
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Re-usable thermally reversible crosslinked adhesives from robust polyester and poly(ester urethane) Diels–Alder networks†
Laxmisha M.
Sridhar
a,
Murielle O.
Oster‡
a,
Donald E.
Herr§
a,
Jonathan B. D.
Gregg
b,
James A.
Wilson
 b and
Andrew T.
Slark
*b
b and
Andrew T.
Slark
*b
aHenkel Corporation, 1 Henkel Way, Rocky Hill, CT 06067, USA
bDepartment of Chemistry, University of Sheffield, Brook Hill, Sheffield S3 7HF, UK. E-mail: a.slark@sheffield.ac.uk
First published on 20th November 2020
Abstract
The sustainable design of polymers for applications requires careful consideration of how they can be re-used or recycled at the end of service life. There has been considerable interest in covalent adaptable networks (CANs) which offer the potential of the properties of crosslinked polymers but where the materials can be reprocessed like thermoplastics. Although there have been advances in CAN chemistry, materials tend to creep and industrial applications are limited. Here we show thermally reversible crosslinked adhesives from dissociative Diels–Alder networks which can be re-used repeatedly with versatile adhesion and creep resistance. Monomer and isocyanate-free polyester and poly(ester urethane) prepolymers were successfully synthesized by facile techniques with high atom efficiency and the resulting CANs are easy to apply in bulk from the melt. Mechanical properties can be tuned depending on the prepolymer design with the networks providing versatile adhesion to different substrates and creep resistance to 70–80 °C, above both the Tg and Tm of the networks. The adhesives are thermally stable during application and can be re-used repeatedly by simple heating/cooling cycles in bulk, without solvents or additional process steps, providing the same level of performance. Our results demonstrate that these Diels–Alder networks are robust in mechanical performance up to the temperature where significant dissociation begins to occur. This opens the possibility for the considered design of prepolymer architecture and reversible chemistry to meet the performance requirements of different applications in a truly sustainable fashion via scalable, efficient, industrially facile methodologies – where materials are free of solvents or monomers in their synthesis, processing, application and re-use.
Introduction
Plastics have many benefits and are essential for everyday life but they are under scrutiny due to environmental contamination, with only around 10% of all plastics ever produced having been recycled.1 The waste hierarchy has been recognised internationally as a tool to promote progressive waste management.2 This prioritises waste prevention, followed by re-use, recycling, recovery and disposal as a last resort. Thermoplastics can be mechanically recycled but must be separated to be reprocessed effectively3 and chemical recycling via depolymerization of plastic waste to monomer feedstocks is a future option, especially if effective catalysts can be developed.4,5 However, there is a pressing need for effective re-use strategies which require redesign of chemistry, materials and processes.A wide range of polymeric adhesives are used to join a variety of substrates for consumer, packaging, construction, transport and electronics applications.6 Irreversibly crosslinked polymer networks provide the highest bond strength and durability but have a negative environmental impact, since permanent adhesion prevents easy separation at end-of-life which does not facilitate re-use or recycling of materials. Polyurethanes (PU) are a popular choice of adhesives on account of their high performance, durability and versatility in joining a range of substrates enabling manufacture of composite materials.6 One component PU reactive hot melt adhesives (PUR) comprise isocyanate functional prepolymers which crosslink irreversibly via moisture cure providing versatile adhesion, heat resistance and durability. However, the prepolymers are made using an excess of diisocyanate and typically contain a few percent of monomer, typically 4,4′-methylene-diphenyl diisocyanate (MDI), which is volatile at processing temperatures. Conventional PUR is classified as hazardous with a potential sensitising effect on the skin and respiratory organs7 which partly results from the potential hydrolysis of aromatic isocyanates to make highly toxic aromatic amines. Responsible polymer research in the modern world requires the design of materials that are non-hazardous and not only provide the benefits of high-performance in-service but also enable reprocessing and re-use at their end-of-life, thereby reducing the overall environmental burden.
There is a growing interest in CANs which comprise chemical crosslinks that are dynamic under specific stimuli, potentially enabling recyclability.8–10 CANs offer the possibility of the beneficial properties of a crosslinked thermoset combined with the processability of thermoplastic polymers. Methods of making CANs are summarized in several recent excellent reviews of the subject.11–14 Typically, bond exchange proceeds via an associative or dissociative process. In associative CANs, the reaction proceeds via an addition–elimination pathway where flow occurs at high temperature but network integrity is maintained. This is a characteristic of vitrimers which were pioneered by Leibler15via transesterification in epoxies using ester/hydroxyl groups. Other notable examples of associative CANs include transamination of vinylogous urethanes,16,17 transesterification using boronic esters18 and the dynamic exchange of thioesters with thiols.19 For dissociative CANs, the reaction proceeds via an elimination–addition pathway with a temperature dependent equilibrium between covalent bond formation and dissociation. At a sufficiently high temperature there is a sol–gel transition and loss of network integrity. While Diels–Alder cycloadditions are well known,20,21 other notable systems include triazolinedione Alder-ene reactions22,23 and hindered urea exchange.24,25 While the chemistry platforms for CANs are developing well, a recent perspective has highlighted the need for better understanding of materials behaviour.12 Despite the promise of CANs, they commonly exhibit creep deformation which is an important drawback.26,27 There is a need to demonstrate creep resistance under load, especially for high performance applications requiring long term dimensional stability.
The (retro-) Diels–Alder reaction between maleimide and furan groups has been used extensively in polymer chemistry to make a variety of polymer networks and architectures,28 including masking the maleimide group for conjugation to biomolecules.29 The landmark work by Wudl30 reported the use of trifunctional maleimide and tetrafunctional furan monomers to form heat-healable networks. There are some reports on reversible adhesives using maleimide and furan groups including epoxy,31 acrylic32–34 and polyurethane35 matrices. However, most of these networks are formed using the low molecular weight monomer N,N′-(4,4′-methylene diphenyl) bismaleimide (BMI) which is highly toxic and unsuitable for practical consumer or industrial applications. The Diels–Alder reaction between maleimide and furan groups has also been used to make reversible non-isocyanate polyurethanes36,37 although no detail has been reported on their material properties. A very recent study38 has highlighted that flexible packaging films bonded with crosslinked polyurethane adhesives can be separated effectively, however this requires immersion of the bonded laminates in hot dimethyl sulfoxide to dissociate the maleimide-furan adducts. Hetero Diels–Alder reactions between cyclopentadiene and thioesters in acrylic networks have also been used for reversible adhesion.39 However, re-use was not demonstrated, and the chemistry is unlikely to be facile enough for broad application.
Our approach was motivated by knowledge of PUR adhesive products where mechanical performance not only comes from covalent crosslinks (urea via moisture cure) but also urethane functional groups and polyester segment crystallinity in prepolymer backbones. Our approach reported here involves a range of polyester and poly(ester urethane) dissociative CANs made from telechelic maleimides and polyfunctional furans (Scheme 1). At ambient temperature network formation is favoured via Diels–Alder cycloaddition reactions, whereas at higher temperatures the equilibrium shifts to the dissociated prepolymers. The chemistry is facile from maleimide and furan functional groups attached to prepolymer backbones with remarkably high atom efficiency and conversion, without needing purification.40 The polymer networks are simply prepared by mixing of heated prepolymers in the bulk and curing under ambient conditions after melt application. We show that mechanical properties can be tuned to be comparable to irreversible PUR, that the adhesion is reversible, versatile and that bonds are resistant to creep deformation above both the Tg and Tm of the networks. We demonstrate that the CANs can be re-used repeatedly producing the same level of performance.
