
Development of an AES based analytical method for the determination of trace metallic impurities in uranium silicide dispersion fuel: from precursors to end products†
Arijit
Sengupta
 *ab,
Rajeswari
B.
a and
R. M.
Kadam
ab
*ab,
Rajeswari
B.
a and
R. M.
Kadam
ab
aRadiochemistry Division, Bhabha Atomic Research Centre, India. E-mail: arijita@barc.gov.in; arijitbarc@gmail.com
bHomi Bhabha National Institute, India
First published on 8th November 2019
Abstract
An analytical method was developed based on the D.C. arc carrier distillation atomic emission spectrometric (D.C. Arc AES) technique for determination of trace metallic impurities (Al, Ca, Co, Cr, Cu, Fe, Mg, Mn, Mo, Na, Ni, Si, Sn, Ti, V and Zn) in uranium silicide samples. Chemical quality control was also carried out for the precursor materials in order to track the source of impurities in the final fuel materials. The method was validated using synthetic samples. To minimize the spectral interference, a suitable carrier (5% AgCl) was used to sweep away the trace constituents into the arc preferentially, leaving the major matrix as a refractory material. The use of a charge coupled device (CCD) detector was found to improve the overall analytical performance in terms of detection limits, precision, linear dynamic range and most importantly providing an opportunity to choose additional analytical lines, which is highly desirable, especially for trace level determination of analytes in an emission-rich matrix. The energy dispersive X-ray fluorescence (EDXRF) spectroscopic technique was employed each time for comparative evaluation of the precursors and the product.
Introduction
In the modified Apsara reactor in India, uranium silicide (U3Si2) with low uranium enrichment (less than 20%) has been employed as a fuel material to achieve its desired performance in increasing the thermal and fast neutron flux as well as enhancement in power from 1 MW to 2 MW.1–3 Highly enriched uranium either as a U–Al alloy or U3O8 dispersed in Al was exploited earlier for Apsara reactor fuel materials. These advanced fuel materials not only decrease the challenges associated with uranium enrichment, but can also address some important issues associated with the original fuels, together with providing some advantageous features.4–6 The poor stability of the alpha phase of uranium at lower temperature, the thermodynamic instability of the low swelling gamma phase, and the poor solid solubility of uranium are some of the important drawbacks that can be resolved using these advanced fuel materials. Out of several U–Si based compounds, U3Si2 has been chosen for this purpose due to its moderate density and low swelling behaviour.7,8 Though U2Si is one of the potential candidates having higher density compared to the other candidates, the higher swelling behaviour is the main reason for its rejection.9,10To achieve the expected performance for these advanced fuel materials, suitable and stringent chemical quality control is a must in different stages of fuel fabrication. This not only certifies the quality of the fuel materials, but can also track the origin of impurities if present in the materials. Analytes like B and Cd have very low specification limits to maintain good neutron economy in the reactor core. Low melting point analytes like Zn can modify the mechanical property degradation of the fuel by liquid-metal embrittlement, while high melting analytes like Mo and W can cause creep resistance. To keep the thermo-chemical and mechanical properties intact during its lifetime in the reactor, an efficient, reliable, precise, and less time consuming analytical method is desirable to determine multiple elements simultaneously. Atomic emission spectrometry is one of such analytical techniques that have been used worldwide for simultaneous multi-elemental determination of metallic analytes at minor, trace and even ultra-trace levels in nuclear material matrices.11–13
U being a multielectronic system has a rich emission spectrum. Therefore, the presence of such element can have a detrimental effect on the determination of analytes especially when they are present at very low levels (trace or ultratrace).14–16 So, it is necessary to suppress its introduction into the excitation source. In the case of D.C. arc carrier distillation, physical separation based on volatility is achieved by choosing a suitable carrier, while chemical separation is desirable for a plasma based excitation source.17,18 Solid state analysis is very valuable especially in the case of nuclear materials, since it requires less sample handling, resulting in less personal exposure, a smaller chance of process pick-up, and no need for dissolution of samples.19,20 Since the dissolution of U3Si2 is challenging, D.C. arc carrier distillation has been preferably chosen for trace metal assay in the U3Si2 matrix. With the use of a charge coupled device (CCD) as a detection system, the instrument can provide a greater choice of analytical lines compared to the photomultiplier tube based detector system. Moreover, this CCD based detector21–23 can provide better analytical performance including two dimensional determination, which otherwise cannot be achieved by PMT based systems.
