. D. Vila, M. E. Charochak, S. Prabhakar, W. J. Bradley, C. Jaye, D. A. Fischer, S. T. Hayashi, S. A. Bradley and J. J. Rehr, ACS Catal., 2017, 7, 1452–1461 CrossRef CAS.
- K. Y. Cheah, N. Chiaranussati, M. P. Hollewand and L. F. Gladden, Appl. Catal., A, 1994, 115, 147–155 CrossRef CAS.
- J. L. Bonardet, M. C. Barrage and J. Fraissard, ACS Symp. Ser., 1996, 634, 99–116 CrossRef CAS.
- J. P. Janssens, R. M. de Deugd, A. D. van Langeveld, S. T. Sie and J. A. Moulijn, Stud. Surf. Sci. Catal., 1997, 111, 283–294 CrossRef CAS.
- A. Pachulski, R. Schödel and P. Claus, Appl. Catal., A, 2011, 400, 14–24 CrossRef CAS.
- F. Robault, A. Crisci, M. Durrand-Charre and D. Jouanne, Microsc. Microanal., 2006, 12, 331–334 CrossRef PubMed.
- J. P. Hofmann, D. Mores, L. R. Aramburo, S. Teketel, M. Rohnke, J. Janek, U. Olsbye and B. M. Weckhuysen, Chem.–Eur. J., 2013, 19, 8533–8542 CrossRef CAS PubMed.
- E. de Smit, I. Swart, J. F. Creemer, G. H. Hoveling, M. K. Gilles, T. Tyliszczak, P. J. Kooyman, H. W. Zandbergen, C. Morin, B. M. Weckhuysen and F. M. F. De Groot, Nature, 2008, 456, 222–225 CrossRef CAS PubMed.
- L. Jolivet, M. Leprince, S. Moncayo, L. Sorbier, C.-P. Lienemann and V. Motto-Ros, Spectrochim. Acta, Part B, 2019, 151, 41–53 CrossRef CAS.
- R. Noll, C. Fricke-Begemann, S. Connemann, C. Meinhardt and V. Sturm, J. Anal. At. Spectrom., 2018, 33, 945–956 RSC.
- P. Lucena, L. M. Cabalin, E. Pardo, F. Martin, L. J. Alemany and J. J. Laserna, Talanta, 1998, 47, 143–151 CrossRef CAS PubMed.
- P. Lucena, J. M. Vadillo and J. J. Laserna, Anal. Chem., 1999, 71, 4385–4391 CrossRef CAS PubMed.
- P. Lucena, J. M. Vadillo and J. J. Laserna, Appl. Spectrosc., 2001, 55, 262–272 CrossRef.
- P. Lucena, J. M. Vadillo and J. J. Laserna, J. Anal. At. Spectrom., 2002, 17, 548–551 RSC.
- F. Trichard, L. Sorbier, S. Moncayo, Y. Blouët, C.-P. Lienemann and V. Motto-Ros, Spectrochim. Acta, Part B, 2017, 133, 45–51 CrossRef CAS.
- L. Sorbier, F. Trichard, S. Moncayo, C. P. Lienemann and V. Motto-Ros, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 304, 12016 Search PubMed.
- F. Trichard, F. Gaulier, J. Barbier, D. Espinat, B. Guichard, C.-P. Lienemann, L. Sorbier, P. Levitz and V. Motto-Ros, J. Catal., 2018, 363, 183–190 CrossRef CAS.
- L. Sorbier, E. Rosenberg, C. Merlet and X. Llovet, Mikrochim. Acta, 2000, 132, 189–199 CrossRef CAS.
- J. Barbier, F. Gaulier, B. Guichard, P. Levitz and D. Espinat, Energy Fuels, 2017, 31, 7426–7437 CrossRef CAS.
- F. Gaulier, J. Barbier, B. Guichard, P. Levitz and D. Espinat, Energy Fuels, 2015, 29, 6250–6258 CrossRef CAS.
- J. Ancheyta, F. Trejo and M. S. Rana, Asphaltene - Chemical Transformation during hydroprocessing of heavy oils, CRC Press (Taylor & Francis Group), Boca Raton, 2009 Search PubMed.
- J. F. Le Page and S. G. Chatila, Raffinage et conversion des produits lourds du pétrole, Technip, Paris, 1990 Search PubMed.
- Produits pétroliers - Détermination de la teneur en asphaltènes (insolubles heptane), AFNOR, NF T 60-115, 2000.
- V. Motto-Ros, E. Negre, F. Pelascini, G. Panczer and J. Yu, Spectrochim. Acta, Part B, 2014, 92, 60–69 CrossRef CAS.
- M. Gaft, Y. Raichlin, F. Pelascini, G. Panzer and V. Motto Ros, Spectrochim. Acta, Part B, 2019, 151, 12–19 CrossRef CAS.
- V. Motto-Ros, S. Moncayo, F. Trichard and F. Pelascini, Spectrochim. Acta, Part B, 2019, 155, 127–133 CrossRef CAS.
- P. W. Tamm, H. F. Harnsberger and A. G. Bridge, Ind. Eng. Chem. Process Des. Dev., 1981, 20, 262–273 CrossRef CAS.
- J.-M. Mermet, Spectrochim. Acta, Part B, 2008, 63, 166–182 CrossRef.
- E. Negre, V. Motto-Ros, F. Pelascini, S. Lauper, D. Denis and J. Yu, J. Anal. At. Spectrom., 2015, 30, 417–425 RSC.
- I. Merdrignac, C. Truchy, E. Robert, I. Guibard and S. Kressmann, Pet. Sci. Technol., 2004, 22, 1003–1022 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |