
Highly effective capture and subsequent catalytic transformation of low-concentration CO2 by superbasic guanidines†
Hui
Zhou
 *,
Wei
Chen
,
Ji-Hong
Liu
,
Wen-Zhen
Zhang
and
Xiao-Bing
Lu
*
*,
Wei
Chen
,
Ji-Hong
Liu
,
Wen-Zhen
Zhang
and
Xiao-Bing
Lu
*
State Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, China. E-mail: zhouhui@dlut.edu.cn; xblu@dlut.edu.cn
First published on 17th October 2020
Abstract
Herein, we present a highly efficient and convenient approach for carbon dioxide (CO2) capture and catalytic transformation under mild conditions using N,N′-bis(imidazolyl)guanidines (BIGs, organoguanidine-based strong superbases) as the organocatalyst, even from simulated flue gas (10% CO2/90% N2, v/v) or directly from dry air (∼400 ppm CO2). The zwitterionic BIG–CO2 adducts were successfully isolated and characterized. X-ray single crystal analysis revealed the bent geometry of the binding CO2 in the BIG–CO2 adduct with an O–C–O angle of 129.7° and increased C–O bond distances (1.253 and 1.237 Å) in comparison with free CO2. Notably, the resulting BIG–CO2 adducts were found to be capable of catalyzing the novel cycloaddition of various propiolamidines with simulated flue gas to generate functionalized (4E,5Z)-4-imino-5-benzylideneoxazolidine-2-ones in good yields and excellent selectivity.
Introduction
Nitrogen base-involving carbon dioxide (CO2) capture and sequestration (CCS), as a kind of climate change mitigation technology, has attracted so much global attention with the aim of reducing anthropogenic CO2 emissions.1 Understanding the interaction pattern of nitrogen bases and CO2 is critical to boost the development of CCS technology (Scheme 1). As is well known, primary and secondary amines could react with CO2 by rapid nucleophilic attack to form zwitterionic nitrogen base–CO2 adducts (N–CO2 adducts). Due to the inherent instability, N–CO2 adducts easily react with another molecule of an amine via a proton transfer process to form stable ammonium carbamate salts (Scheme 1, I). This method has been widely applied to remove CO2 from highly concentrated and stationary CO2 emission sources, such as power plants and industrial sectors.2 In 2010, the first N–CO2 adduct derived from 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) was successfully isolated and characterized by the Villiers group.3 Note that X-ray single-crystal diffraction data indicate that the intramolecular hydrogen bonding leads to an increased stability of TBD–CO2 adducts (Scheme 1, II). With the assistance of Lewis acids, N–CO2 adducts could also be stabilized, thus generating cyclic frustrated Lewis pair (FLP)–CO2 adducts (Scheme 1, III).4 Furthermore, N-heterocyclic imines (NHI) as more electron-rich nitrogen donors were recently developed to activate the pressured CO2 by the Dielmann group and the corresponding stable NHI–CO2 adducts were obtained through a nucleophilic attack process (Scheme 1, IV).5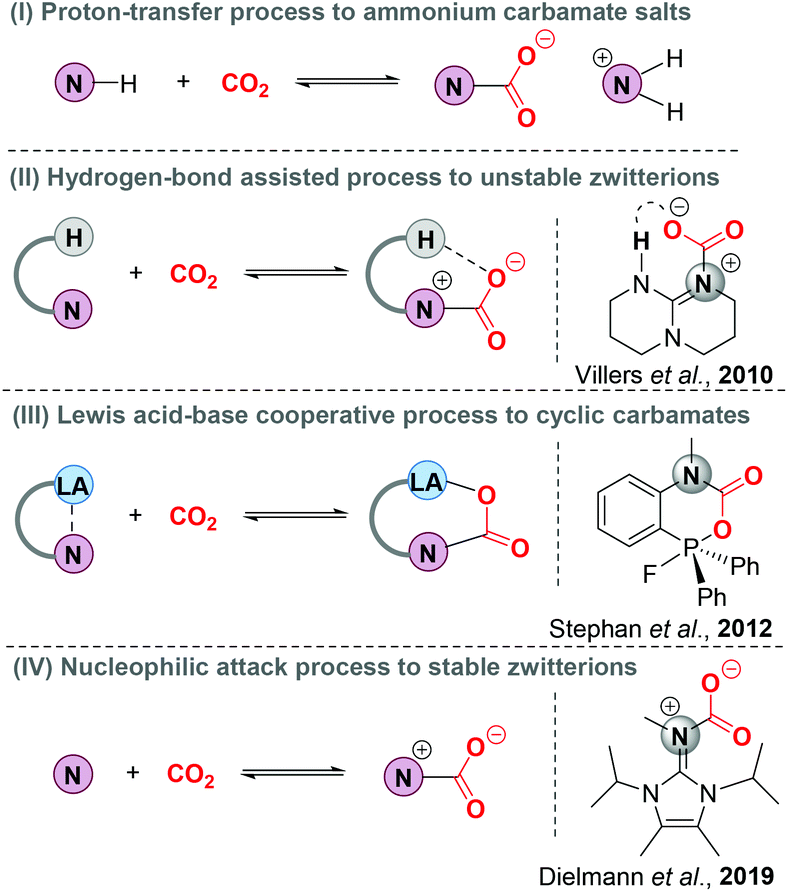 | ||
| Scheme 1 Representative methods for CO2 capture and sequestration by nitrogen base derivatives. | ||
Obviously, the donor strength of Lewis bases is a vital factor for CO2 activation and sequestration. As a consequence, a series of Lewis base–CO2 adducts have been synthesized,6 employing strong basic carbon,7 phosphine,8 and oxygen bases.9N,N′-Bis(imidazolyl)guanidine (BIG) bases, formed by direct attachment of imidazolyl substituents to guanidine derivatives, have emerged as a new class of nitrogen bases.10 Importantly, the pKa values of the resulting BIG bases were determined to be between 26.1 and 29.3 in THF. Although commonly known organoguanidines have widely been applied for CO2 capture, activation, and chemical transformation,11 no literature regarding the use of strong basic BIG systems for CO2 capture and sequestration has been reported. Herein, we report the synthesis, isolation and structural characterization of zwitterionic BIG–CO2 adducts via a nucleophilic addition process. More importantly, this system is even effective in extracting CO2 from the ambient air. Additionally, we demonstrate that BIG–CO2 adducts also have the ability to catalyze the novel cycloaddition of propiolamidines with simulated flue gas to selectively form functionalized (4E,5Z)-4-imino-4-imino-5-benzylideneoxazolidine-2-ones with high activity.
