
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The preparation and applications of amides using electrosynthesis
Peter W.
Seavill
 * and
Jonathan D.
Wilden
*
* and
Jonathan D.
Wilden
*
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: pseavill@gmail.com; j.wilden@ucl.ac.uk
First published on 2nd November 2020
Abstract
The synthesis of amides is extremely important given the ubiquitous presence of this motif in biological systems, as well as in the pharmaceutical industry, where it is estimated that amide preparation is the most common chemical reaction employed. As such, greener routes to this motif have long been a vital research goal. Amide groups are also synthetically versatile, capable of taking part in a wide range of different transformations, especially when subjected to electrochemical conditions. The current resurgence of electrosynthesis has led to new and more sustainable syntheses of (and using) amides to be published in recent times. As such, this tutorial review aims to highlight these reactions. Given the vital importance of amides and the growing interest in electrosynthesis, the combination of these two topics is an exciting and interesting field of research that holds a lot of promise in the future of synthetic chemistry.
 Peter W. Seavill | Peter Seavill obtained his MSci from University College London (UCL) in 2016, undertaking a fourth-year research project in the synthesis of chiral alkynyl sulfoxides supervised by Dr Jonathan Wilden. He then continued his studies at UCL as a Ph.D. student in the groups of Dr Jonathan Wilden and Prof. Katherine Holt in the interdisciplinary field of Electro-Organic Synthesis. During his Ph.D., Peter's research centred around developing new electrochemical methodology for use in synthesis, particularly through efficient copper-catalysed processes. For this work, he was awarded the Davies Prize and the Ramsay Medal in 2019, before obtaining his Ph.D. in 2020. |
 Jonathan D. Wilden | Jon Wilden is currently Associate Professor in organic chemistry at University College London (UCL) and from 2005 was Assistant Professor (lecturer) at the same institution. With a previous interest in organic synthesis and electron transfer reactions, the Wilden group has been focused on Electro-Organic Synthesis in recent times and has embraced the green and sustainable qualities of this technique. Prior to this, he worked as a postdoctoral researcher with Prof. Stephen Caddick both at the University of Sussex and at UCL. His Ph.D. studies focused on the total synthesis of a complex natural product at the University of Southampton with Prof. David Harrowven. |
1. Introduction
It is difficult to overstate the importance of the amide functionality in molecular sciences. Most notably, this motif is the critical bond that forms the backbone of peptides, proteins and a wealth of other biomolecules.1 Medicinally, this group is also of critical importance, with amide-forming reactions estimated to be the most common reaction carried out in the pharmaceutical industry2 and approximately a quarter of all marketed drugs (and two-thirds of all drug candidates) contain at least one amide bond.3 A consideration of how amides interact with biological targets (particularly with respect to N–H pKa and hydrogen bonding) is also a key aspect of drug discovery.4 Additionally, inorganic chemists have exploited amides with associated anisotropy for their properties in chelation and ligand design and consequently the amide motif appears in a wealth of coordination complexes for various applications.5–7With amides being ubiquitous building blocks throughout the chemical sciences, it is perhaps unsurprising that chemists have expended much research effort on developing synthetic approaches to this important functional group. Conceptually, the most attractive way of achieving this is by the union of a carboxylic acid and an amine in a classic condensation reaction involving the expulsion of a molecule of water.3 This transformation has been so elegantly perfected in biological systems with enzymes being able to achieve this reaction with ease under the mildest of conditions.8 Unfortunately, this seemingly simple transformation presents several problems to the organic chemist hoping to replicate the reaction in vitro. Most strikingly, the innate acidity of the carboxylic acid and basicity of the amine nucleophile mean that both species are often in ionised forms, prohibiting their direct condensation.3 A number of solutions have been developed to address this issue. The earliest solution involves the formation of an acid halide which then reacts with an amine nucleophile to form the desired amide bond.9 Acid halides are, however, highly reactive and consequently incompatible with many of the other functional groups that may be present, particularly in biomolecules (not to mention the harsh conditions and reagents required to prepare them).
Coupling agents have also been developed as a way to mediate amide synthesis, examples of which are shown in Fig. 1: N,N′-dicyclohexylcarbodiimide (DCC), 1-ethyl-3-(3′-dimethylaminopropyl)-carbodiimide hydrochloride (EDC), N-[(dimethylamino)-1H-1,2,3-triazolo[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide (HATU), 1,1′-carbonyldiimidazole (CDI), (benzotriazol-1-yloxy)-tris(dimethylamino)-phosphonium hexafluorophosphate (BOP).9
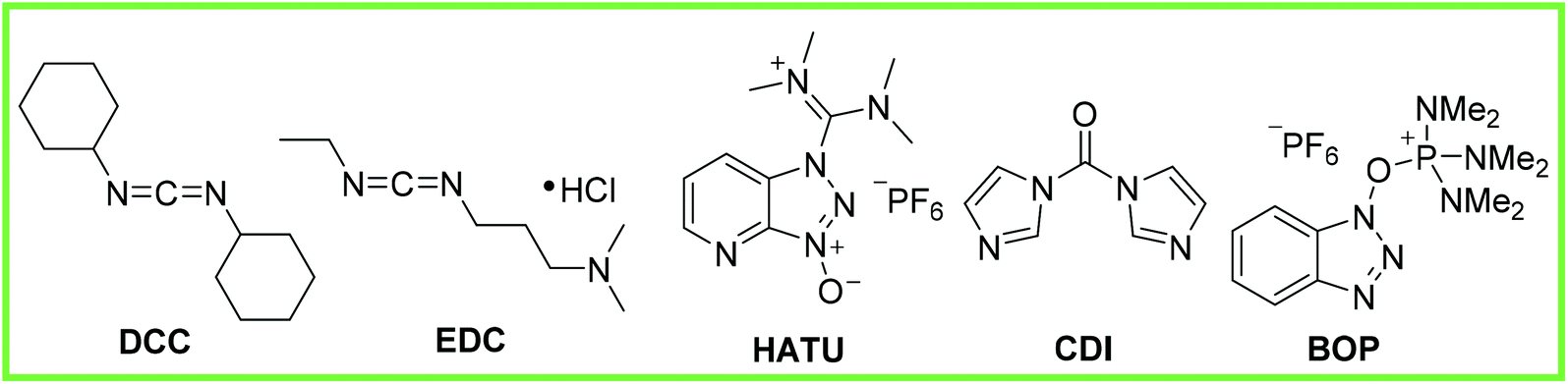 | ||
| Fig. 1 Showing an array of coupling agents used to make amide bonds.9 | ||
The general mechanism of these reagents is to activate the carboxylic acid to the nucleophilic amine. The reaction generally works in high yield, however, without exception these reagents are stoichiometric in nature and in no way could be considered ‘atom efficient’ where the waste products (e.g. ureas and phosphonamides) have high molecular weights and must be separated from the desired products at the end of the reactions.9,10 Furthermore, these coupling agents can be hazardous in terms of toxicity and environmental impact.11
Recent years have seen a much greater appreciation of the need to develop green and sustainable processes both in industrial and academic settings. Consequently, numerous groups have presented alternative and creative approaches to amide bond formation including: Lewis acid catalysts, transition metal catalysis and various redox approaches as greener alternatives.12 Furthermore, with the growing wave of interest in electrosynthesis,13–15 it was perhaps only a matter of time before the widely acknowledged green and sustainable credentials of electrosynthesis were recognized as a tool for amide bond synthesis.
Electrochemical methods offer new modes of reactivity and numerous advantages for the preparation of compounds in a sustainable and green fashion, such as improved safety credentials from hazardous reagents being generated in situ rather than handled directly16,17 (or not being required at all). In addition, there is often no need to add external oxidants, transition metals or mediators,18 which in turn improves the atom economy of such processes.15 These key aspects of sustainable synthesis offered by electrochemistry have been exemplified in many electrochemical amide syntheses, as will be shown in this review, and have led to very mild conditions being used to produce amides when compared to traditional methods. The same is true for electrochemical syntheses that utilise amide starting materials for various applications.
The first part of this tutorial review therefore explores the approaches that have been demonstrated to synthesise amides using electrochemistry, whilst the second part is dedicated to the wide-ranging applications of amides in electrosynthesis. The scope of this review is confined to examples with direct involvement of the amide group in the reaction rather than mentioning all electrosyntheses involving substrates that contain an amide group. For this, extensive reviews of general electrosynthetic chemistry have previously been published.13–15 Recently, excellent ‘beginner's guide’-style reviews for synthetic chemists looking to learn how to carry out reactions electrochemically have also been published.19,20 Therefore, this review focuses exclusively on synthetic procedures used to produce and utilise amides, rather than provide a discussion of electrochemical techniques and equipment.
2. Electrosynthesis of amides
2.1. Anodic oxidation of iodide
A very common reaction in electrosynthesis is the oxidation of halide species to produce halogen radicals or diatomic halogens in situ. This circumvents the need to directly handle hazardous halogen species and is therefore a prime example of the improved safety credentials that electrosynthetic protocols offer over standard syntheses. Iodide salts in particular are often used for this, owing to their significantly lower standard reduction potential when compared to other halogens (I2 = +0.54 V, Br2 = +1.07 V, Cl2 = +1.36 V vs. SHE). In other words, I2 is harder to reduce than Br2 and Cl2, which means that I− is easier to oxidise than Br− and Cl−. For the purpose of electrochemical amide preparation, several synthetic strategies have been derived from iodide salt oxidation and will be discussed in this section.In 2013, Huang and co-workers reported an interesting example of an iodoform reaction carried out in an undivided cell on methyl ketones 1 in the presence of formamides 2 to produce the desired amide products (Scheme 1).21
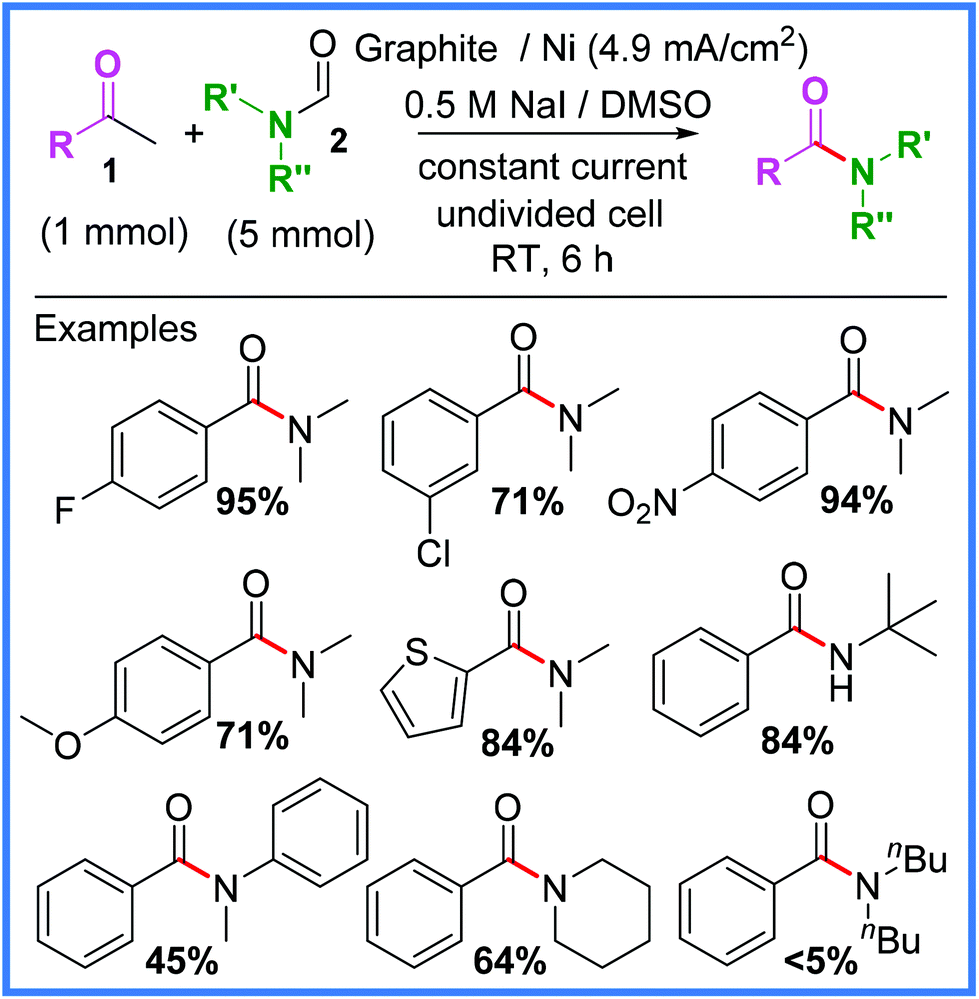 | ||
| Scheme 1 Electrochemical iodoform reaction and scope.21 | ||
It was found that NaI performed best as the iodide source in this reaction because when iodides with different cations were used (NH4+ or nBu4N+), by-products derived from the ammonium cation such as benzamide would form. This was due to the ammonium cations being reduced at the cathode to produce undesired, competitive amines. A range of aryl methyl ketone substituents were tolerated using these conditions, as well as several different formamides. However, it was noted that particularly sterically hindered examples, such as dibutyl formamide, afforded much lower yields.21
The mechanism for this reaction was investigated and proposed as in Scheme 2. Anodic oxidation of NaI produces I2in situ, which allows the methyl ketone starting material 1 to be transformed into the triiodomethyl ketone species 3. Meanwhile, at the nickel cathode, the formamide species 2 is thought to be converted to an amine with the liberation of carbon monoxide. This amine then reacts with 3 to release iodoform and yield the desired amide product.21
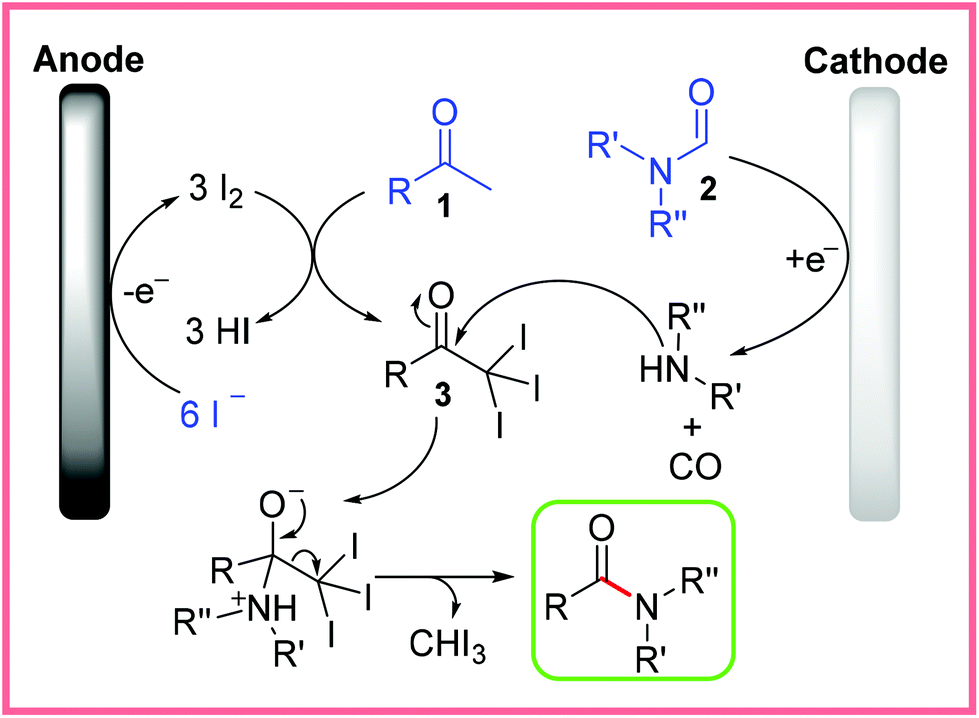 | ||
| Scheme 2 Proposed electrochemical iodoform mechanism.21 | ||
Another example using anodically oxidised iodide and methyl ketones, this time to produce α-ketoamides 4, was reported by Zha and Wang.22 In this work, an O2 balloon was used in conjunction with similarly mild reagents in an undivided cell to deliver a range of amides in moderate to good yields as shown in Scheme 3. A range of amines was tolerated.22
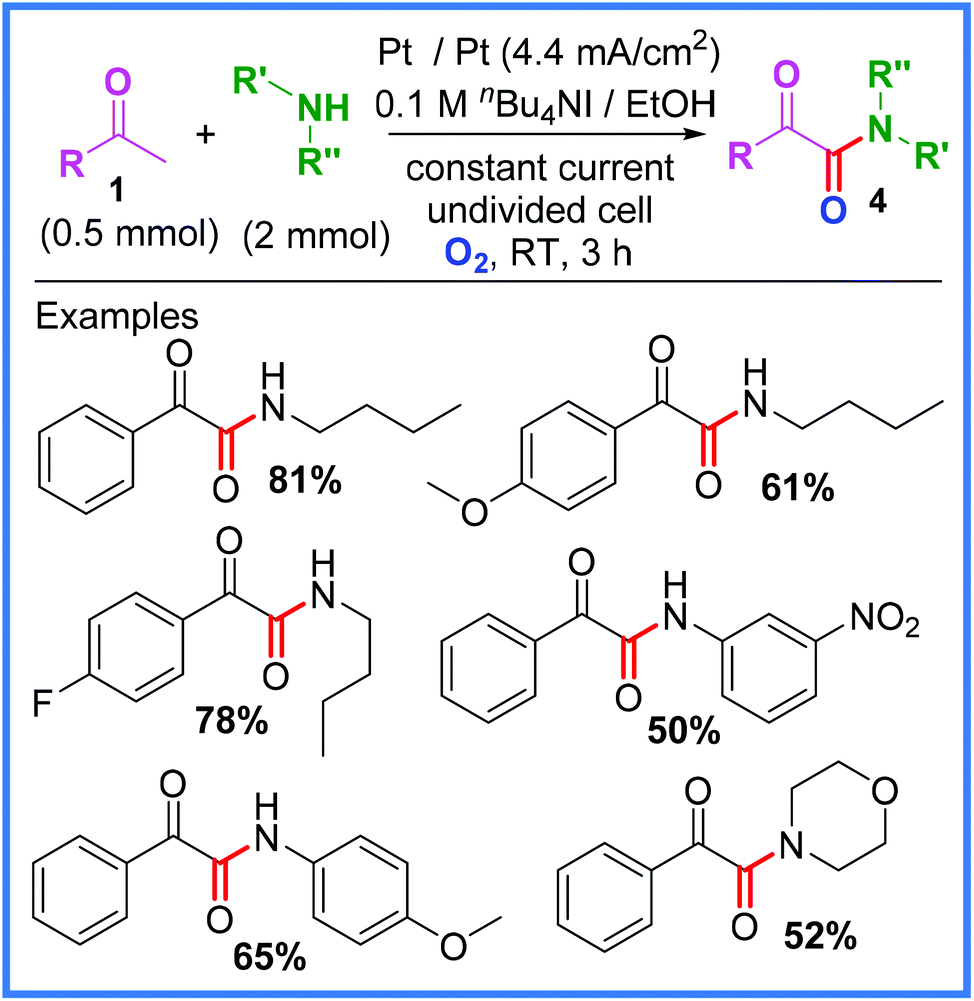 | ||
| Scheme 3 Electrochemical α-ketoamide synthesis and scope.22 | ||
Furthermore, when ammonium acetate was used in place of an amine substrate, as in Scheme 4, α-oxophenylacetamide derivatives 5 could be produced in good yield. In order to increase the solubility of the ammonium acetate, MeOH was used as the solvent instead of EtOH, which in turn led to KI being used as the iodide source. It was also found that a basic environment was crucial to the success of this reaction and so tBuNH2 was added to modulate this. Interestingly, a strong electron-donating substituent on the phenyl group (p-OMe) was found to drastically lower the yield of this reaction, giving only 5% of the desired product.22
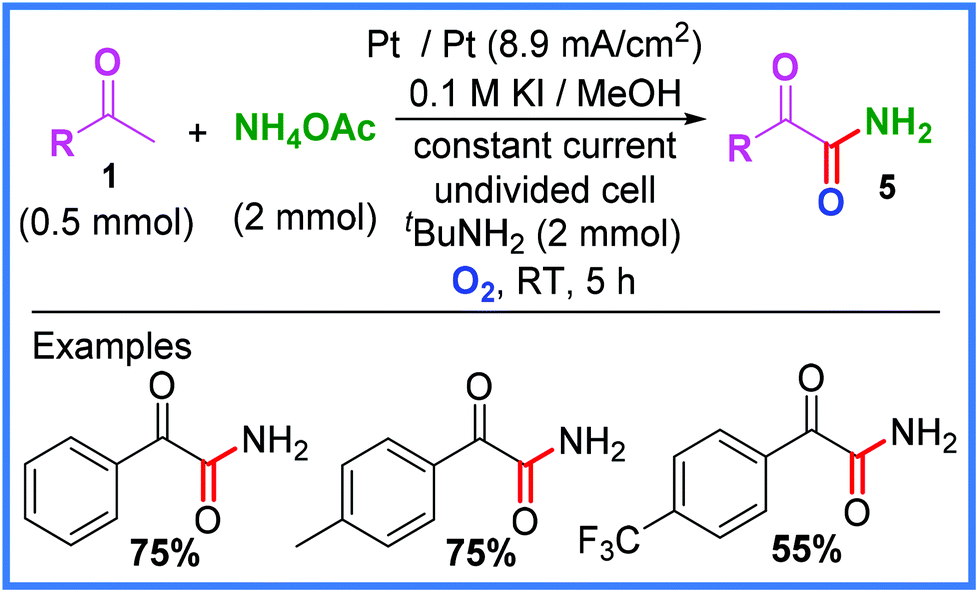 | ||
| Scheme 4 Electrochemical α-ketoamide synthesis using ammonium acetate.22 | ||
The proposed mechanism for this reaction is shown in Scheme 5, with the oxidation of iodide being the first step. Following this, the iodine free radical can react with the methyl ketone substrate 1 to generate the methyl ketone radical 6, which readily accepts O2 to form 7. The unstable species 7 then splits apart to form the oxo acetaldehyde 8 and a hydroxyl radical. The oxo acetaldehyde 8 undergoes nucleophilic attack from amine substrates to yield 9. A final oxidation step of 9 at the anode then produces the desired α-ketoamide product 4. The hydroxyl radical formed during the split of 7 is thought to be reduced at the cathode.22
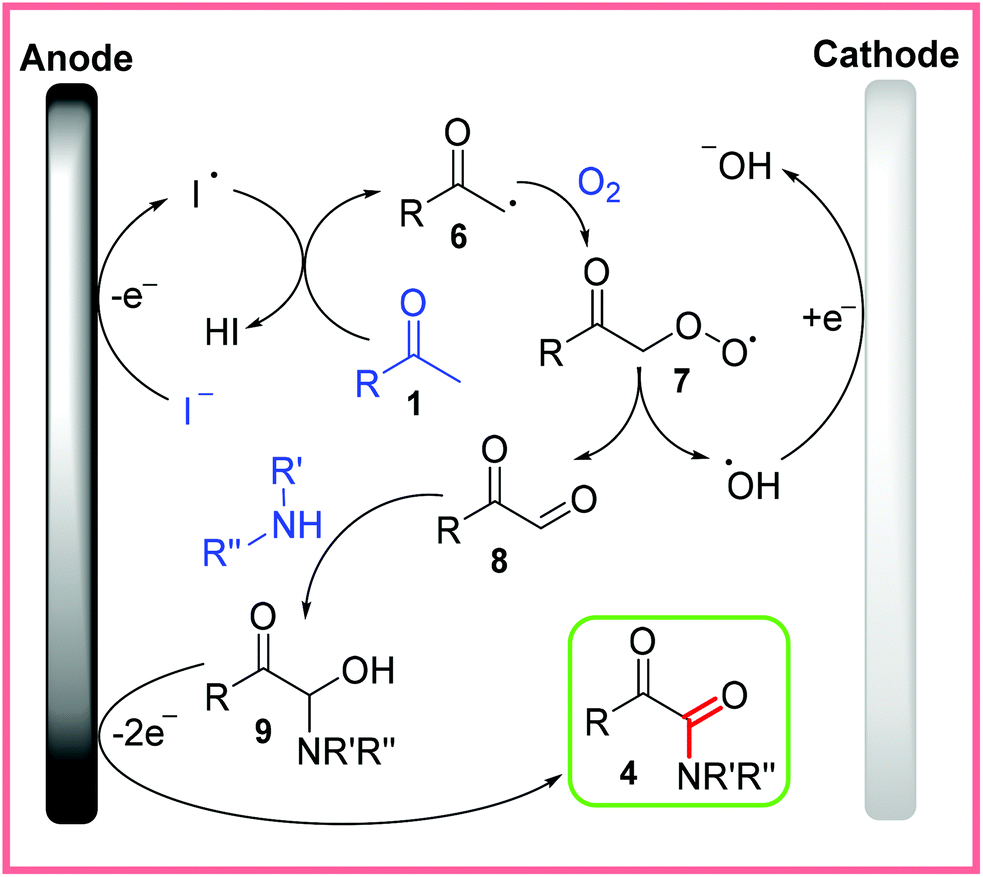 | ||
| Scheme 5 Proposed mechanism for electrochemical α-ketoamide synthesis.22 | ||
It should also be noted that very recently,23 a general method for electrochemical α-ketoamide 4 synthesis has been published which does not require a halide mediator or even an electrolyte, however, this process starts from α-ketoaldehyde starting materials rather than methyl ketones.
Later work by Zha and Wang showed how their approach could be extended to the production of isatins 10, which are important heterocycle skeletons found in various natural products and pharmaceuticals.24 This work was achieved through the coupling of 2-aminoacetophenones 11 in an undivided cell and is shown, with some examples, in Scheme 6. The mechanism for this reaction was proposed to be almost identical to that shown in Scheme 5.24
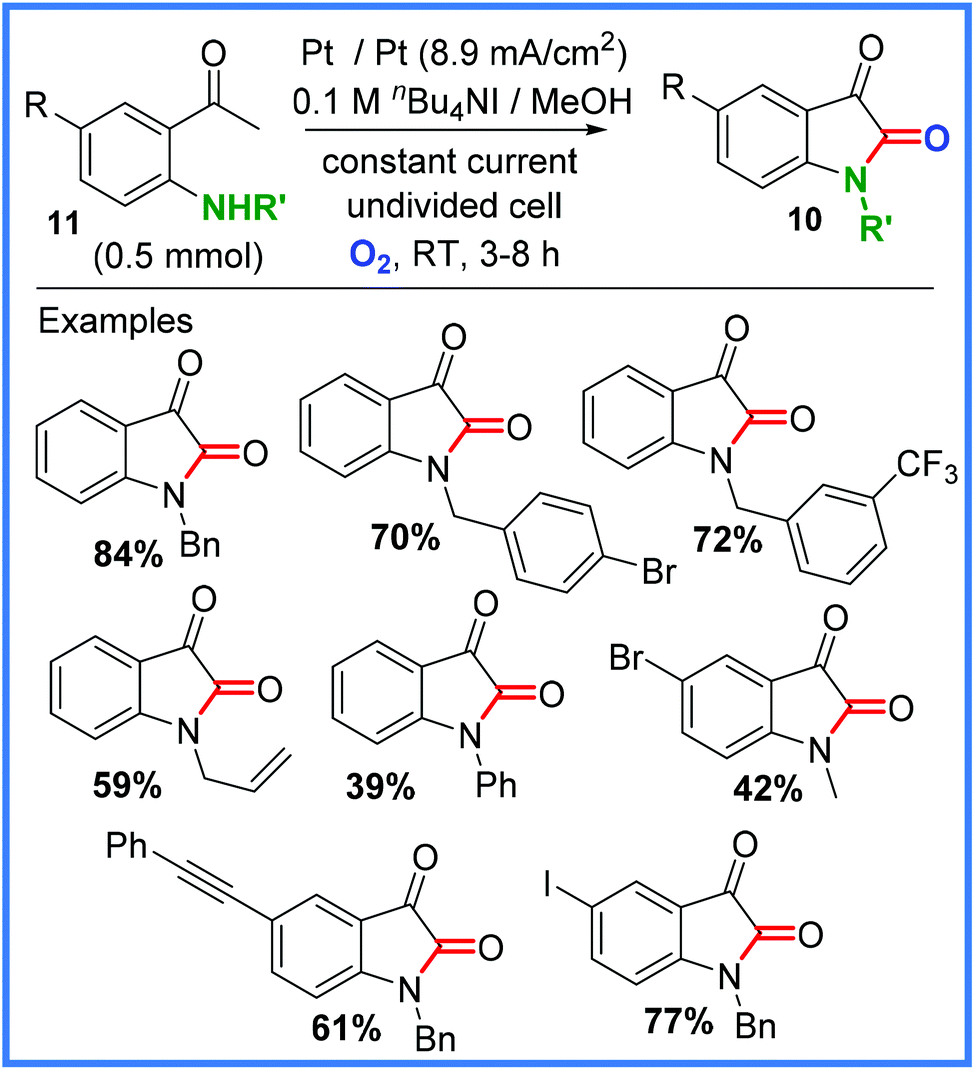 | ||
| Scheme 6 Electrochemical synthesis of isatins.24 | ||
Finally, this group have also shown how iodide oxidation can be utilised to produce phosphinic amides.25
2.2. Nitrile groups as an amide source
Another strategy for electrochemically preparing amides is the use of a nitrile group and water. Often this can be achieved through the use of mixed solvent systems (particularly MeCN/H2O), making these reactions quite efficient and sustainable. Variations of this approach will be shown in this section starting with an early example from Becker and co-workers in 1988 who directly oxidised polyfluoroalkyl iodides in the presence of MeCN.26 This example combines iodo-species oxidation (as seen in the previous section) with a nitrile group acting as the amide source. In the example shown in Scheme 7, 12 was isolated in 67% yield when a glassy carbon working electrode (WE) was used in a divided cell and a potential of +2.3 V (vs. a Ag wire quasi-reference electrode) was applied.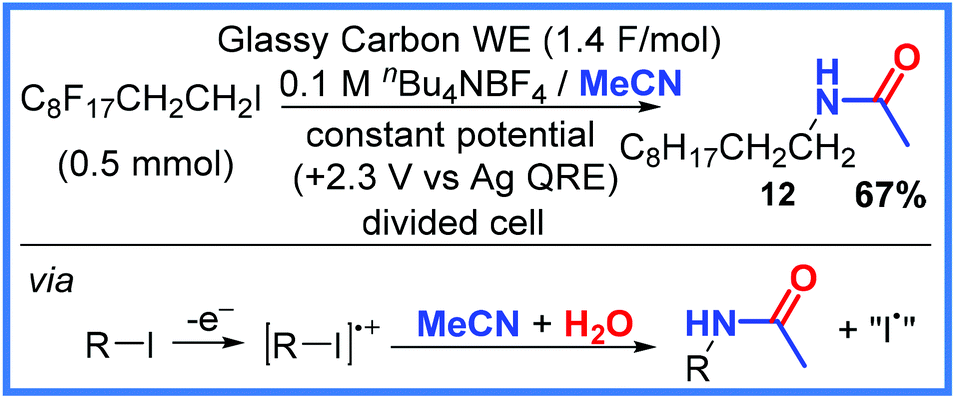 | ||
| Scheme 7 Electrochemical iodide oxidation and reaction with MeCN.26 | ||
A more recent example using a similar approach comes from the Ley group,27 who showed that toluenes could be activated using anodic flow oxidation in the presence of MeCN to effectively yield benzyl amides. This work, shown in Scheme 8, used a Pt anode and a stainless steel (SS) cathode as electrodes in a flow system with MeCN and H2O solvents providing the key amide structure. It was found that the reaction is likely to proceed via carbocation formation (13) at the anode with the removal of 2 electrons and the loss of H+. The resulting carbocation 13 then reacts with MeCN and H2O to generate the desired amide product. It was also found that adding a Brønsted acid to the reaction mixture (methanesulfonic acid) allowed the reaction to be carried out more smoothly and at a lower voltage. The use of a second stream consisting of NH3/MeOH proved beneficial as it fully neutralised the acid to mitigate decomposition of the amide products. The reaction taking place at the cathode was proposed to be the reduction of H+ when nBu4NPF6 was used as electrolyte and the reduction of Li+ when LiBF4 was used. These conditions were tolerated by a range of functional groups to give reasonably good yields.27
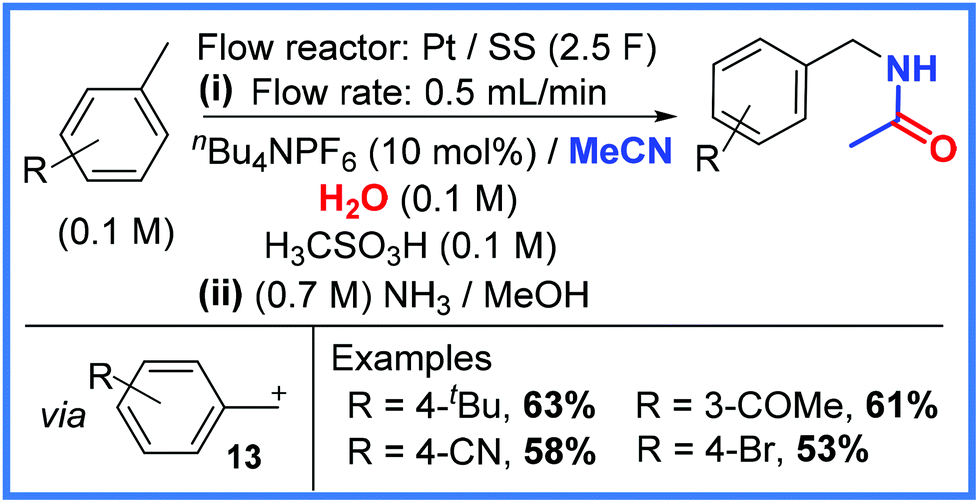 | ||
| Scheme 8 Anodic flow oxidation in the presence of MeCN and H2O to produce amides.27 | ||
There are also examples of amide bonds being formed from nitrile groups other than in MeCN, such as the work of Estrada and Rieker from 1994,28,29 which showed how 2-nitrobenzonitrile 14 could be converted into quinazolinones 15 in the presence of various alcohols. This reaction employed a mercury pool cathode and a carbon anode in an undivided cell and is shown in Scheme 9.
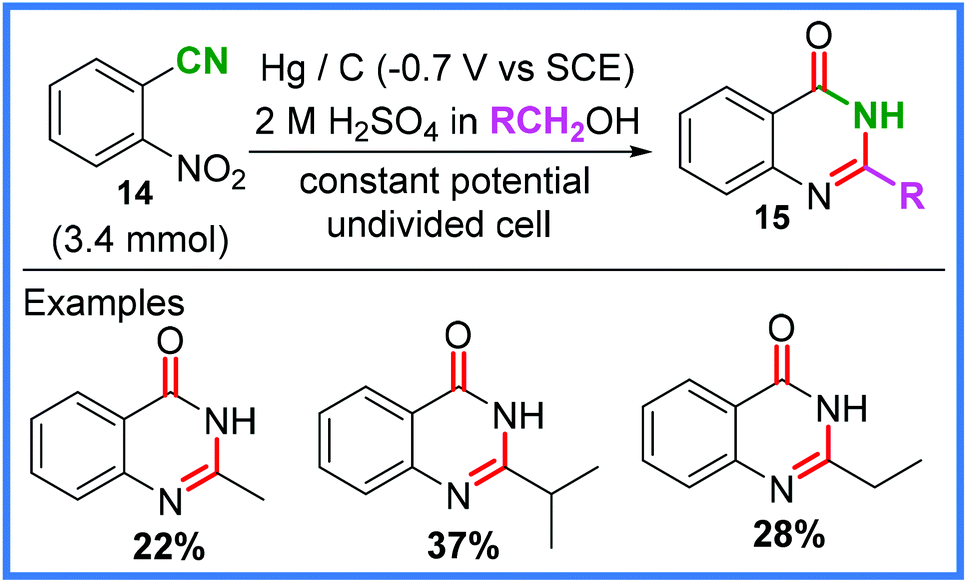 | ||
| Scheme 9 Electrochemical quinazolinone synthesis and scope.28,29 | ||
The use of mercury as an electrode material is rather undesirable from a sustainability standpoint and whilst the yields for this reaction were fairly low, the proposed mechanism is quite intricate and so is shown in Scheme 10. Alcohol solvents were oxidised to aldehydes 16 at the anode. At the same time, the nitro group of 14 was reduced to a hydroxylamine 17, allowing 16 and 17 to react together through a series of dehydration and cycloaddition steps to eventually form the desired quinazolinone product 15.28,29
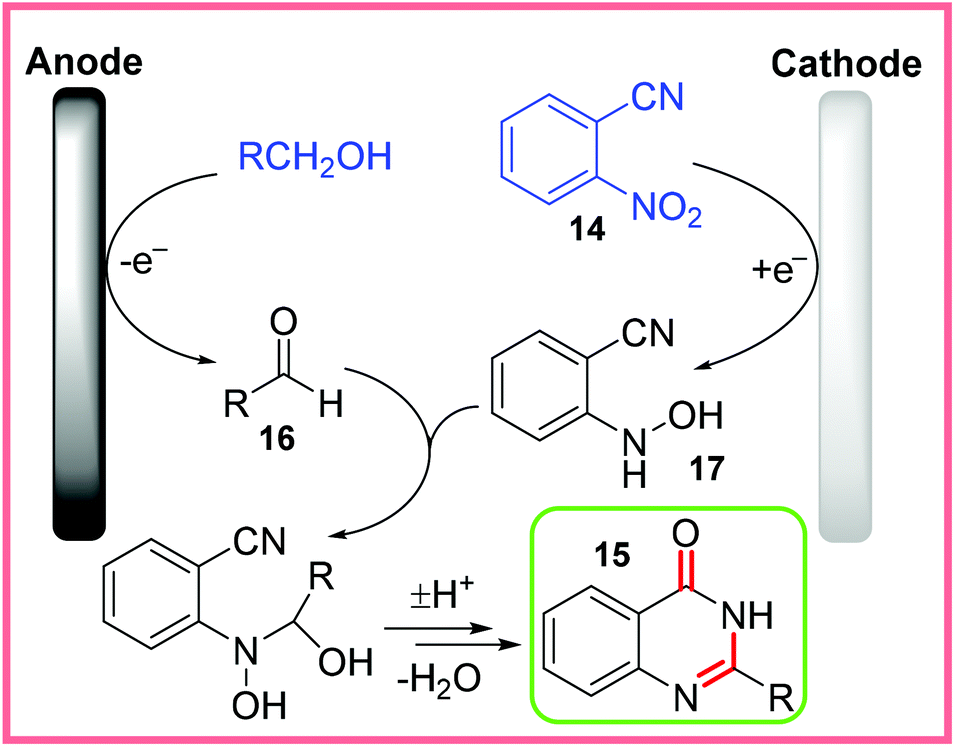 | ||
| Scheme 10 Proposed mechanism for electrochemical quinazolinone synthesis.28,29 | ||
2.3. Rearrangement reactions
Rearrangement reactions have also been utilised to produce amides electrochemically. One such example was reported by Liu and co-workers,30 who used the oxidation of an iodide salt to initiate a Favorskii rearrangement of 1,3-diarylacetones 18 in an undivided cell. This reaction, shown in Scheme 11, was found to be catalysed best by nBu4NI and when other halide salts such as nBu4NCl were used, the reaction was much lower yielding. A range of substituents were tested in this work, including symmetric and non-symmetric ketones, as well as different amines. Most examples tested gave moderate to good yields, however, some exceptions include dibenzyl ketones bearing electron-donating substituents (such as OMe), as these did not undergo the desired Favorskii rearrangement (though did still produce amide products).30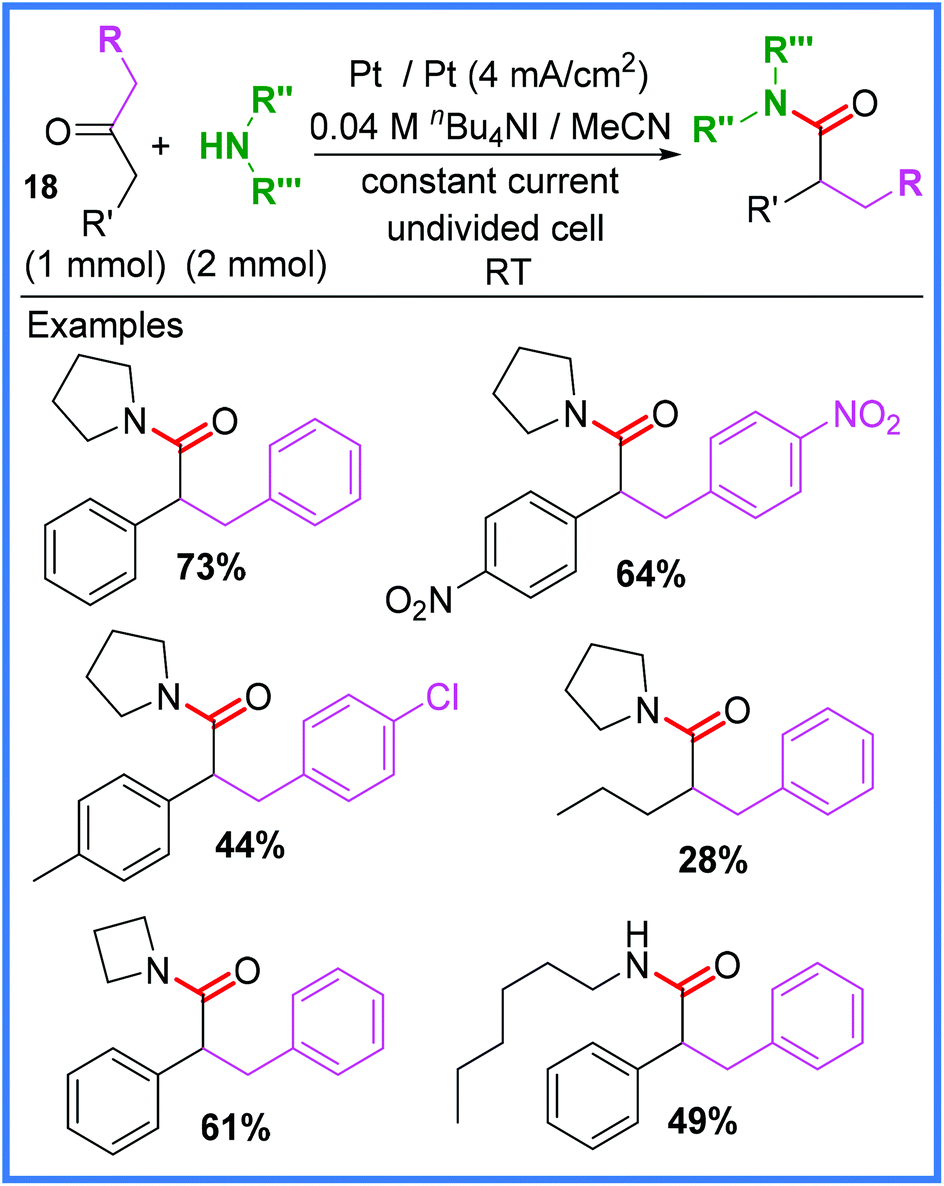 | ||
| Scheme 11 Electrochemical Favorskii rearrangement.30 | ||
The mechanism for this reaction is shown in Scheme 12, starting with iodide oxidation to produce I2, which reacts with 18 to form an α-iodoketone 19. Reaction of 19 with an amine to produce enamine 20 then follows, which allows the cyclopropanone intermediate 21 to form. Attack of 21 with another molecule of amine opens the cyclopropanone up and leads to the carbanion 22, before hydrolysis produces the final, desired product. The reaction taking place at the cathode was proposed to be the reduction of H+ ions.30
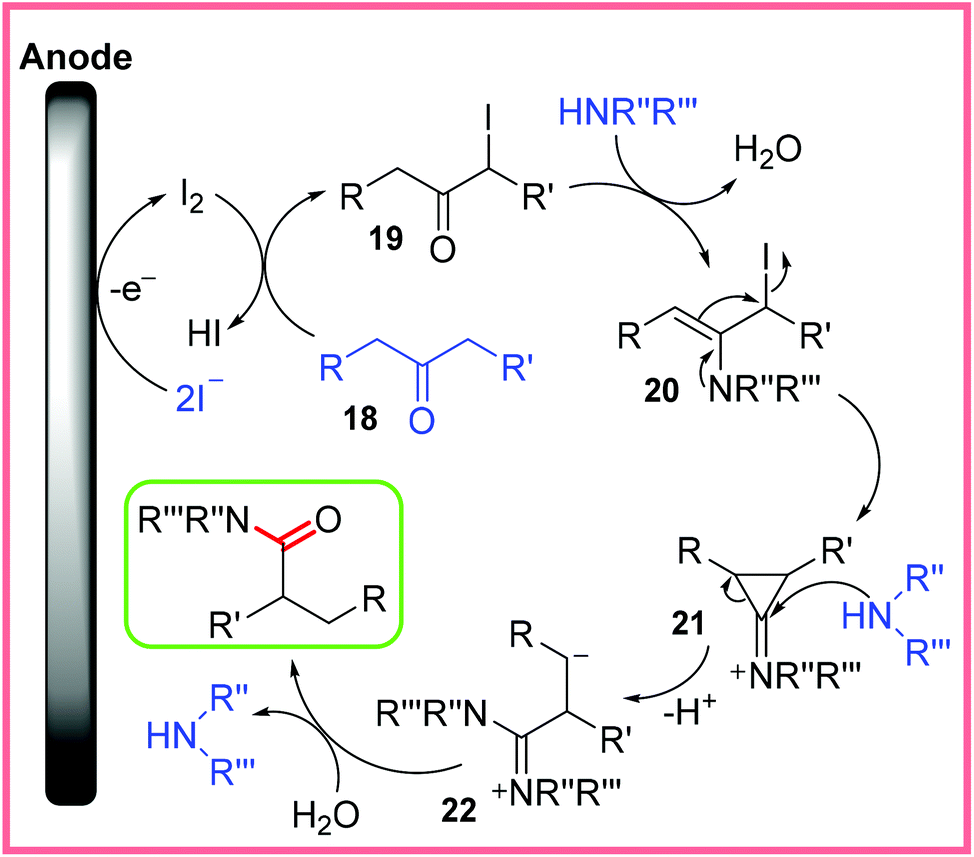 | ||
| Scheme 12 Proposed mechanism for an electrochemical Favorskii rearrangement.30 | ||
Other electrochemically-induced Favorskii rearrangements to produce amides have been reported, including the work of Inesi and co-workers,31 who reduced polyhaloketone substrates to elicit the desired reaction.
Another important rearrangement that has been utilised for electrochemical amide synthesis is the Smiles rearrangement. In 2019, Chang and co-workers developed a base-free, radical variant of this reaction, generating amidyl radicals through the reduction of electron-deficient arenes in an undivided cell.32 Di-nitro arenes 23 proved to be the most effective starting materials to use as they required comparatively low reduction potentials. The conditions for this reaction are shown in Scheme 13.32
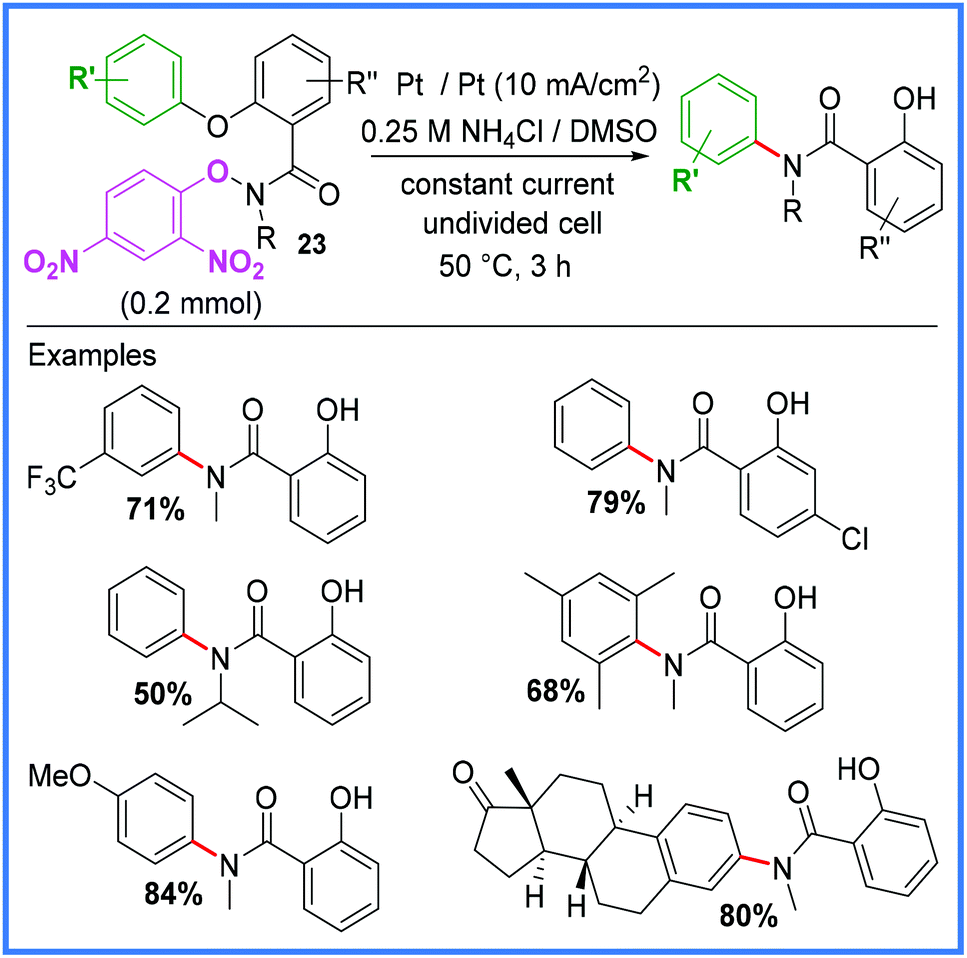 | ||
| Scheme 13 Electrochemical Smiles rearrangement.32 | ||
The scope of this reaction was quite broad, with even an estrone substituent being tolerated, and the reaction was possible on gram scale. The mechanism for this reaction is shown in Scheme 14, where 23 is first reduced to 24, prompting N–O bond fragmentation to produce the amidyl radical 25. This then allows the Smiles rearrangement to take place, forming the O-centred radical intermediate 26. Reduction at the cathode and protonation of 26 then yields the final product. The reaction taking place at the anode was thought to be the oxidation of Cl− ions to Cl2.32
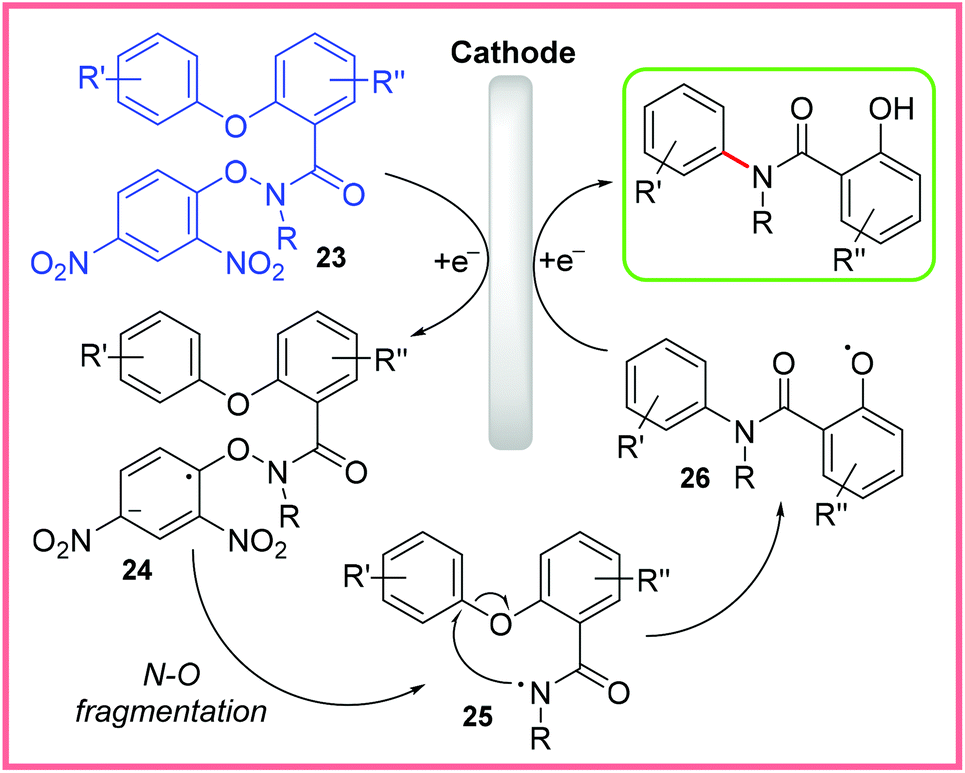 | ||
| Scheme 14 Plausible mechanism for electrochemical Smiles rearrangement.32 | ||
Other types of rearrangements that have been carried out electrochemically to produce amides include the Beckmann rearrangement, as reported by Tang and co-workers,33 and the Mumm rearrangement, as reported by Zhang and co-workers.34
2.4. Other anodic oxidations
As seen in Schemes 13 and 14, the di-nitrobenzene motif has been used in cathodic reduction reactions to yield amides, however, it has also been used in oxidative processes such as the Nucleophilic Aromatic Substitution of Hydrogen (NASH) reaction seen in Scheme 15.35 This process, demonstrated by Gallardo and co-workers, makes use of anodic oxidation to release H+ from the aromatic ring after nucleophilic attack, producing the desired product. This method proved reasonably effective for yielding a range of anilines when amines were used as the nucleophile, as well as the amide 27 shown in Scheme 15 when acetamide was used. The authors noted that careful control of the oxidative potentials applied was key to the success of this reaction.35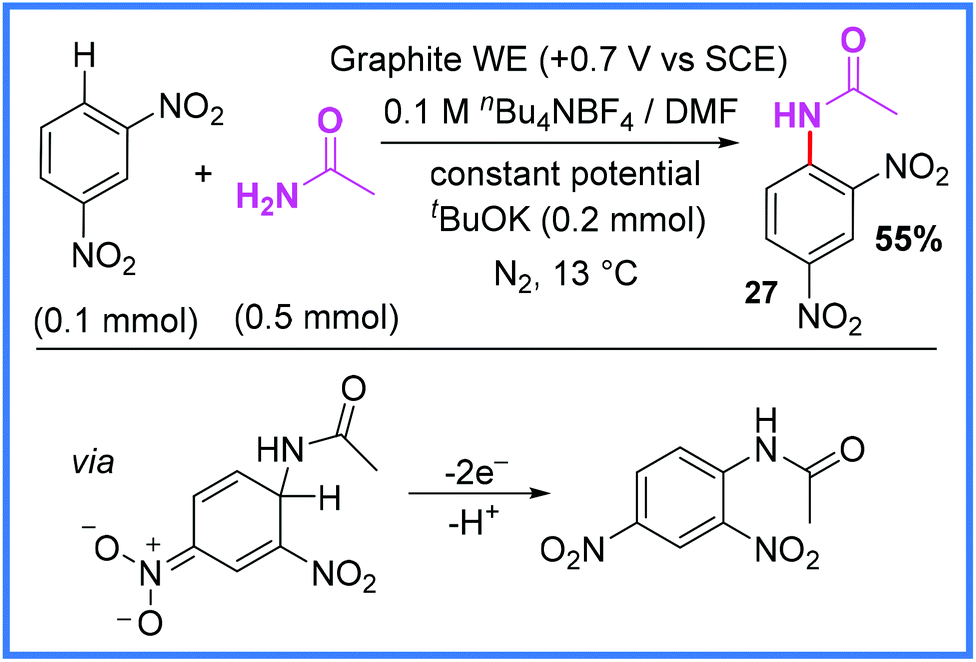 | ||
| Scheme 15 Electrochemical nucleophilic aromatic substitution of hydrogen reaction.35 | ||
Another example of an interesting anodic oxidation approach comes from Tang and co-workers,36 who used potassium thioacetate or thiocarboxylic acids to produce a range of amide products in an undivided cell. The use of thioacids as acyl sources often allows for mild conditions to be employed when synthesising amides and this is well evidenced in the electrochemical protocol shown in Scheme 16. The authors found that direct oxidation of thioacetic acid was quite low yielding, so the deprotonated potassium salt was used instead. It was also found that this problem could be circumvented by adding Et3N along with various thioacetic acids to ensure better yields. A range of substituents were tolerated using these conditions, however it should be noted that electron-rich amines performed better than electron-deficient.36
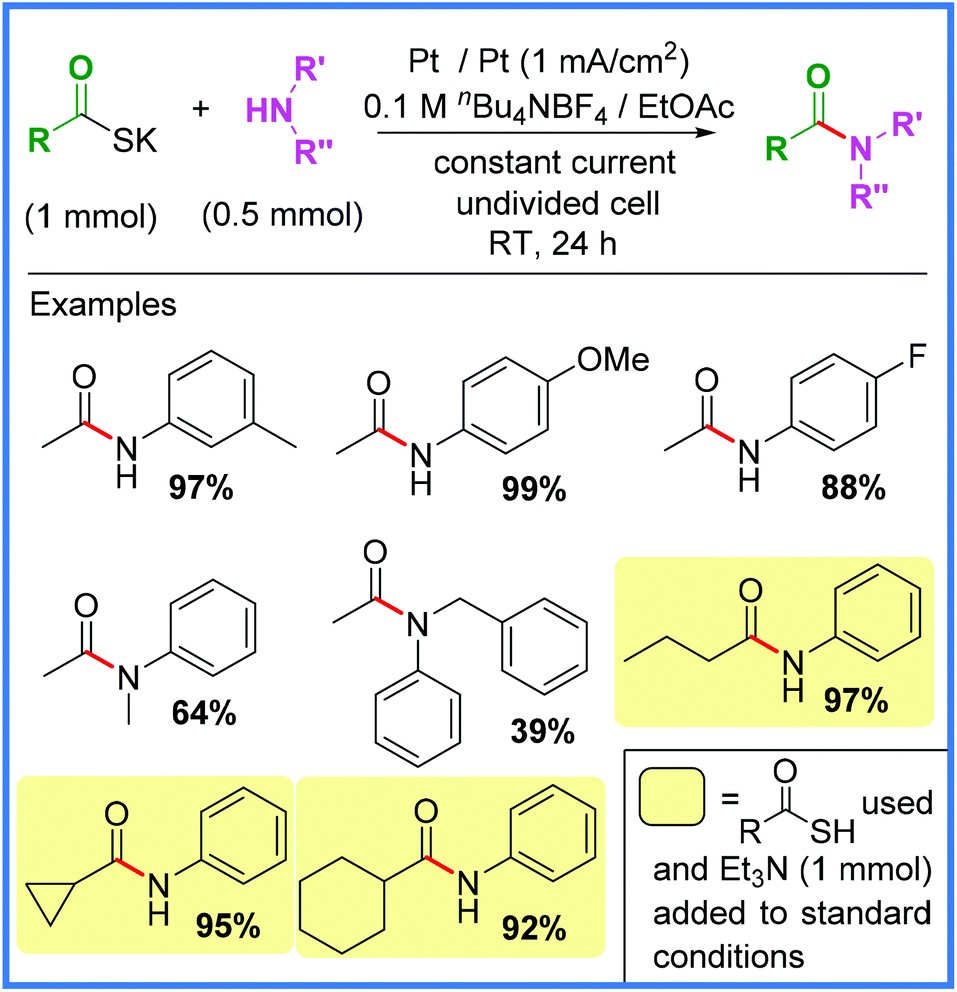 | ||
| Scheme 16 Electrochemical amide synthesis from thiocarboxylic acids.36 | ||
It is believed that this reaction proceeds via oxidation of 28 to give a radical 29, which then dimerises to give the disulfide 30 as shown in Scheme 17. Nucleophilic attack by an amine then leads to the amide product. The proposed reaction taking place at the cathode was the reduction of H+.36
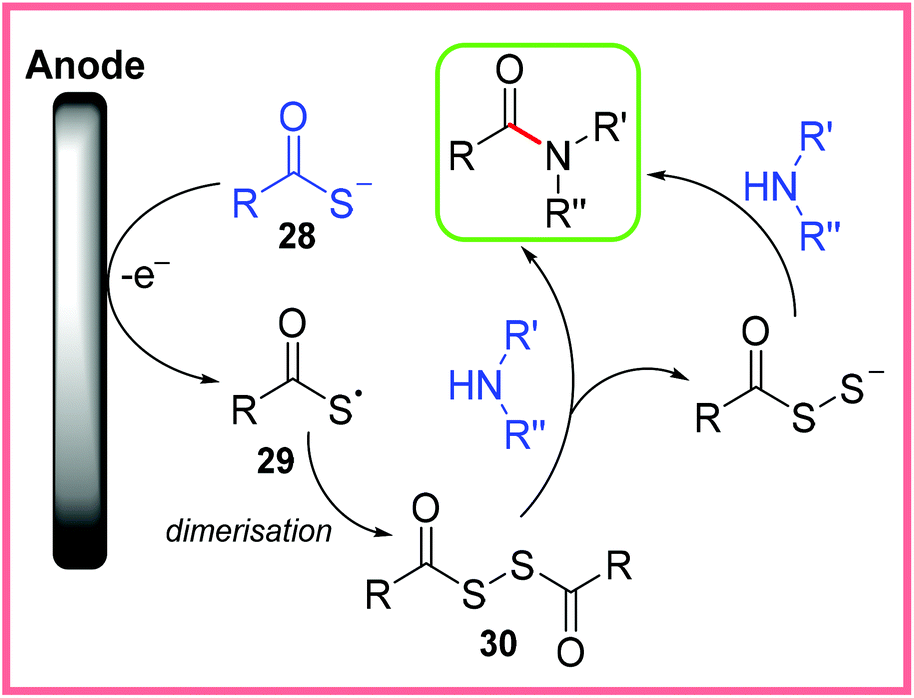 | ||
| Scheme 17 Proposed mechanism for electrochemical amide synthesis from thiocarboxylic acids.36 | ||
Examples of nitroxyl radical mediators being used in electrosynthesis to promote oxidation of N-methylanilines 31 has also been demonstrated as a viable method of amide synthesis. Kashiwagi and Anzai37 used an analogue of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) 32a (Scheme 18) in various oxidation states to produce N-alkylformanilides 33 by incorporating oxygen atoms into 31 from H2O in a divided cell. This was achieved using a controlled oxidative potential and fairly mild conditions.14,37
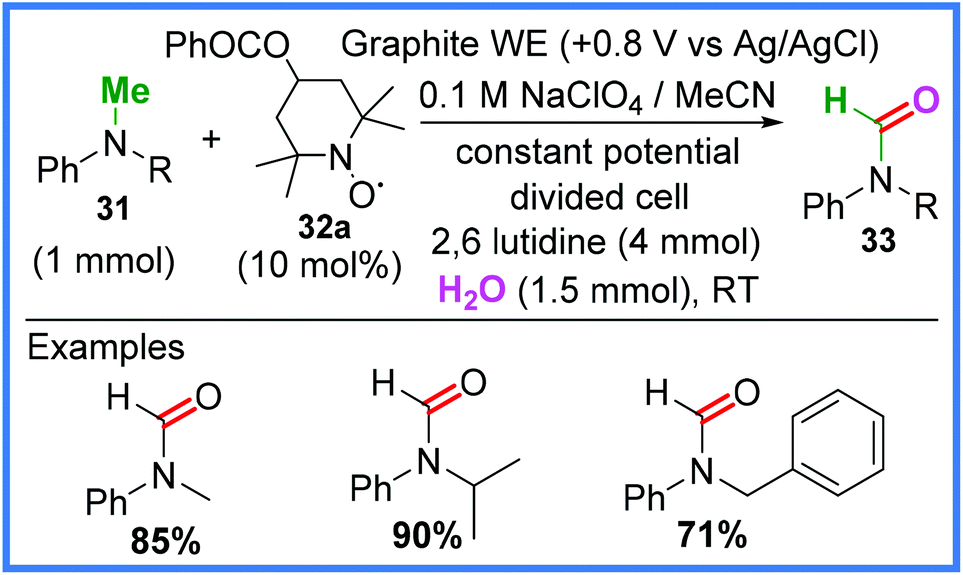 | ||
| Scheme 18 Electrochemical N-alkylformanilide synthesis.14,37 | ||
The mechanism postulated by Kashiwagi and Anzai for this reaction is shown in Scheme 19. It starts with the oxidation of the mediator 32a to 32b at the anode, which then reacts with the N-methylaniline 31 to produce the intermediate 34. This intermediate collapses giving 32c and the iminium ion 35, where 32c can be oxidised at the anode to reform 32b. The iminium ion 35 can react with H2O to form the hemiaminal 36. A final oxidation of 36 by 32b then leads to the final N-alkylformanilide 33.14,37
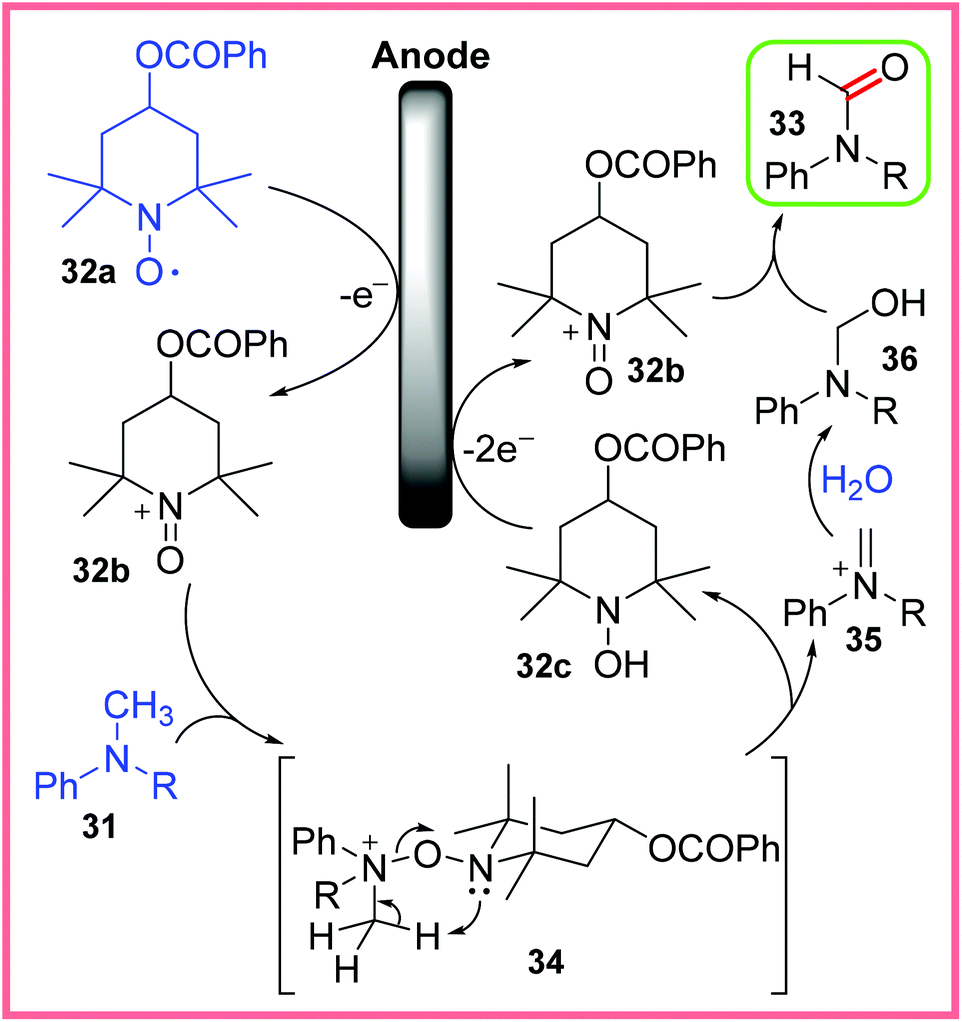 | ||
| Scheme 19 Postulated mechanism for the oxidation of N-methylanilines.14,37 | ||
Electrochemical decarboxylation (Kolbe electrolysis)38 is a well-known type of anodic oxidation and in 2014, Matthessen and co-workers put this reaction to good use when they synthesised a range of nitrile compounds from amino acids in an undivided cell.39 The rationale behind this work was that amino acids could be used as a more sustainable source of reactive nitrogen compounds after decarboxylation than the energy-intensive functionalisation of hydrocarbons with NH3. Whilst the aim of this work was to produce nitrile compounds, the conditions shown in Scheme 20 produced an amide product 37 in very good yield, presumably via the hydration of a nitrile intermediate. The key to this reaction appeared to be the in situ generation of hypobromite at the Pt anode which acted as a mediator to facilitate the decarboxylation reaction. The reduction of H+ ions was the proposed cathodic process.39
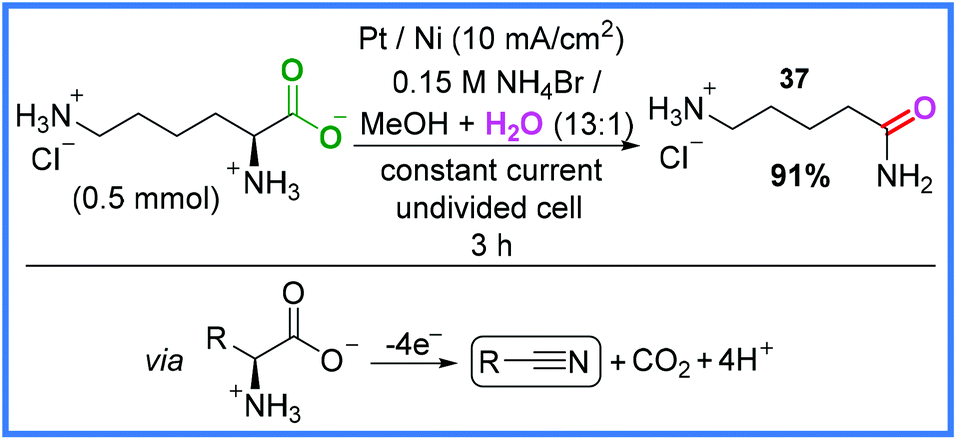 | ||
| Scheme 20 Electrochemical decarboxylation and nitrile hydration to produce an amide.39 | ||
Some of the most common methods of amide synthesis are the reactions of carboxylic acid derivatives (acyl chlorides, anhydrides and esters) with amines,10 which often suffer from low atom economies and high levels of hazardous waste. In addition, methods that employ carboxylic acids directly often require coupling agents or the use of transition metal Lewis acid activators (ZrCl4, AlMe3 and Ti(OiPr)4), therefore, new electrochemical methods that obviate such requirements are of great interest.40 One such example of this has been reported by Ke and co-workers, wherein a bromide salt (used as both the electrolyte and a catalyst) was employed to promote direct amidation of carboxylic acids under aqueous conditions in an undivided cell, as shown in Scheme 21.40
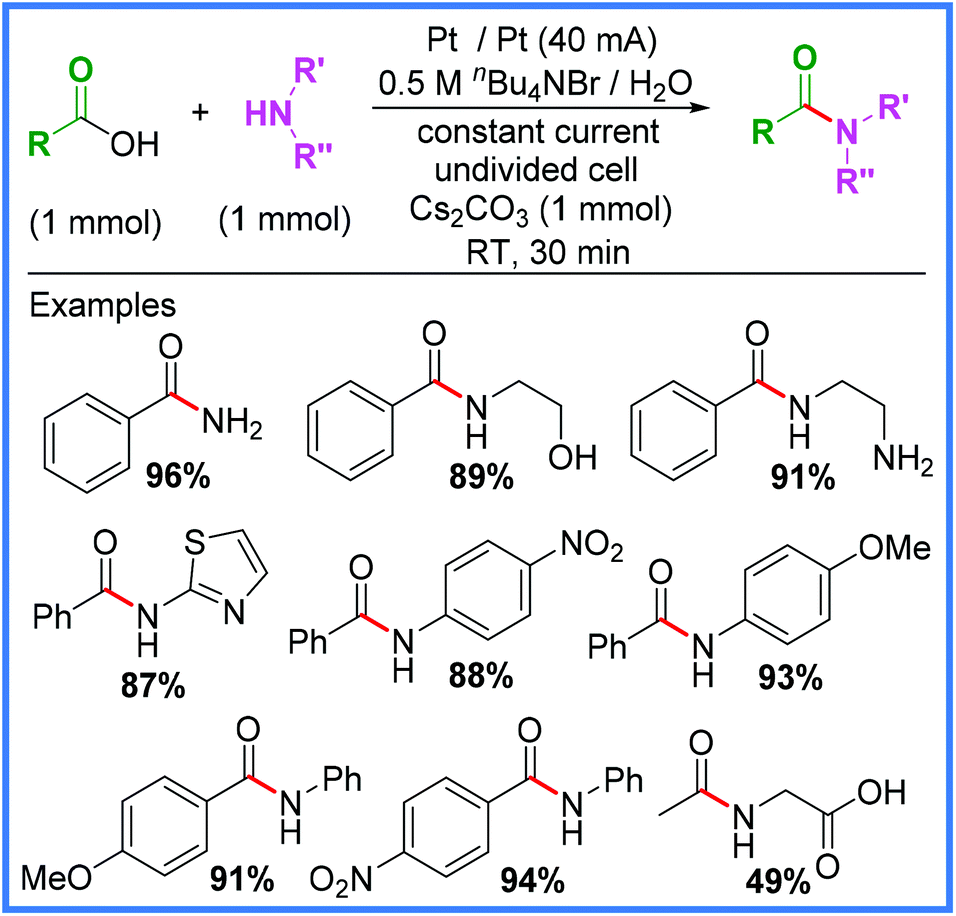 | ||
| Scheme 21 Direct electrochemical amidation of carboxylic acids under aqueous conditions.40 | ||
A wide array of substituents and functional groups were tested using these conditions and generally gave excellent yields, making this a particularly valuable method of electrochemical amide synthesis. Radical trapping experiments suggested the involvement of an acyl radical intermediate, which led the authors to propose the rather unusual mechanism shown in Scheme 22. They proposed the base-catalysed nucleophilic substitution of the carboxylic acid starting material to produce the acyl bromide 38. This species then undergoes homolytic cleavage to yield the acyl radical 39 and a bromine radical. The bromine radical will likely be reduced at the cathode to reform a bromide anion and 39 can react with an amine to produce the desired amide product after anodic oxidation.40
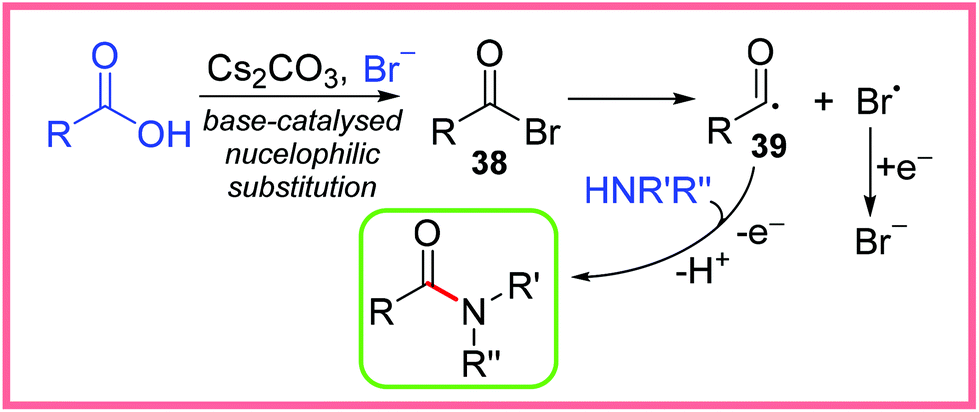 | ||
| Scheme 22 Proposed mechanism for the electrochemical amidation of carboxylic acids.40 | ||
2.5. N-Heterocyclic carbenes as mediators
An important class of mediators for amide synthesis not yet discussed are the N-Heterocyclic Carbenes (NHC). Effective electrochemical protocols using NHCs have been reported, including processes carried out in flow reactors, such as the work of Green and co-workers (Scheme 23).41 This process proved to be high yielding when aldehydes and the thiazolium bistriflimide 40 was used as an NHC precursor in the presence of the base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU).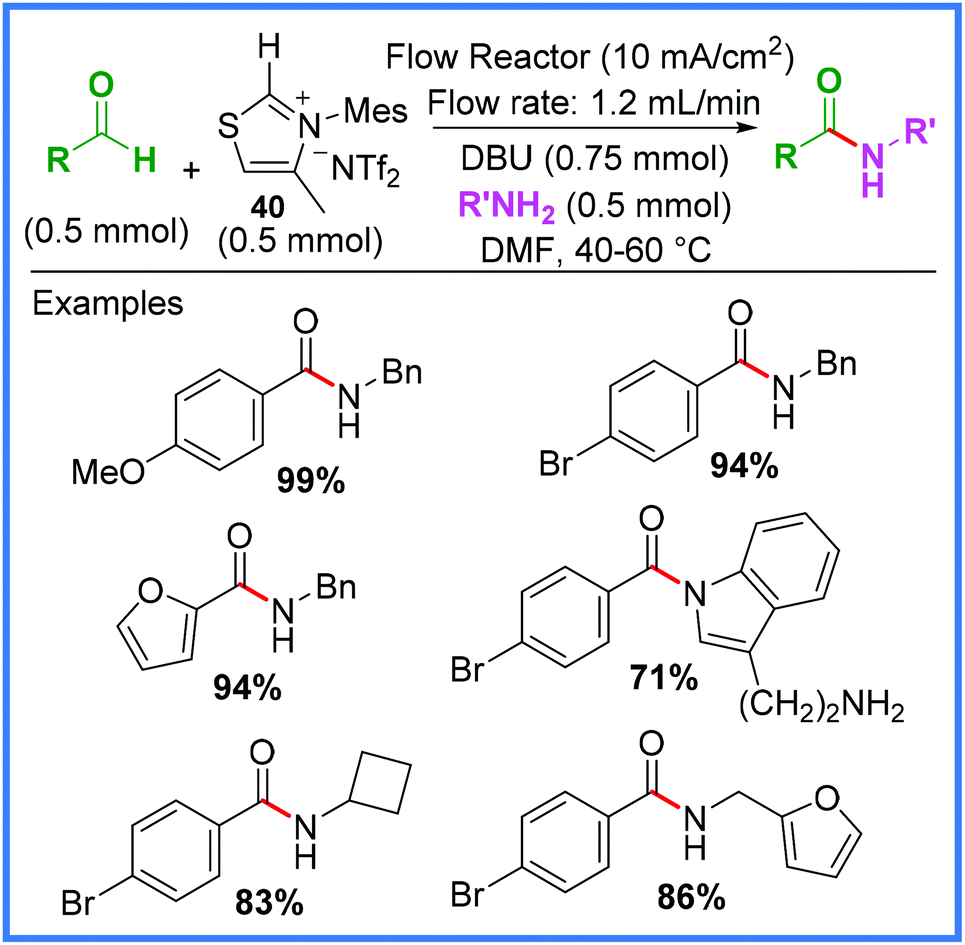 | ||
| Scheme 23 Electrochemical flow process using a N-heterocyclic carbene mediator.41 | ||
It was found that attempting to mix all the reagents together in one go prevented the vital Breslow intermediate 41a/41b from forming, likely because of competing imine formation. Instead, the authors utilised one of the significant benefits of performing reactions in flow, namely the ability to easily control reagent mixing, to introduce the amine after the Breslow intermediate had formed. This greatly enhanced the yield of the reaction. Scheme 24 shows the likely mechanism at work, with 40 being used to produce an NHC which reacts with an aldehyde to produce 41a/41b. The base DBU plays a key role in deprotonating species during these early stages of the mechanism. Anodic oxidation then leads to the acyl thiazolium 42, before reaction with an amine delivers the amide product and regenerates 40. The reduction of 40 or protonated DBU was proposed as the cathodic process in this reaction.41
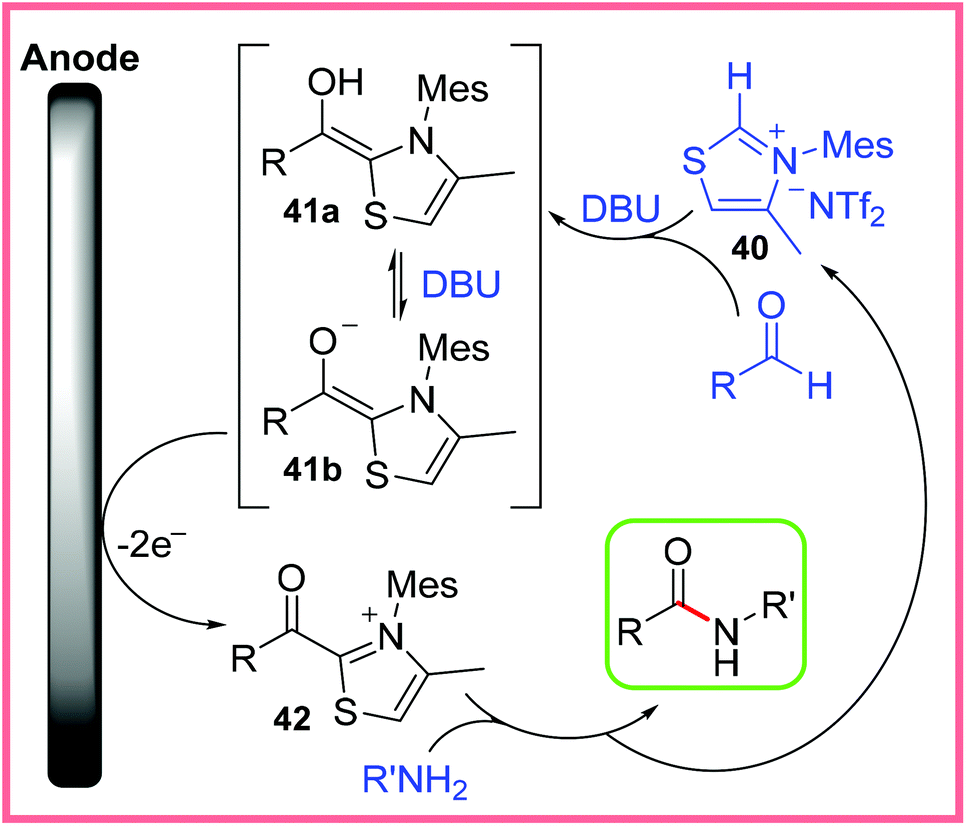 | ||
| Scheme 24 Mechanism for electrochemical N-heterocyclic carbene-mediated amidation.41 | ||
An example of an NHC being generated via cathodic reduction has also been reported, which shows the versatility of these mediators in electrosynthesis. This work, reported by Feroci and co-workers,42 relies upon an ionic liquid 43 (also functioning as an electrolyte) which is reduced at the cathode to the NHC 44in situ in a divided cell. This then reacts with an α,β-unsaturated aldehyde to form the intermediate 45 before reaction with an amine yields the amide product. It is important to note that the alkene functionality is lost in the final product. This stepwise reaction is shown in Scheme 25.42
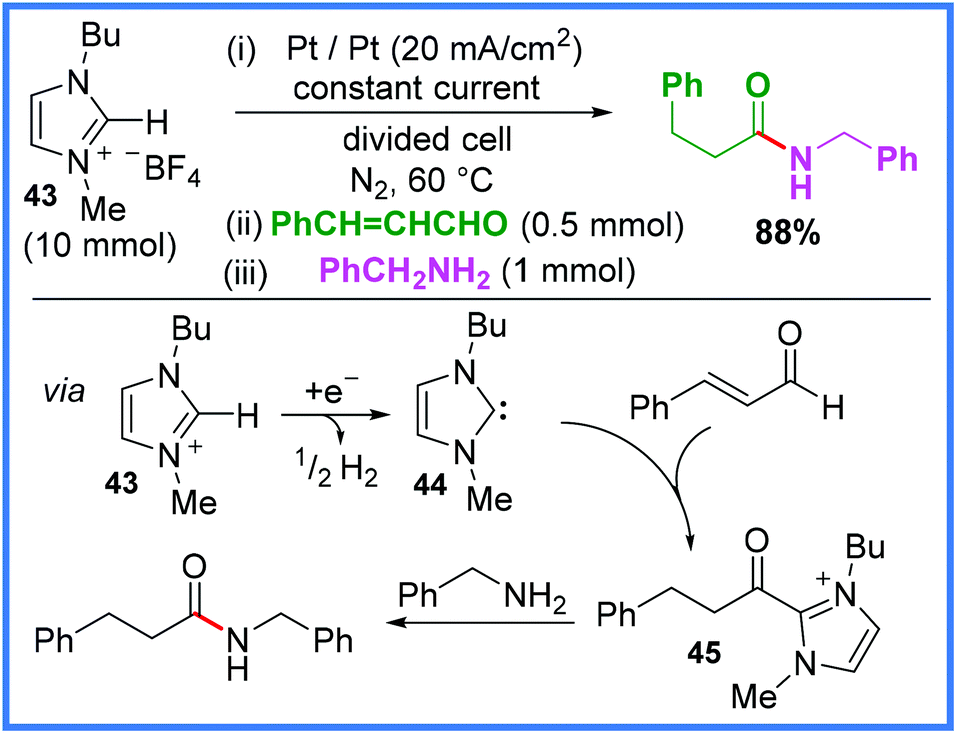 | ||
| Scheme 25 N-Heterocyclic carbene prepared by cathodic reduction of an ionic liquid to mediate amide synthesis.42 | ||
2.6. The anion pool method
The electrochemical reduction/deprotonation of substrates such as amines to make them more nucleophilic (the ‘anion pool method’) has been explored as a means of amide bond formation. Research published by Dissanayake and co-workers in 2019 showed a highly sustainable and atom-efficient approach to amide synthesis using this method in a divided cell as shown in Scheme 26.43 The authors made excellent use of inexpensive Reticulated Vitreous Carbon (RVC) electrodes and anhydrides 46 as the acyl source in this work, ensuring that the carboxylates 47 that are formed after initial reaction of the anhydrides were not wasted by reacting them further with benzyl bromide. This led to a highly atom-efficient process that produced two very useful products from every reaction: an amide and an ester 48. The authors carried out control reactions to show that without any applied current at room temperature, neither product was produced. However, at an elevated temperature of 55 °C the amide was formed in the absence of a current, while the ester was still not produced in this test. It was found that in order for the ester product to be produced in good yield, a continual current had to be applied after amide formation to reduce any adventitious protons in solution, thereby maintaining the nucleophilic nature of the resulting carboxylates 47 long enough for them to react with benzyl bromide.43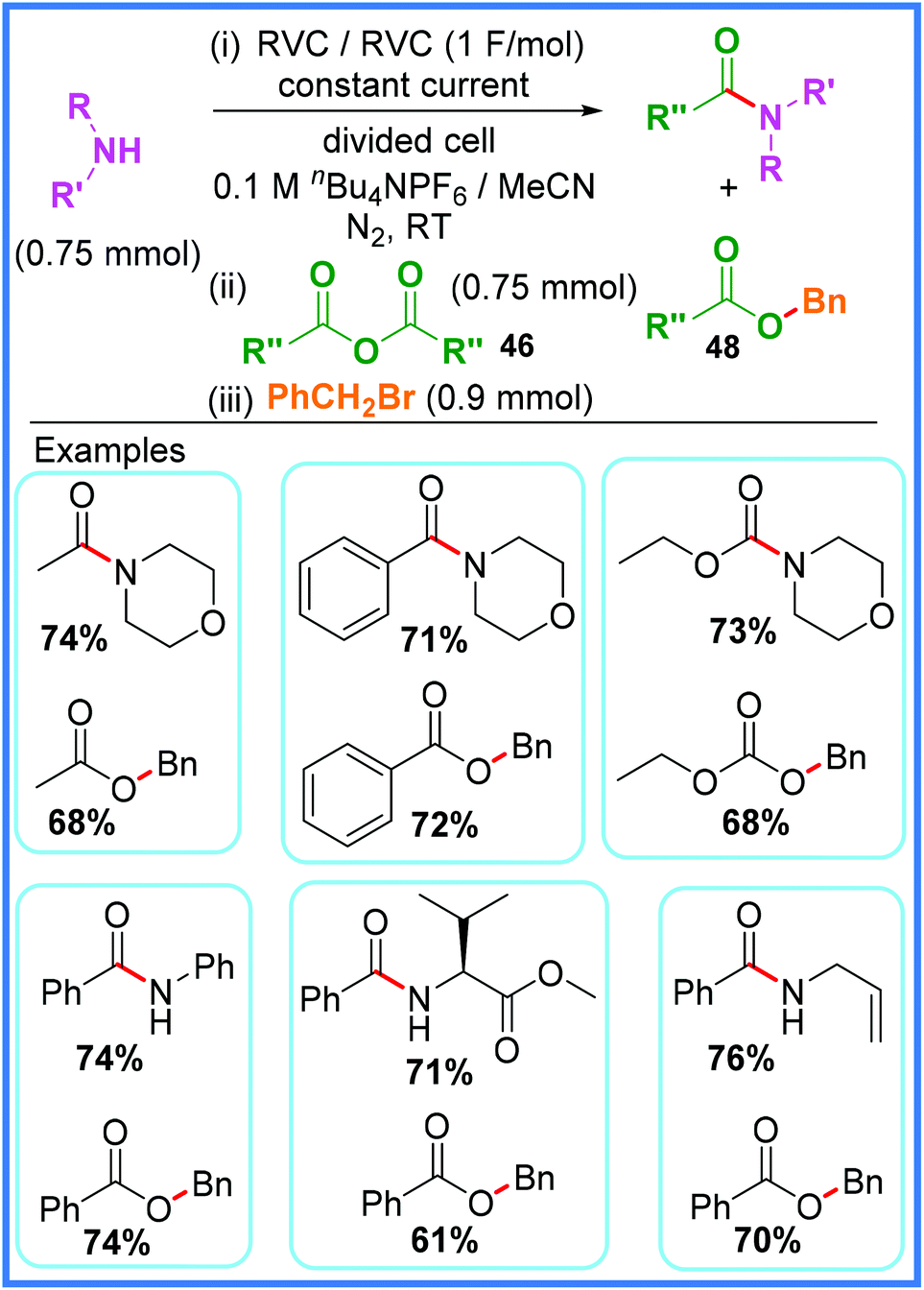 | ||
| Scheme 26 Electrochemical anion pool method for amide and ester synthesis.43 | ||
The yields for products produced using this method were good, for both the amide and the ester products, and the reaction proved viable on gram scale, however it should be noted that on this larger scale there was some disparity between the yield of the amide and that of the benzoic ester. It was found that the amide could be synthesised in high yield, but the benzoic ester proved much lower yielding. This issue was rectified by adding a catalytic amount of K2CO3 base to break up benzoate homoconjugated dimers that were likely forming, thereby bringing the yields for both the amide and the ester to above 80%.43
The mechanism for this reaction is shown in Scheme 27, starting with amine cathodic reduction/deprotonation, which then reacts with the anhydride 46 to give an amide product. The resulting carboxylate 47 is then exposed to benzyl bromide to give the ester product 48.43
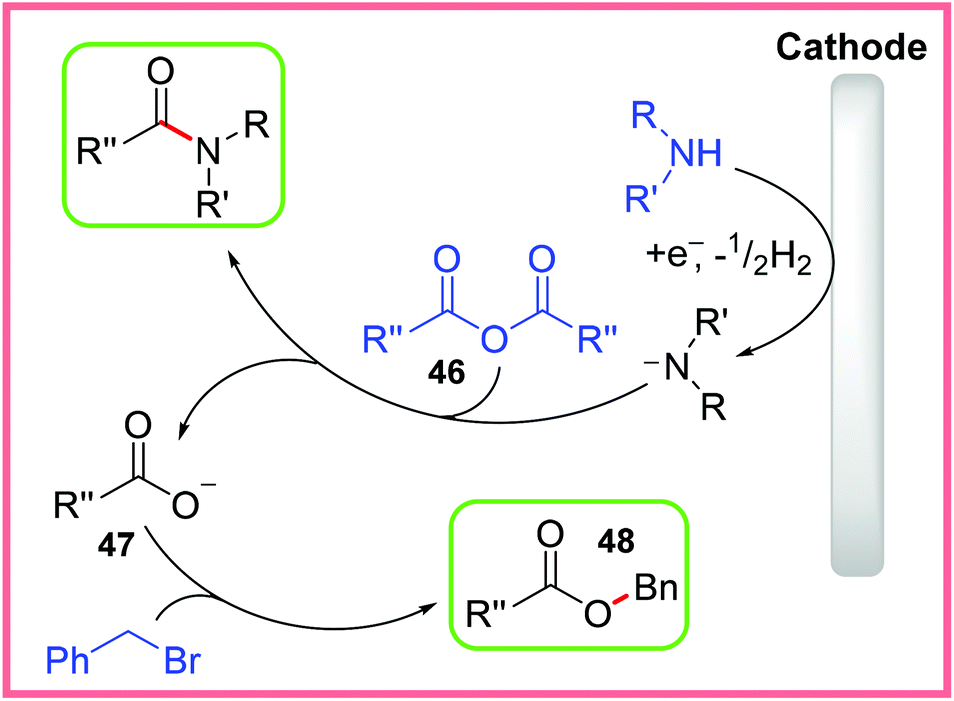 | ||
| Scheme 27 Proposed mechanism for electrochemical amide and ester synthesis.43 | ||
This same group have also had success with producing acylated indazoles 49 using this method as shown in Scheme 28.44 The mechanism for this reaction is the same as in Scheme 27 with the exception that esters were not produced in this work.
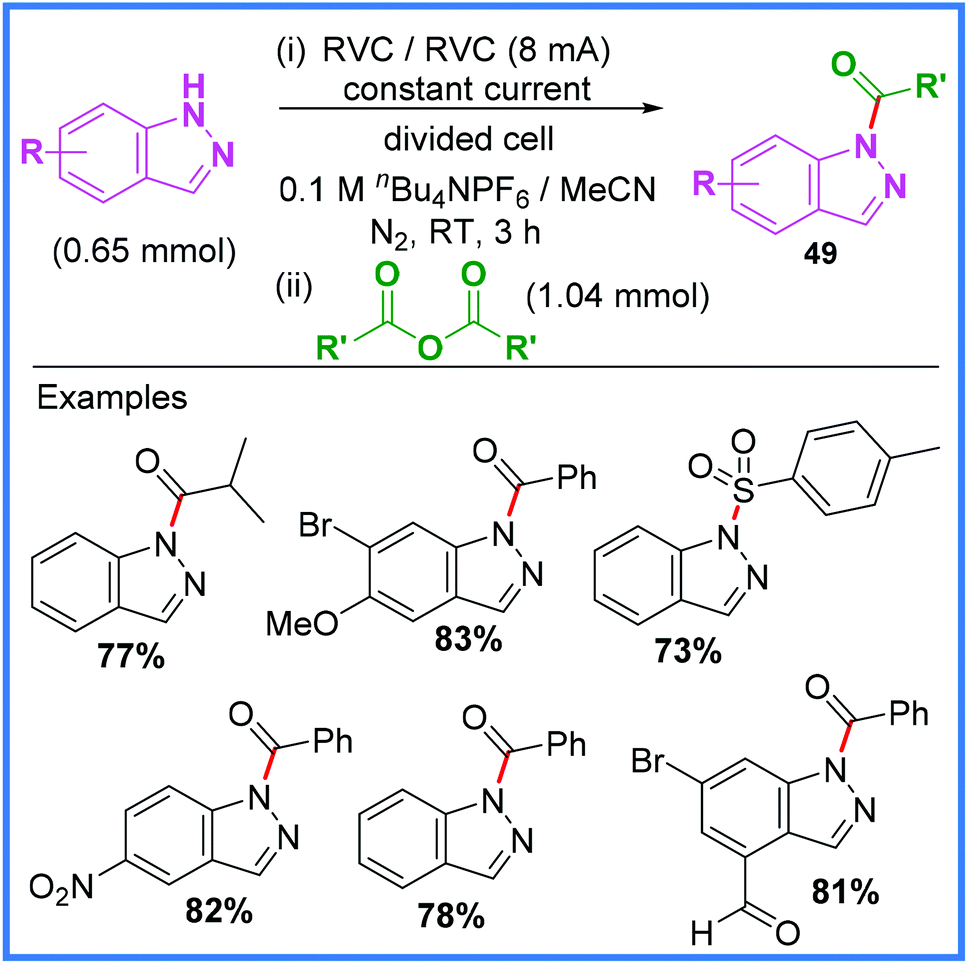 | ||
| Scheme 28 Electrochemical acylation of indazoles using the anion pool method.44 | ||
2.7. Cathodic reduction cleavage reactions
Cathodic reduction has been used for the in situ generation of H2O2 from molecular O2 by Kurose and co-workers to facilitate amide formation.45 Interestingly, in the work of Boujlel and Simonet in 1985, the combination of molecular O2 and cathodic reduction was also used to produce amide products, this time through the cleavage of diimides 50, as in Scheme 29.46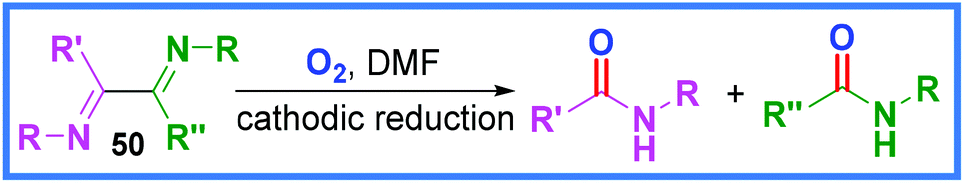 | ||
| Scheme 29 Cathodic reduction of diimines.46 | ||
A more recent example of an electrochemical cleavage reaction to produce amides is the work of Mentel and co-workers published in 2009,47 which demonstrated an environmentally benign reduction of carboxylic acid hydrazides 51 in a Britton–Robinson buffer solution in a divided cell (Scheme 30). The addition of 2 electrons from the cathode and 2 protons allowed the amide products to form along with ammonia. This reaction is often carried out non-electrochemically through the use of a RANEY® nickel catalyst and similar types of electrochemical reductions often employ a mercury pool cathode, so the comparatively mild conditions shown in Scheme 30 are favourable.47 This reaction produced several amides in moderate yield.