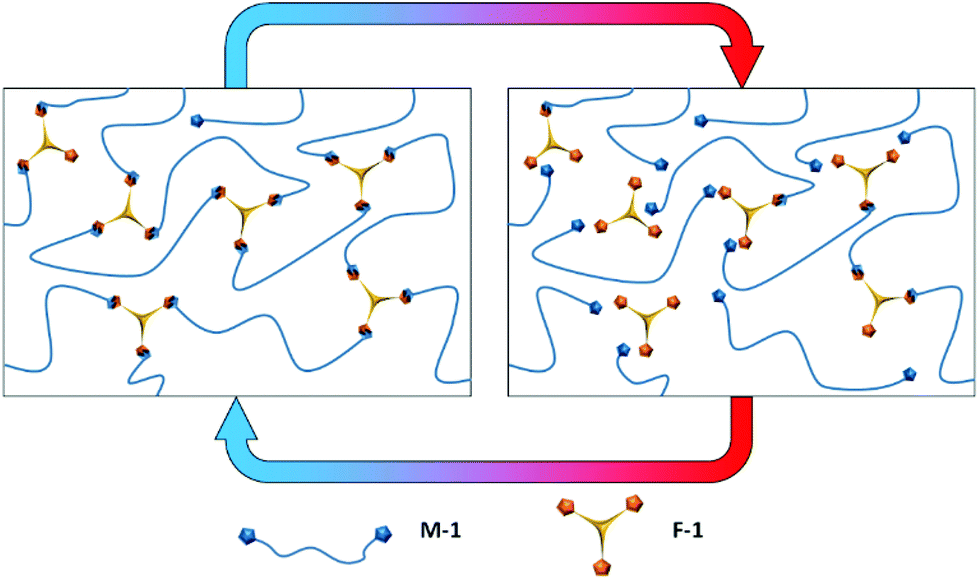 | ||
| Scheme 1 The dynamic equilibrium of Diels–Alder networks based on trifunctional furan (F-1) and telechelic bis-maleimide (M-1). At ambient temperature network formation is favoured via the cycloaddition between furan and maleimide functional groups (left hand side). At higher temperatures network dissociation is favoured (right hand side). | ||
Results & discussion
Polyester CANs from model trifunctional furan crosslinkers and bifunctional maleimides
The trifunctional furan F-1 was synthesized via the reaction of 1,3,5-benzenetricarbonyl chloride with furfuryl alcohol (Scheme 2A). 1H NMR spectroscopy of F-1 indicates the successful incorporation of the furan functional groups via facile, quantitative reaction in high yield (Fig. S1†).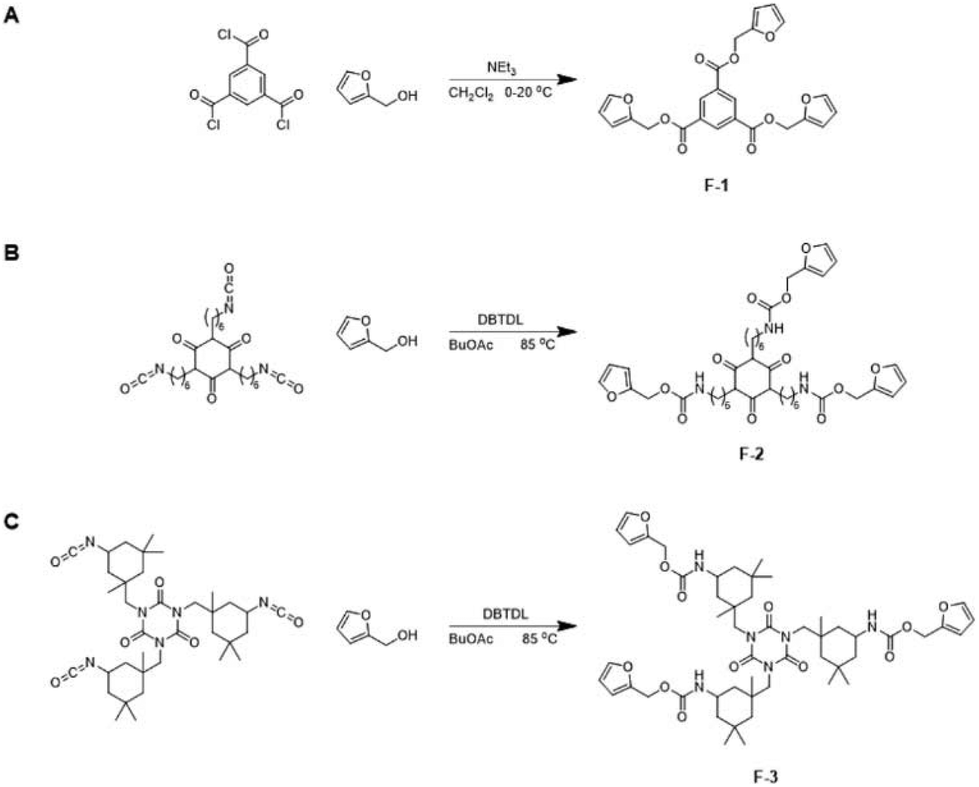 | ||
| Scheme 2 One-step synthesis of multifunctional furans F-1, F-2 and F-3 using furfuryl alcohol. | ||
F-1 was then copolymerized with M-1 to form the network DA-1 by heating a mixture of the two components in bulk followed by coating a film from a completely miscible melt which immediately cooled to ambient temperature (Scheme 3, Table 1). The coated film was left to cure at room temperature and after crosslinking it was visibly transparent and homogeneous.
 | ||
| Scheme 3 Network formation of DA-1 from the reaction of trifunctional furan F-1 with bifunctional maleimide M-1. | ||
The IR spectra of F-1, M-1 and the resulting network DA-1 were analysed. The maleimide absorption is clearly seen in M-1 at σ = 696 cm−1 but extremely low in DA-1 suggesting that the maleimide functional groups have copolymerized successfully (Fig. S2†). The DSC traces show the crystalline nature of the multifunctional furan F-1 (Tm = 101.8 °C) whereas M-1 is amorphous with a glass transition temperature (Tg = −54.1 °C) (Fig. 1A). The resulting network DA-1 has a much higher Tg (47.3 °C), whereas the sharp melting point associated with F-1 disappears and is replaced by a broad endotherm at 145.7 °C which corresponds to the retro Diels–Alder reaction (see later for further discussion).
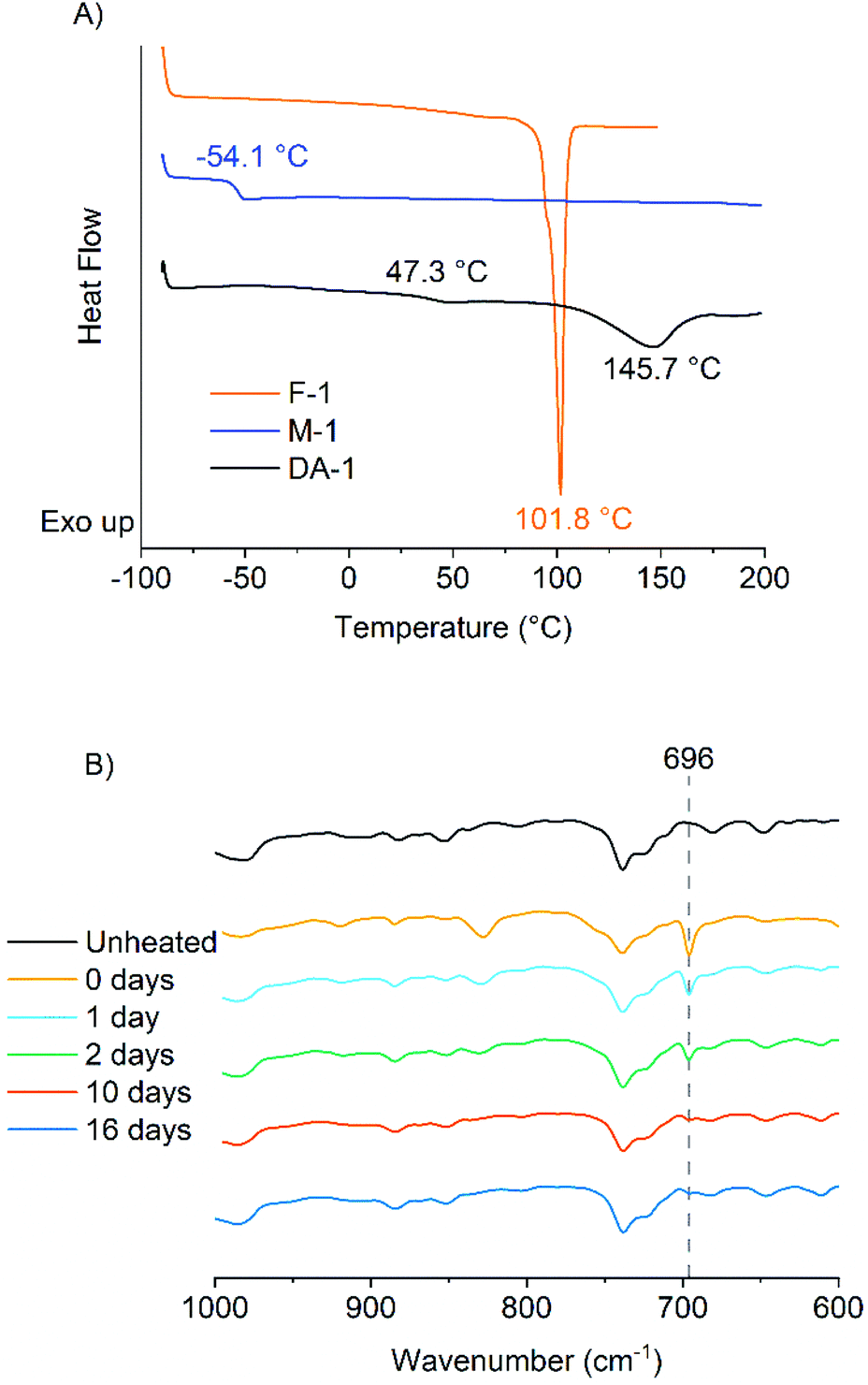 | ||
| Fig. 1 (A) DSC thermograms of F-1, M-1 and DA-1 (first heating cycle). (B) Expanded IR spectra of DA-1 over the range 600–1000 cm−1 before heating and over time at ambient temperature post-deposition from the melt. | ||
FTIR spectroscopy was used to evaluate DA-1 over time at ambient temperature after its immediate deposition from the melt. The maleimide absorption at 696 cm−1 is barely discernible for a film which was stored at ambient temperature (Fig. 1B for expanded spectra and Fig. S3† for full spectra). After heating for 1 hour at 150 °C, this band at 696 cm−1 is initially most prominent immediately after cooling (0 days) but gradually becomes attenuated over time at ambient temperature. This spectral feature decays rapidly over the first 48 hours and continues to weaken over the remaining time resulting in a relatively high final degree of conversion (Fig. 2, left-hand axis). The composition DA-1 was also applied from the bulk melt between beechwood substrates and the bond formed immediately (t = 0). The bond strength increases relatively quickly over a period of 48 hours before tending towards a plateau (Fig. 2, right-hand axis), i.e. the development in bond strength over time inversely correlates to the decrease in the maleimide functional group concentration. Reaching a bond strength of 4 MPa in 24 hours is highly significant since this is the stress at which surface substrate wood fibre tear occurs.
