
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis of aldoximes from alkenes via Rh-catalysed hydroformylation in an aqueous solvent system†
M.
Terhorst
 a,
C.
Plass
b,
A.
Hinzmann
b,
A.
Guntermann
b,
T.
Jolmes
a,
J.
Rösler
a,
D.
Panke
a,
H.
Gröger
b,
D.
Vogt
a,
A. J.
Vorholt
c and
T.
Seidensticker
*a
a,
C.
Plass
b,
A.
Hinzmann
b,
A.
Guntermann
b,
T.
Jolmes
a,
J.
Rösler
a,
D.
Panke
a,
H.
Gröger
b,
D.
Vogt
a,
A. J.
Vorholt
c and
T.
Seidensticker
*a
aTU Dortmund University, Department for Biochemical and Chemical Engineering, Laboratory of Industrial Chemistry, Emil-Figge-Straße 66, 44227 Dortmund, Germany. E-mail: thomas.seidensticker@tu-dortmund.de; Tel: + 49 231 7552310 Web: http://www.tc.bci.tu-dortmund.de
bBielefeld University, Faculty of Chemistry, Chair of Industrial Organic Chemistry and Biotechnology, Universitätsstraße 25, 33615 Bielefeld, Germany
cMPI for Chemical Energy Conversion, Department of Molecular Catalysis, Group Multiphase Catalysis, Stiftstrasse 34-36, 45470 Mülheim an der Ruhr, Germany
First published on 28th October 2020
Abstract
Aldoxime synthesis directly starting from alkenes was successfully achieved through the combination of hydroformylation and subsequent condensation of the aldehyde intermediate with aqueous hydroxylamine in a one-pot process. The metal complex Rh(acac)(CO)2 and the water-soluble ligand sulfoxantphos were used as the catalyst system, providing high regioselectivities in the initial hydroformylation. A mixture of water and 1-butanol was used as an environmentally benign solvent system, ensuring sufficient contact of the aqueous catalyst phase and the organic substrate phase. The reaction conditions were systematically optimised by Design of Experiments (DoE) using 1-octene as a model substrate. A yield of 85% of the desired linear, terminal aldoxime ((E/Z)-nonanal oxime) at 95% regioselectivity was achieved. Other terminal alkenes were also converted successfully under the optimised conditions to the corresponding linear aldoximes, including renewable substrates. Differences of the reaction rate have been investigated by recording the gas consumption, whereby turnover frequencies (TOFs) >2000 h−1 were observed for 4-vinylcyclohexene and styrene, respectively. The high potential of aldoximes as platform intermediates was shown by their subsequent transformation into the corresponding linear nitriles using aldoxime dehydratases as biocatalysts. The overall reaction sequence thus allows for a straightforward synthesis of linear nitriles from alkenes with water being the only by-product, which formally represents an anti-Markovnikov hydrocyanation of readily available 1-alkenes.
Introduction
The direct formation of C–N bonds starting from non-functionalised base chemicals is still a challenging task. The involved reactions are often lacking selectivity or by- as well as side-products are formed whereby unnecessary waste is generated. In this regard, homogeneous catalysis can help to reduce energy costs and waste formation, yet it remains challenging to produce primary aliphatic amines as value products.1,2 One way of increasing selectivity is the initial synthesis of oxygen-containing platform chemicals from base chemicals and the subsequent substitution of oxygen in C–O bonds with nitrogen, as performed for instance in catalytic reactions, like alcohol amination or reductive amination.1 Both types of reaction are based on the condensation of carbonyl compounds with either ammonia, primary or secondary amines. The reactive site is thus predetermined, resulting in excellent regioselectivity. However, chemoselectivity remains a challenge, since, in both reactions, the selective synthesis of primary or secondary amines is complicated by multiple substitutions at the nitrogen centre (Fig. 1). | ||
| Fig. 1 Comparison of reductive amination, alcohol amination, hydroaminomethylation and the objective of this work concerning consecutive reactions. | ||
A potent, but yet underrepresented class of intermediates on the way towards nitrogen-containing platform chemicals are oximes. The C–N bond formation is performed employing a simple condensation reaction between hydroxylamine and a carbonyl compound. With this, high regioselectivity is maintained as well as multiple substitutions at the nitrogen centre are avoided, potentially resulting in excellent chemoselectivity. Moreover, oximes are incredibly versatile in their follow-up chemistry, since, for instance, amines or amides are accessible by hydrogenation3 and rearrangement,4 respectively (Fig. 1). Recently, some of us have shown the very high potential of aldoximes as precursors for the corresponding nitriles via biocatalysis using aldoxime dehydratases with unprecedented selectivities under very mild conditions.5
Aldoximes are typically produced by reacting hydroxylamine (or a corresponding precursor) with the corresponding aldehyde.4,6 Consequently, non-functionalised base chemicals initially have to be converted to aldehydes, which are then typically isolated and purified prior to the subsequent reaction, such as aldoxime formation. Hydroformylation is the state-of-the-art technology for atom-efficient synthesis of aldehydes by reacting readily available alkenes with syngas (CO/H2) to aldehydes employing homogeneous Rh- or Co-catalysts.7
A compelling approach to intensify (catalytic) syntheses and thereby increase sustainability is the merger of synthetic steps, elegantly enabled by in situ formation of one of the reactive intermediates.8 The combination of several reaction steps without purifications of intermediate products has the potential to drastically reduce the number of unit operations and thereby decrease energy consumption and waste generation, while at the same time feedstock utilisation is increased. Homogeneous transition metal-catalysed hydroformylation has proven its ability in many examples to be linked to numerous follow-up reactions, including the formation of C–N-bonds.9 Most importantly, this is due to the rich follow-up chemistry of aldehydes and the ability of homogeneous Rh-complexes to catalyse many different reactions, including hydrogenations, for instance.