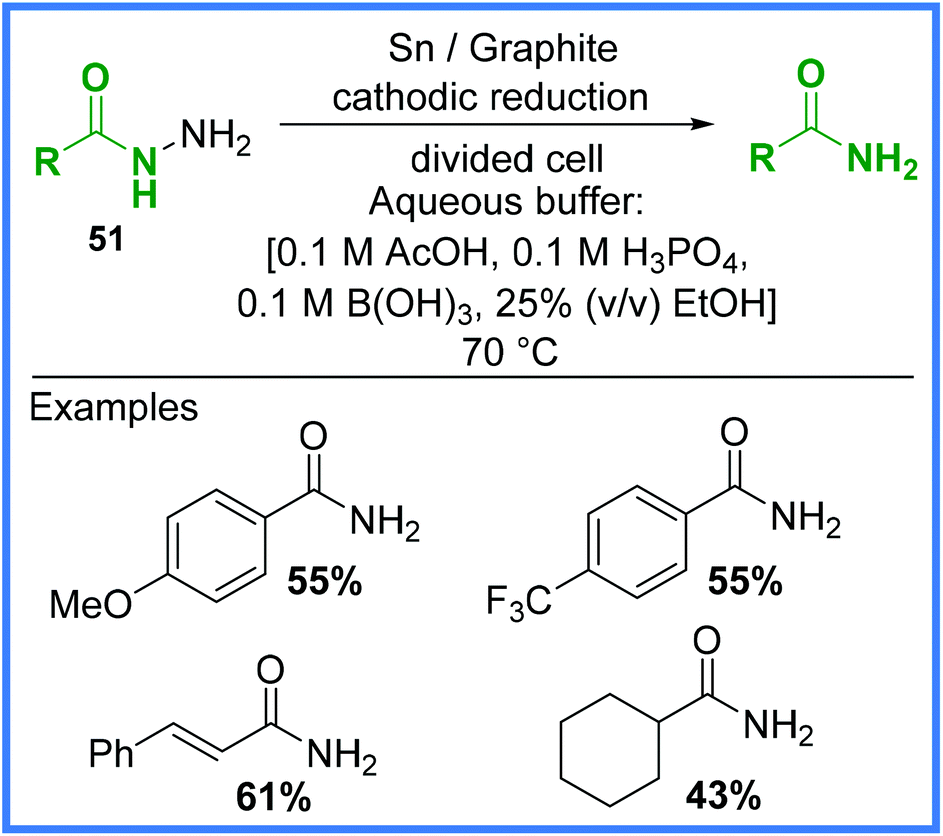 | ||
| Scheme 30 Electrochemical cleavage of carboxylic acid hydrazides.47 | ||
2.8. Synthesis by Shono-type oxidation
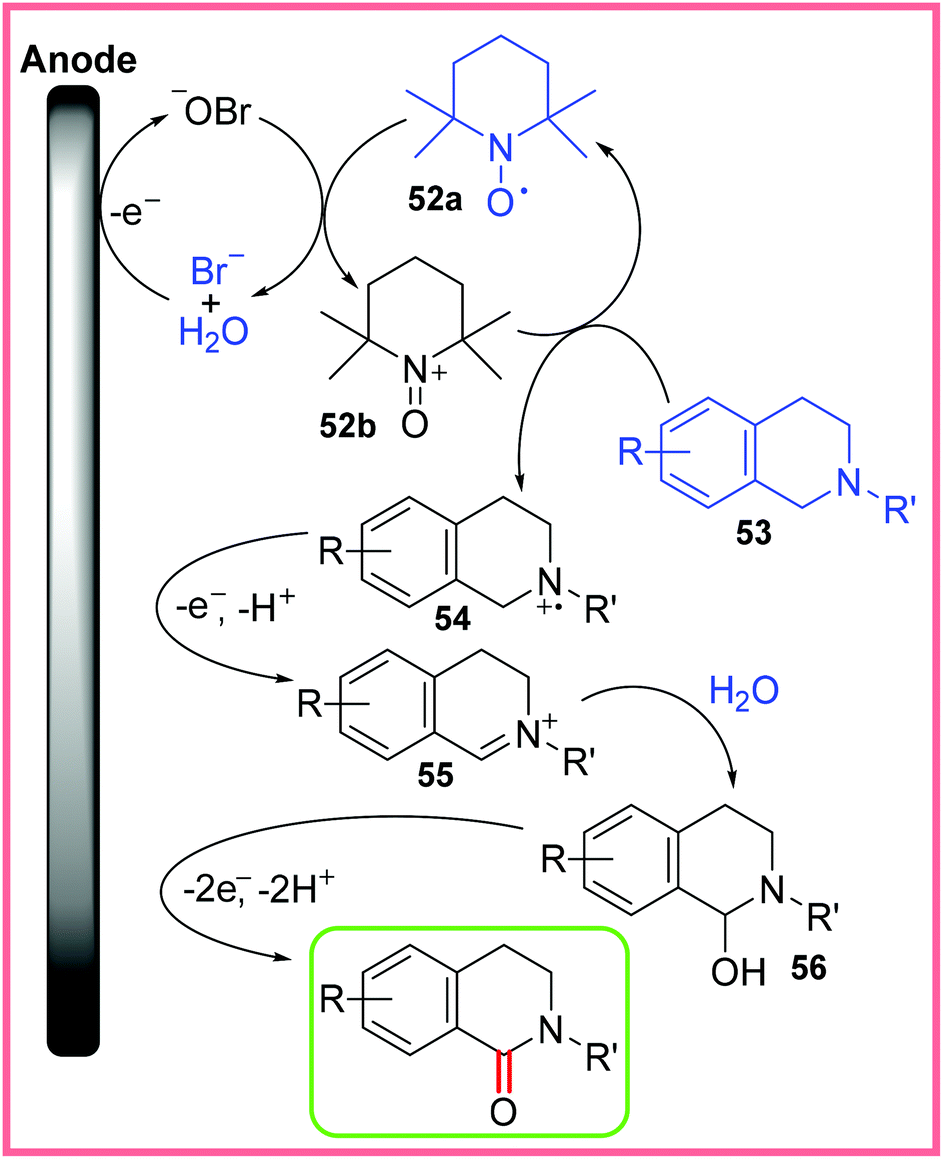
In 1981, Shono and co-workers reported a methoxylation reaction wherein anodic oxidation of an amide substrate led to the formation of an iminium ion that could be attacked by MeOH.48,49 Further uses of amides in Shono-type oxidation will be discussed in the next section, but it is first important to mention the electrochemical synthesis of an amide by this method. Scheme 31 shows the work of Li and co-workers where a mixed hypobromite/TEMPO 52a system was used to oxidise tetrahydroisoquinolines 53 in an undivided cell, allowing amide-containing products to form in moderate yields. This work utilised a biphasic DCM/H2O solvent system.50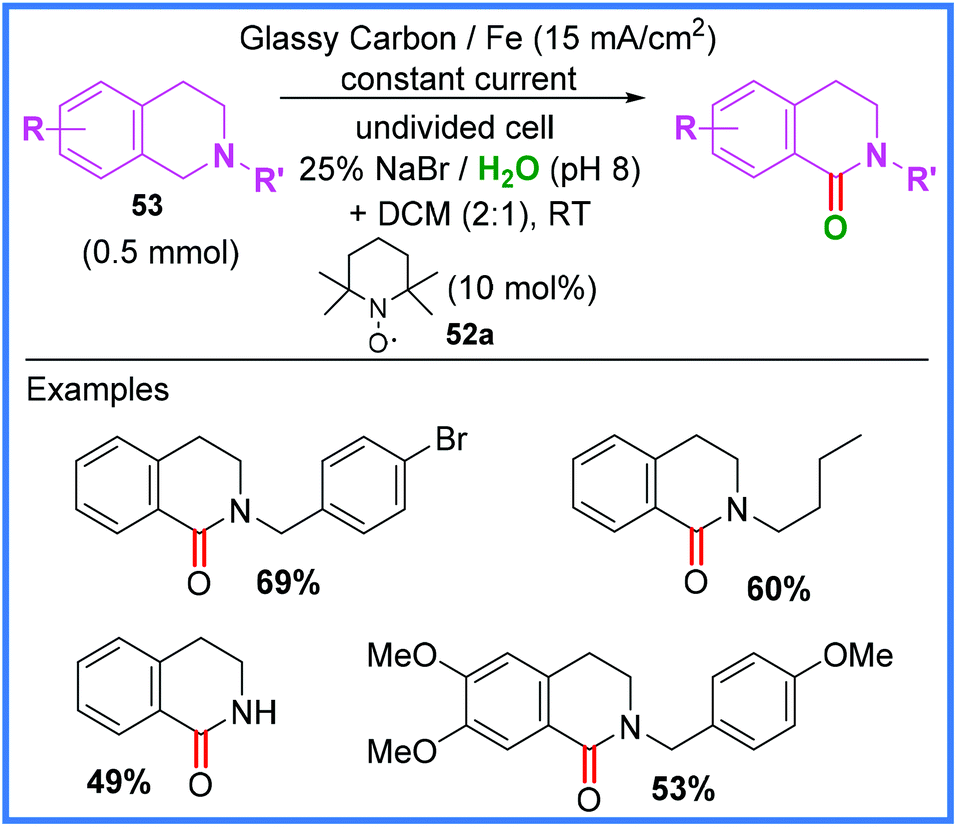 | ||
| Scheme 31 Electrochemical Shono-type oxidation of tetrahydroisoquinolines.50 | ||
It was found that both the bromide and TEMPO 52a reagents were vital in this reaction and a proposed mechanism is shown in Scheme 32. First, anodic oxidation of bromide produces hypobromite in the aqueous phase of this reaction. Hypobromite is capable of oxidising 52a to 52b, which then oxidises 53 into the radical cation 54. This species is oxidised further and deprotonated giving the iminium ion 55. Nucleophilic attack of this iminium ion by water leads to isoquinolinol 56 which is oxidised and deprotonated a final time to yield the desired amide-containing product.50 The oxidations from 54 to 55 and 56 to the product are not depicted touching the anode as it is possible that 52b facilitates these oxidations.
 | ||
| Scheme 32 Plausible mechanism for tetrahydroisoquinoline oxidation.50 | ||
3. Applications of amides in electrosynthesis
3.1. Amides as substrates in Shono-type oxidations
Following on from the previous section, Shono-type oxidation reactions have also been carried out on amide substrates (rather than used to prepare amides) and so these will be discussed in this section. The first example of this is shown in Scheme 33 and represents the classic Shono oxidation reaction whereby an iminium ion 57 is formed electrochemically and then nucleophilically attacked by MeOH.49 This sort of reaction has also been reported when using carbamate substrates.51,52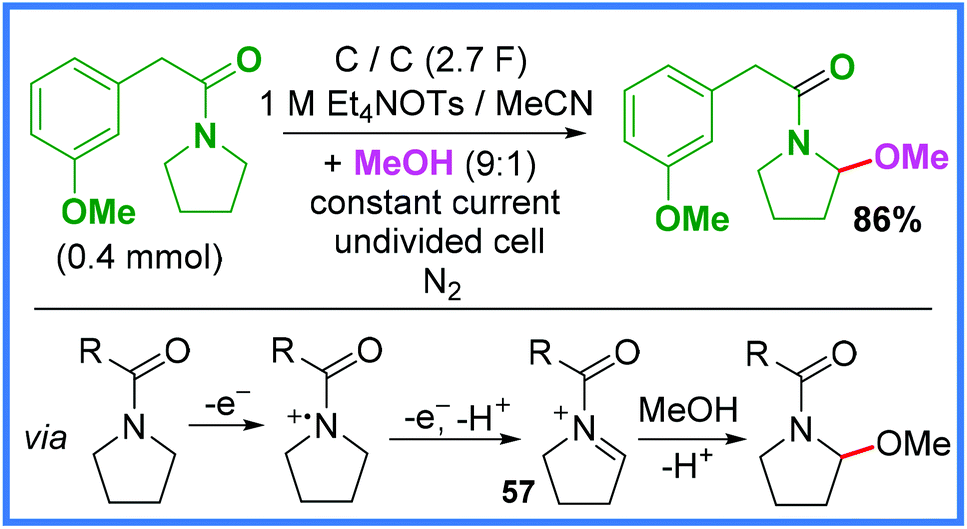 | ||
| Scheme 33 Electrochemical methoxylation reaction via Shono oxidation.49 | ||
In 2016, an example of Shono-type oxidation being used to bond γ-lactams 58 and anilines together was reported by Gong and Huang,53 who used mild and sustainable conditions (i.e. room temperature and no metal catalyst) to produce a range of coupled products in an undivided cell (Scheme 34). In this example, the reduction of NH4+ to produce H2 gas at the cathode was proposed, in addition to the standard Shono-type mechanism taking place at the anode. Whilst the use of 20 equivalents of the lactam 58 is undesirable, this was the first example of anilines being used as the nucleophilic coupling partner in Shono-type oxidation, possibly because the oxidation potential of nucleophiles traditionally employed in this reaction (MeOH and cyanide ions) are rather high, whilst anilines are comparatively lower.53
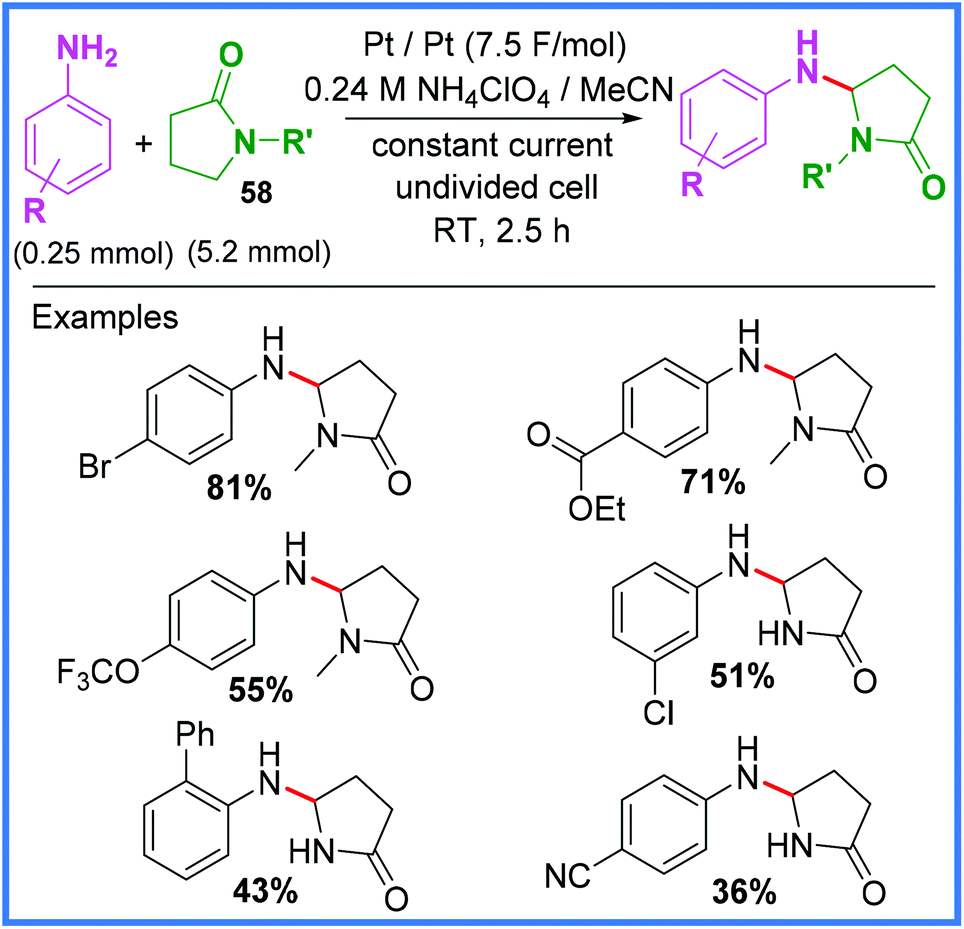 | ||
| Scheme 34 Shono-type oxidation of lactams using anilines.53 | ||
In addition to the intermolecular examples mentioned so far, Siu and co-workers reported an interesting example of intramolecular amide cyclisation using Shono-type oxidation,54 which relies upon tethered alcohols as the nucleophile source. The conditions shown in Scheme 35 proved high yielding for all the substrates that the authors tested, allowing a range of 6- and 7-membered rings to be produced.54 A similar approach has been reported by Lee for the diastereoselective cyclisation of hydroxylamides.55,56
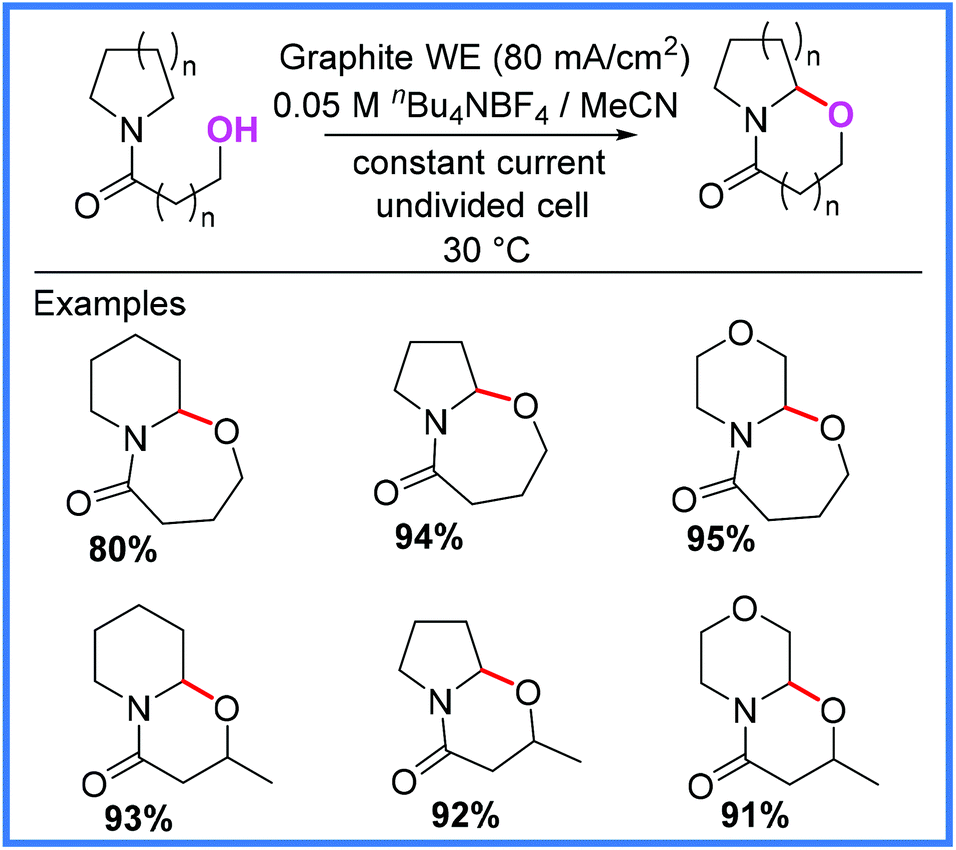 | ||
| Scheme 35 Electrochemical intramolecular amide cyclisation.54 | ||
Finally, the use of Shono-type oxidation has even been extended to the selective deethylation of tertiary amides (a common pharmaceutical moiety) in a metabolism-mimicking protocol, as reported by Bal and co-workers in 2019.57 The N-dealkylation of drug molecules is a common metabolism pathway facilitated by cytochrome P450. This work, carried out in an undivided cell and shown in Scheme 36, demonstrates the potential for late-stage synthesis of drug metabolites directly from the parent drug using electrosynthesis, saving a great deal of intellectual and practical effort if the same metabolites were to be synthesised independently. It was found that the use of a constant current rather than a constant potential was highly important to achieve the desired reaction, as in constant potential conditions, the authors only obtained the expected α-methoxylated Shono product from the iminium ion 59 reacting with MeOH. However, when the authors used a constant current approach, they noted that 59 could react with MeO− or HO− to produce the desired deethylated products along with ester and aldehyde by-products. It was proposed that the reduction of MeOH and H+ ions was taking place at the cathode in this reaction.57
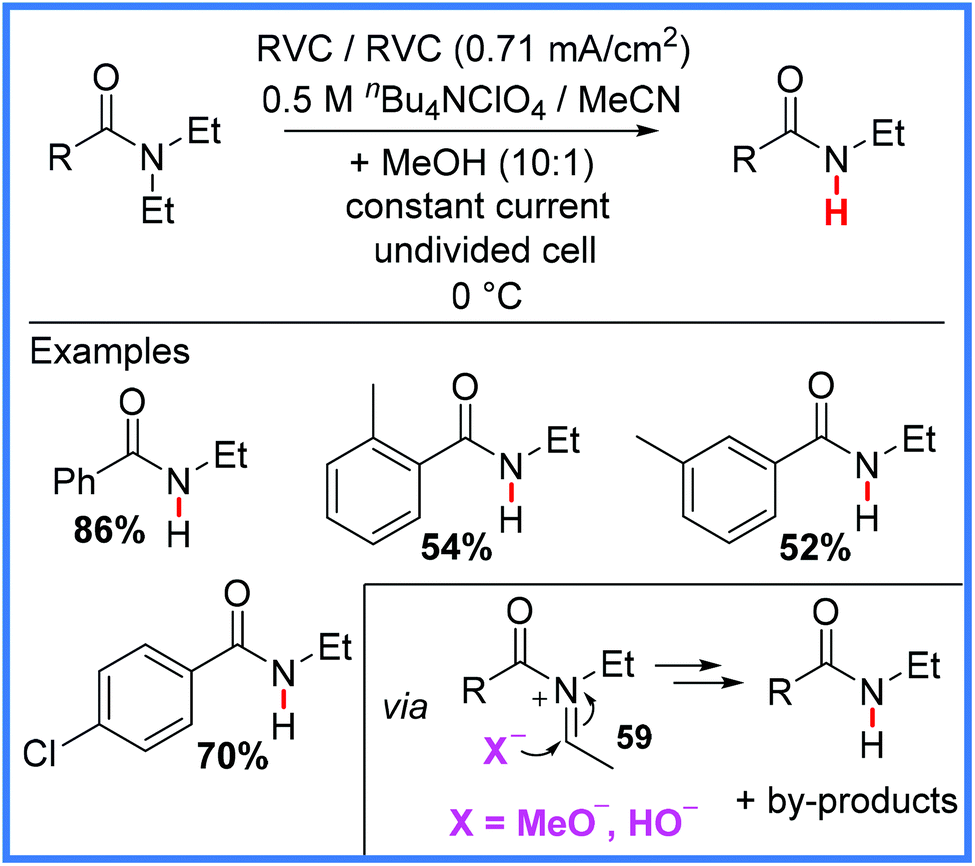 | ||
| Scheme 36 Electrochemical deethylation of tertiary amides.57 | ||
3.2. Electrochemical metal-catalysed C–H activations
Another application of amides in electrosynthesis is to act as ligating directing groups to assist in metal-catalysed C–H activation reactions. Various metals have been employed alongside amide substituents in these reactions as will be discussed in this section. Cobalt has been used to excellent effect by Ackermann and co-workers to facilitate inexpensive C–H oxygenation reactions as shown in Scheme 37.58,59 By using amide starting materials that also bore a pyridine-N-oxide (PyO) group 60, it was found that methanol, ethanol and various other alcohols could be incorporated into the final products in good yields. Notably, this electrochemical method could be carried out under mild conditions at room temperature and avoided the need to add stoichiometric silver(I) oxidants.58,59 Cobalt has also been used for electrochemical C–H activations of amides for reactions with amines, alkenes and alkynes.60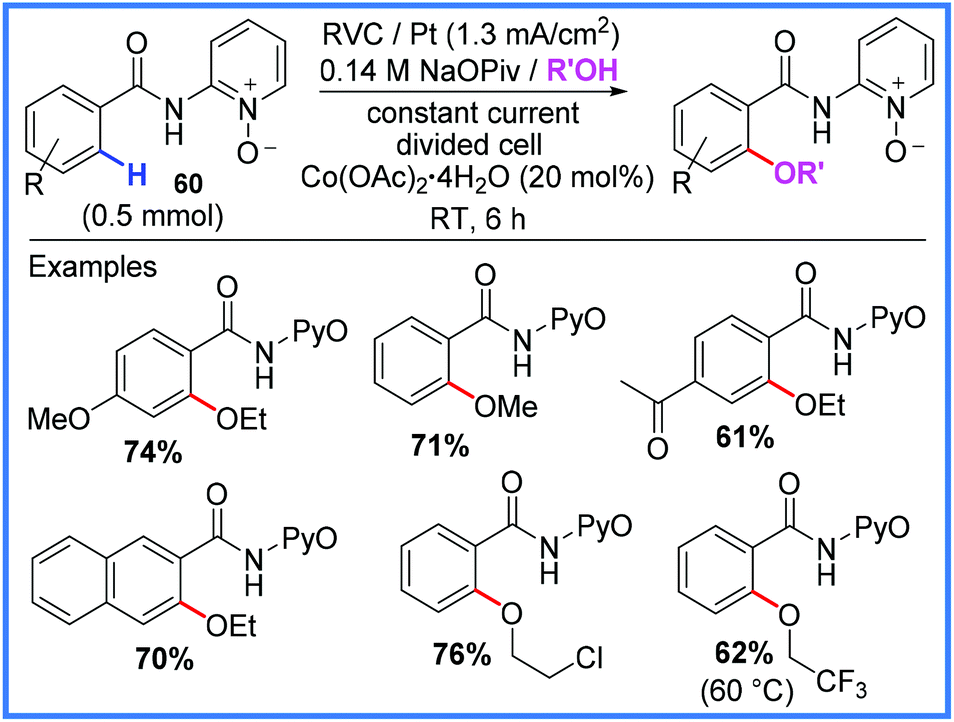 | ||
| Scheme 37 Electrochemical cobalt-catalysed oxygenation.58,59 | ||
The mechanism for this reaction has been investigated and is believed to proceed as shown in Scheme 38, wherein the starting Co(II) catalyst is oxidised to Co(III) at the anode. This allows C–H cleavage to take place of the oxidised starting material 60, forming 61. C–O bond formation then occurs by chelation assistance to give 62, thereby allowing the final product and Co(I) to be released. The Co(I) species is then oxidised back up to Co(III) to begin another cycle.58,59
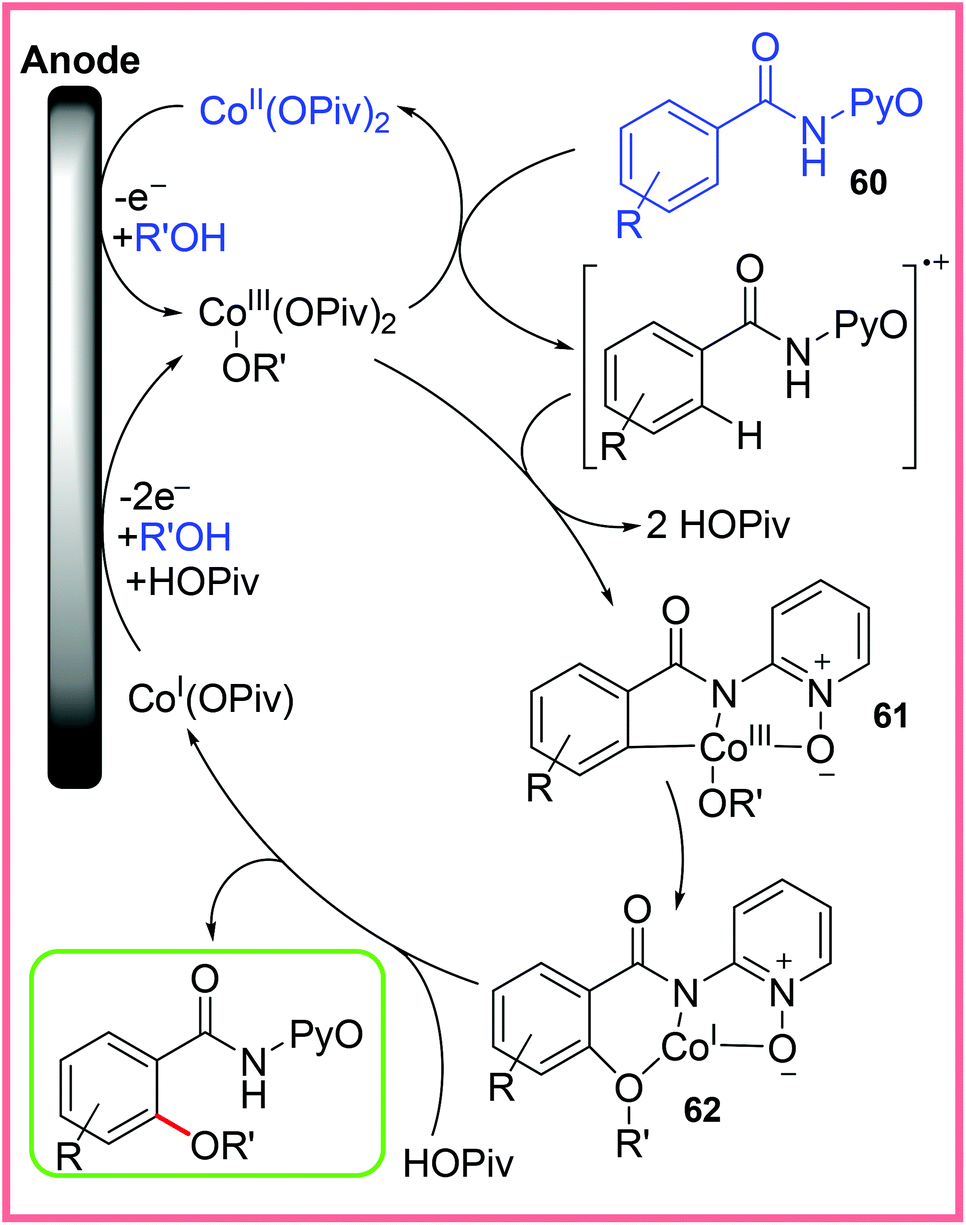 | ||
| Scheme 38 Mechanism for electrochemical Co-catalysed oxygenation.58,59 | ||
Copper has been used in a very similar fashion to the cobalt examples above to facilitate amination reactions in an undivided cell as shown in Scheme 39. In this example from Kathiravan and co-workers,61 the sole oxidiser in the reaction was once again the anode, however, instead of a pyridine-N-oxide group being attached to the amide substrate, a quinoline group was used. These conditions proved effective for a range of substrates.61
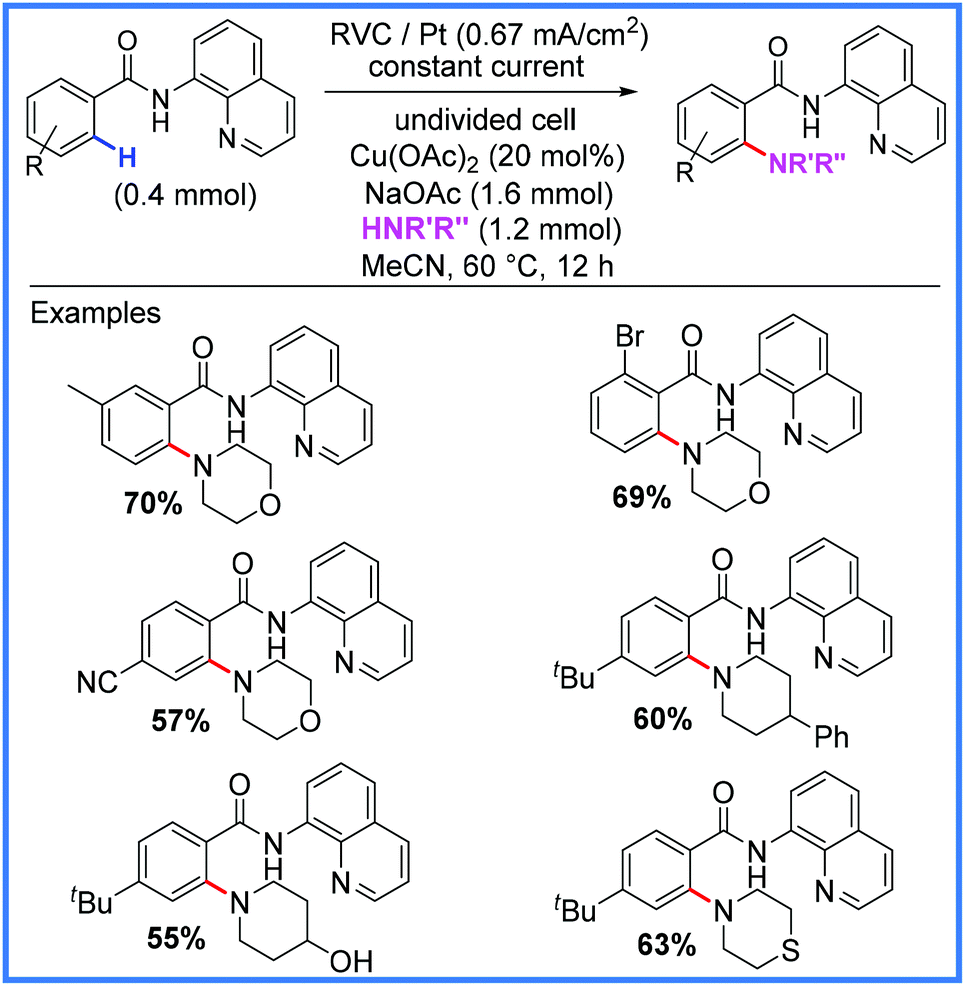 | ||
| Scheme 39 Electrochemical Cu-catalysed amination.61 | ||
Furthermore, copper has been utilised in conjunction with amide direction for the selective electrochemical B–H oxygenation of o-carboranes at room temperature, in work published by Au and co-workers.62
Nickel has recently been shown to be an effective mediator for electrochemical C–H alkylations under mild conditions, as shown by the Ackermann group.63 This work differs from the previous examples in this section as cathodic reduction was thought to be the key to controlling the oxidation state of the metal catalyst rather than anodic oxidation.