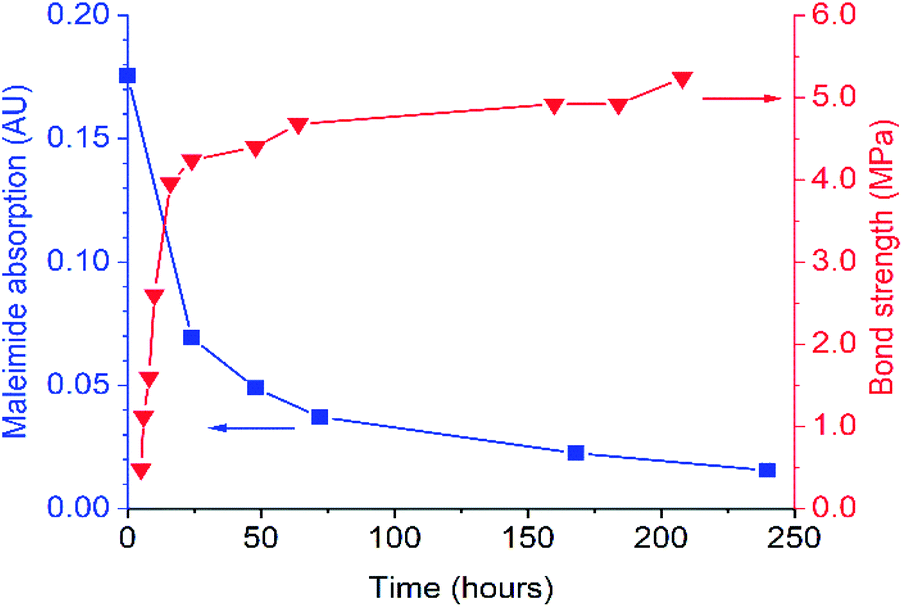 | ||
| Fig. 2 Comparison of the time dependence for both the IR absorbance of maleimide at 696 cm−1 (left axis) and bond strength (right axis) for DA-1 after deposition from the melt (t = 0). | ||
Poly(ester urethane) CANs from multifunctional furan-urethane crosslinkers and bifunctional maleimides
Given these highly promising initial results we investigated the 1-step synthesis of multifunctional furans from the polyfunctional isocyanates HDI trimer and IPDI trimer (Scheme 2B and C, respectively). 1H NMR spectroscopic analysis of the polyfunctional furans F-2 and F-3 (synthesized from the polyfunctional HDI and IPDI isocyanate precursors, respectively) clearly illustrates the incorporation of the furan-urethane groups via successful reaction of the isocyanate functional groups with furfuryl alcohol (Fig. S4 and S5,† respectively). The materials are made in one step with no purification necessary. It is also feasible to make the materials without solvent, e.g.F-2 was made successfully in bulk with the same results.The more rigid F-3 has a higher melting point than F-2 resulting from the cycloaliphatic nature of the former (Fig. S6†). Both F-2 and F-3 were melt-blended separately with M-1 and cooled to room temperature to form crosslinked networks DA-2 and DA-3, respectively. The resulting networks DA-2 and DA-3 both exhibit endotherms at 143.7–145 °C owing to the retro Diels–Alder reaction. DA-2 is amorphous due to complete compatibility between F-2 and M-1. In contrast, DA-3 contains residual crystallinity owing to partial incompatibility of F-3 with M-1 (note the residual melting point at 76.7 °C). Alternative networks were investigated by combining both the more flexible multifunctional furan F-2 and the more rigid multifunctional furan F-3 with M-1. A series of compositions was explored by copolymerizing M-1 with different relative concentrations of the polyfunctional furans F-2 and F-3 (Fig. S7†). A wide range of mechanical properties are achievable (modulus 10–350 MPa and elongation 5–100%) with softer, more flexible compositions resulting from a relatively high concentration of the crosslinker derived from the HDI trimer (F-2).
Poly(ester urethane) CANs from multifunctional furan-urethane crosslinkers and bifunctional maleimide ester-urethane prepolymers
Maleimide terminated prepolymers were made in a facile manner from polyester polyols by consecutive addition reactions in bulk (1) with MDI to form an isocyanate terminated prepolymer and (2) reaction of the terminal isocyanate groups with 2-hydroxyethyl maleimide (Scheme 4). Two telechelic poly(ester urethane) prepolymers were synthesized, semi-crystalline M-2 and amorphous M-3. 1H NMR spectra for the maleimide prepolymers M-2 and M-3 illustrate the aromatic urethane (B, C) and aliphatic maleimide (A) end-groups (Fig. S8 and S9†). The molecular weight distributions of the maleimide terminated prepolymers were determined by GPC (Fig. S10†). Comparison with the starting polyols confirms a modest increase in apparent molecular weight which is a result of the reagent stoichiometries used for reactions 1 and 2 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol. eq. NCO:OH in step 1 and 1:1 mol. eq. OH:NCO in step 2). A minor reaction product (estimated to be approximately 5% based on the GPC RI response) is observed at low molecular weight (apparent Mp approximately 550 g mol−1). This is likely to result from the reaction of residual MDI monomer remaining from step 1 with HEMI in step 2, also incorporating residual very low molecular weight oligomers from the original polyol. Compared to MDI itself, these adducts will be non-volatile (higher molecular weight and polarity) and they will also be incorporated into the Diels–Alder network. This contrasts to conventional moisture curing PUR where toxicity comes from the volatility of the MDI monomer and hydrolysis to toxic aromatic amines. If necessary, it would be feasible to make the prepolymer without this low level of side product by removing free monomer from step 1. This is achieved on an industrial scale using asymmetric isocyanates such as 2,4-MDI instead of 4,4′-MDI and vacuum distillation.7M-2 and M-3 were readily combined with F-3 to make a homogenous melt and crosslinked network DA-4 was formed over time curing at ambient temperature (Scheme 5). At high temperatures the network equilibrium shifts towards dissociated prepolymers (right hand side) whereas crosslinking is favoured at ambient temperature comprising both covalent bonds from the Diels–Alder cycloaddition and association of non-covalent crystalline segments (left hand side). DSC data (Fig. S11†) confirm the crystallinity of the multifunctional furan F-3 (Tm = 73.7 °C), the semi-crystalline nature of the bismaleimide prepolymer M-2 (Tm = 54.5 °C, similar to the original polyol) and the amorphous nature of the bismaleimide prepolymer M-3 (Tg = −33.9 °C, similar to the original polyol). The resulting network DA-4 is a semi-crystalline CAN comprising a sub-ambient Tg (−11.7 °C), a melting point of 50.8 °C and a retro Diels–Alder peak temperature of 140.4 °C. Effectively, the Diels–Alder covalent network is reinforced by the non-covalent interactions of the crystalline polyester segments.
:1 mol. eq. NCO:OH in step 1 and 1:1 mol. eq. OH:NCO in step 2). A minor reaction product (estimated to be approximately 5% based on the GPC RI response) is observed at low molecular weight (apparent Mp approximately 550 g mol−1). This is likely to result from the reaction of residual MDI monomer remaining from step 1 with HEMI in step 2, also incorporating residual very low molecular weight oligomers from the original polyol. Compared to MDI itself, these adducts will be non-volatile (higher molecular weight and polarity) and they will also be incorporated into the Diels–Alder network. This contrasts to conventional moisture curing PUR where toxicity comes from the volatility of the MDI monomer and hydrolysis to toxic aromatic amines. If necessary, it would be feasible to make the prepolymer without this low level of side product by removing free monomer from step 1. This is achieved on an industrial scale using asymmetric isocyanates such as 2,4-MDI instead of 4,4′-MDI and vacuum distillation.7M-2 and M-3 were readily combined with F-3 to make a homogenous melt and crosslinked network DA-4 was formed over time curing at ambient temperature (Scheme 5). At high temperatures the network equilibrium shifts towards dissociated prepolymers (right hand side) whereas crosslinking is favoured at ambient temperature comprising both covalent bonds from the Diels–Alder cycloaddition and association of non-covalent crystalline segments (left hand side). DSC data (Fig. S11†) confirm the crystallinity of the multifunctional furan F-3 (Tm = 73.7 °C), the semi-crystalline nature of the bismaleimide prepolymer M-2 (Tm = 54.5 °C, similar to the original polyol) and the amorphous nature of the bismaleimide prepolymer M-3 (Tg = −33.9 °C, similar to the original polyol). The resulting network DA-4 is a semi-crystalline CAN comprising a sub-ambient Tg (−11.7 °C), a melting point of 50.8 °C and a retro Diels–Alder peak temperature of 140.4 °C. Effectively, the Diels–Alder covalent network is reinforced by the non-covalent interactions of the crystalline polyester segments.
 | ||
| Scheme 4 The synthesis of maleimide terminated poly(ester urethane) prepolymers from polyester polyols. | ||
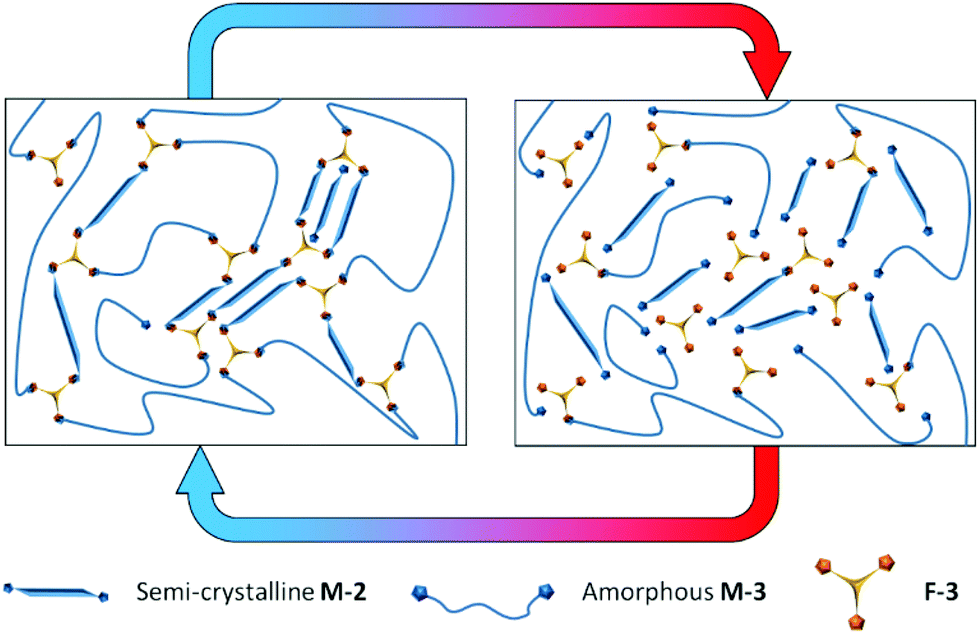 | ||
| Scheme 5 The dynamic equilibrium of Diels–Alder network DA-4 based on multifunctional furan F-3, semi-crystalline bismaleimide M-2 and amorphous bismaleimide M-3. At ambient temperature network formation is favoured via the cycloaddition between furan and maleimide functional groups reinforced by crystalline segments. At higher temperature network dissociation is favoured and crystalline regions melt. | ||
Comparing the network properties of the different CANs DA-1, DA-2 and DA-4
Comparing the thermal properties of DA-4 with DA-1 and DA-2 (Fig. 3 and Table 2) show that the network properties are highly dependent on fg, the molar concentration of maleimide and furan functional groups. The glass transition temperature lowers with a reduction in fg. Although all networks show an endotherm at a higher temperature due to the retro Diels–Alder reaction, the peak position of the endotherm and the size of the enthalpy change both reduce such that DA-1 > DA-2 ≫ DA-4, which is consistent with the relative reduction in the concentrations of the maleimide/furan functional groups. The strong dependence of network properties on fg is similar to that observed by Van Assche et al. on polyether networks based on furan functional prepolymers and BMI monomer cast from solution where materials were investigated in the range fg = 0.69–2.52.41,42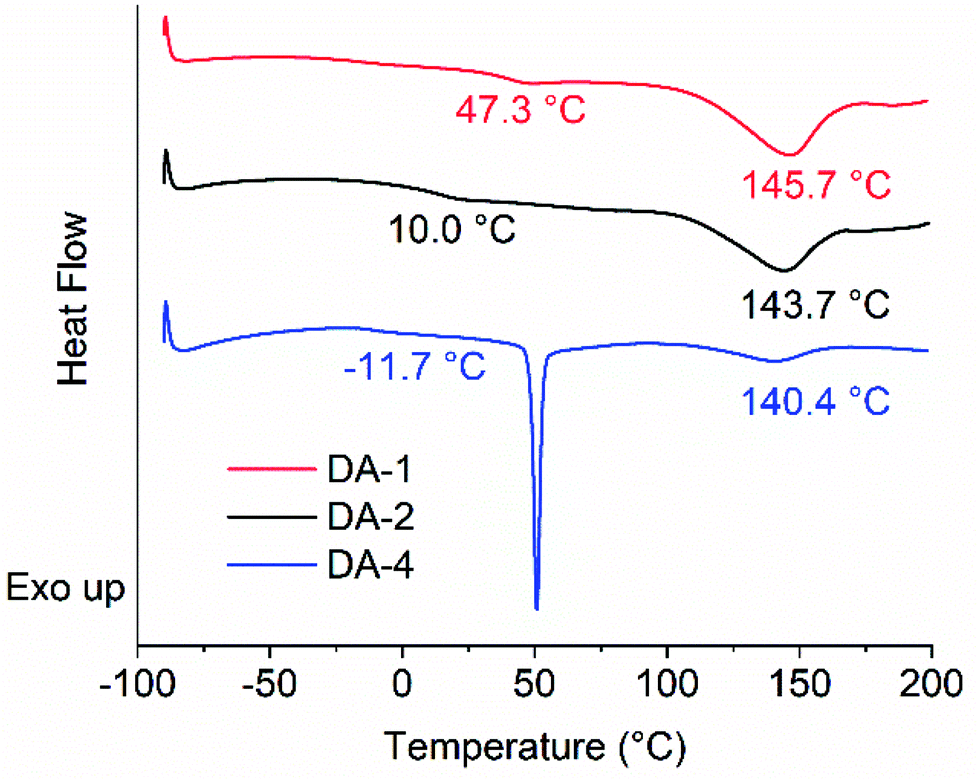 | ||
| Fig. 3 DSC thermograms of DA-1, DA-2 and DA-4 (first heating cycle). | ||
The relative change in concentration of maleimide functional groups was also monitored as a function of time at ambient temperature after deposition from the melt (Fig. 4). The initial absorbance is proportional to fg, i.e.DA-1 > DA-2 ≫ DA-4. Independent of the type of network and functional group concentration, the maleimide concentration reduces relatively quickly over 48 hours and tends towards a limiting value after 7 days. Strikingly similar behaviour is observed despite significant differences in functional group concentration, the molecular weight between reacting functional groups and the functional groups present in the backbones. Thus, each system has sufficient mobility for functional groups to react and for the networks to form at ambient temperature. However, closer inspection of the data reveals a relatively low functional group concentration remaining after 7 days (DA-4 < DA-2 < DA-1) which may result from higher mobility in systems with a lower glass transition temperature (the corresponding Tg values for these three networks are −11.7 °C, 10.0 °C and 47.3 °C, respectively). Comparing the absorbance observed after curing for 7 days at ambient temperature to that recorded immediately after coating indicates that the approximate conversions for DA-1, DA-2 and DA-4 are 82%, 88% and 95%, respectively.
 | ||
| Fig. 4 Comparative IR absorbance at wavelength 696 cm−1 over time for DA-1, DA-2 and DA-4 (compared against absorbance at 793 cm−1). Absorbance values after 7 days are: 0.027 for DA-1 (82% conversion), 0.014 for DA-2 (88% conversion) and 0.003 for DA-4 (95% conversion). | ||
The networks DA-1, DA-2 and DA-4 were also evaluated by DMTA on films of similar thickness used in the adhesion and creep experiments (Fig. 5A). Both DA-1 and DA-2 were optically clear, with DA-4 slightly hazy (semi-crystalline). All three networks have high stiffness below Tg with DA-4 having the highest mechanical properties (>3 GPa) owing to its semi-crystalline nature (Fig. 5B). In all cases the storage modulus reduces above the corresponding Tg. There is also a step-change in modulus for DA-4 above ambient temperature which is attributed to melting of the crystalline phase. After passing through the Tg or Tm, the moduli reduce slowly with temperature in the range 40–80 °C. At higher temperatures, the modulus decreases rapidly while tanδ accelerates through unity, the transition point between a solid and a liquid (Fig. 5C). This coincides with stretching and failure of the film samples being analysed. This is a direct consequence of the temperature dependent equilibrium between the maleimide/furan adducts at lower temperatures and dissociated maleimide/furan functional groups at higher temperatures.
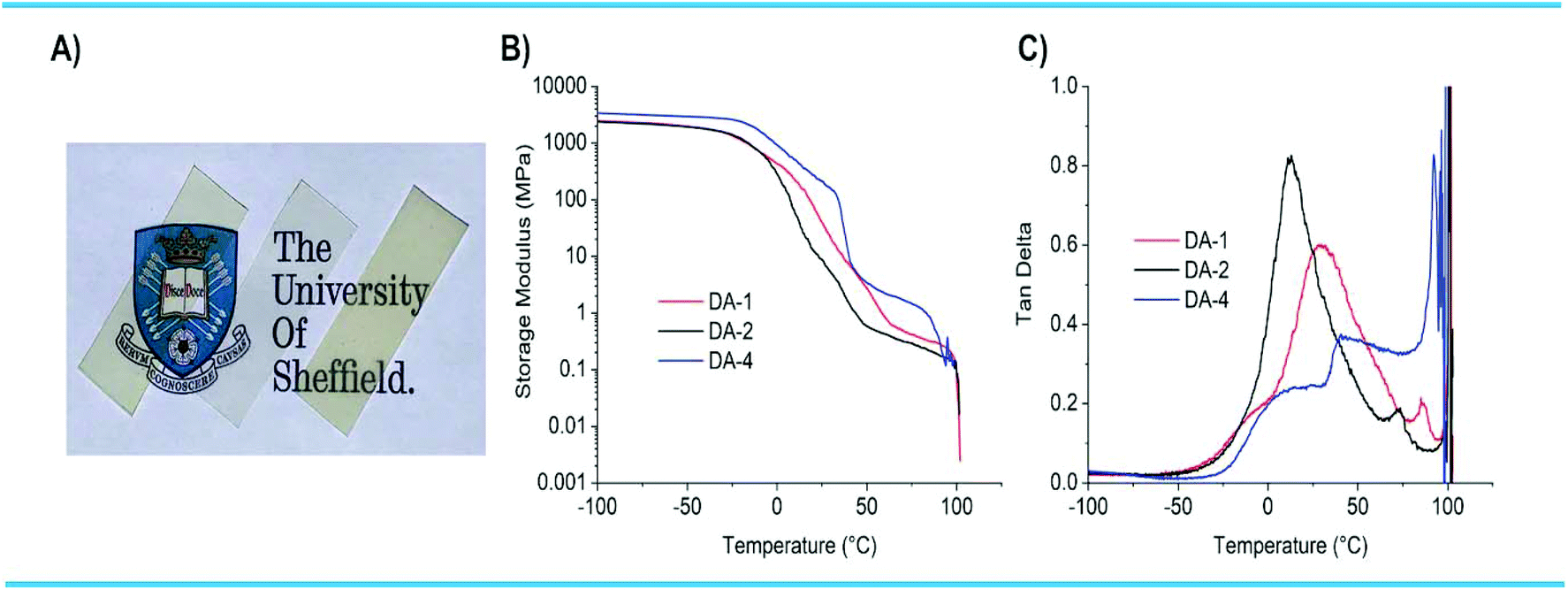 | ||
| Fig. 5 (A) 250 μm films of networks DA-1 (left), DA-2 (middle) and DA-4 (right) used for DMTA and mechanical property evaluation. (B) Storage modulus against temperature from DMTA on films and (C) tanδ against temperature from DMTA on films. | ||
The temperature where rapid changes occur depends on the network type and fg. For DA-1 (high fg), the rapid reduction in G′ starts to occur at 97 °C with the solid–liquid transition at 101 °C (where tanδ = 1). However, for DA-4 (low fg), the rapid change in G′ starts to occur at 85 °C, with the solid–liquid transition at 95 °C. The pronounced reduction in G′ and increase in tanδ at higher temperatures are a consequence of CAN dissociation. The transition between G′ starting to reduce sharply and material flow – where the material behaves as a “strong liquid” – has also been observed for polyether Diels–Alder networks based on maleimides and furans.43
Mechanical properties, adhesion and creep resistance of thermally reversible CANs compared to irreversible crosslinked PU
The mechanical properties of the thermally reversible Diels–Alder networks DA-1 and DA-4 were compared to the irreversible moisture cured PUR which is widely used for various industrial applications (Fig. 6A). DA-1, which has the highest functionality and is obtained from the lowest molecular weight precursors, exhibits the highest break stress and lowest elongation. DA-4, with relatively low maleimide and furan functionality, has similar stress–strain properties to that observed for the PUR – both materials contain semi-crystalline polyester segments.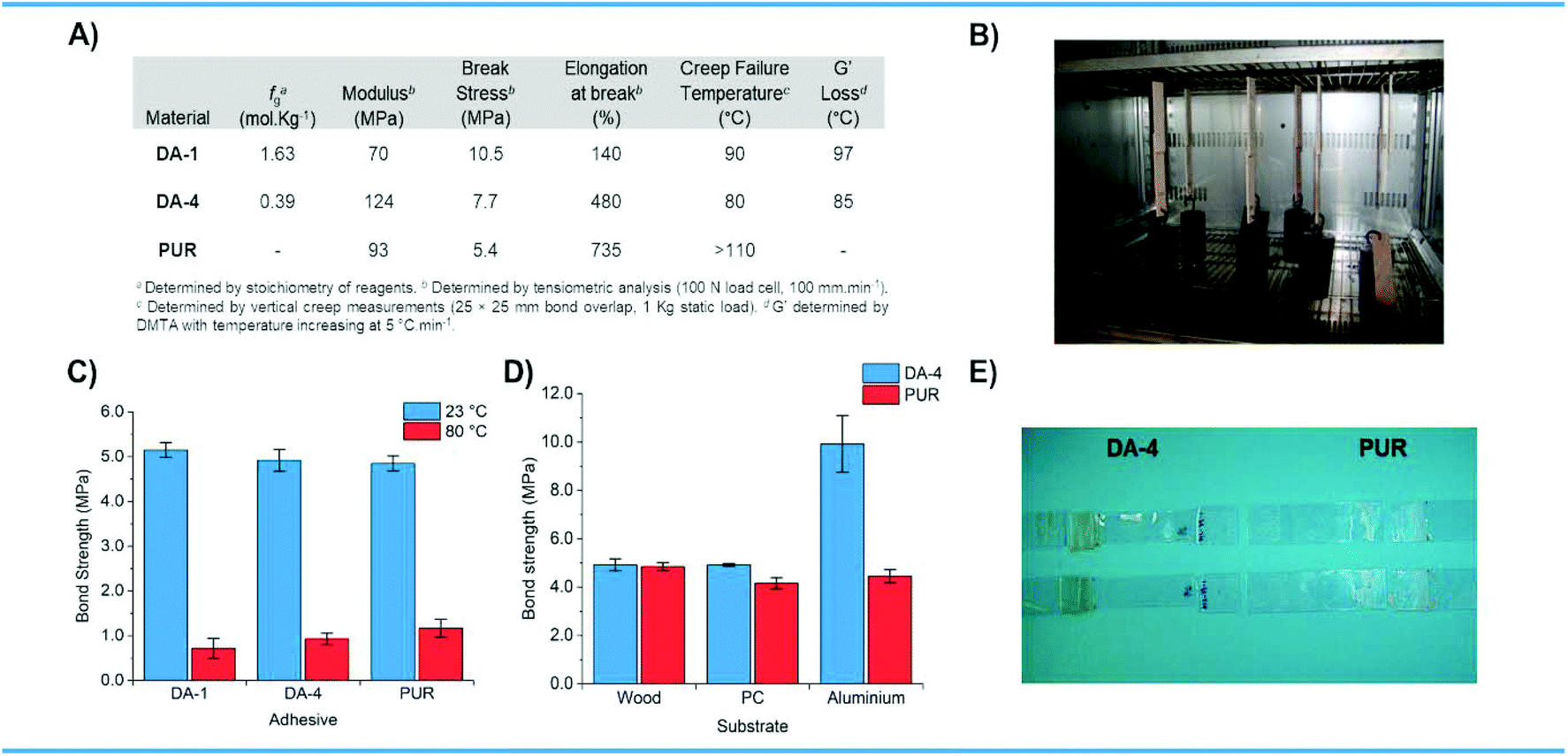 | ||
| Fig. 6 (A) Mechanical properties of films and creep resistance measurements for DA-1 and DA-4 compared to PUR. (B) Creep experiments for bonded joints under static load in an oven for 24 hours. (C) Bond strengths measured at 23 °C and 80 °C for beechwood substrates joined using DA-1, DA-4 and PUR. (D) Bond strengths measured at 23 °C comparing beechwood, polycarbonate and aluminium substrates joined using DA-4 and PUR. (E) Polycarbonate substrates after tensile testing using DA-4 and PUR showing substrate stretching and failure using the CAN (left). | ||
The CANs DA-1 and DA-4 with high and low fg were used as adhesives to bond different substrates and were compared to the PUR reference. For bonding beechwood substrates, the bond strengths for the Diels–Alder systems are similar to the irreversible moisture cured reference at ambient temperature (Fig. 6C). The bond strength achieved using DA-4 is also comparable to that obtained for the PUR reference at 80 °C, with DA-1 being a little lower.
DA-4 was then compared to the PUR reference for bonding three different types of material substrates – wood, plastic and metal (Fig. 6D). Although the bond strength obtained for this CAN is similar to the moisture cured reference for beechwood, it is somewhat higher on polycarbonate (PC). Indeed, DA-4 causes the PC substrates to stretch and break at failure whereas PUR does not (Fig. 6E). The bond strengths obtained on aluminium were very different with the results from DA-4 far superior to the PUR reference by more than a factor of two. Creep resistance (Fig. 6B) for the thermally reversible polyester and poly(ester urethane) Diels–Alder networks was evaluated up to 110 °C, which exceeds the temperature at which these networks undergo a solid-to-liquid transition as indicated by the DMTA studies discussed previously. The data show that the irreversible benchmark PUR reference is resistant to creep over the entire temperature range studied (Fig. 6A). The creep resistance of the thermally reversible systems depends on the type of network with DA-1 (resistance at 80 °C, failure at 90 °C) proving to be significantly more resilient than DA-4 (resistance at 70 °C, failure at 80 °C). This is attributed to the higher crosslink density of DA-1 which is a direct consequence of the higher concentration of furan and maleimide functional groups (fg). Both CANs are resistant to creep deformation in their rubbery state, well above both Tg and Tm. These are exceptional results compared to the general drawback of CAN creep behaviour reported elsewhere.26,27
The relative failure temperatures observed in the creep experiments (performed on bonded samples under high load) correlate with the temperatures in DMTA where there is a substantial change in stiffness observed. These temperatures observed for DA-1 are remarkably close to the gel point temperature of 92 °C reported by Bowman et al.20 for the Diels–Alder network formed from BMI monomer and a trifunctional furan monomer. This system can be considered as a highly relevant reference system based on low molecular weight materials. However, the previous DSC results indicated a peak endotherm assigned to the retro Diels–Alder reaction at much higher temperatures (TrDA = 140.4–145.7 °C). The latter experiments were conducted at standard DSC heating rates of 10 °C min−1. Slower heating rates for DA-1 (high fg) result in a substantial reduction of the characteristic peak endotherm temperature from 145.7 °C to 115.8 °C (Fig. S12A†). A similar effect was observed for DA-4 (low fg) where the peak endotherm temperature reduces from 140.5 °C to 113 °C as the heating rate is lowered (Fig. S12B†). The relationship between the TrDA and heating rate (Fig. S13†) shows that the retro Diels–Alder temperature is systematically higher for DA-1 (high fg). The dependence of TrDA on fg corresponds to the reports by Barner-Kowollik44,45 who demonstrated that the retro Diels–Alder temperature decreases with the molecular weight between crosslinks, resulting from an increasing change in entropy between the associated and dissociated states for longer chains (up to an approximate Mn of 10000 g mol−1). Our materials show a similar trend despite the intrinsic broader molecular weight distributions in these polyester and polyurethane prepolymer chains (ĐM = 2.1–2.4) prepared from step-growth polymerization compared to the more uniform acrylic chains produced by controlled radical polymerisation (ĐM = 1.1–1.2).
It is interesting to compare the creep failure temperatures observed for the adhesive networks reported here to the temperature required for self-healing of coatings from polyether networks recently reported elsewhere41 – also based on the reversible cycloaddition of maleimides and furans. Self-healing of the coatings was possible starting at 70–80 °C depending on cross-link density, quite similar to the upper limits of creep resistance observed here especially considering the different nature of the backbones. This suggests that there is sufficient segmental mobility at these temperatures for flow to occur, enabling coatings to heal or causing adhesive joints to fail under load. This also indicates that the polyester and poly(ester urethane) Diels–Alder networks reported here could self-heal using heat as a stimulus. We intend to evaluate self-healing of these materials in the future.