In view of all this, a D.C. arc carrier distillation based analytical method was developed for the determination of trace metallic constituents in U3Si2 samples. The precursors for such type of fuel are U metal and Al turnings. Chemical quality control has also been carried out for the precursors in order to track the sources of metallic impurities in the final products. Energy dispersive X-ray fluorescence spectroscopy, being another multi-elemental, non-destructive technique, has been used for comparative evaluation.24–26
Experimental
Materials
SpecPure® grade individual metal oxides (SnO2, Mn3O4, CuO, Cr2O3, MgO, V2O5, CaCO3, PbO, Fe2O3, Si powder, TiO2, ZnO, Al2O3, and WO3) and AgCl used in the present study were procured from SPEX Industries, USA. For Mo, Na, Co, and Ni, the precursors used were (NH4)6Mo7O24·4H2O, NaCl, Co powder and Ni sponge, respectively. They were also of the same grade as the metal oxides and were procured from the same source (metal). High purity U3O8 powder obtained by inter-laboratory comparison experiments from different laboratories of the Department of Atomic Energy (DAE), Government of India was used for preparation of standards.20 A standard carrier distillation-type electrode, ASTM designation E-130-66 type S-2 on a type S-1 pedestal, was used in the present study as the anode, while ASTM designation E-130-66 type C-1 was used as the cathode.27 A simplified schematic representation for fabrication of U3Si2 samples and the requirement of chemical quality control (QC) is depicted in Fig. 1. Appropriate amounts of uranium and silicon powder were blended for 4 h to achieve a high degree of homogeneity in the product. Then the mixture was compacted at 600 MPa pressure to a 20 mm diameter. Then annealing was carried out under 1.33 Pa vacuum at 1550 ± 10 °C for 4 h. After cooling, the U3Si2 pellets were ready for analysis.1 The product was found to be highly homogeneous as also reported in the literature.1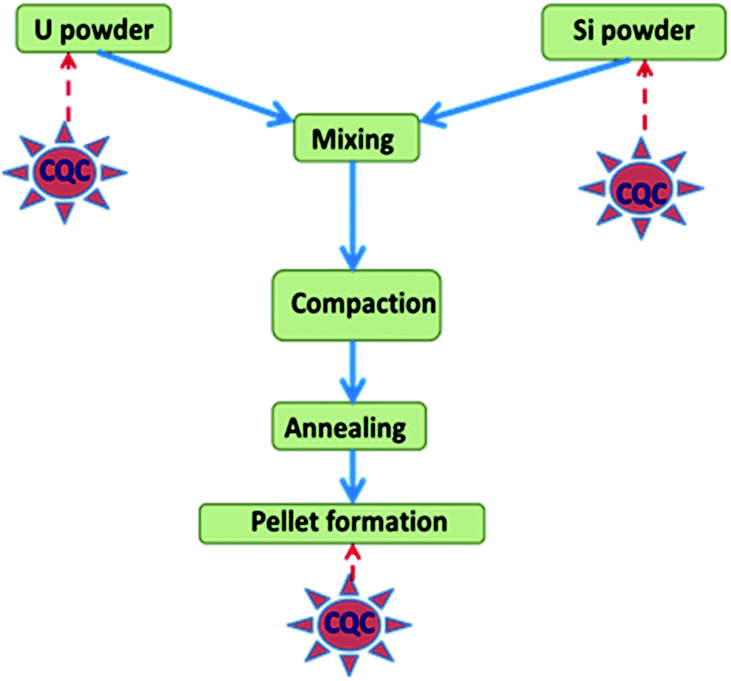 | ||
| Fig. 1 Simplified schematic representation for fabrication of U3Si2 and requirement of QC. | ||
Instrumentation
The D.C. arc carrier distillation technique was implemented using a Spectro-Arcos Atomic Emission Spectrometer (Spectro-Arcos, Germany) having a D.C. arc as the excitation source and a charge coupled device (CCD) as the detector system. Table 1 summarizes the experimental conditions and the instrumental parameters. Linear arrays with 3648 pixels per array of a thermally stabilized CCD detector have been used (ESI Fig. 1†).| Instrumental specifications | |
|---|---|
| Optical design | Paschen–Runge mounting, circular design |
| Focal length | 750 mm |
| Grating | Holographic |
| Groove density | 1800 grooves per mm (1), 3600 grooves per mm (2) |
| Wavelength range | 130–800 nm |
| Entrance slit width | 15 microns |
| Resolution (FWHM) | 0.01 nm from 130–450 nm, 0.02 nm from 450–800 nm |
| Thermal regulation | Controlled to 30 ± 1 °C |
| Detector | Linear CCD arrays (3648 pixels per array) |
A Jordan Valley EX-3600 M EDXRF spectrometer, with a Rh anode along with a Si(Li) detector (with a 12.5 mm thick beryllium window), was used for the present investigation. A solid sample in powder form was used and the measurements were carried out in air. nEXt system software was used for spectral acquisition and analysis.26 A Si(Li) detector with a resolution of 139 eV at Mn-Kα (5.9 keV) and a spectral range in the 1–40 keV energy region was utilized in the present study. In the sample chamber, the samples were placed in Teflon cup assemblies. The bases of the Teflon cups were fitted with Mylar film of 6.3 mm thickness. The optimized parameters for EDXRF analysis are summarized in Table 2.