Results and discussion
Synthesis and characterization of BIG–CO2 adducts
Firstly, BIG hydrotetrafluoroborates 1a–1d were synthesized as previously reported by Ullrich Jahn et al.10a BIG bases were prepared by the deprotonation of 1a–1d with KN(SiMe3)2 in THF solution and further purified by extraction with n-hexane. When the n-hexane solution of BIG bases 2a–2d was placed under an atmosphere of pure CO2 at room temperature, white precipitates of BIG–CO2 adducts (3a–3d) were rapidly formed and isolated in good to excellent yields (Scheme 2).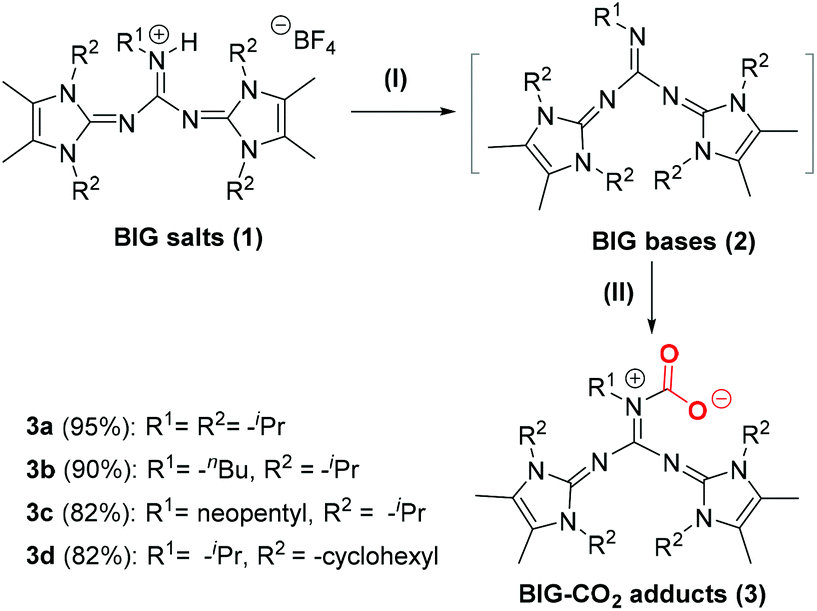 | ||
| Scheme 2 CO2 activation and fixation by BIG bases. Reaction conditions: (I) KN(SiMe3)2, THF, 25 °C, 2 h; (II) CO2 (1 atm), n-hexane, 25 °C, 2 h. | ||
Furthermore, BIG–CO2 adducts (3a–3d) were structurally characterized by 1H NMR, 13C NMR, IR and MS (ESI).†13C NMR spectra of 3a–3d show the chemical shifts of the carbonyl group in the range of 162.1–162.3 ppm, which are quite close to those of reported NHI–CO2 adducts.10a Meanwhile, a 13C isotope labeling experiment was conducted, in which the CO2 resonance of 3a (162.1 ppm) obtained from 13CO2 was enhanced obviously. And also, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequencies of 3a–3d were investigated in the range of 1610–1631 cm−1. Gratifyingly, the single crystal of 3a was obtained and determined by X-ray single crystal diffraction, as shown in Fig. 1. The crystal structure data show that the C1–O1 and C1–O2 bond distances, 1.253(6) and 1.237(4) Å, respectively, are both elongated, in comparison with that of free gaseous CO2 (1.160 Å). A bent geometry of the binding CO2 with an O1–C1–O2 angle of 129.78(7)° was observed in the BIG–CO2 adduct, indicating that CO2 is activated through nucleophilic attack by BIG bases. Moreover, the dihedral angles between the plane of the guanidine core and the two imidazole planes are 109.85(4)° and 119.54(4)°, respectively.
O stretching frequencies of 3a–3d were investigated in the range of 1610–1631 cm−1. Gratifyingly, the single crystal of 3a was obtained and determined by X-ray single crystal diffraction, as shown in Fig. 1. The crystal structure data show that the C1–O1 and C1–O2 bond distances, 1.253(6) and 1.237(4) Å, respectively, are both elongated, in comparison with that of free gaseous CO2 (1.160 Å). A bent geometry of the binding CO2 with an O1–C1–O2 angle of 129.78(7)° was observed in the BIG–CO2 adduct, indicating that CO2 is activated through nucleophilic attack by BIG bases. Moreover, the dihedral angles between the plane of the guanidine core and the two imidazole planes are 109.85(4)° and 119.54(4)°, respectively.
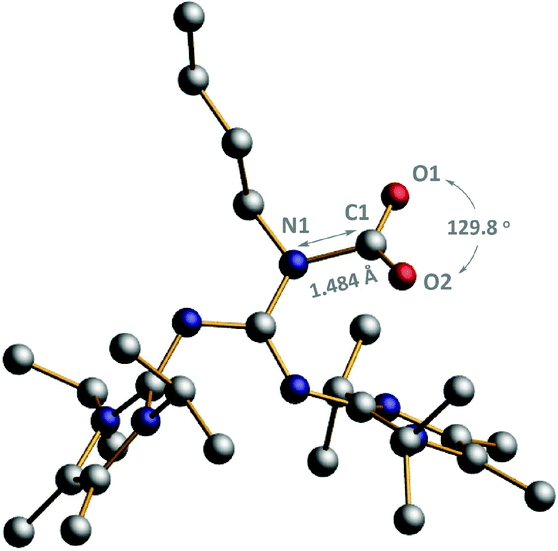 | ||
| Fig. 1 POV-Ray illustrations of the molecular structure of BIG–CO2 adduct 3b. Hydrogen atoms have been omitted for clarity. C: black, O: red, and N: dark blue. Selected bond lengths (Å) and angles (°): N1–C1, 1.484(6); O1–C1, 1.253(6); O2–C1, 1.237(4); O1–C1–O2, 129.78(7). | ||
In addition, the thermal stability of BIG–CO2 adducts 3a–3d was investigated by means of thermogravimetric analysis (TGA). From the results (ESI, Fig. S1–S4†), the reversible decarboxylation of 3a–3d began in the range of 129.2–135.8 °C, and the observed weight losses were matched well with the theoretical content of CO2 in BIG–CO2 adducts.
CO2 capture ability of BIG bases under various concentrations of CO2
Reversible CO2 capture and release of BIG bases was also studied by Density Functional Theory (DFT) calculations (ESI, Fig. S5†). The free energy barriers of BIG bases 2a–2d for CO2 capture are very low (5.5–7.6 kcal mol−1), and the formation of BIG–CO2 adducts 3a–3c is even more exergonic. Based on this observation, BIG bases have the potential to be applied for CO2 activation and capture. Up to now, most of the existing CCS technologies are applied in highly concentrated and stationary CO2 emission sources. Direct CO2 capture from ambient air, by contrast, is becoming more promising to permanently lower the atmospheric CO2 concentration, thus achieving negative carbon emissions.12 As shown in Fig. 2, the CO2 capture ability of BIG bases with 2a as an example was measured under various concentrations of CO2. When the commercially pure CO2 was introduced with a balloon, a white precipitate was formed immediately to obtain 3a in 84% isolated yield within 3 minutes. Moreover, CO2 capture from simulated flue gas (10% CO2/90% N2, v/v) could also be carried out at a flow rate of 100 mL min−1, and the corresponding 3a was formed in 65% yield within 15 minutes. When switching to dry air containing about 400 ppm of CO2, a yield of 52% could be obtained with an enhanced reaction time of 2 hours.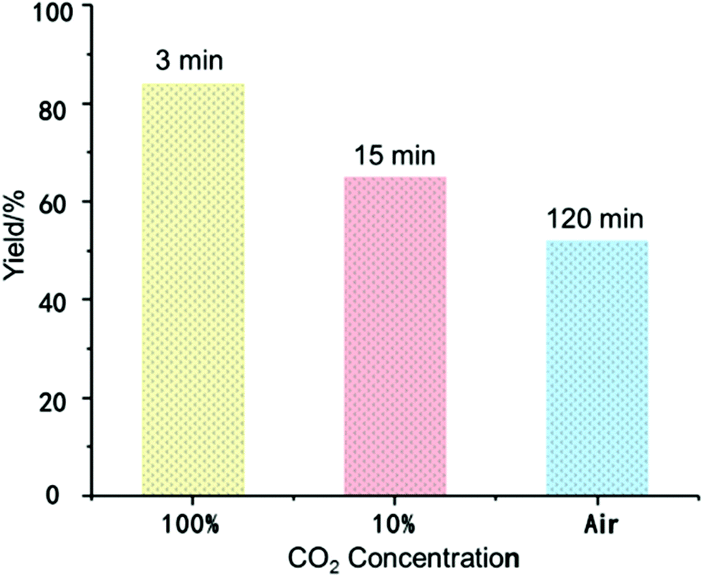 | ||
| Fig. 2 CO2 fixation ability of a BIG base (2a) under various concentrations of CO2. Reaction conditions: BIG base (2a) (0.1 mmol), hexane (5 mL), 25 °C, CO2 (balloon); simulated flue gas (100 mL min−1); dry air (100 mL min−1). | ||
Protonation of BIG–CO2 adducts in the presence of H2O
Most of the known Lewis base–CO2 adducts are sensitive to protic solvents, such as water and alcohols.13 Surprisingly, recently reported NHI–CO2 adducts bearing a methyl group at the exocyclic nitrogen atom showed unprecedented chemical stability towards hydrolysis, probably due to the hydrophobic nature of the CO2 binding site.5 The stability of BIG–CO2 adducts toward water was also evaluated in THF solution at ambient temperature. In the presence of 1.2 equiv. H2O, 2a–2c was rapidly hydrolyzed via a decarboxylation process to form ammonium bicarbonates 4a–c in 90–98% isolated yields. Meanwhile, the X-ray crystal structure of 4a was also determined (ESI, Fig. S7†).Application of BIG for COS and CS2 capture and activation
Because of the structural similarity of COS and CS2 molecules to CO2,14 their capture using BIG base 2a was also investigated under the same reaction conditions. As shown in Scheme 3, the COS and CS2 capture processes could smoothly proceed at room temperature and the corresponding BIG–COS adduct 5 and CS2 adduct 6 were isolated in 97% and 98% yields, respectively. The solid-state structure of the BIG–CS2 adduct 6 shows that CS2 binds to the nitrogen atom with a N1–C1 bond of 1.427(7) Å and a S–C–S angle of 122.96(4)° (Fig. 3). Note that the BIG–CS2 adduct 6 is the first example