Palladium has also been used in these types of reactions, as exemplified by Amatore and co-workers in 2007 (Scheme 40) with a Heck reaction carried out in a divided cell.64 In this work, benzoquinone 63 was used as a convenient mediator and unlike the previous examples in this section, a secondary directing group attached to the amide such as pyridine-N-oxide or quinoline was not required.64
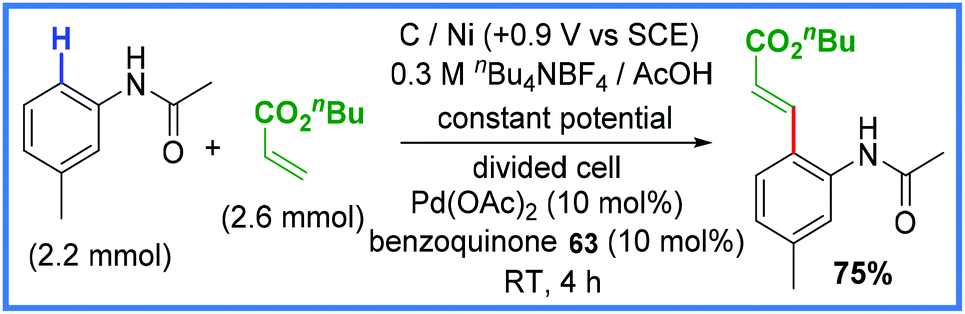 | ||
| Scheme 40 Electrochemical Heck reaction.64 | ||
The proposed mechanism for this reaction broadly follows a traditional Heck reaction with the Pd(II) catalyst initiating C–H activation to produce the complex 64. This species then undergoes carbopalladation with an alkene, followed by a β-hydride elimination step (once rotated into the correct geometry). This yields the desired product. Reductive elimination of the resulting Pd(II) species 65 gives Pd(0), which is oxidised up to Pd(II) again by benzoquinone 63. This oxidation produces hydroquinone 66 which can be oxidised at the anode to reform 63, thereby making this oxidant catalytic as shown in Scheme 41.64
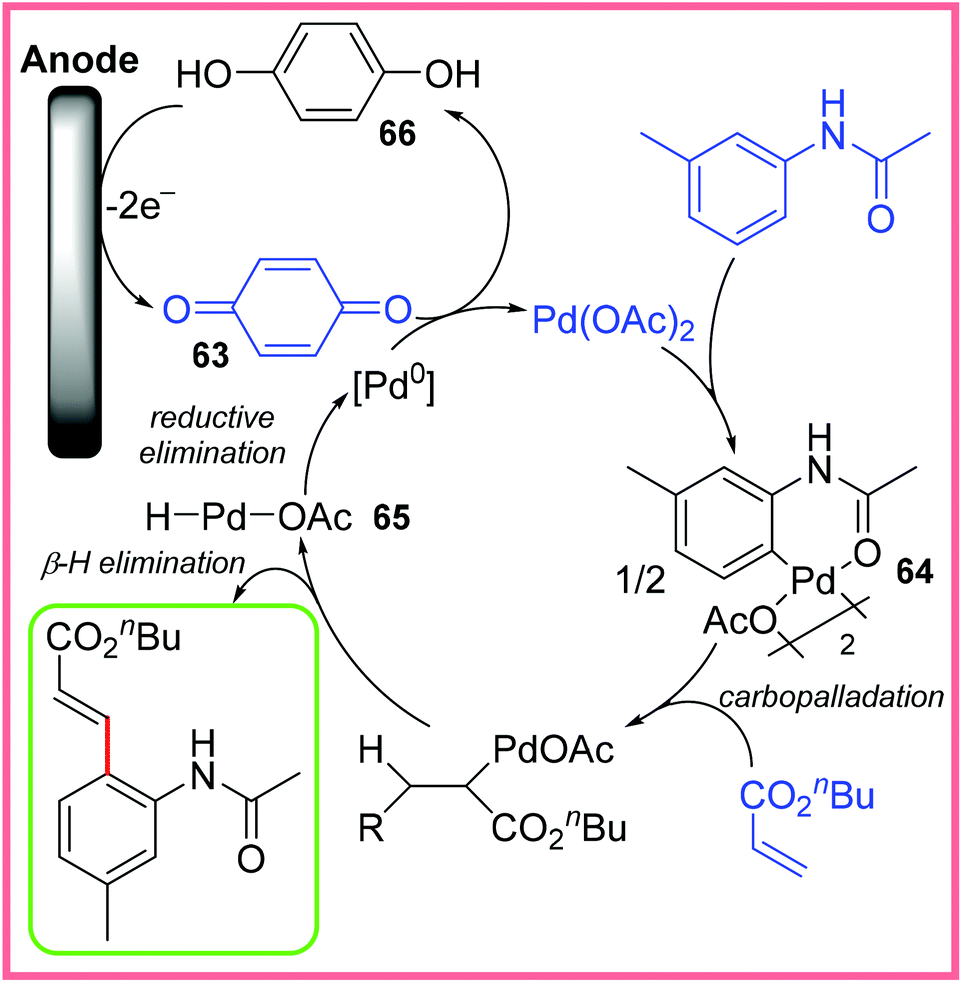 | ||
| Scheme 41 Proposed mechanism for the electrochemical Heck reaction.64 | ||
Aside from Heck reactions, Pd has also been used to facilitate electrochemical amide-directed chlorinations65 and iodinations,66 as shown by Kakiuchi and co-workers.
3.3. Heterocycle formation
There have been numerous examples of electrosynthetic heterocycle formation using amides as will be discussed in this section, however, for the purpose of this review, only examples where the amide motif is directly involved in the formation of the heterocycle will be discussed. The various syntheses have been subdivided into categories based on the type of new bond formed, i.e. C–C, C–N, C–O, etc.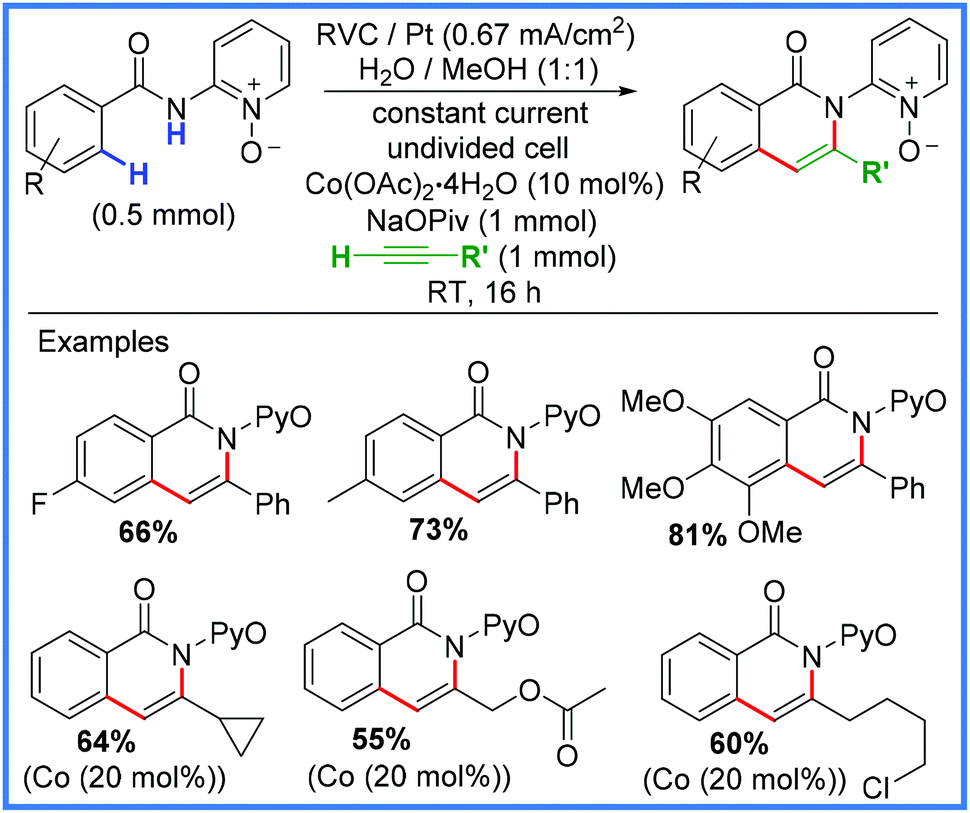 | ||
| Scheme 42 Electrochemical C–H/N–H activation using Co catalysis.67 | ||
In addition to this alkyne protocol, Tang and co-workers have reported the use of alkenes in a very similar Co-catalysed reaction, delivering a method of incorporating gas-phase ethene into fine chemicals.68
Moving away from cobalt chemistry, the synthesis of indoles and azaindoles has also been achieved using amides electrochemically. Hou and co-workers published their robust and widely applicable synthesis of these compounds in 2016,69 making use of a ferrocene mediator to produce amidyl radicals which could efficiently react with tethered alkynes in an undivided cell. The indole synthesis portion of this work is shown in Scheme 43.69
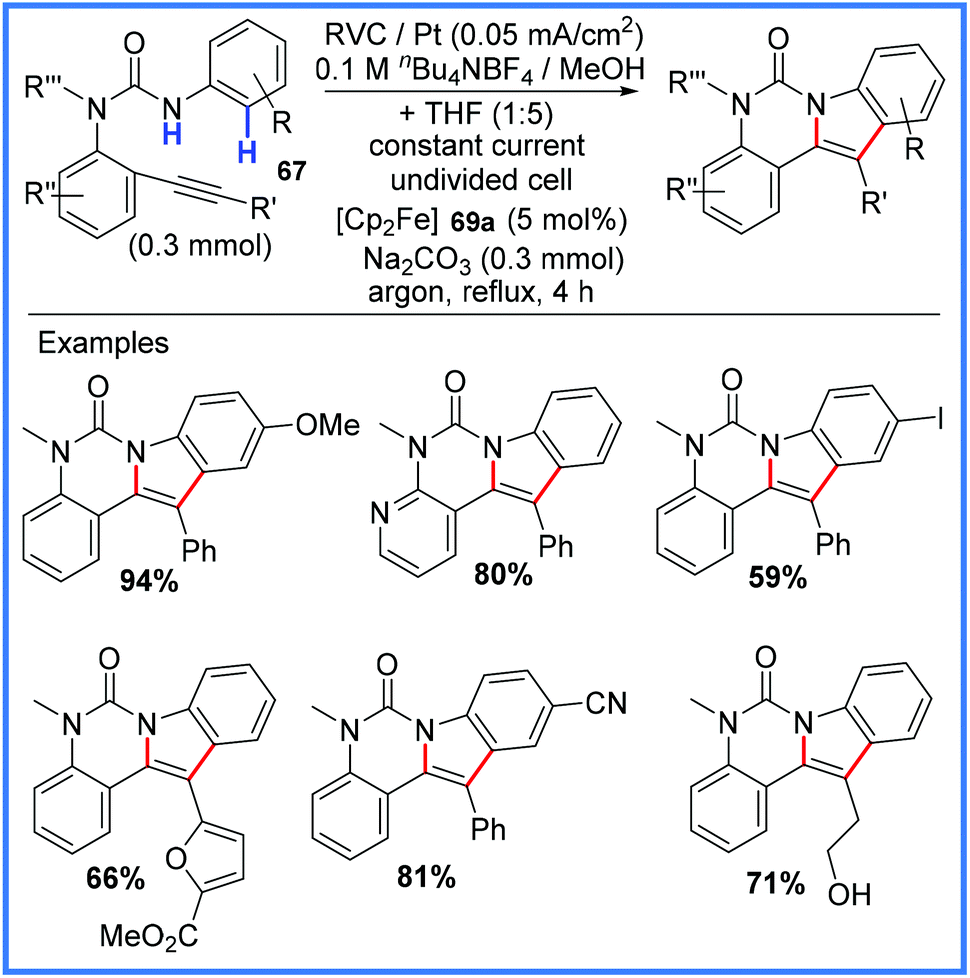 | ||
| Scheme 43 Electrochemical indole synthesis.69 | ||
This reaction demonstrates a significantly different C–H/N–H activation than the Co-catalysed work discussed previously as the authors propose a radical mechanism without a transition metal involved in the bond-forming steps. This mechanism is shown in Scheme 44 starting with deprotonation of the amide starting material 67 by electrogenerated MeO− to produce the anion 68. Meanwhile, ferrocene 69a is oxidised up to the 3+ oxidation state 69b at the anode, allowing 69b to then oxidise the anion 68 to produce the amidyl radical 70. Subsequently, 70 undergoes 6-exo-dig cyclisation to give the vinyl radical 71, which in turn undergoes a second cyclisation to give 72. Finally, 72 is oxidised at the anode and deprotonated to produce the desired indole product.69
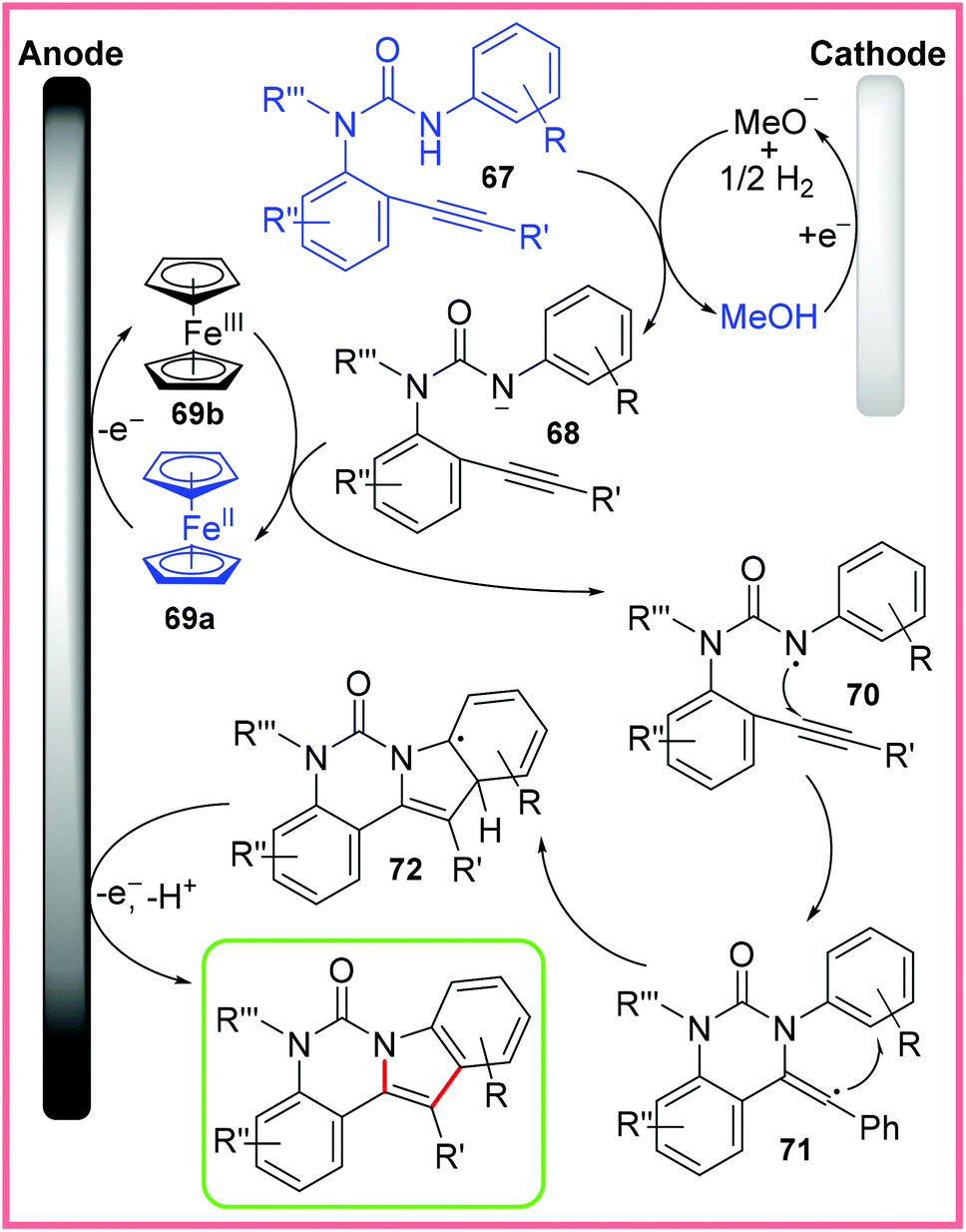 | ||
| Scheme 44 Mechanism for electrochemical indole synthesis.69 | ||
A more recent example of this type of reaction, this time to form isoxazolidine-fused isoquinolinones, was published in 2020 by Zhang and co-workers.70 This work also made use of a tethered alkyne to encourage the desired C–C and C–N bonds to form.