| Parameters | Value |
| Target | Rh |
| Filter | No filter |
| High voltage | 30 kV |
| Emission current | 200 μA |
| Preset time | 100 s |
| Atmosphere | Air |
| Energy range | 40 keV |
| Throughput | High |
Method
D.C. arc carrier distillation
As mentioned earlier, D.C. arc carrier distillation is a technique where physical separation of matrix-rich U occurs from the analytes at a trace level by addition of a suitable amount of carrier. 5% AgCl was chosen as the carrier for the present investigation. The choice of carrier and its composition was made based on the reported carrier suitable for the U3O8 matrix.20,27 This choice is reasonable because after annealing the U3Si2 samples to the most stable refractory form, U3O8 is one of the major components expected.27 Before proceeding to the actual analysis, all the samples were heated at 600 °C for 5 hours for their conversion into the most stable refractory oxides. In the case of U powder microspheres, it is U3O8, for Si, Si powder and for U3Si2 samples, it is a mixture of U3O8 with a very small amount of Si powder. In view of this, for analysing U powder, Si powder and U3Si2 samples, U3O8, Si powder and U3O8 standards were used. Seven point calibration curves were established for all the analytes. In D.C. arc carrier distillation, 100 mg of charge was placed in a graphite anode, while a graphite cathode with a sharp end was used for ignition of the arc. The gap between the two electrodes was kept at 4 mm. To strike the arc a high voltage or current was applied. After ignition of the arc, the electrode from the pointed cathode runs towards the anode containing sample charges and strikes it. As a result of that, the analytes present in the samples get atomized and they are excited. During de-excitation, they emit light of a characteristic wavelength. The role of carrier, i.e. AgCl, in the present case is to sweep away the analytes into the arc, leaving behind the major matrix. Therefore, the carrier should have an intermediate volatility and boiling point and must be obtained in a very pure form. It should also be highly stable and non-hygroscopic. The concentrations of the analytes in the standards are as follows (Table 3). The groups were made based on the specification limits, volatility and compatibility of the analytes. In the case of Si powder standards, Si was not taken into consideration. The sample introduction arrangement in D.C. arc carrier distillation is depicted in Fig. 2.| Element | Std 1 | Std 2 | Std 3 | Std 4 | Std 5 | Std 6 | Std 7 |
|---|---|---|---|---|---|---|---|
| Sn (ppm) | 1 | 2 | 5 | 10 | 20 | 50 | 100 |
| Mn, Na, Cu (ppm) | 2 | 4 | 10 | 20 | 40 | 100 | 200 |
| Co, Cr, Mg, Ni, V, Ca (ppm) | 5 | 10 | 25 | 50 | 100 | 250 | 500 |
| Fe, Zn, Al, Mo, Ti (ppm) | 10 | 20 | 50 | 100 | 200 | 500 | 1000 |
| W (ppm) | 40 | 80 | 200 | 400 | 800 | 2000 | 4000 |
| Si (ppm) | 80 | 120 | 240 | 440 | 840 | 2040 | 4040 |
 | ||
| Fig. 2 Sample introduction arrangement in D.C. arc carrier distillation. | ||
EDXRF
The same standards were used for establishing the calibration curves in EDXRF. The cross-sectional area for the aperture of the X-ray beam at the sampling site is circular in nature with an 8 mm diameter. 100 mg of standards/samples in powder form were placed uniformly in the sample holder having a polyvinyl chloride tubular guide with a 13 mm internal diameter. The analyte peaks were selected carefully in order to avoid spectral overlap. The peak area corresponds to the specified peak that was used for the quantification of the analytes. Standards and samples were counted under identical geometry in all cases. Triplicate measurements were made for each loading.As is well known, analytes like B and Be have very low Z and are hence not suitable for the EDXRF method. Moreover, the requirement for the analysis of B, Be and Cd is at the level of 0.1–5 ppm. EDXRF is also not suitable for such low level of detection. The standards were made using H3BO3, BeO, and CdO procured from Sigma Aldrich, USA at concentrations of 0.1 ppm, 0.2 ppm, 0.5 ppm, 1 ppm, 2 ppm and 5 ppm. These standards were analysed using D.C. arc carrier distillation. The analytical performance including detection limits, linear dynamic range, etc. was evaluated using their most suitable analytical lines. However, EDXRF analysis of these standards was unsuccessful as expected.
The powder samples cannot be handled in open air; this is associated with the risk of samples becoming air-borne and personal exposure. Hence, sample processing was carried out inside a glovebox. The D.C. arc AES system used here is glovebox adapted. The D.C. arc carrier distillation technique has been routinely employed for the trace metal assay of nuclear materials: (U, Pu)O2, PuO2, (U, Pu)C, U–Pu–Zr, Pu–Ga alloy, ZrO2, etc. using these glovebox adapted arrangements.17–20 Sample heating was also carried out inside a glovebox adapted furnace. The glovebox was in negative pressure and total confinement was maintained. The air activity was also monitored regularly. So it was guaranteed that no personal hazard was associated with the reported arrangement.
Results and discussion
For D.C. arc carrier distillation analysis, the samples were heated at 600 °C for their conversion into the most stable refractory oxides. Each 100 mg charge was consisted of 5% AgCl and 95% sample.28 Then it was ground well in a mortar and pestle for complete homogenization with an almost uniform size distribution. Then each charge was put on the anode through a small funnel. Then the electrodes were tapped and vented. The tapping resulted in the homogeneous distribution of charge, while venting resulted in the ejection of gas into the arc without physical throwing of the charge into the arc. The anode was linked with a graphite made pedestal for the homogeneous distribution of heat throughout the electrode. Fig. 3 shows the schematic representation of the processing of the sample for the D.C. arc analysis. The same powder was used for EDXRF analysis too. | ||
| Fig. 3 Schematic representation of the steps involved in D.C. arc carrier distillation AES prior to arcing. | ||
Optimization of the methodology is the most important step for any analytical developmental work. In D.C. arc analysis, the choice of analytical lines in different matrices is very important for getting better results. Though Si does not have a rich emission spectrum, the presence of U either in U powder or U3Si2 requires a judicious choice of analytical lines to minimize any spectral interference without any compromise on the other merits of analytical performance like detection limits, sensitivity, linear dynamic range or even precision.
Table 4 summarizes the best analytical lines of the analytes in the specified matrices and also their analytical performances. The interference-free analytical lines for U were also chosen for Si powder, since the spectral interference for Si is not prominent. Hence, the same analytical lines were also chosen for the U3Si2 matrix for convenience as well as comparison purposes as follows: Ca: 396.8 nm, Co: 242.4 nm, Cr: 357.8 nm, Cu: 324.7 nm, Fe: 259.9 nm, Mg: 285.2 nm, Mn: 257.6 nm, Mo: 202.0 nm, Na: 588.9 nm, Ni: 305.0 nm, Si: 288.1 nm, Sn: 242.1 nm, Ti: 498.1 nm, V: 318.5 nm, W: 239.7 nm and Zn: 213.8 nm. The detection limit was calculated by performing 10 replicate measurements of the blank matrices (without any analytes). The concentration corresponds to the intensity equivalent to the average value of the blank + the standard deviation. The curve fitting and other types of uncertainties have not been included separately. The linear dynamic range in the D.C. arc source was found to be much smaller compared to that in the plasma based AES source due to self-absorption. The arc not being an optically thin source results in self-absorption. The precision of the D.C. arc AES was also found to be very high compared to that of plasma based AES or even EDXRF. arc wandering is one of the predominating reasons for that.
| Elements | U powder | Si powder | U3Si2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Analytical line (nm) | Detection limit (ppm) | Linear dynamic range (ppm) | Analytical line (nm) | Detection limit (ppm) | Linear dynamic range (ppm) | Analytical line (nm) | Detection limit (ppm) | Linear dynamic range (ppm) | |
| a NA: not analyzed. | |||||||||
| Al | 308.2 | 15 | 15–950 | 308.2 | 10 | 10–1000 | 308.2 | 15 | 15–900 |
| Ca | 396.8 | 5 | 5–450 | 396.8 | 5 | 5–450 | 396.8 | 8 | 8–400 |
| Co | 242.4 | 5 | 5–400 | 242.4 | 5 | 5–450 | 242.4 | 8 | 8–400 |
| Cr | 357.8 | 5 | 5–500 | 357.8 | 5 | 5–450 | 357.8 | 9 | 9–450 |
| Cu | 324.7 | 2 | 2–150 | 324.7 | 2 | 2–175 | 324.7 | 4 | 5–150 |
| Fe | 259.9 | 10 | 10–900 | 259.9 | 10 | 10–950 | 259.9 | 10 | 10–800 |
| Mg | 285.2 | 5 | 5–400 | 285.2 | 5 | 5–450 | 285.2 | 6 | 6–400 |
| Mn | 257.6 | 2 | 2–170 | 257.6 | 2 | 2–160 | 257.6 | 2 | 2–150 |
| Mo | 202.0 | 10 | 10–800 | 202 | 10 | 10–900 | 202 | 10 | 10–800 |
| Na | 588.9 | 2 | 2–200 | 588.9 | 2 | 2–200 | 588.9 | 5 | 5–200 |
| Ni | 305.0 | 5 | 5–400 | 305 | 5 | 5–500 | 305 | 7 | 7–400 |
| Si | 288.1 | 80 | 80–2000 | NA | NA | NA | NA | NA | NA |
| Sn | 242.1 | 1 | 1–70 | 242.1 | 1 | 1–100 | 242.1 | 1.5 | 1.5–70 |
| Ti | 498.1 | 5 | 5–400 | 498.1 | 5 | 5–500 | 498.1 | 8 | 8–350 |
| V | 318.5 | 5 | 5–500 | 318.5 | 5 | 5–500 | 318.5 | 8 | 8–450 |
| W | 239.7 | 40 | 40–3500 | 239.7 | 40 | 40–4800 | 239.7 | 50 | 50–3500 |
| Zn | 213.8 | 5 | 5–450 | 213.8 | 5 | 5–490 | 213.8 | 5 | 5–400 |
Moreover, each replicate measurement corresponds to different arcs and hence different associated arc characteristics. The linear dynamic range is the region of concentration where the response varies linearly with the concentration and this is the only region where the determination of analytes is possible. Depending upon the nature of the matrix, it can differ. Sometimes visual changes in the arc can also be observed due to the presence of certain matrix elements. Different analytes can have either the same or different linear dynamic ranges. Therefore, for each matrix and each of the analytes, the linear dynamic range should be investigated prior to the analysis of actual samples. Si being one of the major matrices is unsuitable for D.C. arc carrier distillation AES, as the associated self-absorption can lead to underestimation. For comparison purposes, the EDXRF analysis for Si was also not taken into account.
Table 5 summarizes the analytical performance for the lines in the EDXRF spectra of U powder, Si powder and U3Si2 matrices. For most of the analytes, the Kα line was chosen. Though compared to D.C. arc carrier distillation the detection limits for the analytes are poorer in EDXRF, in some of the cases the linear dynamic range is wider for EDXRF. Moreover, the relative standard deviation in EDXRF was found to be better (less than 10%) compared to that in D.C. arc carrier distillation AES (25–30%).
| Elements | U powder | Si powder | U3Si2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Analytical line (nm) | Detection limit (ppm) | Linear dynamic range (ppm) | Analytical line (nm) | Detection limit (ppm) | Linear dynamic range (ppm) | Analytical line (nm) | Detection limit (ppm) | Linear dynamic range (ppm) | |
| a NA: not analyzed. | |||||||||
| Al | Al-Kα | 20 | 20–900 | Al-Kα | 25 | 25–900 | Al-Kα | 25 | 25–900 |
| Ca | Ca-Kα | 15 | 15–500 | Ca-Kα | 10 | 10–450 | Ca-Kα | 25 | 25–450 |
| Co | Co-Kα | 20 | 20–400 | Co-Kα | 25 | 25–450 | Co-Kα | 25 | 25–400 |
| Cr | Cr-Kα | 20 | 20–500 | Cr-Kα | 15 | 15–500 | Cr-Kα | 20 | 20–500 |
| Cu | Cu-Kα | 5 | 5–170 | Cu-Kα | 10 | 10–200 | Cu-Kα | 5 | 10–150 |
| Fe | Fe-Kα | 10 | 10–900 | Fe-Kα | 15 | 15–1000 | Fe-Kα | 20 | 20–900 |
| Mg | Mg-Kα | 25 | 25–400 | Mg-Kα | 15 | 15–500 | Mg-Kα | 25 | 25–400 |
| Mn | Mn-Kα | 20 | 20–200 | Mn-Kα | 20 | 20–200 | Mn-Kα | 25 | 25–200 |
| Mo | Mo-Lα | 25 | 25–1000 | Mo-Lα | 25 | 25–950 | Mo-Lα | 30 | 30–800 |
| Na | Na-Kα | 10 | 10–200 | Na-Kα | 20 | 20–200 | Na-Kα | 25 | 25–200 |
| Ni | Ni-Kα | 20 | 20–450 | Ni-Kα | 25 | 25–500 | Ni-Kα | 25 | 25–400 |
| Si | Si-Kα | 100 | 100–2000 | NA | NA | NA | Si-Kα | 100 | 100–2000 |
| Sn | Sn-Kα | 25 | 25–100 | Sn-Kα | 20 | 20–80 | Sn-Kα | 25 | 25–80 |
| Ti | Ti-Kα | 25 | 25–450 | Ti-Kα | 20 | 20–450 | Ti-Kα | 30 | 30–400 |
| V | V-Kα | 20 | 20–500 | V-Kα | 25 | 20–500 | V-Kα | 30 | 30–500 |
| W | W-Lβ | 50 | 50–4000 | W-Lβ | 50 | 50–3500 | W-Lβ | 30 | 50–3500 |
| Zn | Zn-Kα | 20 | 20–500 | Zn-Kα | 20 | 20–450 | Zn-Kα | 25 | 25–450 |
Based on this analytical performance, a set of synthetic samples were prepared having the analyte concentrations the same as those of standard 5 and they were labelled as synthetic sample 1. Table 6 summarizes the analytical results for synthetic samples by D.C. arc carrier distillation AES and the data are compared with EDXRF results. The results for the two techniques were found to be in good agreement, with the exception that the precision for D.C. arc carrier distillation AES was poorer compared to that for EDXRF.
| Elements | Synthetic sample 1 | |||||
|---|---|---|---|---|---|---|
| U powder | Si powder | U3Si2 | ||||
| AES (ppm) | XRF (ppm) | AES (ppm) | XRF (ppm) | AES (ppm) | XRF (ppm) | |
| a NA: not analyzed; uncertainty evaluated based on k = 1. | ||||||
| Al | 130 ± 25 | 110 ± 8 | 50 ± 8 | 60 ± 5 | 100 ± 20 | 90 ± 8 |
| Ca | 95 ± 10 | 102 ± 4 | 90 ± 15 | 96 ± 5 | 107 ± 15 | 96 ± 5 |
| Co | 102 ± 9 | 96 ± 4 | 90 ± 18 | 101 ± 4 | 98 ± 14 | 99 ± 5 |
| Cr | 104 ± 15 | 98 ± 5 | 96 ± 17 | 103 ± 5 | 110 ± 12 | 98 ± 4 |
| Cu | 38 ± 5 | 37 ± 2 | 45 ± 8 | 41 ± 2 | 39 ± 7 | 40 ± 2 |
| Fe | 192 ± 14 | 197 ± 8 | 220 ± 36 | 190 ± 10 | 210 ± 30 | 195 ± 9 |
| Mg | 105 ± 15 | 97 ± 5 | 89 ± 20 | 104 ± 5 | 95 ± 17 | 98 ± 5 |
| Mn | 45 ± 6 | 38 ± 2 | 46 ± 7 | 40 ± 2 | 38 ± 6 | 43 ± 3 |
| Mo | 190 ± 22 | 204 ± 7 | 180 ± 32 | 204 ± 8 | 210 ± 35 | 185 ± 10 |
| Na | 41 ± 7 | 36 ± 3 | 37 ± 5 | 44 ± 4 | 35 ± 6 | 40 ± 3 |
| Ni | 104 ± 15 | 95 ± 5 | 98 ± 16 | 96 ± 5 | 90 ± 12 | 103 ± 5 |
| Si | 804 ± 90 | 790 ± 25 | NA | NA | NA | NA |
| Sn | 18 ± 2 | 21 ± 1 | 23 ± 4 | 19 ± 1 | 20 ± 4 | 22 ± 2 |
| Ti | 205 ± 30 | 204 ± 8 | 188 ± 33 | 190 ± 10 | 215 ± 30 | 195 ± 10 |
| V | 95 ± 15 | 103 ± 4 | 110 ± 17 | 101 ± 4 | 97 ± 10 | 102 ± 5 |
| W | 770 ± 100 | 819 ± 30 | 820 ± 110 | 790 ± 40 | 750 ± 100 | 830 ± 37 |
| Zn | 180 ± 30 | 207 ± 9 | 220 ± 34 | 190 ± 10 | 185 ± 30 | 195 ± 9 |
Table 7 shows a comparative evaluation of trace metal assay of 16 analytes in a typical U3Si2 sample along with its precursors by AES and XRF. No Sn or Mo was found to be present either in the precursors or in the final product. Though no Ti was found in Si powder, ∼200 ppm of Ti was found in U powder and was found to carry forward in the final U3Si2 matrix. Cu was found to be present in U powder at the 40 ppm level, while almost twice that amount of Cu was present in Si powder. Due to this higher contribution in Si powder, the amount of Cu in U3Si2 was found to be ∼60 ppm. The contribution of Na in U3Si2 was found to be due to the presence of Na in U powder. However, the amount of Na in U3Si2 is to some extent higher. This may be attributed to the process pick-up during fabrication of the fuel. In precursors as well as in products, Ca, Co, Cr, Fe, Mg and Zn were found to be in the range of 100–200 ppm, while Mn, V and W were more than 200 ppm. In most of the cases the concentrations of analytes are in the same range as seen in precursors. This also indicates that there is almost no significant process pick-up either during the fabrication of fuel or during processing of the samples during analysis. It should also be noted that D.C. arc and EDXRF data were found to match well. In this regard it is also worth mentioning that B and Cd are two crucial analytes having very low specification limits in nuclear materials. For B and Cd, 249.6 nm and 228.8 nm analytical lines, respectively, were chosen by D.C. arc carrier distillation AES. For these 2 analytes the calibration curves were established in the range of 0.1–10 ppm. Keeping other parameters optimized, these two analytes were analyzed in a synthetic sample and an actual sample as well. The analysis of the synthetic sample was found to be satisfactory. In this typical case, 0.5 ppm, 0.6 ppm and 0.8 ppm B were found to be in U powder, Si powder and U3Si2, respectively, while 0.8 ppm, 0.7 ppm and 0.9 ppm Cd were present in U powder, Si powder and U3Si2, respectively. No B or Cd peaks were observed in EDXRF analysis of the samples. This was attributed to the fact that B is a light element and EDXRF is not sensitive at very low concentration (<10 ppm).
| Elements | U powder | Si powder | U3Si2 | |||
|---|---|---|---|---|---|---|
| AES (ppm) | XRF (ppm) | AES (ppm) | XRF (ppm) | AES (ppm) | XRF (ppm) | |
| a NA: not analyzed, BDL: below detection limit. | ||||||
| Al | 220 ± 40 | 230 ± 17 | 70 ± 8 | 80 ± 7 | 180 ± 30 | 200 ± 20 |
| Ca | 120 ± 10 | 116 ± 5 | 130 ± 20 | 146 ± 7 | 140 ± 20 | 126 ± 6 |
| Co | 109 ± 7 | 96 ± 5 | 180 ± 30 | 171 ± 9 | 180 ± 30 | 199 ± 9 |
| Cr | 200 ± 25 | 193 ± 9 | 100 ± 17 | 103 ± 4 | 140 ± 10 | 138 ± 4 |
| Cu | 40 ± 7 | 37 ± 2 | 90 ± 10 | 91 ± 4 | 65 ± 9 | 58 ± 2 |
| Fe | 170 ± 20 | 167 ± 9 | 190 ± 32 | 170 ± 8 | 170 ± 30 | 185 ± 9 |
| Mg | 180 ± 22 | 197 ± 10 | 100 ± 22 | 104 ± 5 | 190 ± 25 | 208 ± 10 |
| Mn | 220 ± 30 | 238 ± 10 | 185 ± 27 | 170 ± 8 | 210 ± 30 | 193 ± 10 |
| Mo | BDL | BDL | BDL | BDL | BDL | BDL |
| Na | 60 ± 9 | 73 ± 3 | BDL | BDL | 90 ± 11 | 80 ± 4 |
| Ni | 230 ± 30 | 245 ± 15 | 110 ± 21 | 96 ± 5 | 290 ± 40 | 303 ± 15 |
| Si | 120 ± 20 | 110 ± 5 | NA | NA | NA | NA |
| Sn | BDL | BDL | BDL | BDL | BDL | BDL |
| Ti | 200 ± 25 | 204 ± 10 | BDL | BDL | 215 ± 30 | 200 ± 10 |
| V | 260 ± 30 | 243 ± 14 | 110 ± 15 | 108 ± 4 | 265 ± 30 | 272 ± 10 |
| W | 200 ± 25 | 189 ± 10 | 120 ± 20 | 99 ± 5 | 220 ± 30 | 230 ± 10 |
| Zn | 105 ± 10 | 107 ± 9 | 155 ± 30 | 140 ± 7 | 140 ± 20 | 155 ± 9 |
The accuracy and precision can be combined in order to determine the standard uncertainties or method uncertainties for the trace metallic impurities using certified reference materials. The combined standard uncertainty (u) can be expressed as29
 | (1) |
| U = ku | (2) |
 | (3) |
Using the optimized methodology, 10 U3Si2 samples were analyzed for these analytes by both the techniques. Fig. 4 shows the spread of different analytes in those U3Si2 samples. The ranges of concentration for theses analytes were found to be as follows: Ca 100–150 ppm, Co 170–270 ppm, Cr 125–180 ppm, Cu 30–75 ppm, Fe 100–175 ppm, Mg 200–250 ppm, Mn 150–300 ppm, Mo less than 50 ppm, Na 75–150 ppm, Ni 250–350 ppm, Ti 120–180 ppm, Sn less than 5 ppm, V 225–300 ppm, Zn 110–150 ppm and W 175–225 ppm. Based on specification limits, these four fuel samples can be sent to the reactor for their use. The overall precision for D.C. arc carrier distillation was within 20%, while that for EDXRF was found to be less than 6%.
 | ||
| Fig. 4 Concentration ranges for the analytes in ten U3Si2 samples. | ||
Table 8 summarizes the D.C. arc carrier distillation results for the analytes B, Be and Cd along with their analytical performance in a U3Si2 matrix. Though these elements were able to be analyzed by the D.C. arc carrier distillation methodology, EDXRF analysis was found to be unsuccessful. However, these analytes are very critical due to their very high neutron absorption cross-section. Hence, their tolerance limits in an actual fuel sample are less than 5 ppm, which cannot be achieved by EDXRF. The most suitable analytical lines for B, Be and Cd were found to be 249.7 nm, 234.8 nm and 228.8 nm, respectively. In D.C. arc AES, their detection limits were found to be 0.1–0.2 ppm. The method was validated using a synthetic sample. Furthermore, five real life samples (S1, S2, S3, S4 and S5) were analyzed. Four of them have concentrations below 0.4 ppm and hence can be accepted as a fuel; however, for S4, the concentrations of the critical analytes are too high for it to be considered as a nuclear grade fuel material.
| Elements | B | Be | Cd |
| Analytical lines (nm) | 249.7 | 234.8 | 228.8 |
| Detection limit (ppm) | 0.15 | 0.2 | 0.1 |
| Linear dynamic range (ppm) | 0.15–5 | 0.2–5 | 0.1–4.5 |
| Synthetic sample | |||
| Amount added (ppm) | 0.3 | 0.7 | 0.25 |
| Amount estimated (ppm) | 0.28 ± 0.05 | 0.73 ± 0.10 | 0.21 ± 0.05 |
| Actual samples | |||
| S1 (ppm) | 0.17 ± 0.02 | 0.21 ± 0.02 | 0.12 ± 0.01 |
| S2 (ppm) | 0.3 ± 0.03 | 0.36 ± 0.08 | 0.15 ± 0.02 |
| S3 (ppm) | 0.16 ± 0.01 | 0.3 ± 0.07 | 0.1 7 ± 0.03 |
| S4 (ppm) | 4.0 ± 0.7 | 3.3 ± 0.6 | 2.4 ± 0.3 |
| S5 (ppm) | 0.19 ± 0.02 | 0.22 ± 0.04 | 0.26 ± 0.04 |
The advantages of the D.C. arc carrier distillation based methodology over the EDXRF based methodology are as follows.
• The detection limits for any analytes are poorer in EDXRF compared to the D.C. arc carrier distillation technique. Elements like Ca, Co, Cr, Cu, Mg, Na, Ni, V, Mn, and Zn have detection limits below 10 ppm for D.C. arc AES, while for EDXRF they are more than or equal to 10 ppm. So any U3Si2 samples having any of these analytes at concentrations below 10 ppm cannot be analyzed by EDXRF. Though the tolerance level for these analytes in the U based sample matrix is well above 10 ppm, there may be some samples where any of these analytes can be present at concentrations less than 10 ppm. Thus, D.C. arc AES is a better choice.
• For analytes like B and Be, the tolerance limits are less than 2.5 ppm. Therefore, for these analytes EDXRF is not applicable. However, the determination of these analytes is very critical due to their very high neutron absorption cross-section.
• Though Cd can be determined by EDXRF, the specification limit in nuclear materials for Cd is less than 3 ppm from the neutron economy point of view. Hence, EDXRF is also not useful for this critical analyte.
Additionally, D.C. arc AES is not only a simultaneous multi-element method facilitating the determination of 20 analytes (Al, B, Be, Ca, Cd, Cu, Cr, Co, Fe, Mn, Mo, Sn, Si, Ti, U, V, W, Ni, Na and Mg) at a stretch in a U3Si2 matrix, it also has the following advantages that are worth mentioning.
• Compared to other techniques like ICP-AES or ICP-MS, where samples are analyzed in solution form and not in solid form, dissolution is a time-consuming and cumbersome step required for analysis. Though U metal can easily be dissolved in HNO3 medium (3–4 M and above), dissolution of Si samples and U3Si2 samples is very difficult.31–36 Alkali dissolution is mostly effective. However, such sample solution in alkali medium is not recommended for the instrumental parts including the ICP torch and tubing.
• To avoid interference, suitable ligands are required for preferential separation of the major matrix without any loss of these analytes even at a trace level. Finding such an efficient ligand is very difficult. There is a high probability of other analytes being co-extracted with the major matrix at a ppm level, which can lead to underestimation.
• Determination of lighter elements like B and Be in a heavier matrix like U at less than 1 ppm level is not practically possible by MS.
• Since the U used in U3Si2 samples is enriched, the radiotoxic hazard associated with such sample handling also needs to be monitored. More steps in sample processing lead to more personal exposure.
• More steps and more added reagent lead to a greater chance of process pick-up or contamination.
• Separation of the major matrix by solvent extraction will generate lots of radiotoxic organic waste, which is an additional burden to be managed after analysis.
Conclusions
A D.C. arc carrier distillation based AES spectrometric technique has been developed for the determination of Al, Ca, Co, Cr, Cu, Fe, Mg, Mn, Mo, Na, Ni, Sn, Si, Ti, V, W and Zn at a trace level in U3Si2, U powder and Si powder. The total chemical quality control of the final product as well as the precursor is important in order to determine the sources of impurities. 5% AgCl was used for physical separation of these analytes from the emission-rich major matrix to minimize spectral interference. The method was validated using synthetic samples. A comparative evaluation of the chemical quality control was also carried out using the EDXRF technique for UO2, Si powder and U3Si2 along with an evaluation of the analytical performances. Though D.C. arc carrier distillation has poorer precision compared to EDXRF, EDXRF is not useful for determination of low Z elements including B and analytes at low concentration (less than 10 ppm).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge Dr P. K. Pujari, AD & Head, Radiochemistry Division, and Dr Nimai Pathak, Radiochemistry Division, Bhabha Atomic Research Centre, Mumbai, India.References
- V. P. Sinha, G. P. Mishra, S. Pal, K. B. Khan, P. V. Hegde and G. J. Prasad, J. Nucl. Mater., 2008, 383, 196–200 CrossRef CAS.
- S. Nazaré, J. Nucl. Mater., 1984, 124, 14 CrossRef.
- G. L. Hofman, J. Nucl. Mater., 1986, 140, 256 CrossRef CAS.
- M. R. Finlay, G. L. Hofman and J. L. Snelgrove, J. Nucl. Mater., 2004, 325, 118 CrossRef CAS.