By combining hydroformylation with subsequent aldoxime formation, C–N bond formation from unfunctionalised alkenes in a one-pot fashion would be possible, similar to a trifluoromethyloximation.10 The, to the presented work, related hydroaminomethylation (HAM), which is the tandem-catalytic combination of hydroformylation with reductive amination in the presence of respective amines, also forms C–N linkages from alkenes in a single preparative step.11 In comparison to HAM, hydroformylation/aldoxime formation may open broader follow-up chemistry. Nitriles, primary amines and amides are readily accessible from aldoximes through dehydration,5,12 hydrogenation3,13 and Beckmann-rearrangement,14 respectively. Furthermore, HAM with ammonia and primary amines typically produces mixtures of primary, secondary and tertiary amines and neither amides nor nitriles can be synthesised by HAM.
To the best of our knowledge, hydroformylation has, however, never been combined with subsequent aldoxime formation in a one-pot approach. This might be because, in conventional organic syntheses, hydroxylamine is applied as a salt, e.g. chloride or hydroxylammonium sulfate, and thus requires activation by bases.4,6 The latter may harm the catalytic hydroformylation and lead to undesired consecutive reactions of the intermediate aldehyde. In addition, the application of the hydrochloride is disadvantageous in terms of sustainability and atom-efficiency, since stoichiometric amounts of salt are formed as waste, which may also cause issues with corrosion of the autoclave. Aqueous hydroxylamine solution, which is also commercially available, thus seems superior for producing (ald)oximes in a more sustainable and atom efficient manner.
As we have gained experience in both, aqueous biphasic hydroformylation15,16 and in combining two or more catalytic transformations in a tandem (catalytic) system,17 we now strive for the connection of hydroformylation and oxime formation in a one-pot approach (Scheme 1) using aqueous hydroxylamine. To achieve sufficient mixing and to ensure contact of the organic and the aqueous phase, the use of short-chain alcohols proved to be effective.15,18,19 To provide high regioselectivity towards the commonly more valuable linear product together with effective immobilisation of the catalyst in the aqueous phase, sulfoxantphos appears to be a viable option as a ligand. The immobilisation possibly enables spontaneous separation of the two phases after the reaction and, thus, simple isolation of the aldoxime seems feasible. 1-Octene was chosen as the model substrate for this one-pot reaction (Scheme 1), as it was used in a similar investigation of our groups for the synthesis of nonanitrile and 2-methyl octanenitrile.20
 | ||
| Scheme 1 One-pot approach for the synthesis of aldoximes from alkenes by combining hydroformylation with the condensation of the intermediate aldehyde with hydroxylamine and possible consecutive reactions of the aldoxime. | ||
Results and discussion
Initial investigations
We first checked whether both reaction steps could be carried out independently from each other to achieve the general objective of combining two reaction steps in a one-pot reaction. Thus, we studied, if the condensation of the aldehyde and hydroxylamine in the green solvent system21 water/1-butanol is practicable. Therefore, we applied 1.5 eq. aqueous hydroxylamine solution, based on previous investigations on oxime formation,3,22 and 1 eq. of nonanal. Within 20 min, the oxime was obtained in nearly quantitative yield, confirming that even in the presence of water condensation takes place almost instantaneously (cf. ESI†).Next, hydroformylation of 1-octene in water/1-butanol solvent was performed as an individual reaction (without the addition of NH2OH (aq.)). Towards this end, reaction conditions developed for the hydroformylation of methyl 10-undecenoate were applied.19 Within 3 h, a yield of 80% of the linear nonanal was achieved. This proofs that both reactions, hydroformylation and aldoxime formation, can be carried out independently in the chosen green solvent mixture.