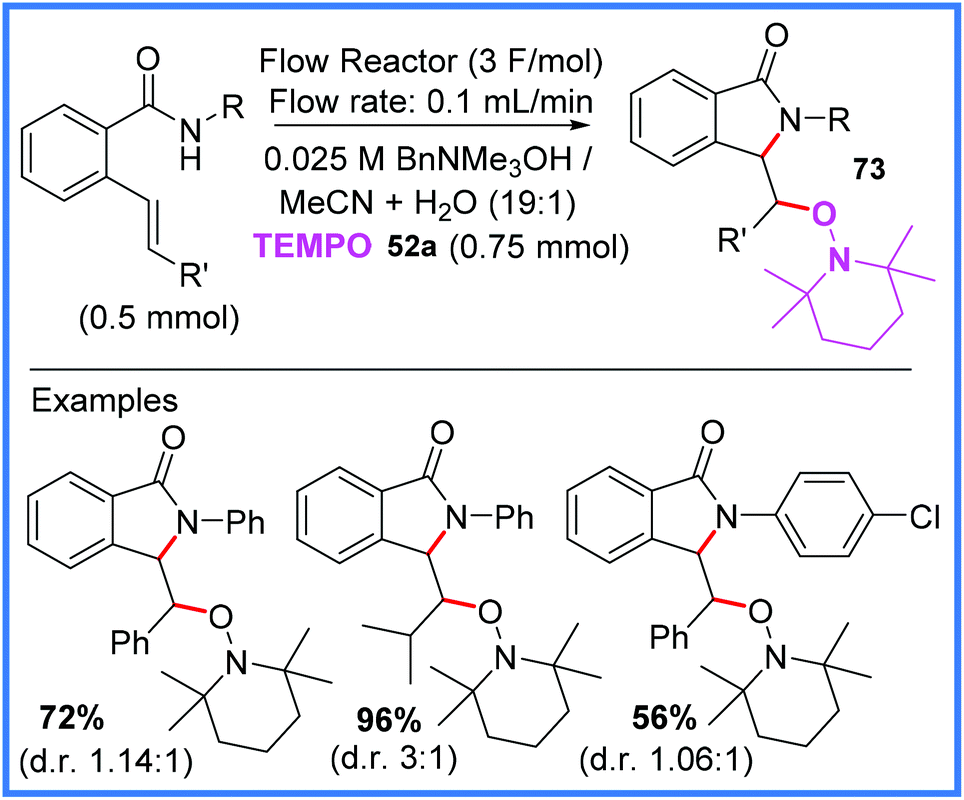 | ||
| Scheme 45 Flow electrosynthesis of isoindolinones.71 | ||
Scheme 46 shows the proposed mechanism for this reaction wherein TEMPO 52a is first oxidised at the anode to produce 52b. This mediator then oxidises the amide starting material, after it has been deprotonated by electrogenerated hydroxide, to produce the amidyl radical 74. This radical then cyclises and is then trapped by 52a to produce the isoindolinone product 73.71
 | ||
| Scheme 46 Mechanism for flow electrosynthesis of isoindolinones.71 | ||
 | ||
| Scheme 47 Electrochemical pyrrolidine synthesis.76 | ||
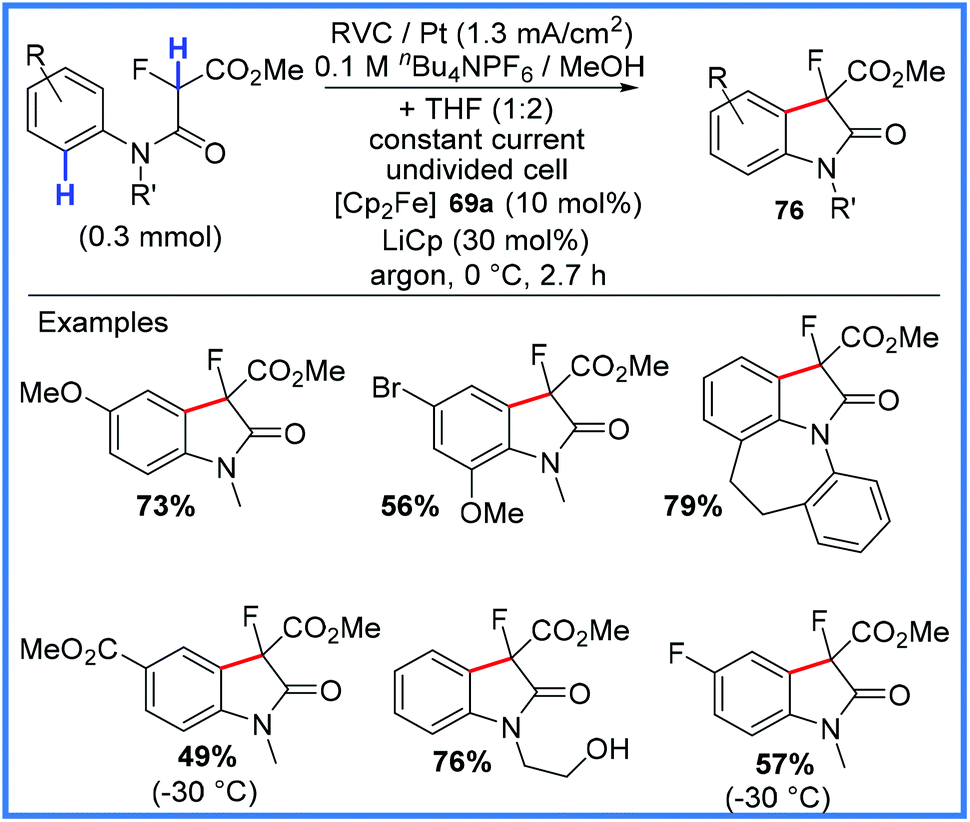 | ||
| Scheme 48 Electrochemical synthesis of 3-fluorooxindoles.77 | ||
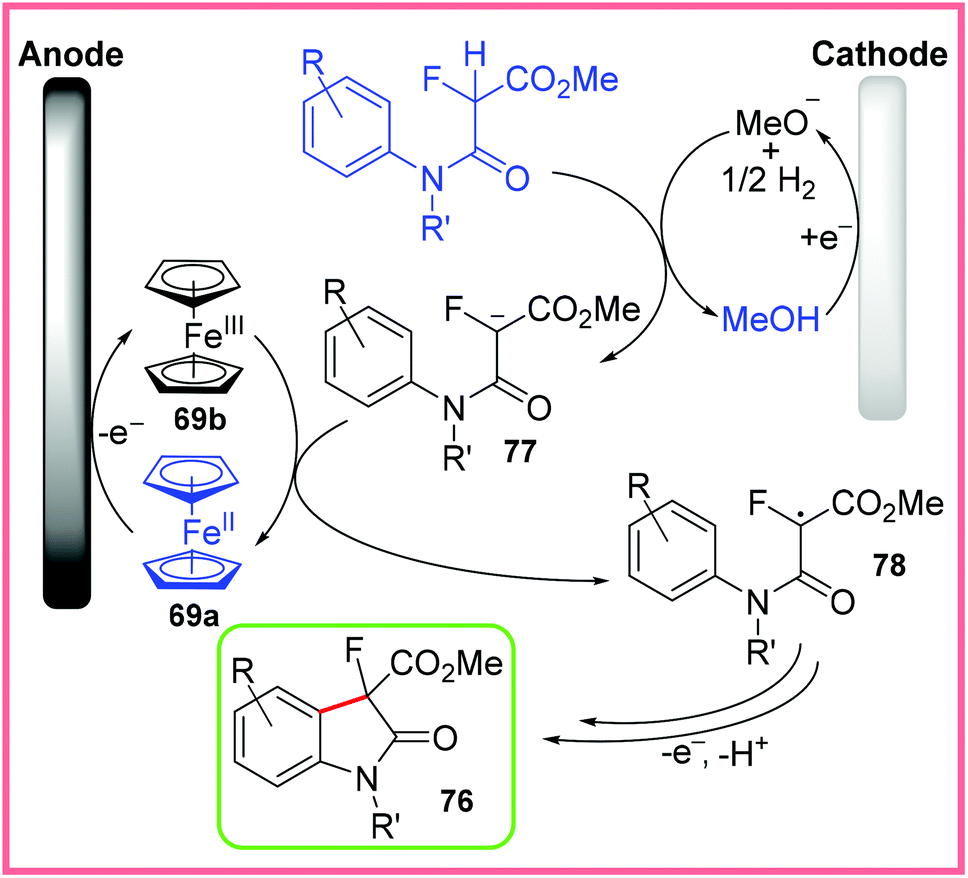
The proposed mechanism for this reaction is given in Scheme 49, with the starting amide first being deprotonated by electrogenerated methoxide to give 77. Ferrocene 69a, used as a mediator, is oxidised to 69b at the anode, allowing 77 to be oxidised to the radical 78. This carbon-centred radical then undergoes cyclisation with oxidation and deprotonation to yield the desired 3-fluorooxindole 76.77
 | ||
| Scheme 49 Mechanism for electrochemical 3-fluorooxindole synthesis.77 | ||
Kolbe-type anodic decarboxylation has also been carried out on amide-containing substrates, this time for the sustainable synthesis of 2-pyrrolidinones via the formation of new C–C bonds, as reported by Quertenmont and co-workers.78
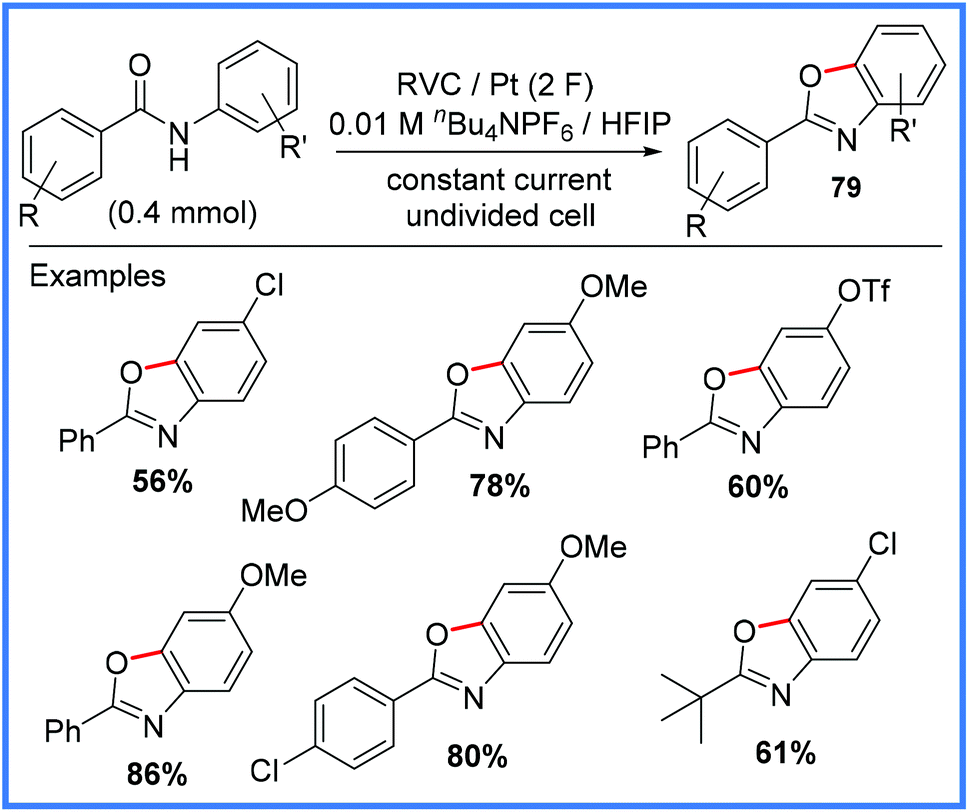 | ||
| Scheme 50 Electrochemical synthesis of benzoxazoles.79 | ||
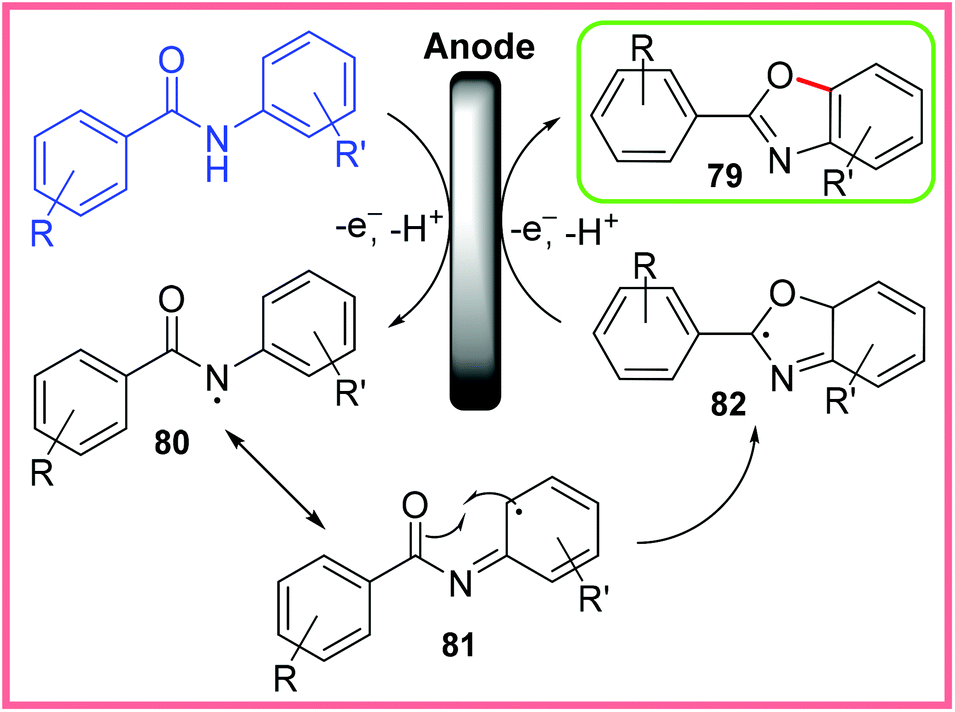
The proposed mechanism for this reaction is shown in Scheme 51, where amidyl radicals 80 are first generated at the anode. It is thought that this radical receives stabilisation through resonance with the neighbouring aromatic ring 81, opening up the possibility for radical cyclisation to take place producing 82. This heterocyclic radical then undergoes a second anodic oxidation to produce the desired benzoxazole 79. The initial oxidation step is possibly facilitated by deprotonation of the amide by HFIP anions (generated at the cathode).79
 | ||
| Scheme 51 Proposed mechanism for electrochemical benzoxazole synthesis.79 | ||
Protocols for the formation of selenylated oxazolines 83 from diselenides also exist, as reported recently by Mallick and co-workers in 2020 and shown in Scheme 52.80 This method proved highly effective as a large scope of substituents were tolerated and yields obtained were very good. It is also a highly sustainable method being transition metal-free, not requiring an external oxidant and carried out at room temperature in an undivided cell. This approach also proved viable for synthesising selenylated isoxazolines from oximes, making it very versatile.80
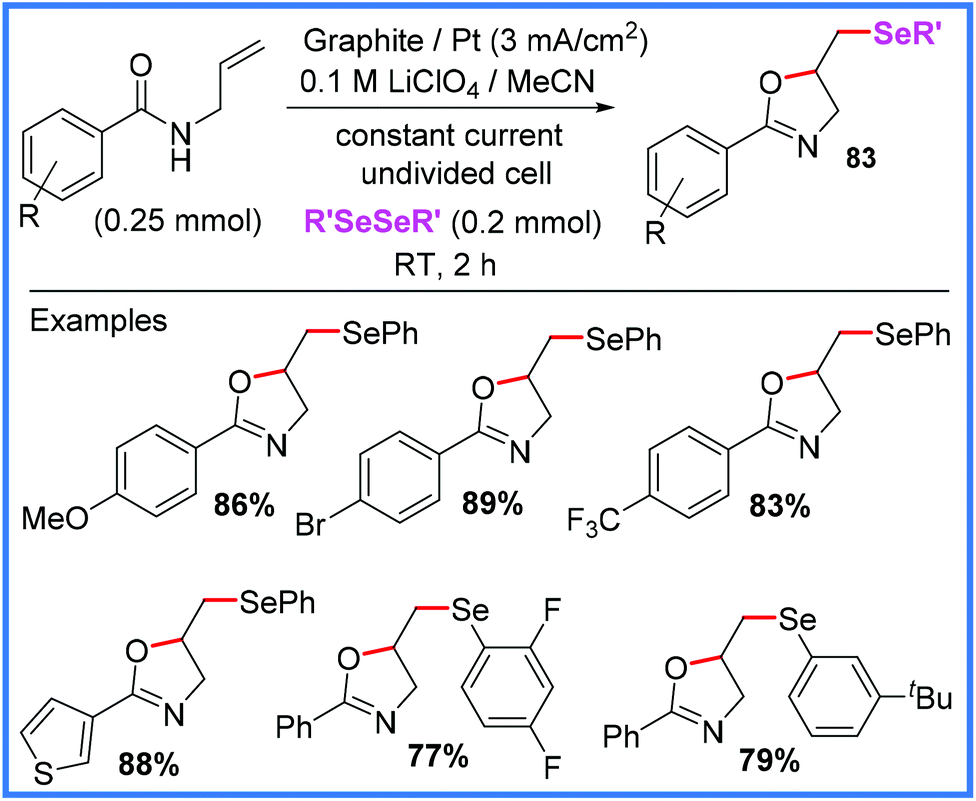 | ||
| Scheme 52 Electrochemical selenylated oxazoline synthesis.80 | ||
The authors proposed two potential pathways for this reaction to proceed. The first pathway is ionic cyclisation as shown in Scheme 53, whereby the diselenide starting material is oxidised at the anode giving selenium cations 84. These cations can react with the alkene of the starting material to produce 85. Nucleophilic cyclisation can then proceed to produce 83.80
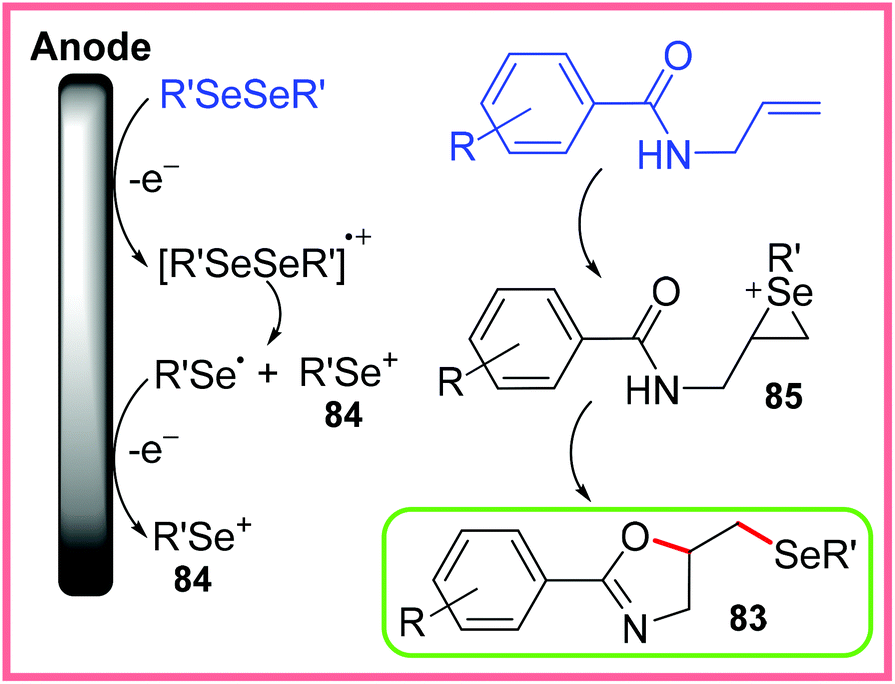 | ||
| Scheme 53 Ionic cyclisation pathway for the formation for selenylated oxazolines.80 | ||
The other proposed pathway for this reaction is radical cyclisation, shown in Scheme 54. In this mechanism, the diselenide is first cathodically reduced, causing fragmentation, before oxidation at the anode produces a pair of selenide radicals 86. Oxidation of the amide starting material then produces the delocalised radical 87, which allows radical cyclisation and coupling with 86 to occur producing 83. In both pathways, the reduction of H+ ions was thought to occur at the cathode.80
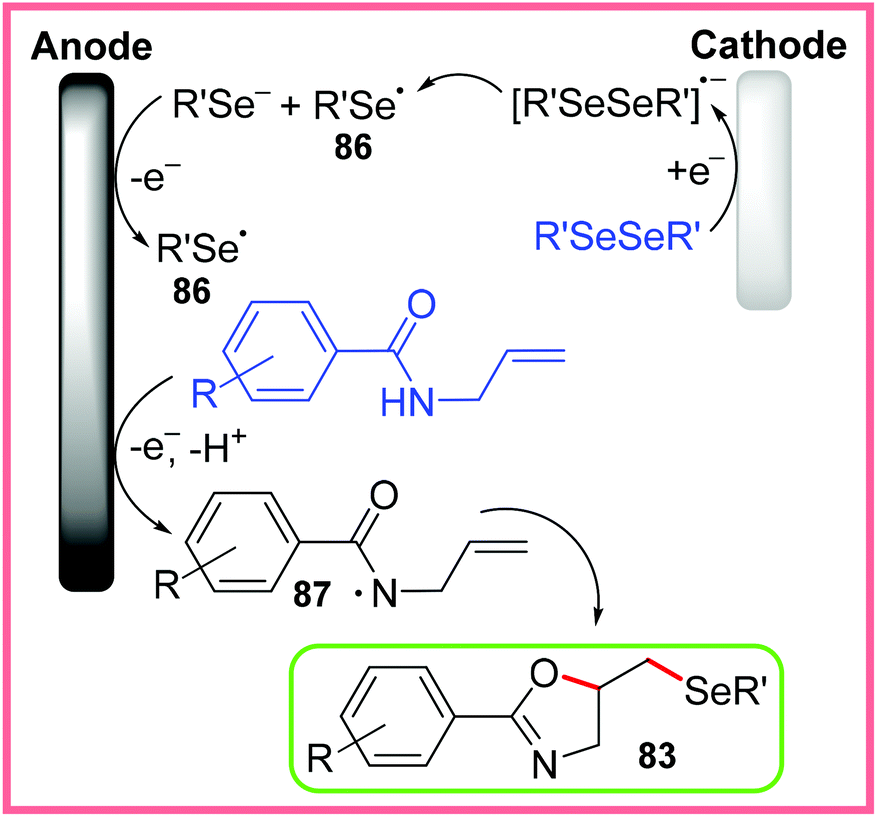 | ||
| Scheme 54 Radical cyclisation pathway for the formation of selenylated oxazolines.80 | ||
Other methods for electrochemical oxazoline synthesis from amides include the cathodic reduction of dichloroethylamides as reported by Guirado and co-workers.81,82
Syntheses of related compounds such as oxazoles have also been reported. The Waldvogel group successfully generated hypervalent ArIF2 species via anodic oxidation of iodoarenes which proved capable of mediating fluorocyclisation of N-propargylamides to 5-fluoromethyl-2-oxazoles.83 Additionally, a broad range of benzoxazines have been synthesised efficiently by anodic oxidation protocols from the Xu84 and Yu85 groups. Sulfonated benzoxazines have also recently been produced by electrochemical radical cascade cyclisation as reported by He and co-workers.86
Such a wealth of highly sustainable and green electrosynthetic methods for producing heterocycles from amides provides a very promising foundation for future preparations of valuable pharmaceuticals and agrochemicals, which are largely comprised of heterocyclic compounds.
3.4. Precursors to other functional groups
Another important mode of reactivity for amides is their conversion into other useful functional groups. One interesting example of this is shown in Scheme 55 as reported by Li and co-workers in 2018,87 demonstrating an elegant use of the Hofmann rearrangement to convert amides into methyl carbamates 88 in an undivided cell. The authors utilised some of the key advantages of electrosynthesis in this work, namely the use of benign NaBr as a mediator, thereby avoiding the handling of hazardous halogens, to produce a wide range of carbamates in excellent yields. This reaction also proved possible on the gram scale.87 | ||
| Scheme 55 Electrochemical Hofmann rearrangement to produce carbamates.87 | ||
The mechanism proposed by the authors for this reaction is shown in Scheme 56, where bromide is oxidised into bromine at the anode and methanol is converted into methoxide at the cathode, providing both a mediator and an effective base electrochemically. The starting amide then reacts under these conditions to produce the N-bromo species 89, which is further converted into an isocyanate intermediate 90 through deprotonation followed by R group migration. In the presence of methoxide, 90 can be nucleophilically attacked to produce the desired methyl carbamates 88.87
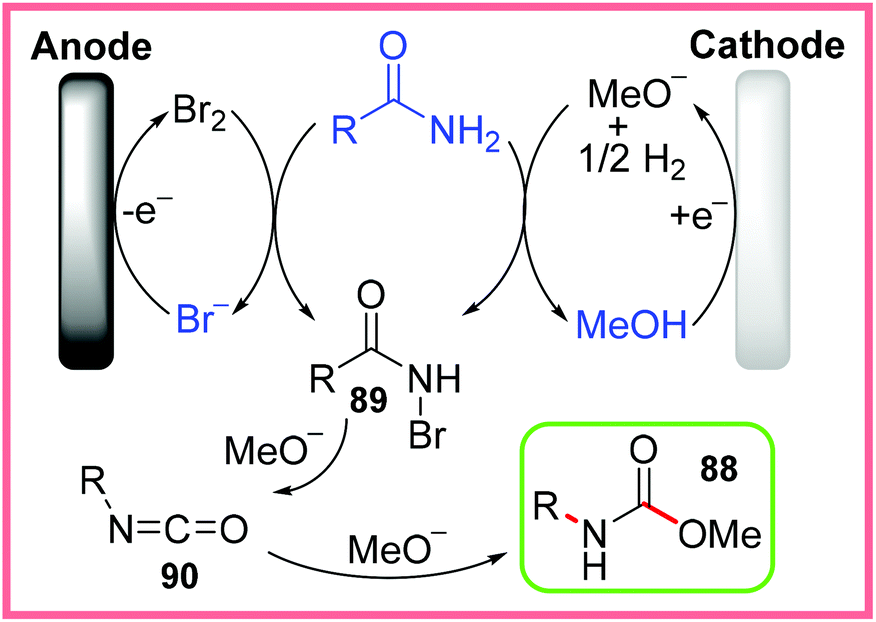 | ||
| Scheme 56 Proposed mechanism for electrochemical Hofmann rearrangement.87 | ||
Another important example of amide functional group conversion is represented in Scheme 57, which shows amides being cathodically reduced to amines by deoxygenation. This work, reported by Edinger and Waldvogel,88 made use of a Pb cathode in a divided cell and an acidic medium. Interestingly, it was noted that if alkylammonium ions 91 were not present in the reaction mixture, the dominant cathodic processes that took place were corrosion of the Pb electrode and proton reduction leading to the evolution of hydrogen. It was postulated by the authors that the alkylammonium ions, when they were present, decorated the surface of the Pb electrode, preventing corrosion and the evolution of hydrogen, whilst allowing the desired deoxygenation reactions to take place. This method also proved effective for producing sulfides from sulfoxides.88
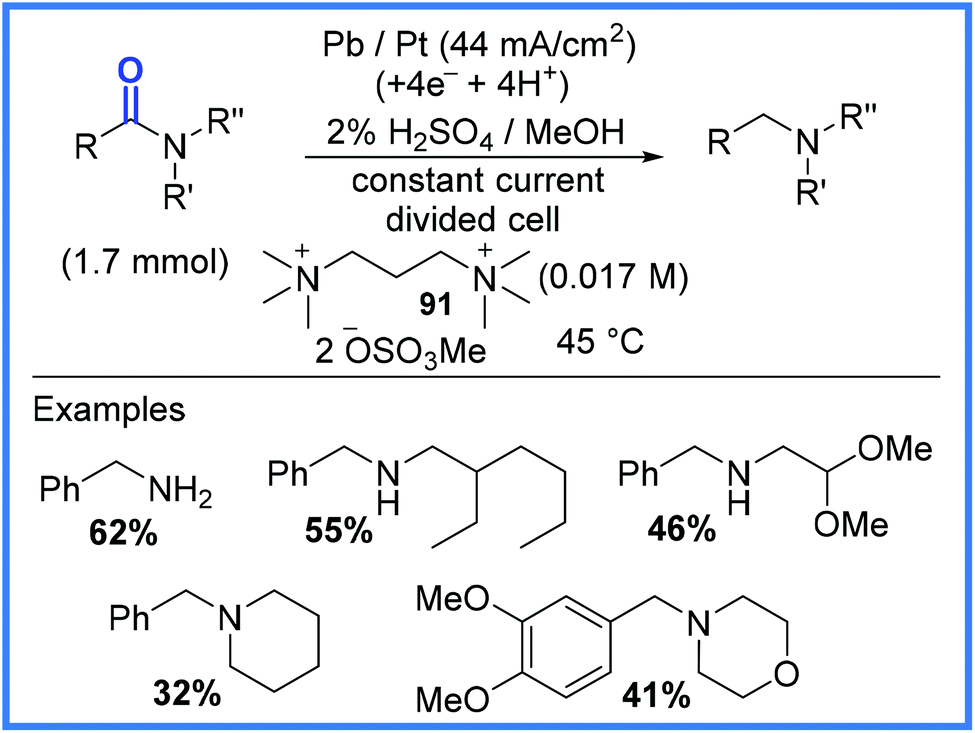 | ||
| Scheme 57 Electrochemical deoxygenation of amides to produce amines.88 | ||
There have also been reports of N-alkoxyamides 92 being converted into esters in the presence of iodide salts in an undivided cell as shown in Scheme 58. In this work, reported by Subramanian and co-workers,89 the reaction proceeded with the release of N2 gas, a clean by-product, under mild conditions.
 | ||
| Scheme 58 Electrochemical conversion of N-alkoxyamides into esters.89 | ||
This reaction is thought to proceed via anodic iodide oxidation which initiates the formation of the radical species 93 from 92. This radical then dimerises to produce 94, which undergoes thermal rearrangement. In this process, N2 is released and the desired ester product is produced. This mechanism is shown in Scheme 59. It is thought that H+ ion reduction at the cathode was largely suppressed by the presence of nBu4N+ ions, which got adsorbed onto the cathode surface.89
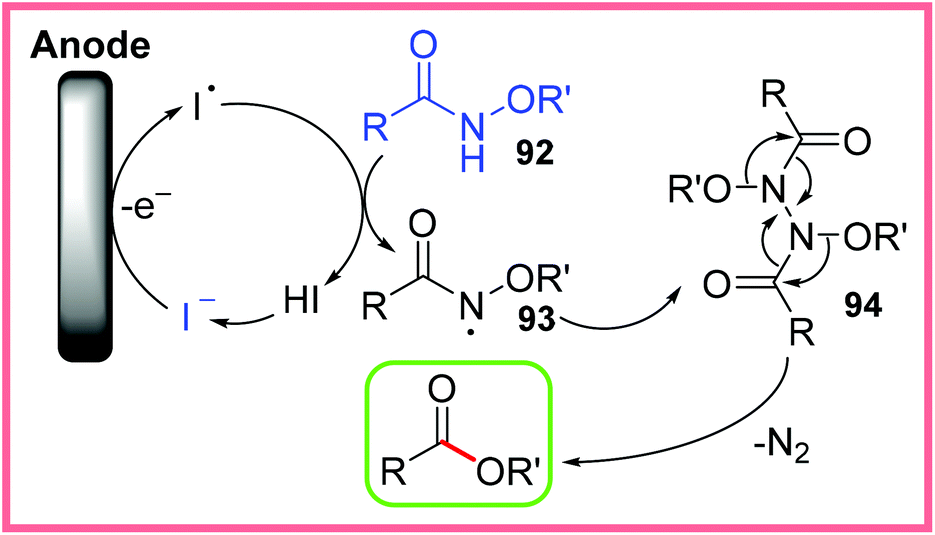 | ||
| Scheme 59 Plausible mechanism for the electrochemical conversion of amides to esters.89 | ||
Electrosynthetic methods for the cathodic reduction of amides to produce aldehydes90 and alcohols91 also exist.