of a nitrogen-based zwitterionic adduct. The groups of Vlasse15 and Jessop16 independently reported the reaction of CS2 with cyclic amidines to form cyclic carbamic carboxylic trithioanhydride rings, while acyclic acetamidines were cleaved by CS2 at room temperature to give an isothiocyanate and a thioacetamide. In addition, Cantat et al. showed that TBD reacted with CS2 and the guanidinium dithiocarbamate was selectively synthesized via a proton transfer process.17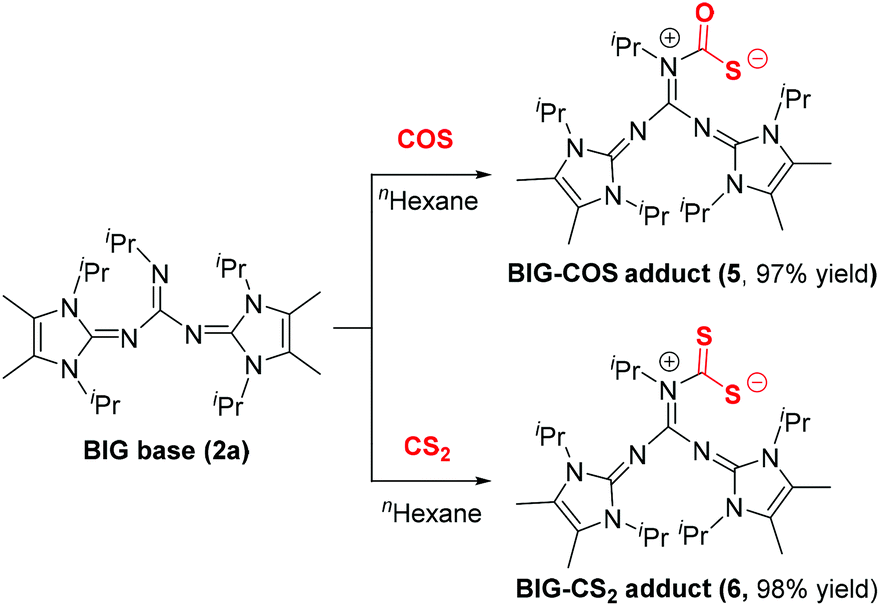 | ||
| Scheme 3 COS/CS2 activation and fixation by BIG bases. | ||
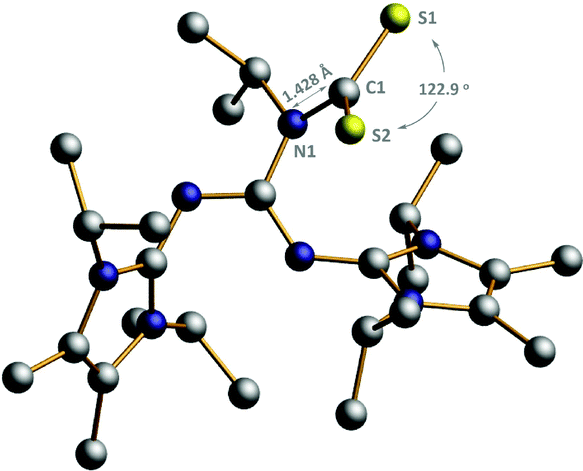 | ||
| Fig. 3 POV-Ray illustrations of the molecular structure of 6. Hydrogen atoms have been omitted for clarity. C: black, S: yellow, and N: dark blue. Selected bond lengths (Å) and angles (°): N1–C1, 1.427(7); S1–C1, 1.703(6); S2–C1, 1.680(9); S1–C1–S2, 122.96(4). | ||
Application of BIG–CO2 adducts as organocatalysts for CO2 catalytic transformation
As an additional CO2-mitigation strategy to CCS, CO2 capture and utilization (CCU) is attracting global interest.18 Recently, Lewis base–CO2 adducts,6,19 as a new class of organic catalysts, have exhibited unique reactivity and selectivity for CO2 transformation to value-added chemicals, which inspires us to further investigate the application of BIG–CO2 adducts in the CCU process. Gratifyingly, BIG–CO2 adduct 3b could efficiently catalyze the novel cycloaddition of propargylamidine 7a with simulated flue gas at 80 °C in 24 hours (for detailed optimized conditions, see ESI, Table S1†), thus generating 4-imino-5-benzylideneoxazolidine-2-one 8a in 90% isolated yield. The reactions of propargylamidines (7b–7f) bearing methyl, methoxyl or halogen groups (–F, –Cl, and –Br) on the aryl ring gave the corresponding products 8b–8f in moderate to excellent yields. When the R2 group was transformed to the cyclohexyl group, the corresponding substrate 7g was converted to 8g with 90% yield. Meanwhile, the absolute stereostructures of (4E,5Z)-8a and 8g were clearly confirmed by single-crystal X-ray diffraction study (Table 1).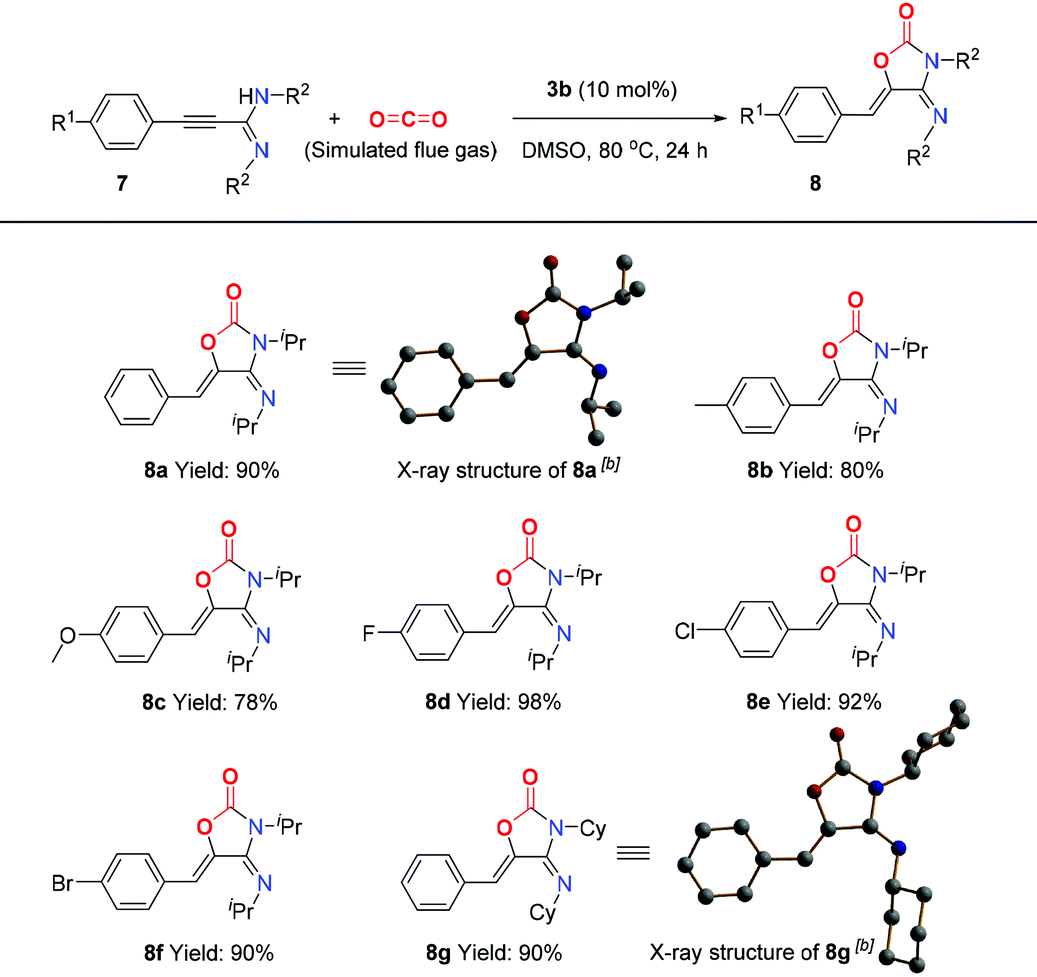
To demonstrate the synthetic utility of this transformation, the gram-scale synthesis and further transformations of the products were next elucidated, as shown in Scheme 4. Under the standard conditions, the reaction of 7f (1.2 g) with simulated flue gas proceeded smoothly, isolating the corresponding product 8f in 81% yield (Scheme 4, I). Moreover, the carbon–carbon double bond of product 8a was selectively reduced by H2 in the presence of Pd/C catalyst at room temperature, and the reduced product 9 was isolated in 95% yield (Scheme 4, II). Oxazolidine-2,4-diones, as a significant class of heterocyclic scaffolds, are frequently found in biologically active and medicinally useful molecules.20 Note that the hydrolysis of 8f could take place smoothly in an aqueous solution of hydrochloric acid, thus affording oxazolidine-2,4-dione 10 in 98% yield (Scheme 4, III).
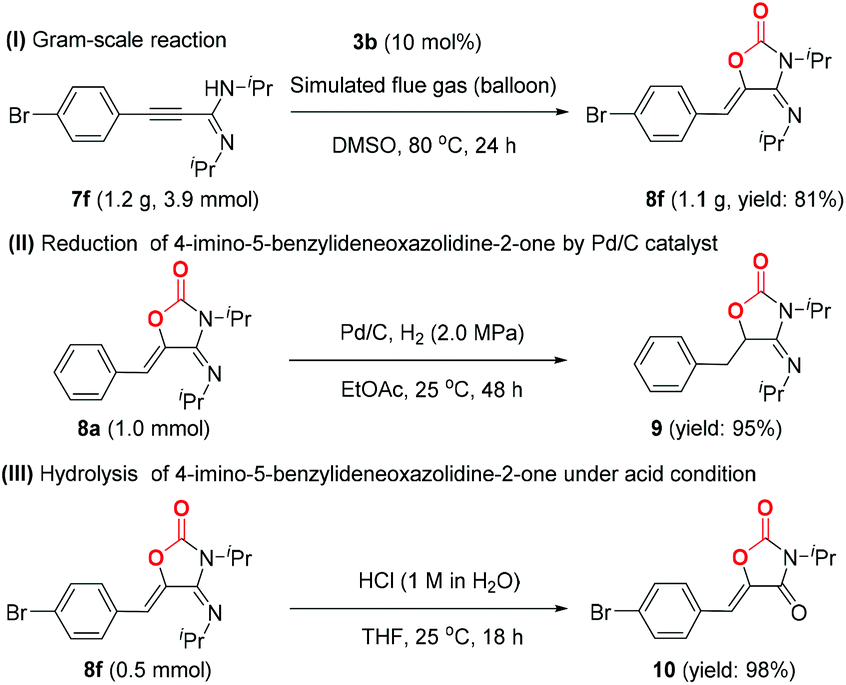 | ||
| Scheme 4 Gram-scale synthesis and synthetic applications of products. | ||
Conclusions
In summary, we have demonstrated the high efficiency of superbasic guanidines for reversible capture of low-concentration CO2 at room temperature and atmospheric pressure, affording the corresponding CO2 adducts in good and excellent yields. Moreover, the resulting BIG–CO2 adducts were demonstrated to be efficient organocatalysts for the cyclization of CO2 (10% CO2/90% N2, v/v) and propiolamidines to produce various functionalized (4E,5Z)-4-imino-5-benzylideneoxazolidine-2-ones in high yields and excellent selectivity. The present study provides an alternative method for CO2 capture and subsequent catalytic transformation of low-concentration CO2 under mild conditions. Further explorations regarding the applications of organocatalytic systems for the synthesis of various heterocyclic chemicals are now in progress.Experimental
Representative experimental procedure for the synthesis of BIG–CO2 adducts (3a–3d)
In a nitrogen-filled glove box, BIG salt 1a (545 mg, 1.0 mmol) was added to a suspension of KHMDS (199 mg, 1.0 mmol) in THF (10 mL) and the mixture was stirred at 25 °C for 2 h. Then the solvent was removed in vacuo and the residues were extracted with n-hexane (10 mL). After filtration to remove the inorganic salt, the filtrate was exposed to 1.0 atm of CO2 at room temperature for 2 h. The resulting precipitates were collected via filtration, washed with n-hexane (3 × 5 mL) and then dried in vacuo to afford BIG–CO2 adduct 3a as a white solid (476 mg, 95% yield). 1H NMR (400 MHz, CDCl3) δ 4.52 (dt, J = 14.1, 7.0 Hz, 4H), 4.06 (dt, J = 13.1, 6.5 Hz, 1H), 2.23 (s, 12H), 1.47 (d, J = 7.1 Hz, 24H), 1.21 (d, J = 6.5 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 162.1, 156.2, 147.6, 120.2, 47.9, 44.0, 23.1, 21.3, 10.1. IRvCO: 1650 cm−1. HRMS (ESI): calcd for C27H47N7O2: 458.3966 [M − CO2 + H]+. Found: 458.3962 [M − CO2 + H]+.
3b. White solid (464 mg, 90% yield). 1H NMR (500 MHz, CDCl3) δ 4.49 (dt, J = 14.1, 7.0 Hz,4 H), 3.60–3.07 (m, 2H), 2.19 (s, 12H), 1.54 (dt, J = 15.0, 7.5 Hz, 2H), 1.44 (d, J = 7.0 Hz, 24H), 1.31 (dt, J = 15.0, 7.4 Hz, 2H), 0.86 (dt, J = 14.1, 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 162.1, 157.4, 147.9, 120.1, 47.9, 42.3, 32.5, 21.3, 20.2, 13.9, 10.1. IRvCO: 1647 cm−1. HRMS (ESI): calcd for C28H49N7O2: 472.4122 [M − CO2 + H]+. Found: 472.4155 [M − CO2 + H]+.
3c. White solid (434 mg, 82% yield). 1H NMR (400 MHz, CDCl3) δ 4.76–4.24 (m, 4H), 3.23 (s, 2H), 2.25 (s, 12H), 1.48 (d, J = 7.1 Hz, 24H), 0.92 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 162.2, 157.3, 147.6, 120.3, 53.6, 47.9, 32.2, 27.4, 21.4, 10.1. IRvCO: 1654 cm−1. HRMS (ESI): calcd for C29H51N7O2: 486.4279 [M − CO2 + H]+. Found: 486.4271 [M − CO2 + H]+.