It was expected that besides the typical side-products of hydroformylation (hydrogenated substrate, isomerised substrate, aldol condensates), also products from consecutive reactions of the oxime group (nitriles, amides, amines) would be observed. Besides, hydroxylamine might unfavourably affect the regioselectivity in the hydroformylation step. Despite all these concerns, the initial one-pot reaction already showed a promising yield of the linear oxime of 20% after 2 h by simply adding 1.5 eq. NH2OH (aq.) (for further information, see ESI†). This yield is significantly lower than the yield in the hydroformylation reaction without addition of hydroxylamine carried out before (cf. ESI,† 80%). However, it is still very promising, taking into account that the conditions were not yet optimised for the intended one-pot reaction. All side-products observed in this initial one-pot reaction were analysed and quantified and revealed the overall reaction network (Scheme 2).
 | ||
| Scheme 2 Reaction network of the one-pot hydroformylation/aldoxime formation of 1-octene (1a) for nonanal oxime (3a) formation including all observed by-products. | ||
Besides the linear and branched hydroformylation products nonanal (2a) and 2-methyl octanal (4a), respectively, all typical side-products of the hydroformylation of 1-octene (1a), i.e. octene isomers (7a), octane (6a) and aldol products (8a), were observed. It is noteworthy that the hydroxylamine shows no influence on the regioselectivity of carbonylation, even under these non-optimised conditions. The ratio of linear to branched products (aldoximes 3a + 5a and aldehydes 2a + 4a) is 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, comparable to the ratio reported in the literature for sulfoxantphos in hydroformylation reactions.23 Additionally, small amounts of products from consecutive reactions of nonanal oxime (3a) were identified, namely nonanitrile (10a) and nonanamide (11a).
:5, comparable to the ratio reported in the literature for sulfoxantphos in hydroformylation reactions.23 Additionally, small amounts of products from consecutive reactions of nonanal oxime (3a) were identified, namely nonanitrile (10a) and nonanamide (11a).
Optimisation via design of experiments (DoE)
The complexity of the reaction network shows that optimisation is necessary to guide the reaction towards the desired linear nonanal oxime 3a. Hence, we carried out a Design of Experiments (DoE) to determine critical parameters, influencing the rate and selectivity of the reaction (Table 1).| Parameter | Lowest value | Middle value | Highest value | Optimised value |
|---|---|---|---|---|
| Constant parameters: n1-octene = 6.7 mmol, mwater = 1.8 g, m1-butanol = 1.8 g, t = 1.5 h. | ||||
| Temperature [°C] | 90 | 120 | 150 | 100 |
| Pressure [bar] | 20 | 40 | 60 | 60 |
| CO/H2 ratio [−] | 1/2 | 1/1 | 2/1 | 1/1 |
| Hydroxylamine/1-octene ratio [−] | 1 | 2 | 3 | 2.5 |
| Rhodium/sulfoxant-phos ratio [−] | 1/1 | 1/5 | 1/10 | 1/5 |
| Rhodium loading (based on 1-octene) [mol%] | 0.05 | 0.275 | 0.5 | 0.5 |
We herein focused on parameters known to cause significant changes in the catalytic performance of the hydroformylation step,24 since both regioselectivity and rate of the desired reaction sequence depend on this step.
Furthermore, the amount of hydroxylamine used was varied because it remains to be seen whether hydroxylamine has a significant influence on the catalysis in terms of activity and selectivity. The mass of water and 1-butanol (both 1.8 g) and the amount of 1-octene (6.7 mmol) was kept constant throughout this investigation. A central composite face-centred (CCF) model was chosen, resulting in 68 reactions, that were each carried out twice (centre point 5 times), giving a total count of 141 reactions.
All reactions were carried out for 1.5 h, allowing us to identify significant changes in the reaction rate and selectivity at medium conversion. After careful evaluation of the results (cf. ESI†), it became apparent that low temperatures favour the chemoselectivity of the reaction by reducing the isomerisation. High pressures affect the regioselectivity as well as the yield of the reaction to a reasonable extent. A possible explanation could be the increased availability of the gases in the liquid phase.
To validate the results of the optimisation, we performed a reaction on a small scale (25 mL) over 4 h under these conditions. We observed a 95% conversion of 1-octene (1a) with a yield of the desired nonanal oxime (3a) of 82%. Regioselectivity towards the linear products was 95%, and chemoselectivity (yield of oximes 3a + 5a) for the one-pot hydroformylation/aldoxime formation was 88%. Side reactions, like aldol condensation (Y8a = 3%), hydrogenation (Y6a = 2%) or isomerisation (Y7a = 7%) of 1-octene, as well as consecutive reactions of the oxime (Y9a = < 1%, Y10a = < 1%) were effectively suppressed or reduced. We assumed, based on the initial results, that the hydroformylation is the rate-determining step of the overall reaction. The reaction was scaled-up to a 350 mL autoclave, equipped with a gas-impeller stirrer and baffles to validate this hypothesis, (cf. ESI†).