It is well known that two stereoisomeric cycloadducts can be formed between furan and maleimide functional groups – the endo isomer is kinetically favoured whereas the exo isomer is more thermodynamically favoured. Van Assche et al.46 recently performed an intriguing study in bulk on low Tg polyether networks which demonstrated that the relative concentration of the two isomers was dependent on both the temperature and time of network formation. Typically, the endo isomer is formed preferentially initially and with time the balance is shifted towards the exo isomer. Since the process of coating from the melt (very fast cooling) and subsequent ambient curing will define the network structure, the first heating runs in the DSC are the most relevant to the adhesive application. For completion, we have performed cooling and reheating in the DSC for the polyfunctional furans, telechelic bis-maleimides and the Diels–Alder networks (Fig. S14, S15 and S16,† respectively). During the second heating run the endotherms for TrDA change qualitatively and quantitively. This results from the networks only being able to partly reform during the short timescale of the DSC experiment (i.e. more kinetically controlled) which contrasts to the network formed after melt coating and subsequent ambient curing. We speculate that the much stronger non-covalent dipolar and hydrogen bonding interactions in the polyester and poly(ester urethane) backbones of DA-1 and DA-4 are likely to play an additional, important role. A separate study would be necessary to understand these effects. Recent studies have been reported on dual networks comprising supramolecular and reversible covalent linkages, involving hydrogen bonds and Diels–Alder bonds.47,48 It was found that mechanical properties can be tuned through polymer architecture comprising ureidopyrimidinone (UPy) and maleimide/furan functional groups, with interpenetrating networks producing stronger materials than single networks. It may also be feasible to incorporate other supramolecular functional groups which have low association constants since they also provide networks with interesting properties.49–52
Thermal stability, reversibility and re-use of CANs
Since adhesives must be stable during application and the maleimide functional group can be reactive towards radicals and nucleophiles,29 we evaluated the thermal stability of the Diels–Alder CAN DA-4 at different temperatures (Fig. 7). It should be noted that this assessment of stability correlates to real applications where hot adhesive materials are in contact with the open air. As expected, the initial melt viscosity decreases with temperature. Remarkably, the data show that the viscosity of DA-4 remains relatively stable when exposed to air at 135–150 °C over a period of six hours. It should be noted that the viscosity profile of the poly(ester urethane) DA-4 at these temperatures is similar to moisture-curing polyurethane products used commercially. In addition, TGA analysis performed on DA-1 and DA-4 shows that both materials are thermally stable up to 200 °C with no weight loss (Fig. S17†). This behaviour far exceeds the thermal stability reported for epoxy adhesives from Diels–Alder networks comprising maleimide/furan functional groups31 and acrylic polymers from hetero Diels–Alder networks based on thioesters.21 The former are unstable after 1 hour at 90 °C due to side reactions whereas the latter are unstable above 120 °C due to thioester decomposition. However, in Fig. 7DA-4 does become very unstable after only one hour at 180 °C. Studies on polyether based maleimide/furan Diels–Alder networks comprising BMI showed that a maximum of 150 °C was acceptable.53 In addition, Hopewell showed that BMI monomer will homopolymerize at temperatures above 150 °C.54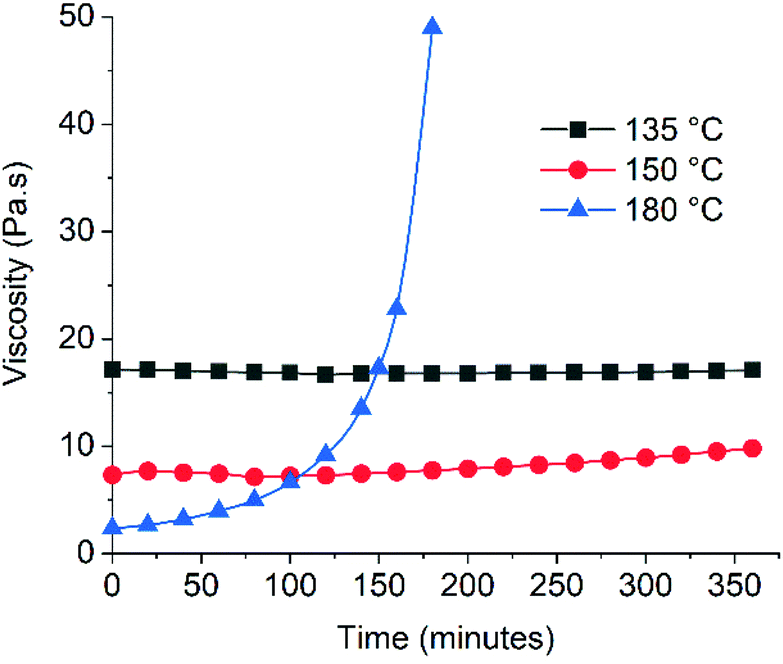 | ||
| Fig. 7 Thermal stability of DA-4 determined by evaluating the time dependence of melt viscosity at a constant temperature using a Brookfield viscometer. | ||
For materials to be considered truly sustainable, their properties should be robust and not vary significantly with repeated reprocessing or recycling. Accordingly, the reversibility of the Diels–Alder CANs was monitored by FT-IR spectroscopy. Cured networks were re-heated to 150 °C for 1 hour to form a liquid and then allowed to cool and cure for 7 days at 20 °C. This thermal cycle was repeated four times over 28 days with each CAN analysed by FT-IR spectroscopy throughout this time period. The networks DA-1 and DA-4 appear to be relatively robust after repeated reheating & curing, since the profiles of maleimide functional groups over time are reasonably consistent over all four cycles (Fig. 8A). There is some variation owing to the differing degrees of contact of the CANs with the IR prism at different time and temperatures. Examination of the complete IR spectral range shows that there is no change in either DA-1 or DA-4 throughout this recycling process (Fig. S18†), suggesting there is very little or no change in chemical composition.
 | ||
| Fig. 8 The effect of re-use on the properties of DA-1 and DA-4. (A) IR absorbance at 696 cm−1 of maleimide functional groups as a function of repeated re-use where the material is heated to dissociate the network and IR spectra recorded at ambient temperature for 7 days after immediate cooling. The cycle is repeated 3 times. (B) Bond strengths obtained after re-using bonded aluminium coupons. Initial bond – formed from film adhesive heated between aluminium coupons, cured for 7 days at ambient temperature and evaluated for adhesive strength. 1st, 2nd, 3rd and 4th re-use – the same joints are reassembled by realigning the broken joint and re-heating. Joints allowed to cure for 8–10 days at ambient temperature, evaluated for adhesive strength and pulled apart. (C) Aluminium substrates after testing following the 4th re-use. | ||
Encouraged by these results, we investigated the effect of repeated re-use on performance by evaluating the adhesion strength achieved during multiple, consecutive bond assembly and disassembly of the same substrates and adhesive. Lap shear joints were prepared with aluminium substrates using crosslinked films of DA-1 and DA-4, heated in order to dissociate the Diels–Alder CAN to lower molecular weight prepolymers and enable wetting of the substrates. After allowing the assembly to crosslink for eight days at ambient temperature, the bond strength was evaluated, and substrates separated during tensile mechanical testing. Several days later, the separated aluminium substrates comprising the fractured polymer layers were then directly re-assembled at 150 °C for 1 hour. The above protocols were repeated four times, i.e. heating to reassemble, curing at ambient temperature, and evaluating the adhesion strength each time before heating to re-bond the separated substrates. Remarkably, the initial bond strength for DA-1 is maintained after four re-uses without any cleaning or modification of the surfaces (Fig. 8B). For DA-4 there appears to be some reduction in bond strength although this is relatively small (approximately −5% after each re-use). The results are achieved despite the likelihood of some adhesive migrating out of the bond line during re-heating and reassembly for each re-use experiment (the separated bonds after the 4th re-use shown in Fig. 8C). Examination of the fractured, cured adhesives after their fourth re-use shows that the IR spectra of DA-1 and DA-4 are very similar to the initial cured film before the first adhesive application and use (Fig. S19†). In addition, DSC shows there appears to be little change of the DA-1 or DA-4 networks after multiple recycling (Fig. S20†). The more resilient behaviour of DA-1 may result from the higher crosslink density although the differences in the chemistry of the backbones may also contribute. Further studies are required to elucidate these effects. Retention of properties after multiple re-bonding and re-use cycles without using solvents contrasts with the reported single debonding of polyurethane Diels–Alder networks using hot dimethyl sulfoxide.38 The results also differ to acrylic hetero Diels–Alder networks based on thioesters where a single bond cleavage was permanent since a Lewis acid catalyst was necessary for network formation at ambient temperature.39 Naito et al.34 reported thermo-resettable acrylic Diels–Alder but the approach is not sustainable since furan functional acrylic polymers were applied from toxic chloroform solution and the toxic BMI monomer was used as a crosslinker. Strong adhesion required materials with Tg values above 20 °C and these are unlikely to be flexible enough in application for bonding substrates with different thermal expansion coefficients.
Our demonstration of achieving a similar level of performance after multiple adhesive re-use is obtained by:
(1) Atom efficient chemistry providing materials in high yields without the need for purification.
(2) Application in the bulk without solvents or toxic monomers followed by curing of bonded materials under ambient conditions.
(3) Repeated debonding steps without the use of solvents, material modification or additional surface preparation.
We aim to build on this promising proof of concept by investigating a range of polymer architectures, enabling sustainable thermally reversibly networks with different properties based not only on robust dynamic covalent bonds but also dynamic non-covalent bonds, via methods that are industrially viable.