3d. White solid (542 mg, 82% yield). 1H NMR (400 MHz, CDCl3) δ 4.19 (dt, J = 11.6, 5.6 Hz, 1H), 4.13–3.97 (m, 4H), 2.24 (s, 12H), 1.79 (dd, J = 62.7, 9.0 Hz, 28H), 1.44–0.99 (m, 18H); 13C NMR (126 MHz, CDCl3) δ 162.3, 155.3, 147.8, 120.6, 56.8, 44.1, 31.0, 26.2, 25.1, 23.5, 10.9. IRvCO: 1639 cm−1. HRMS (ESI): calcd for C39H63N7O2: 618.5218 [M − CO2 + H]+. Found: 618.5205 [M − CO2 + H]+.
Representative experimental procedure for the cycloaddition of propiolamidines with simulated flue gas to (4E,5Z)-4-imino-5-benzylideneoxazolidine-2-ones
In a nitrogen-filled glove box, a 10 mL Schlenk flask equipped with a magnetic stirring bar was charged with propiolamidine 7a (57.1 mg, 0.25 mmol), Cat. 3b (12.9 mg, 0.025 mmol, 10 mol%) and DMSO (1.0 mL). Then the Schlenk flask was immediately transferred from the glovebox, and exchanged with CO2 (10% CO2/90% N2, v/v) using a balloon. The reaction was stirred at 80 °C for 24 h. The crude reaction mixture was purified by column chromatography on silica gel (eluent: petroleum ether/ethyl acetate = 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give the desired (4E,5Z)-4-imino-5-benzylideneoxazolidine-2-one 8a (61.2 mg, 90%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.77–7.63 (m, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.32 (t, J = 7.3 Hz, 1H), 6.35 (s, 1H), 4.10 (tt, J = 12.3, 3.9 Hz, 1H), 3.84 (t, J = 9.1 Hz, 1H), 2.21 (dq, J = 12.5, 3.3 Hz, 2H), 1.85 (d, J = 9.7 Hz, 6H), 1.68 (t, J = 11.8 Hz, 4H), 1.61–1.13 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 152.7, 142.8, 136.1, 132.8, 130.5, 129.0, 128.9, 113.1, 57.0, 52.0, 33.9, 28.6, 26.1, 26.0, 25.3, 24.5. IR: 2930, 2855, 1797, 1670, 1647, 1450, 1367, 1331, 1232, 1201, 1089. HRMS (ESI): calcd for C22H28N2O2: 353.2224 [M + H]+. Found: 353.2212 [M + H]+.
:1) to give the desired (4E,5Z)-4-imino-5-benzylideneoxazolidine-2-one 8a (61.2 mg, 90%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.77–7.63 (m, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.32 (t, J = 7.3 Hz, 1H), 6.35 (s, 1H), 4.10 (tt, J = 12.3, 3.9 Hz, 1H), 3.84 (t, J = 9.1 Hz, 1H), 2.21 (dq, J = 12.5, 3.3 Hz, 2H), 1.85 (d, J = 9.7 Hz, 6H), 1.68 (t, J = 11.8 Hz, 4H), 1.61–1.13 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 152.7, 142.8, 136.1, 132.8, 130.5, 129.0, 128.9, 113.1, 57.0, 52.0, 33.9, 28.6, 26.1, 26.0, 25.3, 24.5. IR: 2930, 2855, 1797, 1670, 1647, 1450, 1367, 1331, 1232, 1201, 1089. HRMS (ESI): calcd for C22H28N2O2: 353.2224 [M + H]+. Found: 353.2212 [M + H]+.
8b. White solid (80%). 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 7.9 Hz, 2H), 7.20 (d, J = 7.9 Hz, 2H), 6.40 (s, 1H), 4.52 (hept, J = 6.8 Hz, 1H), 4.17 (hept, J = 6.1 Hz, 1H), 2.37 (s, 3H), 1.43 (d, J = 6.9 Hz, 6H), 1.28 (d, J = 6.1 Hz, 6H).13C NMR (101 MHz, CDCl3) δ 152.5, 143.0, 139.4, 135.6, 130.5, 129.9, 129.6, 113.5, 49.0, 44.1, 24.1, 21.5, 19.0. IR: 2971, 2935, 1794, 1670, 1647, 1407, 1385, 1349, 1330, 1313, 1252, 1178, 1024. HRMS (ESI): calcd for C17H22N2O2: 287.1754 [M + H]+. Found: 287.1752 [M + H]+.
8c. White solid (78%). 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 8.9 Hz, 2H), 6.91 (d, J = 8.9 Hz, 2H), 6.38 (s, 1H), 4.69–4.36 (m, 1H), 4.32–4.08 (m, 1H), 3.84 (s, 3H), 1.42 (d, J = 6.9 Hz, 6H), 1.27 (d, J = 6.2 Hz, 6H).13C NMR (126 MHz, CDCl3) δ 160.3, 152.6, 143.1, 134.8, 132.2, 125.5, 114.4, 113.3, 55.5, 49.0, 44.1, 24.1, 19.1. IR: 2957, 2927, 1793, 1670, 1646, 1604, 1513, 1408, 1385, 1256, 1178, 1026. HRMS (ESI): calcd for C17H22N2O3: 303.1703 [M + H]+. Found: 303.1693 [M + H]+.
8d. White solid (98%). 1H NMR (400 MHz, CDCl3) δ 7.87–7.54 (m, 2H), 7.21–6.87 (m, 2H), 6.38 (s, 1H), 4.52 (hept, J = 7.0 Hz, 1H), 4.31–4.01 (m, 1H), 1.42 (d, J = 7.0 Hz, 6H), 1.27 (d, J = 6.2 Hz, 6H). IR: 2972, 1797, 1673, 1650, 1602, 1509, 1408, 1386, 1252, 1163, 1023. HRMS (ESI): calcd for C16H19FN2O2: 291.1503 [M + H]+. Found: 291.1493 [M + H]+.
8e. White solid (82%). 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.6 Hz, 2H), 7.35 (d, J = 8.6 Hz, 2H), 6.36 (s, 1H), 4.51 (m, J = 6.9 Hz, 1H), 4.30–4.00 (m, 1H), 1.42 (d, J = 7.0 Hz, 6H), 1.27 (d, J = 6.2 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 152.1, 142.6, 136.4, 134.9, 131.6, 131.3, 129.1, 111.9, 49.2, 44.3, 24.1, 19.0. IR: 2972, 1794, 1673, 1648, 1490, 1409, 1385, 1349, 1251, 1180, 1080, 1022. HRMS (ESI): calcd for C16H19ClN2O2: 307.1208 [M + H]+. Found: 307.1198 [M + H]+.
8f. White solid (90%). 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 8.5 Hz, 2H), 7.43 (d, J = 8.5 Hz, 2H), 6.27 (s, 1H), 4.44 (m, J = 6.9 Hz, 1H), 4.08 (m, J = 6.1 Hz, 1H), 1.35 (d, J = 6.9 Hz, 6H), 1.20 (d, J = 6.2 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 152.1, 142.6, 136.5, 132.1, 131.9, 131.7, 123.3, 112.0, 49.2, 44.2, 24.1, 19.0. IR: 2970, 2938, 1794, 1671, 1646, 1486, 1408, 1385, 1348, 1307, 1250, 1176, 1074, 1021. HRMS (ESI): calcd for C16H19BrN2O2: 351.0703 [M + H]+. Found: 351.0693 [M + H]+.
8g. White solid (90%). 1H NMR (400 MHz, CDCl3) δ 7.77–7.63 (m, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.32 (t, J = 7.3 Hz, 1H), 6.35 (s, 1H), 4.10 (tt, J = 12.3, 3.9 Hz, 1H), 3.84 (t, J = 9.1 Hz, 1H), 2.21 (qd, J = 12.5, 3.3 Hz, 2H), 1.85 (d, J = 9.7 Hz, 6H), 1.68 (t, J = 11.8 Hz, 4H), 1.61–1.13 (m, 10H). 13C NMR (101 MHz, CDCl3) δ 152.7, 142.8, 136.1, 132.8, 130.5, 129.0, 128.9, 113.1, 57.0, 52.0, 33.9, 28.6, 26.1, 26.0, 25.3, 24.5. IR: 2930, 2855, 1797, 1670, 1647, 1450, 1367, 1331, 1232, 1201, 1089. HRMS (ESI): calcd for C22H28N2O2: 353.2224 [M + H]+. Found: 353.2212 [M + H]+.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 91856108), the Fundamental Research Funds for the Central Universities (DUT18LK55) and the Program for Changjiang Scholars and Innovative Research Team in University (IRT-17R14).Notes and references
- (a) K. S. Lackner, Science, 2003, 300, 1677–1678 CrossRef CAS; (b) P. G. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102 CrossRef CAS; (c) S. H. Kim, K. H. Kim and S. H. Hong, Angew. Chem., Int. Ed., 2014, 53, 771–774 CrossRef CAS; (d) D. M. Reiner, Nat. Energy, 2016, 1, 15011 CrossRef.
- (a) Q. Yan and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 16300–16303 CrossRef CAS; (b) F. Inagaki, C. Matsumoto, T. Iwata and C. Mukai, J. Am. Chem. Soc., 2017, 139, 4639–4642 CrossRef CAS.
- C. Villiers, J. P. Dognon, R. Pollet, P. Thuery and M. Ephritikhine, Angew. Chem., Int. Ed., 2010, 49, 3465–3468 CrossRef CAS.
- (a) M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 13559–13568 CrossRef CAS; (b) L. J. Hounjet, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 4714–4717 CrossRef CAS; (c) C. Das Neves Gomes, E. Blondiaux, P. Thuery and T. Cantat, Chem. – Eur. J., 2014, 20, 7098–7106 CrossRef CAS; (d) A. Adenot, N. von Wolff, G. Lefevre, J. C. Berthet, P. Thuery and T. Cantat, Chem. – Eur. J., 2019, 25, 8118–8126 CrossRef CAS.
- L. F. B. Wilm, T. Eder, C. Muck-Lichtenfeld, P. Mehlmann, M. Wunsche, F. Buss and F. Dielmann, Green Chem., 2019, 21, 640–648 RSC.
- (a) L. J. Murphy, K. N. Robertson, R. A. Kemp, H. M. Tuononen and J. A. C. Clyburne, Chem. Commun., 2015, 51, 3942–3956 RSC; (b) H. Zhou and X. Lu, Sci. China: Chem., 2017, 60, 904–911 CrossRef CAS.
- (a) H. A. Duong, T. N. Tekavec, A. M. Arif and J. Louie, Chem. Commun., 2004, 112–113 RSC; (b) H. Zhou, W.-Z. Zhang, C.-H. Liu, J.-P. Qu and X.-B. Lu, J. Org. Chem., 2008, 73, 8039–8044 CrossRef CAS; (c) Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem., Int. Ed., 2009, 48, 4194–4197 CrossRef CAS; (d) Y. B. Wang, Y. M. Wang, W. Z. Zhang and X. B. Lu, J. Am. Chem. Soc., 2013, 135, 11996–12003 CrossRef CAS; (e) H. Zhou, R. Zhang and X.-B. Lu, Adv. Synth. Catal., 2019, 361, 326–334 CrossRef CAS.
- (a) F. Buss, P. Mehlmann, C. Muck-Lichtenfeld, K. Bergander and F. Dielmann, J. Am. Chem. Soc., 2016, 138, 1840–1843 CrossRef CAS; (b) P. Rotering, L. F. B. Wilm, J. A. Werra and F. Dielmann, Chem. – Eur. J., 2020, 26, 406–411 CrossRef CAS.
- (a) Y. Tsutsumi, K. Yamakawa, M. Yoshida, T. Ema and T. Sakai, Org. Lett., 2010, 12, 5728–5731 CrossRef CAS; (b) Y.-B. Wang, D.-S. Sun, H. Zhou, W.-Z. Zhang and X.-B. Lu, Green Chem., 2014, 16, 2266–2272 RSC.
- (a) K. Vazdar, R. Kunetskiy, J. Saame, K. Kaupmees, I. Leito and U. Jahn, Angew. Chem., Int. Ed., 2014, 53, 1435–1438 CrossRef CAS; (b) E. D. Nacsa and T. H. Lambert, J. Am. Chem. Soc., 2015, 137, 10246–10253 CrossRef CAS.
- (a) C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuery, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef CAS; (b) S. Zhang and L.-N. He, Aust. J. Chem., 2014, 67, 980–988 CrossRef CAS; (c) Z. Xin, C. Lescot, S. D. Friis, K. Daasbjerg and T. Skrydstrup, Angew. Chem., Int. Ed., 2015, 54, 6862–6866 CrossRef CAS; (d) N.-K. Kim, H. Sogawa, K. Yamamoto, Y. Hayashi, S. Kawauchi and T. Takata, Chem. Lett., 2018, 47, 1063–1066 CrossRef CAS; (e) G. Li, J. Chen, D. Y. Zhu, Y. Chen and J. B. Xia, Adv. Synth. Catal., 2018, 360, 2364–2369 CrossRef CAS; (f) C. Zhang, Y. Lu, R. Zhao, W. Menberu, J. Guo and Z. X. Wang, Chem. Commun., 2018, 54, 10870–10873 RSC.
- (a) M. Yamashita, K. Goto and T. Kawashima, J. Am. Chem. Soc., 2005, 127, 7294–7295 CrossRef CAS; (b) S. Choi, J. H. Drese, P. M. Eisenberger and C. W. Jones, Environ. Sci. Technol., 2011, 45, 2420–2427 CrossRef CAS; (c) A. Goeppert, M. Czaun, G. K. Surya Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 RSC; (d) T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS; (e) A. Kumar, D. G. Madden, M. Lusi, K. J. Chen, E. A. Daniels, T. Curtin, J. J. t. Perry and M. J. Zaworotko, Angew. Chem., Int. Ed., 2015, 54, 14372–14377 CrossRef CAS; (f) E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS; (g) F. M. Brethomé, N. J. Williams, C. A. Seipp, M. K. Kidder and R. Custelcean, Nat. Energy, 2018, 3, 553–559 CrossRef; (h) C. J. E. Bajamundi, J. Koponen, V. Ruuskanen, J. Elfving, A. Kosonen, J. Kauppinen and J. Ahola, J. CO2 Util., 2019, 30, 232–239 CrossRef; (i) X. Shi, H. Xiao, H. Azarabadi, J. Song, X. Wu, X. Chen and K. S. Lackner, Angew. Chem., 2019, 59, 6984–7006 CrossRef; (j) S. P. Singh, P. Hao, X. Liu, C. Wei, W. Q. Xu, N. Wei, X. Li, H. Lu and A. Y. Ku, Joule, 2019, 3, 2154–2164 CrossRef CAS.
- (a) D. J. Heldebrant, P. G. Jessop, C. A. Thomas, C. A. Eckert and C. L. Liotta, J. Org. Chem., 2005, 70, 5335–5338 CrossRef CAS; (b) B. R. Van Ausdall, J. L. Glass, K. M. Wiggins, A. M. Aarif and J. Louie, J. Org. Chem., 2009, 74, 7935–7942 CrossRef CAS; (c) C. A. Seipp, N. J. Williams, M. K. Kidder and R. Custelcean, Angew. Chem., Int. Ed., 2017, 56, 1042–1045 CrossRef CAS.
- (a) M. Luo, X. H. Zhang and D. J. Darensbourg, Acc. Chem. Res., 2016, 49, 2209–2219 CrossRef CAS; (b) J. Ying, H. Wang, X. Qi, J.-B. Peng and X.-F. Wu, Eur. J. Org. Chem., 2018, 688–692 CrossRef CAS; (c) J. Ying, C. Zhou and X.-F. Wu, Org. Biomol. Chem., 2018, 16, 1065–1067 RSC.
- M. Vlasse, S. Giandinoto, S. T. Attarwala, Y. Okamoto and T. J. Emge, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 487–490 CrossRef.
- M. T. C. Ang, L. Phan, A. K. Alshamrani, J. R. Harjani, R. Y. Wang, G. Schatte, N. J. Mosey and P. G. Jessop, Eur. J. Org. Chem., 2015, 7334–7343 CrossRef CAS.
- N. von Wolff, C. Villiers, P. Thuéry, G. Lefèvre, M. Ephritikhine and T. Cantat, Eur. J. Org. Chem., 2017, 676–686 CrossRef CAS.
- (a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS; (b) Z.-Z. Yang, L.-N. He, Y.-N. Zhao, B. Li and B. Yu, Energy Environ. Sci., 2011, 4, 3971–3975 RSC; (c) Z.-Z. Yang, L.-N. He, J. Gao, A.-H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602–6639 RSC; (d) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS; (e) C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef; (f) J. Wang and Y. Zhang, ACS Catal., 2016, 6, 4871–4876 CrossRef CAS; (g) B. Grignard, S. Gennen, C. Jerome, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC; (h) Z. Zhang, X.-Y. Zhou, J.-G. Wu, L. Song and D.-G. Yu, Green Chem., 2019, 22, 28–32 RSC; (i) S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS.
- X. F. Liu, X. Y. Li, C. Qiao, H. C. Fu and L. N. He, Angew. Chem., Int. Ed., 2017, 56, 7425–7429 CrossRef CAS.
- (a) J. W. Clark-Lewis, Chem. Rev., 1958, 58, 63–99 CrossRef CAS; (b) R. L. Dow, B. M. Bechle, T. T. Chou, D. A. Clark, B. Hulin and R. W. Stevenson, J. Med. Chem., 1991, 34, 1538–1544 CrossRef CAS; (c) Y. Momose, T. Maekawa, T. Yamano, M. Kawada, H. Odaka, H. Ikeda and T. Sohda, J. Med. Chem., 2002, 45, 1518–1534 CrossRef CAS; (d) K. Evason, C. Huang, I. Yamben, D. F. Covey and K. Kornfeld, Science, 2005, 307, 258–262 CrossRef CAS; (e) J. M. Cox, H. D. Chu, C. Yang, H. C. Shen, Z. Wu, J. Balsells, A. Crespo, P. Brown, B. Zamlynny, J. Wiltsie, J. Clemas, J. Gibson, L. Contino, J. Lisnock, G. Zhou, M. Garcia-Calvo, T. Bateman, L. Xu, X. Tong, M. Crook and P. Sinclair, Bioorg. Med. Chem. Lett., 2014, 24, 1681–1684 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, NMR spectra. CCDC No. 1997581 (3b), 1997583 (4a), 1997582 (6), 1997584 (8a), 1997585 (8g). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0gc03009k |
| This journal is © The Royal Society of Chemistry 2020 |