The reaction profile (Fig. 2) reveals that the condensation takes place immediately after the aldehyde is formed since no aldehyde intermediates were detectable. Aldol condensates (8a) and nonanamide (10a) were only formed in traces (<0.5%). After 3 h reaction time, no significant changes in the yields of the products occur. The conversion at this point is at 95% with a yield of the linear nonanal oxime 3a of 87%. Excellent chemoselectivity towards the desired one-pot reaction of 95% was achieved. Consequently, a scale-up of the reaction is possible without losses in selectivity or reaction rate. It is noteworthy that the baffles and gas-impeller stirrer used did not positively affect the rate of the reaction.
 | ||
| Fig. 2 Reaction profile of the one-pot hydroformylation/aldoxime formation of 1-octene (1a) for nonanal oxime (3a) formation after optimisation via DoE. Conditions: Rh(acac)(CO)2 (0.5 mol%), sulfoxantphos (2.5 mol%), 134 mmol 1-octene, 335 mmol hydroxylamine (2.5 eq., as 50 w% aqueous solution), cRh,aq = 0.015 mol L−1, cligand,aq = 0.074 mol L−1, T = 100 °C, p = 60 bar, Vtotal = 110 mL. aConversion/yield determined via GC-FID using n-decane as internal standard. | ||
Small scale substrate variation
By application of other 1-alkenes of different chain lengths (1-hexene (1b), 1-decene (1c) and 1-dodecene (1d)) under the optimised reaction conditions in small scale (cf. ESI†), the yields decrease with increasing chain length. This fact was expected, considering that the reaction rates depend on the solubility of the substrate in the polar aqueous phase. Internal alkenes such as cyclooctene (1e) and 4-octene (1f) were not converted with reasonable rates as expected for the Rh/sulfoxantphos catalyst system. The substitution of sulfoxantphos with TPPTS (Tri(sodium-3-sulfonatophenyl)phosphane) might enhance the hydroformylation of internal alkenes.15 The application of TPPTS, however, increases the polarity of the aqueous phase (large excess of the monodentate, trisulfonated ligand to be applied for reasonable regioselectivity and to avoid Rh-losses into the organic phase). Neither cyclooctene (1e) nor 4-octene (1f) were converted successfully (cf. ESI†). Presumably, this is caused by the reduced solubility of the alkene in the aqueous phase due to its increased polarity.On the contrary, the non-sulfonated TPP (triphenyl phosphane) catalyses the one-pot hydroformylation/aldoxime formation effectively (cf. ESI†), since the reaction takes place in the organic phase without mass transfer or solubility limitations. However, after the reaction, the catalyst and the product are both in the organic product phase. Thus, separation of the catalyst from the aldoxime for straightforward isolation is hampered.
The chemoselectivity of the Rh/sulfoxantphos system for terminal double bonds was used to selectively convert only the terminal double bond of 4-vinylcyclohexene (1g) to the linear oxime in 82% yield on a small scale (cf. ESI†). Styrene (1h) also gave high yields of the aldoximes in small scale, with the expected regioselectivity of linear and iso-oximes (34% linear, 53% iso) that was earlier observed for the parent ligand xantphos.25 The iso-oxime 5h is the main product, but the selectivities indicate that sulfoxantphos can shift the regioselectivity towards the linear hydroformylation product. The selectivity towards the linear product 3h was improved from 4% with TPP26 to 34% with sulfoxantphos.
Further extending the scope, substrates with oxygen-containing functional groups were tested. Difunctional products as potential polymer precursors are thus accessible. Depending on the consecutive chemistry of the oxime group, even different classes of polymers seem possible. In this regard, functionalised molecules based on renewables, like the terpenol dihydromyrcenol (1i) and the oleochemical methyl 10-undecenoate (1k) are of interest since they are already produced on a large scale.27 Dihydromyrcenol, containing a tertiary alcohol function, yielded 68% of the linear oxime, which is comparable to the results achieved with 1-decene. The reaction well tolerates the tertiary alcohol function. For methyl 10-undecenoate, a yield of 41% to the linear oxime was observed. Herein, consecutive reactions of the oxime group to the corresponding nitrile in 34% yield and the amide in 12% yield were observed, resulting in a cumulated yield of 87%, concerning the hydroformylation step. Hence, the carboxylic acid ester group is well tolerated; however, apparently favours the formation of consecutive products. Eugenol (1j) can be used as pharmaceutical active compound28 and modification with nitrogen-containing functional groups may improve its effectiveness. Surprisingly, a yield of the linear oxime of only 15% was obtained. Again, consecutive reactions to the corresponding nitrile and amide were observed in a yield of 41% and 27%, respectively. The combined hydroformylation yield (3j + 5j + 9j + 10j) is, as with the other functionalised substrates, still high at 83%.
Consequently, functional groups are well tolerated by hydroformylation. However, the reaction conditions developed for 1-octene do not necessarily provide the best results for all applied substrates. Nevertheless, the increased formation of nitrile or amide shows high potential to integrate a subsequent reaction step into the designed reaction sequence.
Large scale substrate variation (monitoring of gas consumption)
Reaction profiles were recorded by monitoring the gas consumption in a 350 mL autoclave to get a more detailed insight into the different reactivities and to minimise the possibility for consecutive reactions (see ESI† for details on the experimental setup). As soon as no significant gas consumption was detected, the reaction was stopped to minimise aldoxime conversion to nitriles and amides. After the reaction, the organic phase was separated from the aqueous catalyst phase by decantation and analysed via GC-FID. The gas consumption in the gas reservoir was then normalised to the summarised yield of all hydroformylation products (2 + 3 + 4 + 5 + 8 + 9 + 10). The turnover frequency (TOF) at 20% conversion towards hydroformylation products was determined (TOF20) to compare the reaction rate of the various substrates. In Fig. 3, the gas consumption curves of 1-octene (1a), 1-hexene (1b), 1-decene (1c), 1-dodecene (1d), 4-vinylcyclohexene (1g), styrene (1h), dihydromyrcenol (1i), eugenol (1j) and methyl-10-undecenoate (1k) are shown. As expected, the reaction rate decreases with increasing chain lengths of the different 1-alkenes. The maximum TOF20 was observed for 1-hexene (1518 h−1), while for 1-dodecene TOF20 was only half as high (855 h−1). The time to reach 90% yield of hydroformylation products is 1 h for 1-hexene and 1-octene, whereas 1-decene and 1-dodecene require reaction times above 2 h and 3 h, respectively. The yield of the linear oxime exceeds 80% only for 1-octene. This finding is a result of the optimisation by DoE, since conditions were optimised only for this specific substrate. Hence, the reaction conditions are not in the optimal range for other substrates and consecutive reactions of the oxime group, hydrogenation and isomerisation are more likely to happen. Longer reaction time and lower reaction rates in case of 1-decene and 1-dodecene also contribute to this, since it increases the probability of consecutive reactions and side-reactions. We now assume that the consecutive and side reactions take place simultaneously to the hydroformylation/aldoxime formation. Another factor in support of this assumption is that side products were observed to a much smaller extend for 4-vinylcyclohexene (1f) and styrene (1g) as substrates (Fig. 3, middle curves). The TOF20 exceeds 2000 h−1 in both cases, and the reaction time required to reach 90% yield of hydroformylation products is <1 h. | ||
| Fig. 3 Conversion of selected substrates in the combined hydroformylation/aldoxime formation one-pot-reaction, evaluated by gas consumption curves. Conditions: Rh(acac)(CO)2 (0.5 mol%), sulfoxantphos (2.5 mol%), 134 mmol substrate, 335 mmol hydroxylamine (2.5 eq., as 50 w% aqueous solution), cRh,aq = 0.015 mol L−1, cligand,aq = 0.074 mol L−1, T = 100 °C, pconst. = 60 bar, Vtotal = 105–120 mL. aCombined yield of all hydroformylations products measured via GC-FID using decane as internal standard after reaction normalised to the measured gas consumption. | ||
The consumption curves for dihydromyrcenol (1h) and eugenol (1i) differ from the previously obtained results. At first, a high conversion rate towards the hydroformylation products is observed, changing into an almost linear progression after 1 h. A further flattening of the consumption curves after 2.5 h was observed in both cases. The TOF20 for dihydromyrcenol (1179 h−1) is slightly higher than the TOF20 for 1-decene (1041 h−1). The reaction rate for the conversion of eugenol is the highest of all three oxygen functionalised substrates, which may be attributed to the comparatively good water solubility (2.46 g Laq−1).29 Methyl 10-undecenoate on the other hand (1j) is an oleochemical and thus not well miscible with water. Hence, the TOF20 observed (727 h−1) is comparatively low. Since all three reactions were conducted over a relatively long time, the simultaneously happening, consecutive reactions of the aldoxime group occur to a much greater extent. Control of the selectivity towards a single product (oxime, nitrile or amide) bears enormous potential that will be unravelled in ongoing research.
Application of oximes in biocatalytic nitrile synthesis
Aromatic, as well as aliphatic nitriles, already have a broad range of chemical and industrial application areas.30 The selective synthesis of linear nitriles through simple dehydration of aldoximes has substantial advantages considering sustainability aspects compared to conventional methods, like hydrocyanation (poor regioselectivities, toxic substrate) or their synthesis starting from carboxylic acids and ammonia (high temperatures and pressures needed). Recently, the sustainable, selective and energy-efficient nitrile synthesis using oxime-dehydratases gained considerable attention.31–33 Some of us already showed the benefit of combining transition metal catalysis and biocatalysis for various processes.34 Hence, we wanted to demonstrate in a conclusive investigation that the generated aldoximes can be converted into the corresponding nitriles by utilisation of enzymes as biocatalysts.Therefore, selected aldoximes were synthesised using previously described reaction conditions for the one-pot hydroformylation/aldoxime formation. After the reaction, the organic product phase was easily separated from the aqueous catalyst phase containing the excess of NH2OH by decantation. After evaporation of 1-butanol and column chromatography of the residue, we dehydrated the aldoximes in the presence of an aldoxime dehydratase, following the same procedure applied in a previous publication of our joint research, in which the formation of nonanitrile and 2-methyloctanenitrile was focussed upon (Fig. 4).20
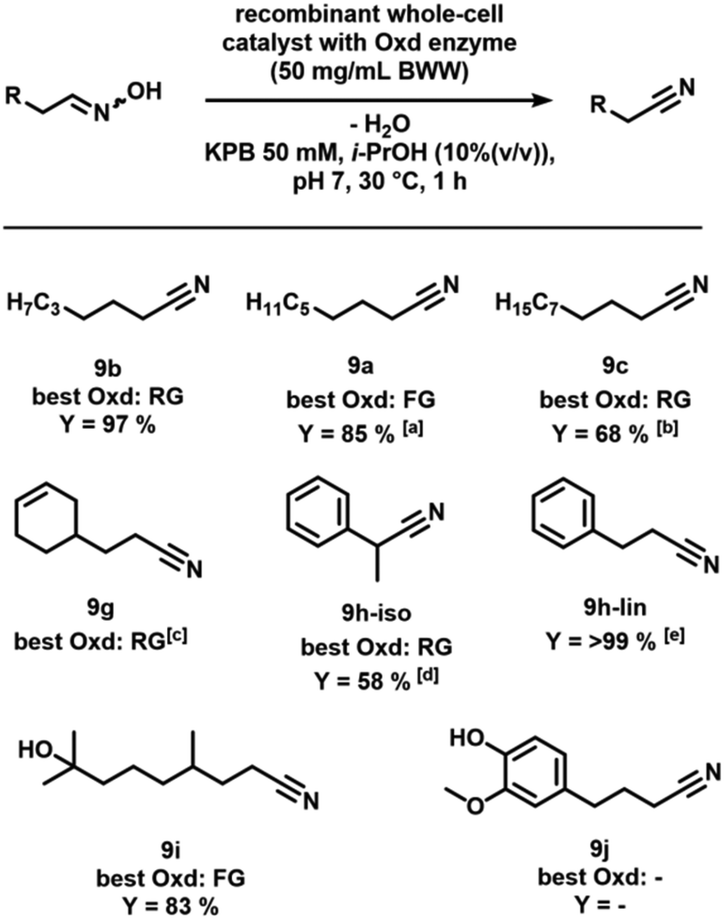 | ||
| Fig. 4 Conversion of selected aldoximes into nitriles using oxime dehydratases (Oxds). Conditions: KPi buffer 50 mM, pH = 7, 33 mg mL−1 whole cell suspension (BWW, bio wet weight), i-PrOH (10% (v/v)), substrate concentration 10 mM, 30 °C, 15 min, extraction with EtOAc, yield determined via GC-FID, aliterature: >99%, t = 1 h (ref. 13), b>99% at 50 mM, t = 3 h, cno quantitative measurement, best enzyme selected by GC area dliterature: >99%, 5 mM, t = 3 h (ref. 32), eliterature-known, OxdFG, 200 mM, t = 2 h (ref. 33). | ||
All terminal oximes derived from 1-alkenes were effectively transformed into the corresponding linear nitriles. The oxime derived from eugenol could not be converted, whereas the oxime generated from dihydromyrcenol was successfully converted, and a yield of the nitrile of 83% was observed. Remarkably, the nitriles can be produced under conditions optimised for nonanal oxime in very short reaction time. In general, when utilising enzymes as biocatalysts, the activity is highly dependent on the 3-dimensional structure of the substrate. Hence, structures similar to the already known aliphatic terminal oximes show the best activities. Literature values indicate that many of the substrates can be converted quantitatively by choosing longer reaction times.20,32,33
We also tried higher substrate loading of 100 mM and did not observe inhibition of the enzymes (for more detailed information, see ESI†). We are convinced that this additional investigation underlines the enormous potential of oxime chemistry.
Thus, the combination of homogeneous transition metal catalysis, biocatalysis and the combination of two reactions within a one-pot process, running in a green solvent system, allows linear nitriles to be generated in a sustainable way directly from terminal alkenes. For instance, 1-octene can be transformed to nonanitrile in 85% overall yield (Fig. 5), whereby nonanal oxime was isolated as intermediate.
 | ||
| Fig. 5 Anti-Markovnikov hydrocyanation of 1-octene through one-pot hydroformylation/aldoxime formation and subsequent biocatalytic dehydration. | ||
Conclusions
For the first time, the effective combination of hydroformylation and aldoxime formation in a one-pot reaction was achieved, yielding aldoximes directly from alkenes in a single preparative step. An excessive salt formation has been bypassed by application of aqueous hydroxylamine. The utilisation of sulfoxantphos as the ligand was found to be purposeful, as it allows the selective synthesis of linear products and immobilises the catalyst in an aqueous phase. A solvent system, composed of the green solvents water and 1-butanol, was key to success, as it enables sufficient contact of the aqueous catalyst phase and organic substrate phase. Using 1-octene as the model substrate, conditions were successfully optimised via Design of Experiments (DoE), yielding >85% of the desired, linear oxime. The reaction conditions were successfully transferred to 8 different substrates, while high linear selectivities of above 90% were maintained at yields above 85%. Varying reaction rates of the substrates and turnover frequencies of over 2000 h−1 were observed by the application of constant pressure and monitoring the gas consumption. Even functionalised renewable substrates were effectively converted to multifunctional compounds, although the consecutive reactions of the aldoxime group dominated and yields of the nitrile over 40% were observed. The additional implementation of a further reaction step and selectivity control towards aldoximes, nitriles or amides seems promising and will be part of ongoing research. In a concluding investigation, we successfully transformed the terminal oximes to the corresponding linear nitriles using oxime-dehydratases in a sustainable, highly selective and energy-efficient reaction. Thus, the overall reaction sequence formally represents an anti-Markovnikov hydrocyanation of readily available 1-alkenes. Concerning 1-octene, nonanitrile was synthesised over three reaction steps (two of them in a one-pot process) in 85% overall yield and 95% regioselectivity in only 2 h, with water as the only by-product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Gefördert durch die Deutsche Forschungsgemeinschaft (DFG) – TRR 63 “Integrierte chemische Prozesse in flüssigen Mehrphasensystemen” (Teilprojekt A11) – 56091768. Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – TRR 63 “Integrated Chemical Processes in Liquid Multiphase Systems” (subproject A11) – 56091768. We thankfully acknowledge Umicore AG & Co. KG for donation of the rhodium precursors. The authors thank Paulo Wohlfahrt and Aoi Itakura for the synthesis of substrates.References
- P. Roose, K. Eller, E. Henkes, R. Rossbacher and H. Höke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2015, pp. 1–55 Search PubMed.