Conclusions
Maleimide functional telechelic prepolymers and polyfunctional furan crosslinkers were successfully synthesized via facile techniques with high atom efficiency. Monomer and solvent-free polyester and poly(ester urethane) CANs were readily prepared in bulk by simply melt blending and coating at moderate temperatures followed by curing in ambient conditions.Quantitative conversion of maleimide functional group concentrations was obtained independent of the backbone chemical composition, Tg/Tm or functional group concentration. TrDA was proportional to the maleimide/furan functional group concentration. The mechanical properties of the CANs can be tuned depending on the compositions of bismaleimide prepolymer and polyfunctional furan. The CANs provided versatile adhesion on wood, polycarbonate and aluminium and are creep resistant under load well above Tg and Tm – up to 80 °C depending on the functional group concentration and crosslink density. This correlated with measurements from DMTA.
The CANs were shown to be thermally stable up to 150 °C. The adhesive and bonded substrates could be re-used repeatedly without the use of solvents by separation of substrates during mechanical testing, reheating to re-bond the original substrate/adhesive combination and curing at ambient temperature. The bond strengths achieved after repeated re-use were the same as the initial level of performance obtained for networks with higher crosslink density.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. T. S. thanks the EPSRC for a Manufacturing Fellowship (EP/R012121/1) and a Doctoral Training Partnership CASE award for J. B. D. G. We thank Andrew Broadhurst (Intertek Wilton) for his expertise providing the DMTA measurements. We are also grateful to Prof. Steve Armes and Prof. Tony Ryan (both University of Sheffield) for useful discussions during the preparation of this manuscript.Notes and references
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, 25–29 CrossRef.
- J. Hultman and H. Corvellec, Environ. Plan A, 2012, 44, 2413–2427 CrossRef.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS.
- H. Sardon and A. P. Dove, Science, 2018, 360, 380–381 CrossRef CAS.
- C. Jehanno, I. Flores, A. P. Dove, A. J. Müller, F. Ruipérez and H. Sardon, Green Chem., 2018, 20, 1205–1212 RSC.
- G. Habenicht, Applied Adhesive Bonding, Wiley VCH, Weinheim, 2008 Search PubMed.
- M. Krebs, Adhesion, Adhesives & Sealants Extra, 2004/2005, 30–33 Search PubMed.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- G. M. Scheutz, J. J. Lessard, M. B. Sims and B. S. Sumerlin, J. Am. Chem. Soc., 2019, 141, 16181–16196 CrossRef CAS.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- W. Denissen, M. Droesbeke, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 14857 CrossRef CAS.
- M. Röttger, T. Domenech, R. Van Der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef.
- B. T. Worrell, S. Mavila, C. Wang, T. M. Kontour, C. H. Lim, M. K. McBride, C. B. Musgrave, R. Shoemaker and C. N. Bowman, Polym. Chem., 2018, 9, 4523–4534 RSC.
- B. J. Adzima, H. A. Aguirre, C. J. Kloxin, T. F. Scott and C. N. Bowman, Macromolecules, 2008, 41, 9112–9117 CrossRef CAS.
- A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515–5520 CrossRef CAS.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS.
- H. A. Houck, K. De Bruycker, S. Billiet, B. Dhanis, H. Goossens, S. Catak, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Chem. Sci., 2017, 8, 3098–3108 RSC.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef.
- L. Zhang and S. J. Rowan, Macromolecules, 2017, 50, 5051–5060 CrossRef CAS.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS.
- Y. Oz and A. Sanyal, Chem. Rec., 2018, 18, 570–586 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS.
- J. H. Aubert, J. Adhes., 2003, 79, 609–616 CrossRef CAS.
- A. A. Kavitha and N. K. Singha, ACS Appl. Mater. Interfaces, 2009, 1, 1427–1436 CrossRef CAS.
- M. Wouters, M. Burghoorn, B. Ingenhut, K. Timmer, C. Rentrop, T. Bots, G. Oosterhuis and H. Fischer, Prog. Org. Coat., 2011, 72, 152–158 CrossRef CAS.
- S. Das, S. Samitsu, Y. Nakamura, Y. Yamauchi, D. Payra, K. Kato and M. Naito, Polym. Chem., 2018, 9, 5559–5565 RSC.
- D. H. Turkenburg, H. van Bracht, B. Funke, M. Schmider, D. Janke and H. R. Fischer, J. Appl. Polym. Sci., 2017, 134, 1–11 CrossRef.
- E. Dolci, G. Michaud, F. Simon, B. Boutevin, S. Fouquay and S. Caillol, Polym. Chem., 2015, 6, 7851–7861 RSC.
- E. Dolci, V. Froidevaux, G. Michaud, F. Simon, R. Auvergne, S. Fouquay and S. Caillol, J. Appl. Polym. Sci., 2017, 134, 1–11 CrossRef.
- K. M. A. Kaiser, J. Appl. Polym. Sci., 2020, 1–12 Search PubMed.
- A. M. Schenzel, C. Klein, K. Rist, N. Moszner and C. Barner-Kowollik, Adv. Sci., 2016, 3, 1500361 CrossRef.
- Henkel Corporation, 2010US38270, 2010.
- J. Brancart, R. Verhelle, J. Mangialetto and G. Van Assche, Coatings, 2018, 9, 13 CrossRef.
- G. Scheltjens, M. M. Diaz, J. Brancart, G. Van Assche and B. Van Mele, React. Funct. Polym., 2013, 73, 413–420 CrossRef CAS.
- M. M. Diaz, G. Van Assche, F. H. J. Maurer and B. Van Mele, Polymer, 2017, 120, 176–188 CrossRef CAS.
- N. K. Guimard, J. Ho, J. Brandt, C. Yeh Lin, M. Namazian, D. G. Nocera, K. K. Oehlenschlaeger, S. Hilf, A. Lederer, F. G. Schmidt, M. L. Coote and C. Barner-Kowollik, Chem. Sci., 2013, 4, 2752–2759 RSC.
- K. Pahnke, J. Brandt, G. Gryn'ova, P. Lindner, R. Schweins, F. G. Schmidt, A. Lederer, M. L. Coote and C. Barner-Kowollik, Chem. Sci., 2015, 6, 1061–1074 RSC.
- A. Cuvellier, R. Verhelle, J. Brancart, B. Vanderborght, G. Van Assche and H. Rahier, Polym. Chem., 2019, 10, 473–485 RSC.
- B. Zhang, Z. A. Digby, J. A. Flum, E. M. Foster, J. L. Sparks and D. Konkolewicz, Polym. Chem., 2015, 6, 7368–7372 RSC.
- B. Zhang, J. Ke, J. R. Vakil, S. C. Cummings, Z. A. Digby, J. L. Sparks, Z. Ye, M. B. Zanjani and D. Konkolewicz, Polym. Chem., 2019, 10, 6290–6304 RSC.
- P. Woodward, D. H. Merino, I. W. Hamley, A. T. Slark and W. Hayes, Aust. J. Chem., 2009, 62, 790–793 CrossRef CAS.
- D. H. Merino, A. T. Slark, H. M. Colquhoun, W. Hayes and I. W. Hamley, Polym. Chem., 2010, 1, 1263–1271 RSC.
- D. H. Merino, A. Feula, K. Melia, A. T. Slark, I. Giannakopoulos, C. R. Siviour, C. P. Buckley, B. W. Greenland, D. Liu, Y. Gan, P. J. Harris, A. M. Chippindale, I. W. Hamley and W. Hayes, Polymer, 2016, 107, 368–378 CrossRef CAS.
- D. Hermida-Merino, B. O'Driscoll, L. R. Hart, P. J. Harris, H. M. Colquhoun, A. T. Slark, C. Prisacariu, I. W. Hamley and W. Hayes, Polym. Chem., 2018, 9, 3406–3414 RSC.
- G. Scheltjens, J. Brancart, I. De Graeve, B. Van Mele, H. Terryn and G. Van Assche, J. Therm. Anal. Calorim., 2011, 105, 805–809 CrossRef CAS.
- J. L. Hopewell, D. J. T. Hill and P. J. Pomery, Polymer, 1998, 39, 5601–5607 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02938f |
| ‡ Present address: Corbion Biomaterials, Arkelsedijk 46, 4206 AC Gorinchem, NL. |
| § Present address: Sartomer Americas, 502 Thomas Jones Way, Exton, PA19341, USA. |
| This journal is © The Royal Society of Chemistry 2020 |