- R. Chang-Kyu, P. Su-II and K. II-Hiun, J. Nucl. Mater., 1991, 184, 161 CrossRef.
- J. Marín, J. Lisboa, J. Ureta, L. Olivares, H. Contreras and J. C. Chávez, J. Nucl. Mater., 1996, 228, 61 CrossRef.
- J. Rosales, I. J. van Rooyen and C. J. Parga, J. Nucl. Mater., 2019, 518, 117 CrossRef CAS.
- T. Barani, G. Pastore, D. Pizzocri, D. A. Andersson, C. Matthews, A. Alfonsi, K. A. Gamble, P. Van Uffelen, L. Luzzi and J. D. Hales, J. Nucl. Mater., 2019, 522, 97 CrossRef CAS.
- R. E. Hoggan, K. R. Tolman, F. Cappia, A. R. Wagner and J. M. Harp, J. Nucl. Mater., 2018, 512, 199 CrossRef CAS.
- E. Jossou, Md J. Rahman, D. Oladimeji, B. Beeler, B. Szpunar and J. Szpunar, J. Nucl. Mater., 2019, 521, 1 CrossRef CAS.
- C.-F. Wang, T. T. Miau, J. Y. Perng, S. J. Yeh, P. C. Chiang, H. T. Tsai and M. H. Yang, Analyst, 1989, 114, 1067 RSC.
- A. Sengupta, V. C. Adya, M. Kumar, S. K. Thulasidas, S. V. Godbole and V. K. Manchanda, At. Spectrosc., 2011, 32(2), 49 CAS.
- A. Sengupta, B. Rajeswari, R. M. Kadam and R. Acharya, At. Spectrosc., 2011, 32(5), 200 CAS.
- A. Sengupta, M. J. Kulkarni and S. V. Godbole, J. Radioanal. Nucl. Chem., 2011, 289(3), 961 CrossRef CAS.
- A. Sengupta, B. Rajeswari, R. M. Kadam and S. V. Godbole, At. Spectrosc., 2012, 33(2), 48 CAS.
- R. H. Scott and A. Strasheim, Anal. Chim. Acta, 1976, 82, 67 CrossRef CAS.
- N. Pathak, V. C. Adya, S. K. Thulasidas, A. Sengupta, T. K. Seshagiri and S. V. Godbole, At. Spectrosc., 2014, 35(1), 17 CAS.
- N. K. Porwal, A. A. Argekar, P. J. Purohit, A. G. Page and M. D. Sastry, Fresenius' Z. Anal. Chem., 1990, 338, 255 CrossRef CAS.
- A. Sengupta, V. C. Adya, T. K. Seshagiri, S. K. Thulasidas, S. V. Godbole and V. Natarajan, J Res Spectrosc, 2014 DOI:10.5171/2014.757111.
- A. G. Dalvi, C. S. Deodhar, T. K. Seshagiri, M. S. Khalap and B. D. Joshi, Talanta, 1978, 25(11–12), 665 CrossRef CAS.
- A. Sengupta, Y. Airan, S. K. Thulasidas and V. Natarajan, At. Spectrosc., 2015, 36(2), 82 CAS.
- Y. Airan, A. Sengupta, S. K. Thulasidas and V. Natarajan, At. Spectrosc., 2015, 36(1), 30 CAS.
- Y. Airan, A. Sengupta, S. K. Thulasidas and V. Natarajan, At. Spectrosc., 2015, 36(1), 15 CAS.
- M. L. Carvalho, T. Magalhães, M. Becker and A. von Bohlen, Spectrochim. Acta, 2007, 62, 1004 CrossRef.
- E. I. Obiajunwa, D. A. Pelemo, S. A. Owolabi, M. K. Fasasi and F. O. Johnson-Fatokun, Nucl. Instrum. Methods Phys. Res., 2002, 194, 61 CrossRef CAS.
- J. L. Ferrero, C. Roldn a, D. Juanes a, J. Carballo a, J. Pereira a, M. Ardid a, J. L. Lluch a and R. Vives, Nucl. Instrum. Methods Phys. Res., 2004, 213, 729 CrossRef CAS.
- B. F. Scribner and R. M. Harold, J. Res. Natl. Bur. Stand., 1946, 37, 379 CrossRef CAS.
- A. G. Page, K. H. Madraswala, S. V. Godbole, M. J. Kulkarni, V. S. Mallapurkar and B. D. Joshi, Fresenius' Z. Anal. Chem., 1983, 315(1), 38 CrossRef CAS.
- M. Schappert, D. Montoya, S. Aragon, M. Rearick, N. Xu and K. J. Mathew, J. Radioanal. Nucl. Chem., 2018, 318, 323–330 CrossRef CAS.
- S. Bürger, K. J. Mathew, P. Mason and U. Narayanan, J. Radioanal. Nucl. Chem., 2009, 279, 659–673 CrossRef.
- R. B. Georg, B. C. Reynolds, M. Frank and A. N. Halliday, Chem. Geol., 2006, 235, 95–104 CrossRef CAS.
- C. L. De La Rocha and M. A. Brzezinski, Anal. Chem., 1996, 68(21), 3746–3750 CrossRef CAS.
- T. Ding, D. Wan, C. Wang and F. Zhang, Geochim. Cosmochim. Acta, 2004, 68(2), 205–216 CrossRef CAS.
- https://inis.iaea.org/collection/NCLCollectionStore/_Public/35/015/35015774.pdf?r=1%26r=1 .
- https://www.osti.gov/servlets/purl/10119587#page=441 .
- https://inis.iaea.org/search/search.aspx?orig_q=RN:14791572 .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ja00321e |
| This journal is © The Royal Society of Chemistry 2020 |