- S. A. Lawrence, Amines. Synthesis, properties, and applications, Cambridge University Press, Cambridge, 2004 Search PubMed.
- E. Gebauer-Henke, W. Leitner, A. Prokofieva, H. Vogt and T. E. Müller, Catal. Sci. Technol., 2012, 2, 2539–2548 RSC.
- J. F. Liebman and Z. Rappoport, The Chemistry of Hydroxylamines, Oximes and Hydroxamic Acids, John Wiley & Sons Ltd, Chichester, England, 2009 Search PubMed.
- T. Betke, J. Higuchi, P. Rommelmann, K. Oike, T. Nomura, Y. Kato, Y. Asano and H. Gröger, ChemBioChem, 2018, 19, 768–779 CrossRef CAS.
- T. Sahyoun, A. Arrault and R. Schneider, Molecules, 2019, 24, 2470 CrossRef CAS.
- A. Behr and P. Neubert, Applied Homogeneous Catalysis, Wiley-VCH, Weinheim, 2012 Search PubMed.
- D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS.
- (a) D. Crozet, M. Urrutigoïty and P. Kalck, ChemCatChem, 2011, 3, 1102–1118 CrossRef CAS; (b) P. Eilbracht, L. Bärfacker, C. Buss, C. Hollmann, B. E. Kitsos-Rzychon, C. L. Kranemann, T. Rische, R. Roggenbuck and A. Schmidt, Chem. Rev., 1999, 99, 3329–3366 CrossRef CAS.
- Y. Wu, Y. Zhang, Z. Yang, J. Jiao, X. Zheng, W. Feng, M. Zhang, H. Cheng and L. Tang, ChemSusChem, 2019, 12, 3960–3966 CrossRef CAS.
- P. Kalck and M. Urrutigoïty, Chem. Rev., 2018, 118, 3833–3861 CrossRef CAS.
- (a) D. Sun, E. Kitamura, Y. Yamada and S. Sato, Green Chem., 2016, 18, 3389–3396 RSC; (b) W. Zhang, J.-H. Lin, P. Zhang and J.-C. Xiao, Chem. Commun., 2020, 56, 6221–6224 RSC; (c) K. Hyodo, S. Kitagawa, M. Yamazaki and K. Uchida, Chem. – Asian J., 2016, 11, 1348–1352 CrossRef CAS.
- Y. Liu, Z. Quan, S. He, Z. Zhao, J. Wang and B. Wang, React. Chem. Eng., 2019, 4, 1145–1152 RSC.
- (a) F. Xu, Y.-Y. Song, Y.-J. Li, E.-L. Li, X.-R. Wang, W.-Y. Li and C.-S. Liu, ChemistrySelect, 2018, 3, 3474–3478 CrossRef CAS; (b) P. Crochet and V. Cadierno, Chem. Commun., 2015, 51, 2495–2505 RSC.
- N. Herrmann, J. Bianga, T. Gaide, M. Drewing, D. Vogt and T. Seidensticker, Green Chem., 2019, 21, 6738–6745 RSC.
- H. W. F. Warmeling, D. Janz, M. Peters and A. J. Vorholt, Chem. Eng. J., 2017, 330, 585–595 CrossRef CAS.
- (a) J. Bianga, K. U. Künnemann, L. Goclik, L. Schurm, D. Vogt and T. Seidensticker, ACS Catal., 2020, 6463–6472 CrossRef CAS; (b) T. Seidensticker, D. Möller and A. J. Vorholt, Tetrahedron Lett., 2016, 57, 371–374 CrossRef CAS; (c) T. Gaide, J. Bianga, K. Schlipköter, A. Behr and A. J. Vorholt, ACS Catal., 2017, 7, 4163–4171 CrossRef CAS.
- (a) R. M. Deshpande, P. Purwanto, H. Delmas and R. V. Chaudhari, J. Mol. Catal. A: Chem., 1997, 126, 133–140 CrossRef CAS; (b) L. C. Matsinha, S. Siangwata, G. S. Smith and B. C. E. Makhubela, Catal. Rev. Sci. Eng., 2019, 61, 111–133 CrossRef CAS; (c) J. Bianga, N. Herrmann, L. Schurm, T. Gaide, J. M. Dreimann, D. Vogt and T. Seidensticker, Eur. J. Lipid Sci. Technol., 2020, 122, 1900317 CrossRef CAS; (d) T. A. Faßbach, F. O. Sommer, A. Behr, S. Romanski, D. Leinweber and A. J. Vorholt, Catal. Sci. Technol., 2017, 7, 1650–1653 RSC; (e) T. A. Faßbach, F. O. Sommer and A. J. Vorholt, Adv. Synth. Catal., 2018, 360, 1473–1482 CrossRef; (f) N. Herrmann, J. Bianga, M. Palten, T. Riemer, D. Vogt, J. M. Dreimann and T. Seidensticker, Eur. J. Lipid Sci. Technol., 2020, 122, 1900166 CrossRef CAS.
- T. Gaide, J. M. Dreimann, A. Behr and A. J. Vorholt, Angew. Chem., Int. Ed., 2016, 55, 2924–2928 CrossRef CAS.
- C. Plass, A. Hinzmann, M. Terhorst, W. Brauer, K. Oike, H. Yavuzer, Y. Asano, A. J. Vorholt, T. Betke and H. Gröger, ACS Catal., 2019, 9, 5198–5203 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- R. M. Southam and M. C. Whiting, J. Chem. Soc., Perkin Trans. 2, 1982, 597 RSC.
- (a) M. Schreuder Goedheijt, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Mol. Catal. A: Chem., 1998, 134, 243–249 CrossRef CAS; (b) H. Nowothnick, A. Rost, T. Hamerla, R. Schomäcker, C. Müller and D. Vogt, Catal. Sci. Technol., 2013, 3, 600–605 RSC.
- B. Cornils, W. A. Herrmann, M. Beller and R. Paciello, Applied homogeneous catalysis with organometallic compounds. A comprehensive handbook in four volumes, Wiley-VCH, Weinheim, Germany, 2018 Search PubMed.
- M. Schmidt, B. Blom, T. Szilvási, R. Schomäcker and M. Driess, Eur. J. Inorg. Chem., 2017, 2017, 1284–1291 CrossRef CAS.
- (a) E. Boymans, M. Janssen, C. Müller, M. Lutz and D. Vogt, Dalton Trans., 2013, 42, 137–142 RSC; (b) R. Lazzaroni, A. Raffaelli, R. Settambolo, S. Bertozzi and G. Vitulli, J. Mol. Catal., 1989, 50, 1–9 CrossRef CAS.
- (a) D. McGinty, C. S. Letizia and A. M. Api, Food Chem. Toxicol., 2010, 48, S70–S75 Search PubMed; (b) D. S. Ogunniyi, Bioresour. Technol., 2006, 97, 1086–1091 CrossRef CAS.
- H. Tsuchiya, Molecules, 2017, 22, 1369 CrossRef.
- S. H. Yalkowsky, Y. He and P. Jain, Handbook of Aqueous Solubility Data, CRC Press, Boca Raton, 2019 Search PubMed.
- P. Pollak, G. Romeder, F. Hagedorn and H.-P. Gelbke, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed.
- (a) T. Betke, M. Maier, H. Gruber-Wölfler and H. Gröger, Nat. Commun., 2018, 9, 5112 CrossRef; (b) T. Betke, P. Rommelmann, K. Oike, Y. Asano and H. Gröger, Angew. Chem., Int. Ed., 2017, 56, 12361–12366 CrossRef CAS; (c) Y. Kato and Y. Asano, Appl. Microbiol. Biotechnol., 2006, 70, 92–101 CrossRef CAS.
- R. Metzner, S. Okazaki, Y. Asano and H. Gröger, ChemCatChem, 2014, 6, 3105–3109 CrossRef CAS.
- S. X. Xie, Y. Kato and Y. Asano, Biosci. Biotechnol. Biochem., 2001, 65, 2666–2672 CrossRef CAS.
- (a) F. Kühn, S. Katsuragi, Y. Oki, C. Scholz, S. Akai and H. Gröger, Chem. Commun., 2020, 56, 2885–2888 RSC; (b) J. Löwe, K.-J. Dietz and H. Gröger, Adv. Sci., 2020, 7, 1902973 CrossRef; (c) F. Uthoff, H. Sato and H. Gröger, ChemCatChem, 2017, 9, 555–558 CrossRef CAS; (d) S. Wedde, P. Rommelmann, C. Scherkus, S. Schmidt, U. T. Bornscheuer, A. Liese and H. Gröger, Green Chem., 2017, 19, 1286–1290 RSC; (e) N. Zumbrägel and H. Gröger, Bioengineering, 2018, 5, 60 CrossRef; (f) For a general review about this research field of combining chemo- and biocatalysis, see: F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03141k |
| This journal is © The Royal Society of Chemistry 2020 |