3.5. Other applications
Finally, this section will cover some of the other electrosynthetic applications of amides that do not fit well into the other sections covered in this review. One of these applications is their use as effective aryl radical traps, as shown in Scheme 60. The Little group published this work in 2015, making use of enamides 95 and tris(p-bromophenyl)amine (TBPA) 96a as a mediator under mild constant potential conditions in a divided cell.92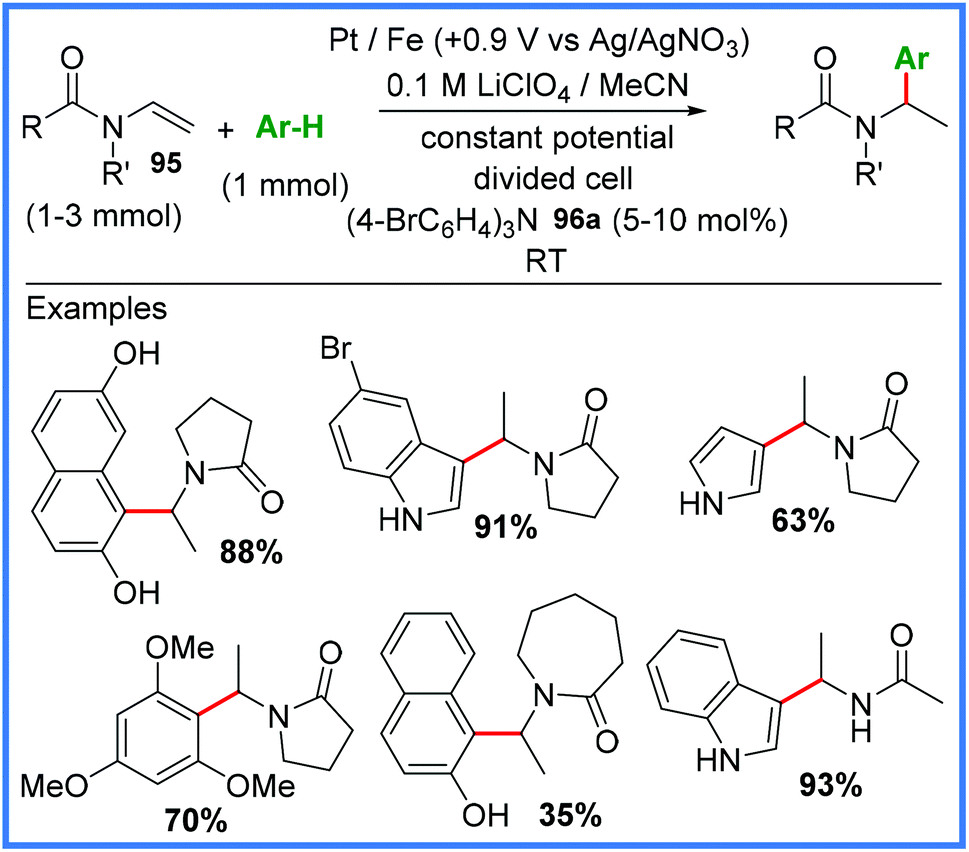 | ||
| Scheme 60 Electrochemical aryl radical trapping using enamides.92 | ||
This protocol was applicable to a range of aryl groups and enamides in good yield. The proposed mechanism for this work is shown in Scheme 61, starting with the oxidation of the TBPA 96a mediator to 96b. This oxidised species then facilitates aromatic oxidation, producing 97. This radical cation formation increases the acidity of the aryl species which leads to the protonation of 95 to form an iminium ion. This is followed by attack by the aromatic group to give the radical cation 98. A final reduction step via reaction with another molecule of the aromatic starting material (generating more 97) produces the final, desired product.92
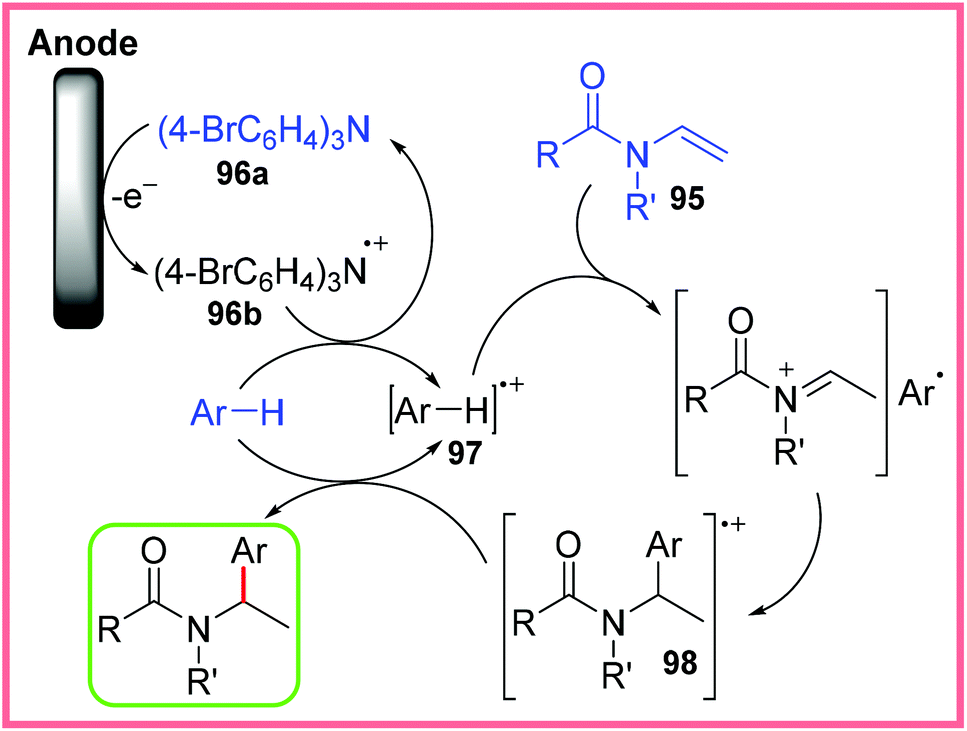 | ||
| Scheme 61 Plausible mechanism for the electrochemical trapping of aryl radicals.92 | ||
Further synthetic applications include the work of Sainsbury and Wyatt in which electrochemical coupling reactions of alkoxylated amide species were reported and studied in 1979.93 The electrochemical decarboxylation of oligopeptides has also been investigated as in the work of Seebach and co-workers from 1989.94 Additionally, there have been reports of amide polymers enhancing the electrochemical reduction of carbon dioxide when electrodeposited onto the surface of Cu electrodes. This recent work, reported by Ahn and co-workers in 2018,95 provides an interesting development in the conversion of CO2 to ethylene (an important topic in CO2 capture and utilisation), as well as showing again the vast range of applications of amides in electrosynthesis.
4. Conclusions and outlook
In summary, this review has explored the creative and sustainable electrochemical syntheses of amides, as well as provide an overview of the types of electrosynthetic reactions that have been carried out using amide substrates. We began by discussing the popular electro-oxidation of iodide salts to produce effective mediators in solution, before moving on to show how nitrile groups and rearrangement reactions had been harnessed to produce amides electrochemically. Methods such as the electro-generation of N-heterocyclic carbenes and the anion pool method were also mentioned, followed by examples of how Shono-type oxidation had been utilised, not just to produce amides, but also to use amides as substrates. This led on to how amides had been employed in electrochemical C–H activations, before the numerous examples of heterocycle synthesis were demonstrated. Finally, the review finished with a brief look at how amides could be converted into other useful functional groups using electrochemistry.The benefits of electrosynthesis compared to traditional synthetic methods have been outlined, showing that external oxidants and transition metals can often be avoided, as well as the commonly used stoichiometric coupling agents frequently employed in amide syntheses. As a result, the methods described in this review are very attractive from a green chemistry perspective, being highly atom-efficient and using overall very mild conditions. Furthermore, the fascinating range of reactivity of the amide motif when exposed to electrochemical conditions shows great promise for future research, including in the synthesis of crucially important heterocycles and pharmaceuticals.
Additionally, given the current growing popularity of electrosynthesis, which shows no signs of slowing down, and the wealth of recent literature in this area, it seems that we are only witnessing the beginning of this highly promising research topic. As more researchers turn their hands towards electrosynthesis, in the coming years it is likely that yet more interest will be garnered in amide synthesis using electrochemical techniques. Finally, given the relatively small number of examples that employ very easily scalable flow electrochemistry currently, it seems widespread adoption in industrial settings is likely still a few years away, however, in time this could become a key topic of research on much larger scales.
Abbreviations
| BOP | (Benzotriazol-1-yloxy)-tris(dimethylamino)-phosphonium hexafluorophosphate |
| CDI | 1,1′-Carbonyldiimidazole |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| DCC | N,N′-Dicyclohexylcarbodiimide |
| DCM | Dichloromethane |
| DMF | N,N-Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| EDC | 1-Ethyl-3-(3′-dimethylaminopropyl)-carbodiimide hydrochloride |
| HAT | Hydrogen atom transfer |
| HATU | N-[(Dimethylamino)-1H-1,2,3-triazolo[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexafluorophosphate N-oxide |
| HFIP | Hexafluoroisopropanol |
| NASH | Nucleophilic aromatic substitution of hydrogen |
| NHC | N-Heterocyclic carbene |
| QRE | Quasi-reference electrode |
| RT | Room temperature |
| RVC | Reticulated vitreous carbon |
| SCE | Saturated calomel electrode |
| SHE | Standard hydrogen electrode |
| SS | Stainless steel |
| TBPA | Tris(p-bromophenyl)amine |
| TEMPO | (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl |
| THF | Tetrahydrofuran |
| WE | Working electrode |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank UCL for financial support.Notes and references
- N. Sewald and H.-D. Jakubke, Peptides: Chemistry and Biology, Wiley-VCH, Weinheim, 2002 Search PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS.
- X. Wang, Nat. Catal., 2019, 2, 98–102 CrossRef.
- B. L. Bray, Nat. Rev. Drug Discovery, 2003, 2, 587–593 CrossRef CAS.
- A. Greenberg, C. M. Breneman and J. F. Liebman, The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science, Wiley-Interscience, New York, 2000 Search PubMed.
- W. E. Bull, S. K. Madan and J. E. Willis, Inorg. Chem., 1963, 2, 303–306 CrossRef CAS.
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385–426 CrossRef CAS.
- M. R. Petchey and G. Grogan, Adv. Synth. Catal., 2019, 361, 3895–3914 CrossRef CAS.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS.
- K. J. McKnelly, W. Sokol and J. S. Nowick, J. Org. Chem., 2020, 85, 1764–1768 CrossRef CAS.
- E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org. Chem., 2020, 4641–4651 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS.
- M. D. Karkas, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- A. Wiebe, T. Gieshoff, S. Mohle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS.
- D. Li, P. W. Seavill and J. D. Wilden, ChemElectroChem, 2019, 6, 5829–5835 CrossRef CAS.
- P. W. Seavill, K. B. Holt and J. D. Wilden, RSC Adv., 2019, 9, 29300–29304 RSC.
- D. Li, T.-K. Ma, R. J. Scott and J. D. Wilden, Chem. Sci., 2020, 11, 5333–5338 RSC.
- G. Hilt, ChemElectroChem, 2020, 7, 395–405 CrossRef CAS.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC.
- H. Huang, G. Yuan, X. Li and H. Jiang, Tetrahedron Lett., 2013, 54, 7156–7159 CrossRef CAS.
- Z. Zhang, J. Su, Z. Zha and Z. Wang, Chem. Commun., 2013, 49, 8982–8984 RSC.
- J.-Y. Chen, H.-Y. Wu, Q.-W. Gui, X.-R. Han, Y. Wu, K. Du, Z. Cao, Y.-W. Lin and W.-M. He, Org. Lett., 2020, 22, 2206–2209 CrossRef CAS.
- P. Qian, J.-H. Su, Y. Wang, M. Bi, Z. Zha and Z. Wang, J. Org. Chem., 2017, 82, 6434–6440 CrossRef CAS.
- Y. Wang, P. Qian, J.-H. Su, Y. Li, M. Bi, Z. Zha and Z. Wang, Green Chem., 2017, 19, 4769–4773 RSC.
- J. Y. Becker, B. E. Smart and T. Fukunaga, J. Org. Chem., 1988, 53, 5714–5720 CrossRef CAS.
- M. A. Kabeshov, B. Musio and S. V. Ley, React. Chem. Eng., 2017, 2, 822–825 RSC.
- E. Estrada and A. Rieker, Z. Naturforsch., B, 1994, 49, 1566–1568 CAS.
- T. Wirtanen, E. Rodrigo and S. R. Waldvogel, Adv. Synth. Catal., 2020, 362, 2088–2101 CrossRef.
- W. Liu, W. Huang, T. Lan, H. Qin and C. Yang, Tetrahedron, 2018, 74, 2298–2305 CrossRef CAS.
- A. Inesi, L. Rossi, M. Feroci and M. Rizzuto, New J. Chem., 1998, 57–61 RSC.
- X. Chang, Q. Zhang and C. Guo, Org. Lett., 2019, 21, 10–13 CrossRef CAS.
- L. Tang, Z.-L. Wang, Y.-H. He and Z. Guan, ChemSusChem, 2020, 13, 4929–4936 CrossRef CAS.
- X. Zhang, T. Cui, X. Zhao, P. Liu and P. Sun, Angew. Chem., 2020, 59, 3465–3469 CrossRef CAS.
- I. Gallardo, G. Guirado and J. Marquet, Eur. J. Org. Chem., 2002, 251–259 CrossRef CAS.
- L. Tang, J. H. Matuska, Y.-H. Huang, Y.-H. He and Z. Guan, ChemSusChem, 2019, 12, 2570–2575 CrossRef CAS.
- Y. Kashiwagi and J.-I. Anzai, Chem. Pharm. Bull., 2001, 49, 324–326 CrossRef CAS.
- H. Kolbe, J. Prakt. Chem., 1847, 41, 138 Search PubMed.
- R. Matthessen, L. Claes, J. Fransaer, K. Binnemans and D. E. De Vos, Eur. J. Org. Chem., 2014, 6649–6652 CrossRef CAS.
- F. Ke, Y. Xu, S. Zhu, X. Lin, C. Lin, S. Zhou and H. Su, Green Chem., 2019, 21, 4329–4333 RSC.
- R. A. Green, D. Pletcher, S. G. Leach and R. C. D. Brown, Org. Lett., 2016, 18, 1198–1201 CrossRef CAS.
- M. Feroci, I. Chiarotto and A. Inesi, Electrochim. Acta, 2013, 89, 692–699 CrossRef CAS.
- D. M. M. M. Dissanayake, A. D. Melville and A. K. Vannucci, Green Chem., 2019, 21, 3165–3171 RSC.
- D. M. M. M. Dissanayake and A. K. Vannucci, Org. Lett., 2019, 21, 457–460 CrossRef CAS.
- Y. Kurose, Y. Imada, Y. Okada and K. Chiba, Eur. J. Org. Chem., 2020, 3844–3846 CrossRef CAS.
- K. Boujlel and J. Simonet, Tetrahedron Lett., 1985, 26, 3005–3006 CrossRef CAS.
- M. Mentel, M. J. Beier and R. Breinbauer, Synthesis, 2009, 1463–1468 CAS.
- T. Shono, Y. Matsumura and K. Tsubata, J. Am. Chem. Soc., 1981, 103, 1172–1176 CrossRef CAS.
- K. D. Moeller, S. Tarazi and M. R. Marzabadi, Tetrahedron Lett., 1989, 30, 1213–1216 CrossRef CAS.
- C. Li, C.-C. Zeng, L.-M. Hu, F.-L. Yang, S. J. Yoo and R. D. Little, Electrochim. Acta, 2013, 114, 560–566 CrossRef CAS.
- A. M. Jones and C. E. Banks, Beilstein J. Org. Chem., 2014, 10, 3056–3072 CrossRef.
- T. Wu, B. H. Nguyen, M. C. Daugherty and K. D. Moeller, Angew. Chem., Int. Ed., 2019, 58, 3562–3565 CrossRef CAS.
- M. Gong and J.-M. Huang, Chem. – Eur. J., 2016, 22, 14293–14296 CrossRef CAS.
- T. Siu, W. Li and A. K. Yudin, J. Comb. Chem., 2000, 2, 545–549 CrossRef CAS.
- M. Ghosh, V. S. Shinde and M. Rueping, Beilstein J. Org. Chem., 2019, 15, 2710–2746 CrossRef CAS.
- D.-S. Lee, Tetrahedron: Asymmetry, 2009, 20, 2014–2020 CrossRef CAS.
- M. K. Bal, C. E. Banks and A. M. Jones, ChemElectroChem, 2019, 6, 4284–4291 CrossRef CAS.
- N. Sauermann, T. H. Meyer, C. Tian and L. Ackermann, J. Am. Chem. Soc., 2017, 139, 18452–18455 CrossRef CAS.
- T. H. Meyer, J. C. A. Oliveira, D. Ghorai and L. Ackermann, Angew. Chem., 2020, 59, 10955–10960 CrossRef CAS.
- N. Sauermann, T. H. Meyer and L. Ackermann, Chem. – Eur. J., 2018, 24, 16209–16217 CrossRef CAS.
- S. Kathiravan, S. Suriyanarayanan and I. A. Nicholls, Org. Lett., 2019, 21, 1968–1972 CrossRef CAS.
- Y. K. Au, H. Lyu, Y. Quan and Z. Xie, J. Am. Chem. Soc., 2020, 142, 6940–6945 CrossRef CAS.
- R. C. Samanta, J. Struwe and L. Ackermann, Angew. Chem., 2020, 59, 14154–14159 CrossRef CAS.
- C. Amatore, C. Cammoun and A. Jutand, Adv. Synth. Catal., 2007, 349, 292–296 CrossRef CAS.
- M. Konishi, K. Tsuchida, K. Sano, T. Kochi and F. Kakiuchi, J. Org. Chem., 2017, 82, 8716–8724 CrossRef CAS.
- K. Sano, N. Kimura, T. Kochi and F. Kakiuchi, Asian J. Org. Chem., 2018, 7, 1311–1314 CrossRef CAS.
- C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383–2387 CrossRef CAS.
- S. Tang, D. Wang, Y. Liu, L. Zeng and A. Lei, Nat. Commun., 2018, 9, 798 CrossRef.
- Z.-W. Hou, Z.-Y. Mao, H.-B. Zhao, Y. Y. Melcamu, X. Lu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 9168–9172 CrossRef CAS.
- L.-B. Zhang, R.-S. Geng, Z.-C. Wang, G.-Y. Ren, L.-R. Wen and M. Li, Green Chem., 2020, 22, 16–21 RSC.
- A. A. Folgueiras-Amador, K. Philipps, S. Guilbaud, J. Poelakker and T. Wirth, Angew. Chem., Int. Ed., 2017, 56, 15446–15450 CrossRef CAS.
- F. Xu, L. Zhu, S. Zhu, X. Yan and H.-C. Xu, Chem. – Eur. J., 2014, 20, 12740–12744 CrossRef CAS.
- L. Zhu, P. Xiong, Z.-Y. Mao, Y.-H. Wang, X. Yan, X. Lu and H.-C. Xu, Angew. Chem., Int. Ed., 2016, 55, 2226–2229 CrossRef CAS.
- P. Xiong, H.-H. Xu and H.-C. Xu, J. Am. Chem. Soc., 2017, 139, 2956–2959 CrossRef CAS.
- H.-C. Xu, J. M. Campbell and K. D. Moeller, J. Org. Chem., 2014, 79, 379–391 CrossRef CAS.
- X. Hu, G. Zhang, F. Bu, L. Nie and A. Lei, ACS Catal., 2018, 8, 9370–9375 CrossRef CAS.
- Z.-J. Wu and H.-C. Xu, Angew. Chem., Int. Ed., 2017, 56, 4734–4738 CrossRef CAS.
- M. Quertenmont, I. Goodall, K. Lam, I. Marko and O. Riant, Org. Lett., 2020, 22, 1771–1775 CrossRef CAS.
- T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, Chem. Commun., 2017, 53, 2974–2977 RSC.
- S. Mallick, M. Baidya, K. Mahanty, D. Maiti and S. De Sarkar, Adv. Synth. Catal., 2020, 362, 1046–1052 CrossRef CAS.
- A. Guirado, R. Andreu and J. Galvez, Tetrahedron Lett., 1998, 39, 1071–1074 CrossRef CAS.
- A. Guirado, R. Andreu, B. Martiz and J. Galvez, Tetrahedron, 2004, 60, 987–991 CrossRef CAS.
- J. D. Herszman, M. Berger and S. R. Waldvogel, Org. Lett., 2019, 21, 7893–7896 CrossRef CAS.
- F. Xu, X.-Y. Qian, Y.-J. Li and H.-C. Xu, Org. Lett., 2017, 19, 6332–6335 CrossRef CAS.
- H. Yu, M. Jiao, R. Huang and X. Fang, Eur. J. Org. Chem., 2019, 2004–2009 CrossRef CAS.
- T.-J. He, W.-Q. Zhong and J.-M. Huang, Chem. Commun., 2020, 56, 2735–2738 RSC.
- L. Li, M. Xue, X. Yan, W. Liu, K. Xu and S. Zhang, Org. Biomol. Chem., 2018, 16, 4615–4618 RSC.
- C. Edinger and S. R. Waldvogel, Eur. J. Org. Chem., 2014, 5144–5148 CrossRef CAS.
- K. Subramanian, S. L. Yedage and B. M. Bhanage, J. Org. Chem., 2017, 82, 10025–10032 CrossRef CAS.
- P.-R. Goetz-Schatowitz, G. Struth, J. Voss and G. Wiegand, J. Prakt. Chem., 1993, 335, 230–234 CrossRef CAS.
- R. A. Benkeser, H. Watanabe, S. J. Mels and M. A. Sabol, J. Org. Chem., 1970, 35, 1210–1211 CrossRef CAS.
- L.-J. Li, Y.-Y. Jiang, C. M. Lam, C.-C. Zeng, L.-M. Hu and R. D. Little, J. Org. Chem., 2015, 80, 11021–11030 CrossRef CAS.
- M. Sainsbury and J. Wyatt, J. Chem. Soc., Perkin Trans. 1, 1979, 108–114 RSC.
- D. Seebach, R. Charczuk, C. Gerber, P. Renaud, H. Berner and H. Schneider, Helv. Chim. Acta, 1989, 72, 401–425 CrossRef CAS.
- S. Ahn, K. Klyukin, R. J. Wakeham, J. A. Rudd, A. R. Lewis, S. Alexander, F. Carla, V. Alexandrov and E. Andreoli, ACS Catal., 2018, 8, 4132–4142 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |