
Integrating Z-scheme heterojunction of Co1-C3N4@α-Fe2O3 for efficient visible-light-driven photocatalytic CO2 reduction†
Bing-Cai
He
,
Chao
Zhang
,
Pei-Pei
Luo
,
Yu
Li
 * and
Tong-Bu
Lu
*
* and
Tong-Bu
Lu
*
MOE International Joint Laboratory of Materials Microstructure, Institute for New Energy Materials and Low Carbon Technologies, School of Materials Science & Engineering, Tianjin University of Technology, Tianjin 300384, China. E-mail: yli@email.tjut.edu.cn; lutongbu@tjut.edu.cn
First published on 29th September 2020
Abstract
Photocatalytic CO2 reduction coupled with water oxidation provides a fascinating approach to mitigating the issues of global warming and energy shortage. Herein, a direct Z-scheme heterojunction of Co1-C3N4@α-Fe2O3 comprising a g-C3N4-supported single-atomic Co site catalyst (denoted as Co1-C3N4) and α-Fe2O3 nanorod arrays is fabricated for efficient CO2 reduction. A CO production rate of 14.9 μmol g−1 h−1 with a high CO selectivity (>99%) is achieved under visible-light irradiation without any sacrificial agents other than water. Time-resolved photoluminescence (TRPL) analysis reveals that both the Z-scheme mechanism and the single-atomic Co sites contribute to the prolonged lifetime of the photo-induced excitons. Moreover, the formation of the Z-scheme heterojunction would lead to an altered charge density of the single-atomic Co sites. In situ diffuse reflectance infrared Fourier-transform spectroscopy and anion adsorption measurements reveal that the key intermediate CO2− could be efficiently stabilized by the positively charged Co sites in Co1-C3N4@α-Fe2O3, thus enhancing the CO2 reduction performance. This work offers a new direction for the rational design of single-atomic site catalysts in artificial photosynthesis.
Introduction
The rapidly growing consumption of fossil fuels with excessive emission of CO2 has been contributing to the severe global warming problem and the potential energy shortage.1,2 Utilizing solar energy to convert CO2 into fuels provides a promising approach to solving the above problems, and scientists have devoted tremendous attention to developing various photocatalysts for CO2 reduction.3–9 Recently, graphitic carbon nitride (g-C3N4)-supported single-atomic site catalysts have emerged as remarkable photocatalysts.10–13 By virtue of the lone pair electrons, the N atoms in the framework can capture various transition metal ions, thus forming single-atomic metal sites on g-C3N4. In addition, the charge densities of the coordinated metal atoms would be altered by the neighboring N atoms, which may in turn change the adsorption strength and reaction barriers. Density functional theory (DFT) calculations have suggested that single-atomic Pd or Pt loaded on g-C3N4 can act as effective catalytic sites for photocatalytic CO2 reduction, leading to two different preferential products, HCOOH and CH4, respectively.14 With the aid of triethylamine (TEA), single-atomic Co2+ sites on C3N4 with different loadings have been successfully synthesized, and the optimized sample with a cobalt loading of 0.128 μmol mg−1 exhibits a CO production rate of approximately 25.5 μmol g−1 h−1 under 60 mW cm−2 incident light in the presence of triethanolamine (TEOA) as a sacrificial electron donor.15 Recently, we have developed an efficient photocatalyst for CO2 reduction by implanting single titanium oxide species on g-C3N4.16 In the presence of the Co(bpy)3Cl2 co-catalyst and TEOA, a CO production rate of 283.9 μmol g−1 h−1 has been achieved under visible light irradiation. These results strongly demonstrate the viability of g-C3N4-supported single-atomic site catalysts for photocatalytic CO2 reduction. However, the use of TEOA makes these catalysts less attractive since TEOA is much more expensive than CO. Therefore, it still remains a great challenge to develop low-cost and robust g-C3N4-based photocatalysts with high efficiency for CO2 conversion using water as an electron source.Owing to the wide band gap (∼2.7 eV), g-C3N4 can only utilize a marginal portion of visible light (λ < 460 nm).17 Moreover, the water oxidation capacity of g-C3N4 is poor due to the high energy level of the valence band.18–20 In this situation, integrating g-C3N4 with an appropriate semiconductor possessing a narrower band gap and a more positive valence band to establish a Z-scheme heterojunction has been acknowledged as an effective strategy, because the Z-scheme heterojunction not only facilitates the spatial separation of the photo-induced electron–hole pairs, but also preserves the maximum capacities for reduction and oxidation of the composites.20–23
Herein, we construct a direct Z-scheme heterojunction of Co1-C3N4@α-Fe2O3, which composes by single-atomic Co sites loaded on g-C3N4 (Co1-C3N4) integrated with α-Fe2O3 nanorod arrays, for efficient visible-light-driven CO2 reduction coupled with water oxidation. The light-absorption range of Co1-C3N4 is greatly extended to the entire visible-light region and the lifetime of photo-induced charge carriers is significantly prolonged by the successful incorporation of α-Fe2O3. Moreover, benefiting from the charge transfer induced by the different Fermi levels between α-Fe2O3 and Co1-C3N4, the charge density of single-atomic Co can be further regulated. The results of anion adsorption measurements imply that the stabilization of the critical intermediate CO2− is more efficient on the single-atomic Co sites with a higher positive charge. Compared with Co1-C3N4, the constructed Z-scheme heterojunction shows a substantial improvement for photocatalytic CO2 conversion to CO, with 2.8 times higher than that of Co1-C3N4 under visible light irradiation and almost 100% CO selectivity.
Experimental
Materials
FeCl3·6H2O (99%, AR) and CoCl2·6H2O (99%, AR) were purchased from Aladdin. Urea (99%, AR), NaOH (96%, AR) and Na2SO4 (99%, AR) were purchased from FuChen Chemical Reagent Factory. Nafion 117 solution (∼5% in alcohol/water) was purchased from Sigma-Aldrich. CO2 (99.999%) and Ar (99.999%) were purchased from Tianjin Huanyu Gas company. 13CO2 (99%) was purchased from Sigma-Aldrich and H218O (99%) was purchased from Energy Chemical. All chemicals were used as received without further purification. The ultrapure water (Milli-Q water) with an electrical resistivity of 18.2 MΩ cm was used in all experiments.Synthetic procedures
The α-Fe2O3 nanorod arrays were fabricated by a modified strategy reported previously.24,25 In brief, a piece of fluorine-doped tin oxide (FTO) glass (1 cm × 2.5 cm) was sonicated in acetone, ethanol and water, respectively. Then the FTO glass was leant on the inner side of a Teflon liner with the FTO side facing down. Subsequently, an aqueous solution (15 mL) containing FeCl3·6H2O (0.51 g) and urea (0.17 g) was added, and the liner was sealed in a stainless steel autoclave, and then heated at 100 °C for 4 h. The as-prepared sample was washed with water and dried by N2 flow to remove redundant sediment. The film was annealed at 550 °C for 2 h and subsequently at 660 °C for 20 min in a muffle furnace with a ramping rate of 2 °C min−1, and the α-Fe2O3 nanorod arrays were finally obtained. The loading amount of the α-Fe2O3 nanorod arrays was determined to be 0.3 mg by weighing the FTO glass before and after the synthesis.The g-C3N4-supported Co single-atom catalyst was prepared by using a two-step strategy.26 Specifically, urea (15 g) was put into a crucible with a cover, and calcined at 550 °C for 4 h with a ramping rate of 10 °C min−1. After cooling down naturally, the product was heated again at 500 °C for 2 h to obtain exfoliated g-C3N4. For the synthesis of g-C3N4-supported single-atomic Co site catalyst (Co1-C3N4), g-C3N4 (10 mg) was well dispersed in water (10 mL) by vigorous sonication for 30 min. Next, an aqueous solution (500 μL) containing CoCl2·6H2O (0.336 mM) was dropped into the g-C3N4 suspension under magnetic stirring. Subsequently, the suspension was stirred for 12 h at 80 °C in an oil bath. The above mixture was then frozen by liquid nitrogen and further dried in vacuum for 48 h by using a lyophilizer. The obtained sample was placed in a porcelain boat and heated to 400 °C for 2 h with a ramping rate of 5 °C min−1 under Ar atmosphere. Similar procedures were applied for the synthesis of the g-C3N4-supported CoO nanoparticle catalyst (CoNP-C3N4), except that the concentration of CoCl2·6H2O was increased to 3.36 mM.
To fabricate the Z-scheme system, a well-dispersed aqueous suspension (300 μL) of Co1-C3N4 (1 mg mL−1, 0.3 mg in total) was first drop-coated onto the α-Fe2O3 nanorod arrays under 60 °C. To strengthen the contact between α-Fe2O3 and Co1-C3N4, the sample was transferred to a tube furnace and calcined at 200 °C for 2 h, with a ramping rate of 5 °C min−1 under Ar atmosphere. The obtained sample was denoted as Co1-C3N4@α-Fe2O3. C3N4@α-Fe2O3 and CoNP-C3N4@α-Fe2O3 were fabricated according to the same method as that of Co1-C3N4@α-Fe2O3, except that Co1-C3N4 was replaced by g-C3N4 and CoNP-C3N4, respectively.
Characterization
The powder X-ray diffraction (XRD) patterns were recorded by using a Rigaku SmartLab 9 kW with Cu-Kα radiation (λ = 1.5418 Å). A Thermo scientific iCAP RQ inductively coupled plasma-mass spectrometer (ICP-MS) was used to determine the Co content. Scanning electron microscopy (SEM) was performed using a FEI Verios 460L scanning electron microscope. Transmission electron microscopy (TEM) images and High-Resolution Transmission Electron Microscopy (HRTEM) images were obtained on FEI Tecnai G2 Spirit Twin and FEI Talos F200X transmission electron microscopes, respectively. Atomic-resolution aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and elemental mapping were obtained by using a FEI Titan Themis Cubed G2 60–300 transmission electron microscope. X-ray photoelectron spectroscopy (XPS) measurements were carried out by using a Thermo Scientific ESCALAB250Xi photoelectron spectrometer equipped with Mg Kα (1253.6 eV) as the X-ray source. Ultraviolet–visible (UV–vis) spectroscopic measurements were performed by using a PerkinElmer Lambda 750 UV/VIS/NIR spectrometer. A PerkinElmer Frontier Mid-IR FTIR spectrometer was employed to obtain Fourier-transform infrared spectra. The steady-state photoluminescence spectra (PL) were recorded on a Hitachi F-4600 fluorescence spectrometer. The time-resolved photoluminescence (TRPL) curves were recorded using a PicoQuant MicroTime 200 time-resolved confocal fluorescence instrument. The electron spin resonance (ESR) analysis was conducted on an electron paramagnetic resonance spectrometer (Bruker EMXplus-6/1) with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the trapping agent at 298 K. CO2 adsorption was tested by using a MicrotracBEL BELSORP-Max gas adsorption instrument at 298 K. A Hansatech Chlorolab-2 liquid-phase oxygen measurement system was employed to detect the oxygen evolution during the photocatalytic CO2 reduction.Photocatalytic CO2 reduction measurements
Photocatalytic CO2 reduction tests were conducted in a gas–solid setup. A piece of the sample was placed in a 35 mL quartz tube. High-purity CO2 (99.999%) was first passed through water to carry H2O vapor and then introduced into the quartz tube. The quartz tube was purged with the CO2/H2O gas mixture for 30 min and sealed by using a rubber stopper. A 300 W xenon lamp (CEL-HXF300, CEAULIGHT) with a 400 nm cutoff filter was positioned above the sample as the light source. Gaseous products (CO, CH4, and H2) were analysed by using a SHIMADZU GC-2014 gas chromatograph equipped with TCD and FID dual detectors. The products of the 13CO2 and H218O isotopic experiments were analysed by using a Hiden Analytical HPR20 mass spectrometer.Photoelectrochemical measurements
All measurements were performed on a CHI 760E electrochemical workstation with a three-electrode setup (working electrode: FTO glass-supported sample; reference electrode: Ag/AgCl electrode; counter electrode: Pt foil) in 0.1 M Na2SO4 aqueous solution. To measure the photo-current responses, a 300 W Xe lamp (CEL-HXF300, CEAULIGHT) with a 400 nm cutoff filter was used as the light source. A potential of 0.5 V vs. Ag/AgCl was applied on the sample and the light was chopped every 10 s to simulate light/dark conditions during the test. Electrochemical impedance spectroscopy (EIS) was carried out in the frequency range of 0.1 MHz to 0.1 Hz with an AC voltage amplitude of 5 mV at an open-circuit potential under visible-light irradiation with a power density of 200 mW cm−2.In situ DRIFTS characterization
In situ diffuse reflectance infrared Fourier-transform spectra (DRIFTS) were recorded on a Bruker IFS 66v Fourier-transform spectrometer at the Infrared Spectroscopy and Microspectroscopy Endstation (BL01B) in the National Synchrotron Radiation Laboratory (NSRL), Hefei.27 The sample was placed in a Harrick diffuse reflectance reaction chamber fixed in the light path. The chamber was equipped with two ZnSe windows and two gas channels. Each spectrum was recorded by 128 scans with a resolution of 4 cm−1. The spectrum recorded under Ar atmosphere was set as the background signal.Results and discussion
The composite structure comprising α-Fe2O3 and Co1-C3N4 was fabricated by a multi-step strategy (Fig. 1a). Co1-C3N4 exhibits a two-dimensional morphology without any distinguishable clusters, as shown in Fig. 1b and Fig. S1.† The results of elemental mapping indicate the uniform dispersion of Co species on g-C3N4 (Fig. 1c). The XRD pattern for Co1-C3N4 also shows no peaks other than those corresponding to g-C3N4 (Fig. S2†). The atomic dispersion of Co sites was further verified by using a HAADF-STEM image, in which the bright spots represent Co atoms and no aggregation of the bright spots was observed (Fig. 1d).28 The line-scan profiles also confirm that the Co atoms are well isolated (Fig. S3†). ICP-MS was employed to determine the actual loading of Co. As listed in Table S1,† the mass loading of Co is 0.07% for Co1-C3N4. A sample comprising CoO nanoparticles on g-C3N4 was also prepared, with a Co mass loading of 0.81% (denoted as CoNP-C3N4, Fig. S4†). SEM and TEM images (Fig. 1e and Fig. S5†) show that the α-Fe2O3 nanorods grown on fluorine-doped tin oxide (FTO) glass have an average length of ∼500 nm and an average diameter of ∼50 nm. The HRTEM image (Fig. 1f) displays lattice fringes with a lattice spacing of 0.25 nm, which can be assigned to the (110) planes of crystalline α-Fe2O3. The top view for the composite photocatalyst clearly shows that the α-Fe2O3 nanorod arrays are covered by highly corrugated Co1-C3N4 nanosheets (Fig. S6†). Fourier-transform infrared spectrum for Co1-C3N4 shows a series of characteristic peaks of g-C3N4 as those reported in the literature (Fig. 1g). Notably, the peak at 806 cm−1, which is assigned to the breathing mode of the triazine unit in carbon nitride, is blue-shifted to 808 cm−1 when Co1-C3N4 is integrated with α-Fe2O3 nanorods.29 Similar shifts are also found for the peaks at 1200 to 1650 cm−1, which correspond to the stretching vibration modes characteristic of aromatic g-C3N4 heterocycles.30 The blue shift can be attributed to the charge transfer between Co1-C3N4 and α-Fe2O3, which strongly evidences the successful formation of heterojunctions.21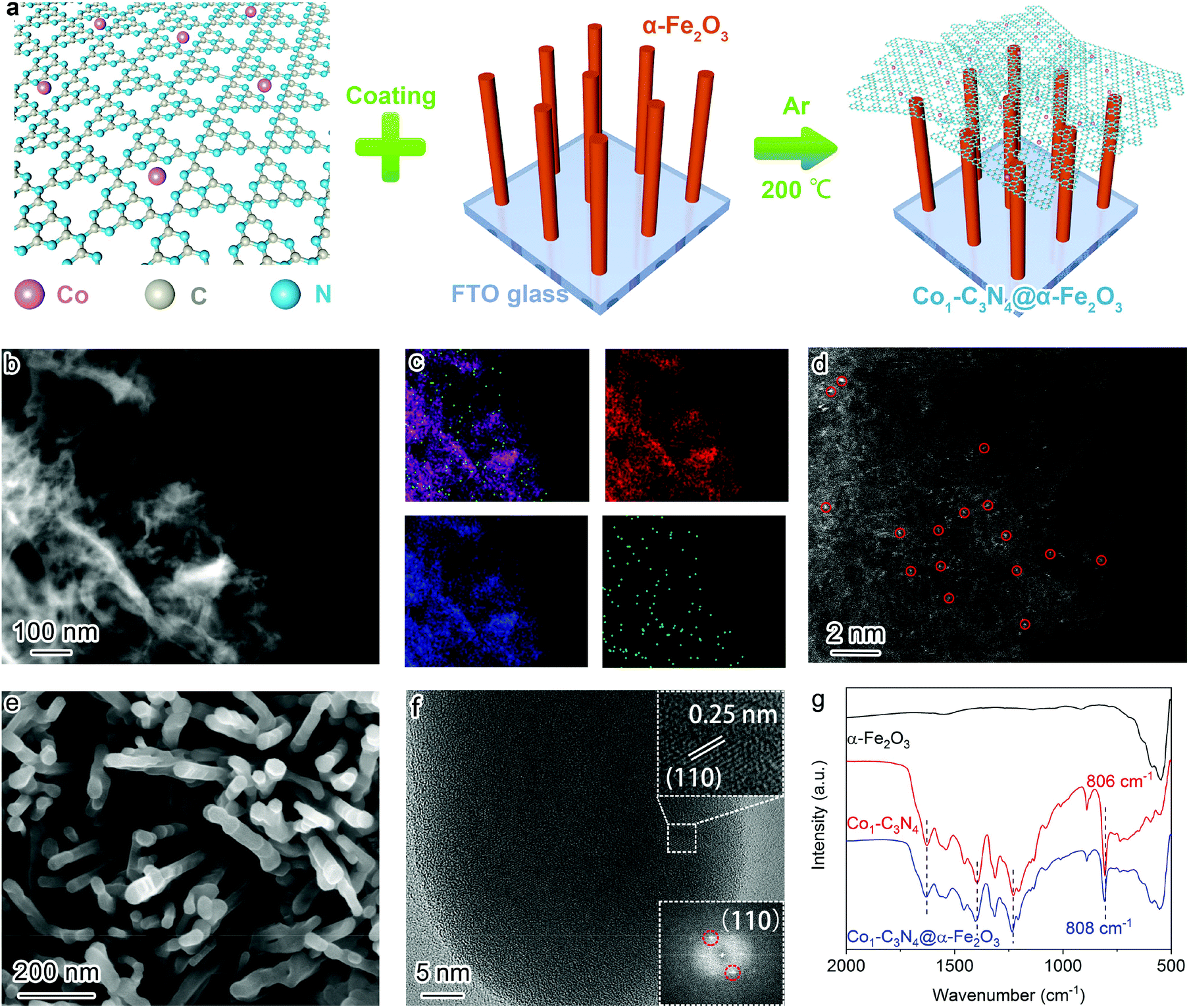 | ||
| Fig. 1 (a) Schematic illustration of the fabrication strategy for Co1-C3N4@α-Fe2O3. (b) Low-resolution HAADF-STEM image for Co1-C3N4 and (c) the corresponding elemental mapping images of C (red), N (blue), and Co (green). (d) Atomic-resolution HAADF-STEM image for Co1-C3N4. The atomically dispersed Co sites are marked in red circles. (e) SEM image and (f) HRTEM image for the α-Fe2O3 nanorod arrays; insets: lattice fringes of a single α-Fe2O3 nanorod (top) and the selected area electron diffraction (SAED) pattern (bottom). (g) Fourier-transform infrared spectra for α-Fe2O3, Co1-C3N4, and Co1-C3N4@α-Fe2O3. | ||
UV–vis diffuse reflectance spectroscopy was employed to explore the light-harvesting ability of the as-prepared photocatalysts (Fig. S7†). The absorption edge for Co1-C3N4@α-Fe2O3 is significantly extended to 700 nm, covering almost the entire visible region. On the basis of the Tauc plots (Fig. S8†), the band gaps of the α-Fe2O3 nanorod and Co1-C3N4 were determined to be 2.12 eV and 2.66 eV, respectively. The energy band structures were further resolved by Mott–Schottky plots (Fig. S9†). The positive slopes of the Mott–Schottky plots indicate the n-type characteristics for both the α-Fe2O3 nanorod and Co1-C3N4.31 The flat band potentials of α-Fe2O3 and Co1-C3N4 were calculated to be 0.36 V and −0.94 V versus the normal hydrogen electrode (NHE), respectively. For the n-type semiconductor, the flat band potential is close to the bottom of the conduction band (CB).31,32 Now we are able to draw a clear band diagram for the composite structure, as shown in Fig. 2c. To validate the Z-scheme route for the photo-induced charge carriers, ESR spectroscopy was employed to detect ˙OH and ˙O2− species with DMPO as the trapping agent (Fig. 2a and b). For pristine α-Fe2O3, only a weak signal attributed to ˙OH was observed under visible-light irradiation; for pristine Co1-C3N4, only the signal attributed to ˙O2− was detected with four identical peaks. These results are understandable by comparing the energy band positions with the reaction potentials of O2/˙O2− and H2O/˙OH (OH−/˙OH). When Co1-C3N4@α-Fe2O3 was employed as the photocatalyst, the signals of both ˙OH and ˙O2− were observed with much stronger intensities than that for each single component. In addition, neither of the two species was observed for Co1-C3N4@α-Fe2O3 under dark conditions (Fig. S10†), suggesting that the reactive oxygen species mentioned above were generated upon incident light. Therefore, we can conclude that the photo-induced charge carriers follow a Z-scheme mechanism rather than a Type II mechanism (Fig. 2c).8,33
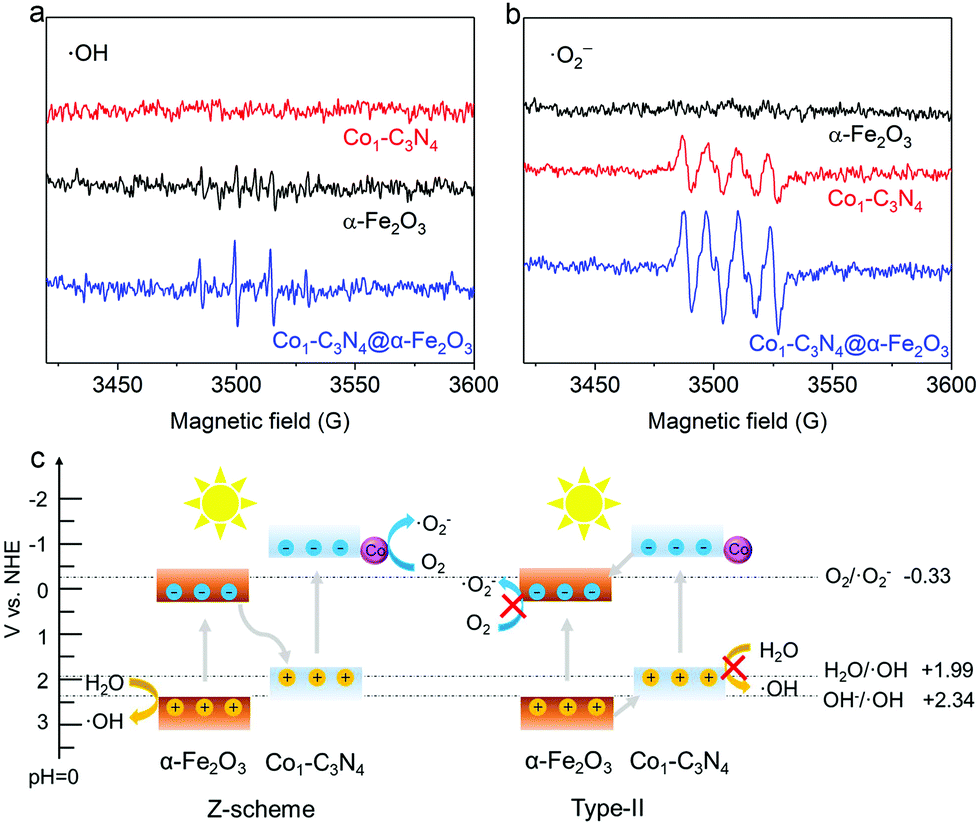 | ||
| Fig. 2 (a and b) DMPO spin-trapping ESR spectra for α-Fe2O3, Co1-C3N4, and Co1-C3N4@α-Fe2O3 under visible-light irradiation. (c) Schematic illustration for the validation of the Z-scheme mechanism rather than Type-II. | ||
With the Z-scheme mechanism for Co1-C3N4@α-Fe2O3 validated, now we are in a position to investigate the efficacy of the composite structure in promoting photocatalytic CO2 reduction. Fig. 3a shows the average CO evolution rates in CO2 overall splitting for the as-prepared catalysts under visible light (λ > 400 nm). No product was detected for α-Fe2O3 nanorod arrays, as the potential of the conduction band is too positive to trigger the reduction of CO2. The CO production rate for Co1-C3N4@α-Fe2O3 was substantially elevated to 14.9 μmol g−1 h−1, 2.9 times as high as that for Co1-C3N4 (5.2 μmol g−1 h−1) and 6.5 times as that for C3N4@α-Fe2O3 (2.3 μmol g−1 h−1). These results imply that the construction of the Z-scheme heterojunction and the introduction of Co favor the photocatalytic reduction of CO2 under visible light. When ultraviolet light was also introduced to the photocatalytic system (by the removal of the cut-off filter), a CO production rate as high as 25.2 μmol g−1 h−1 was achieved, demonstrating the excellent CO2 reduction capability for Co1-C3N4@α-Fe2O3 under full spectrum. To evaluate the utilization efficiency of the solar energy, apparent quantum efficiencies (AQEs) for CO generation were determined under different monochromatic light irradiation (Fig. S11†). The AQEs at 365 nm and 405 nm were determined to be 0.52% and 0.42%, respectively. In addition, it is noteworthy that neither H2 nor CH4 was detected in the gaseous products for a continuous 12-hour photocatalytic test, suggesting the high selectivity for CO (Fig. 3b). To the best of our knowledge, the performance of Co1-C3N4@α-Fe2O3 here is among the top levels in comparison with other g-C3N4-based and single-atomic site catalysts in photocatalytic CO2 coupled with water oxidation (Table S5†). To further examine the essentialness of the single-atomic character of Co in CO2 reduction, CoNP-C3N4@α-Fe2O3 was employed for comparison. The result shows that the CO production rate for CoNP-C3N4@α-Fe2O3 is much lower (4.8 μmol g−1 h−1), suggesting the critical role of single-atomic Co sites in photocatalytic CO2 reduction. A similar irradiation test under Ar atmosphere was conducted on Co1-C3N4@α-Fe2O3, and no CO was detected. To further identify the origin of CO, we traced the carbon source in the reduction process using a 13C isotopic label. Fig. 3c shows the mass spectrum of CO obtained with 13CO2 as a substrate under identical photocatalytic reaction conditions; a distinct 13CO peak (m/z = 29) was observed, indicating that the CO indeed originates from CO2. H218O, instead of H216O, was also adopted to verify the water oxidation half-reaction, and a clear 18O2 peak (m/z = 36) was observed in the mass spectrum. Moreover, molecular oxygen was detected as the product by liquid-phase oxygen measurement system, further confirming that the CO2 reduction is coupled with water oxidation (Fig. S12†). As shown in Fig. 3d and S13,† the production rate and selectivity of CO for Co1-C3N4@α-Fe2O3 remained almost unchanged during the six consecutive cycles (4 h each), and no aggregation of Co species appeared after the stability test, validating the excellent stability for the Z-scheme system under visible light.
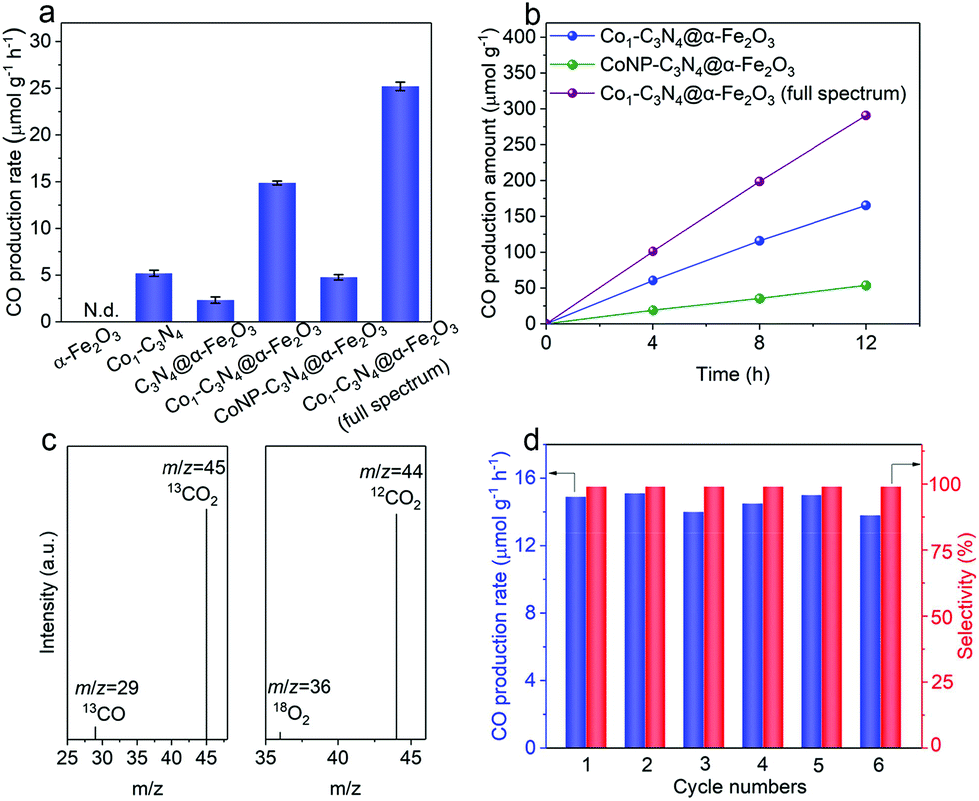 | ||
| Fig. 3 (a) CO production rates of α-Fe2O3, Co1-C3N4, C3N4@α-Fe2O3, Co1-C3N4@α-Fe2O3, and CoNP-C3N4@α-Fe2O3; N.d.: not detected. (b) Time-dependent production of CO by CoNP-C3N4@α-Fe2O3 and Co1-C3N4@α-Fe2O3. (c) Mass spectra of 13CO (m/z = 29) and 18O2 (m/z = 36) produced by Co1-C3N4@α-Fe2O3 in the isotopic experiments. (d) Cycling production of CO for Co1-C3N4@α-Fe2O3 in photocatalytic CO2 reduction with pure water. | ||
Given the above results, a question naturally arises: What role does the Z-scheme play in enhancing the photocatalytic CO2 reduction? In addition to the significantly broadened light-absorption range, the dynamic behaviors of the photo-induced charge carriers are also effectively regulated. The photocurrent responses were recorded under intermittent visible-light irradiation in 0.1 M Na2SO4 aqueous solution (Fig. S14†). The photocurrent density for Co1-C3N4@α-Fe2O3 is higher than those for α-Fe2O3 and Co1-C3N4, suggesting the more efficient photo-induced electron–hole separation for improved CO2 reduction.34 Electrochemical impedance spectroscopy (EIS) was conducted to reflect the charge transfer resistance (Fig. S15†). The Nyquist plots reveal that Co1-C3N4@α-Fe2O3 has the lowest charge transfer resistance, which is beneficial for the separation and migration of the photo-induced charge carriers.24,35 To investigate the lifetime of the electron–hole pairs, we employed PL spectroscopy. As shown in Fig. S16,† the main emission peak located at 442 nm can be ascribed to the inter-band recombination of photo-induced charge carriers. The dramatically quenched PL intensity for Co1-C3N4@α-Fe2O3 indicates the faster electron migration and the efficient suppression of the carrier recombination.29 To gain in-depth information for the carrier dynamics, TRPL decay curves for the as-prepared photocatalysts were recorded. As revealed in Fig. 4a and S17,† Co1-C3N4@α-Fe2O3 shows a significantly slower decay kinetics than α-Fe2O3 and Co1-C3N4. The PL lifetimes are listed in Table S2.† The shorter lifetime τ1 is related to the non-radiative relaxation, and the longer lifetime τ2 is attributed to the inter-band recombination of the photo-induced excitons, which mainly contributes to the photoluminescence.21 Notably, the average lifetime (τ) of Co1-C3N4@α-Fe2O3 is prolonged to 5.17 ns, suggesting the high efficacy of the Z-scheme heterojunction. Specifically, the photo-induced electrons in the conduction band of α-Fe2O3 can efficiently annihilate the holes in the valence band of Co1-C3N4, thus suppressing the recombination of photo-induced excitons inside each component. As a consequence, more photo-induced electrons would be preserved on the conduction band of Co1-C3N4, and more photo-induced holes would be preserved on the valence band of α-Fe2O3, thus leading to superior photocatalytic performances. Furthermore, the incorporation of single-atomic Co sites also results in prolonged average lifetimes with significantly altered τ1 and τ2. The atomically dispersed Co can act as effective trap sites to capture the photo-induced electrons from the conduction band of g-C3N4, thereby inhibiting the recombination of the electrons with the VB holes.36,37 Therefore, for Co1-C3N4@α-Fe2O3, the Z-scheme structure and the single-atomic Co sites collaboratively contribute to the efficient separation of the photo-induced excitons.
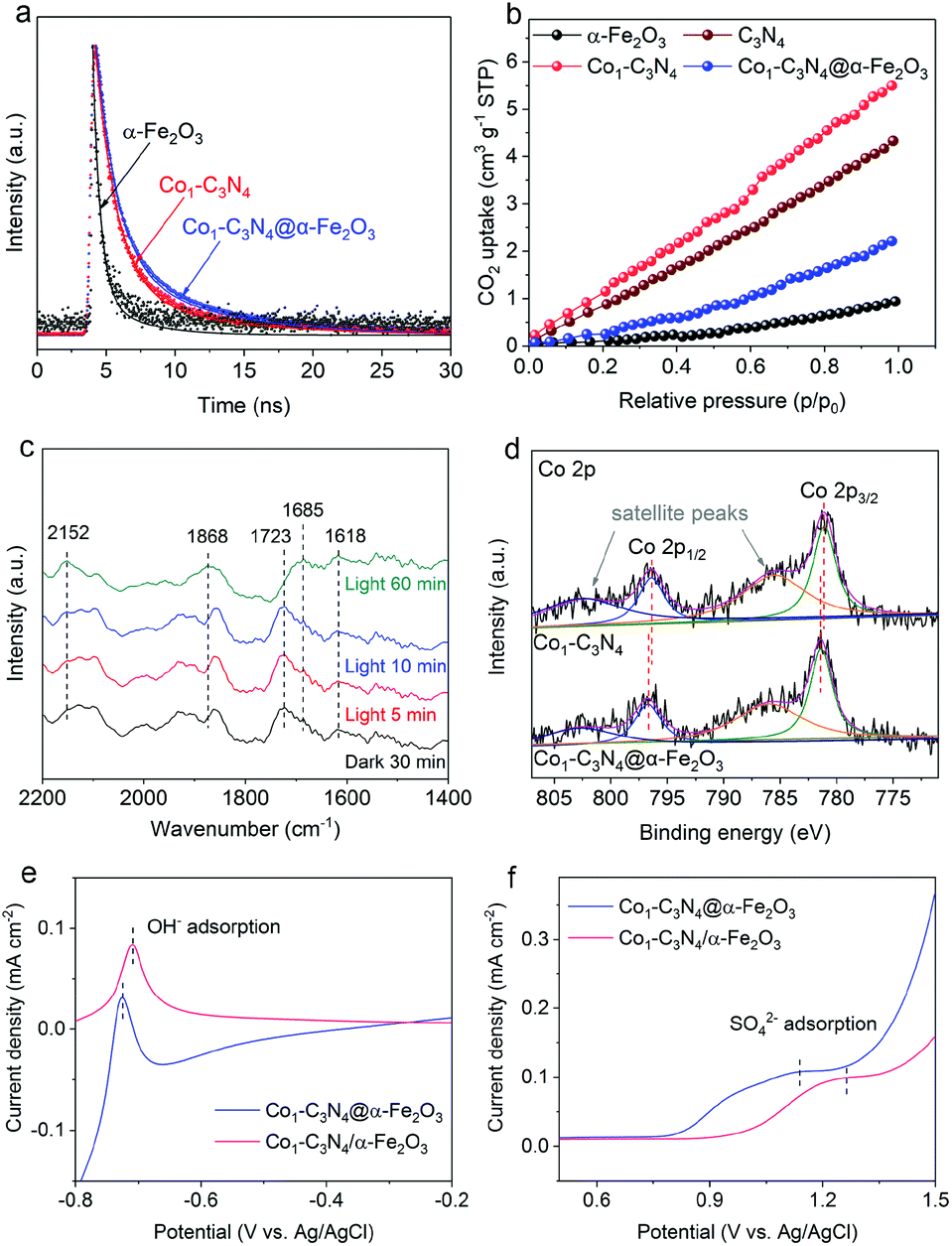 | ||
| Fig. 4 (a) TRPL decay curves for α-Fe2O3, Co1-C3N4, and Co1-C3N4@α-Fe2O3. (b) CO2 adsorption isotherms for α-Fe2O3, g-C3N4, Co1-C3N4, and Co1-C3N4@α-Fe2O3. (c) In situ DRIFTS spectra for Co1-C3N4@α-Fe2O3 under different irradiation conditions with the existence of CO2 and H2O. (d) X-ray photoelectron spectroscopy spectra of the Co 2p core levels for Co1-C3N4 and Co1-C3N4@α-Fe2O3. Single oxidative LSV scans at 100 mV s−1 in (e) Ar-bubbled 0.5 M NaOH and (f) Ar-bubbled 0.1 M Na2SO4 for Co1-C3N4@α-Fe2O3 and Co1-C3N4/α-Fe2O3, respectively. | ||
To shed light on the role of single-atomic Co sites at the catalytic interface during photocatalytic CO2 reduction, CO2 adsorption was employed to determine the chemisorption capacities of these catalysts. As depicted in Fig. 4b, Co1-C3N4 adsorbs a much larger amount of CO2 than bare g-C3N4, suggesting the efficacy of the single-atomic Co sites. It is also worth pointing out that the integration of α-Fe2O3 significantly reduces the CO2 uptake, which can be attributed to the poor CO2 adsorption ability of α-Fe2O3 nanorod arrays. To unravel the possible reaction pathway that occurs on Co1-C3N4@α-Fe2O3, we carried out in situ DRIFTS measurements to explore the key intermediates in photocatalytic CO2 reduction. As illustrated in Fig. 4c, the peaks at 1618 cm−1, 1723 cm−1, and 1868 cm−1 can be assigned to bidentate carbonate (b-CO32−), chelating bridged carbonate (c-CO32−), and multi-bonded CO (m-CO) on the g-C3N4 substrate, respectively.38,39 The carbonate species are formed by the interaction of the surface O atoms of g-C3N4 with adsorbed CO2.38 During the 60 min irradiation, the peak intensity of b-CO32− remained unchanged, whereas the peak intensity of c-CO32− significantly decreased. Meanwhile, an obvious increase was observed in the m-CO peak. These results can be explained by the catalytic effect that c-CO32− is converted into m-CO during the photocatalytic CO2 reduction. More importantly, a peak at 2152 cm−1 emerged and the intensity of the peak gradually increased with the irradiation time. This peak can be assigned to the stretching vibration of CO adsorbed on an isolated Co site.40,41 This result further highlights the key role of the single-atomic Co sites in CO evolution. Moreover, another peak at 1685 cm−1 attributed to CO2− adsorbed on Co sites was observed, the intensity of which noticeably increased after 60 min irradiation.38,42 Therefore, it can be inferred that the CO2-to-CO conversion on the isolated Co sites proceeds via the formation of CO2−, which is generally regarded as the rate-determining step in CO2 reduction.4,43–45 The stabilization of CO2− has a great impact on the overall performance of the photocatalytic CO2 reduction, and largely depends on the local charge density of the active sites.46–48 In our Z-scheme heterojunction, the electrons in Co1-C3N4 with a relatively high Fermi level would spontaneously migrate to α-Fe2O3 to eliminate the disparity between the Fermi levels,34 which was verified by XPS analysis in Fig. S18† and Fig. 4c. The N 1s spectrum for Co1-C3N4 can be deconvoluted into three peaks located at 398.3 eV (C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 400 eV (N–(C)3) and 401.3 eV (C–N–Hx), respectively.13,49 Notably, these peaks shift to higher binding energies after assembly with α-Fe2O3 (Table S3†). Moreover, distinguishable shifts in similar trend were also observed for Co 2p3/2 peaks (Table S3†).13,50 In contrast, in terms of the binding energy of Fe, the Fe 2p peaks of the Z-scheme heterojunction downshift by 0.2 eV relative to those of pristine α-Fe2O3 (Fig. S19 and Table S4†).51 This result is indicative of a partial electron transfer from N and Co to Fe, confirming the electron migration direction between the two components.52,53 Accordingly, the Co species in Co1-C3N4@α-Fe2O3 exhibits a higher oxidation state than that in Co1-C3N4. In this case, the negatively charged CO2− can be better stabilized by the electrostatic interaction with the more positively charged Co sites.45 To confirm the superiority of Co1-C3N4@α-Fe2O3 in CO2− stabilization, the adsorption of OH− and SO42− (as proper surrogates for CO2−) was conducted.54–56 In order to eliminate any possible disturbance, we prepared a non-heterojunction mixture comprising α-Fe2O3 nanorod arrays and Co1-C3N4 with the assistance of Nafion solution rather than via the subsequent thermal treatment (denoted as Co1-C3N4/α-Fe2O3). The oxidative linear sweep voltammograms (LSV) in NaOH aqueous solution reveal that the potential of OH− adsorption for Co1-C3N4@α-Fe2O3 is 19 mV lower than that for Co1-C3N4/α-Fe2O3 (Fig. 4e). This difference becomes more prominent in the adsorption of SO42−. As shown in Fig. 4f, a potential of 1.14 V vs. Ag/AgCl is required for the adsorption of SO42− on Co1-C3N4@α-Fe2O3, 120 mV lower than that on Co1-C3N4/α-Fe2O3 (1.26 V vs. Ag/AgCl). Since the lower potential implies a stronger binding of the anion, these results strongly indicate that the Z-scheme Co1-C3N4@α-Fe2O3 can efficiently stabilize the key intermediate CO2−, thereby facilitating CO evolution.
C), 400 eV (N–(C)3) and 401.3 eV (C–N–Hx), respectively.13,49 Notably, these peaks shift to higher binding energies after assembly with α-Fe2O3 (Table S3†). Moreover, distinguishable shifts in similar trend were also observed for Co 2p3/2 peaks (Table S3†).13,50 In contrast, in terms of the binding energy of Fe, the Fe 2p peaks of the Z-scheme heterojunction downshift by 0.2 eV relative to those of pristine α-Fe2O3 (Fig. S19 and Table S4†).51 This result is indicative of a partial electron transfer from N and Co to Fe, confirming the electron migration direction between the two components.52,53 Accordingly, the Co species in Co1-C3N4@α-Fe2O3 exhibits a higher oxidation state than that in Co1-C3N4. In this case, the negatively charged CO2− can be better stabilized by the electrostatic interaction with the more positively charged Co sites.45 To confirm the superiority of Co1-C3N4@α-Fe2O3 in CO2− stabilization, the adsorption of OH− and SO42− (as proper surrogates for CO2−) was conducted.54–56 In order to eliminate any possible disturbance, we prepared a non-heterojunction mixture comprising α-Fe2O3 nanorod arrays and Co1-C3N4 with the assistance of Nafion solution rather than via the subsequent thermal treatment (denoted as Co1-C3N4/α-Fe2O3). The oxidative linear sweep voltammograms (LSV) in NaOH aqueous solution reveal that the potential of OH− adsorption for Co1-C3N4@α-Fe2O3 is 19 mV lower than that for Co1-C3N4/α-Fe2O3 (Fig. 4e). This difference becomes more prominent in the adsorption of SO42−. As shown in Fig. 4f, a potential of 1.14 V vs. Ag/AgCl is required for the adsorption of SO42− on Co1-C3N4@α-Fe2O3, 120 mV lower than that on Co1-C3N4/α-Fe2O3 (1.26 V vs. Ag/AgCl). Since the lower potential implies a stronger binding of the anion, these results strongly indicate that the Z-scheme Co1-C3N4@α-Fe2O3 can efficiently stabilize the key intermediate CO2−, thereby facilitating CO evolution.
Conclusions
In summary, we have constructed a Z-scheme heterojunction of Co1-C3N4@α-Fe2O3 comprising Co1-C3N4 and α-Fe2O3 nanorod arrays for efficient visible-light-driven CO2 reduction coupled with water oxidation. The introduction of the Z-scheme heterojunction into a single-atomic catalyst of Co1-C3N4 not only promotes the separation of the photo-induced charge carriers for benefiting CO2 reduction, but also enhances the capability of water oxidation. XPS analysis reveals that the single-atomic Co sites in Co1-C3N4 become more positively charged after the formation of the heterojunction, which contributes to the efficient stabilization of CO2− and hence facilitates the CO2 reduction, as verified by the in situ DRIFTS spectra and anion adsorption test. As a result, a CO generation rate of 14.9 μmol g−1 h−1 with over 99% CO selectivity is achieved under visible-light irradiation using water as an electron source. Our findings here underline the importance of the charge regulation on single-atomic sites by the Z-scheme heterojunction for enhancing catalytic performances, and offer a new approach to developing high-efficiency single-atomic site catalysts for artificial photosynthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2017YFA0700104), the National Natural Science Foundation of China (21931007, 21905204, 21805207) and 111 Project of China (D17003). In situ DRIFTS measurements were performed at the Infrared Spectroscopy and Microspectroscopy Endstation (BL01B) in the National Synchrotron Radiation Laboratory (NSRL) in Hefei, China.References
- C. F. Shih, T. Zhang, J. Li and C. Bai, Joule, 2018, 2, 1925–1949 CrossRef CAS.
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS.
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef.
- J. Di, C. Zhu, M. Ji, M. Duan, R. Long, C. Yan, K. Gu, J. Xiong, Y. She, J. Xia, H. Li and Z. Liu, Angew. Chem., Int. Ed., 2018, 57, 14847–14851 CrossRef CAS.
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690–699 CrossRef CAS.
- Z. Jiang, W. Wan, H. Li, S. Yuan, H. Zhao and P. K. Wong, Adv. Mater., 2018, 30, 1706108 CrossRef.
- Y. Jiang, J.-F. Liao, H.-Y. Chen, H.-H. Zhang, J.-Y. Li, X.-D. Wang and D.-B. Kuang, Chem, 2020, 6, 766–780 CAS.
- Y. Li, Z. Wang, T. Xia, H. Ju, K. Zhang, R. Long, Q. Xu, C. Wang, L. Song, J. Zhu, J. Jiang and Y. Xiong, Adv. Mater., 2016, 28, 6959–6965 CrossRef CAS.
- X. Li, W. Bi, L. Zhang, S. Tao, W. Chu, Q. Zhang, Y. Luo, C. Wu and Y. Xie, Adv. Mater., 2016, 28, 2427–2431 CrossRef CAS.
- Y. Cao, S. Chen, Q. Luo, H. Yan, Y. Lin, W. Liu, L. Cao, J. Lu, J. Yang, T. Yao and S. Wei, Angew. Chem., Int. Ed., 2017, 56, 12191–12196 CrossRef CAS.
- W. Liu, L. Cao, W. Cheng, Y. Cao, X. Liu, W. Zhang, X. Mou, L. Jin, X. Zheng, W. Che, Q. Liu, T. Yao and S. Wei, Angew. Chem., Int. Ed., 2017, 56, 9312–9317 CrossRef CAS.
- G. Gao, Y. Jiao, E. R. Waclawik and A. Du, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS.
- P. Huang, J. Huang, S. A. Pantovich, A. D. Carl, T. G. Fenton, C. A. Caputo, R. L. Grimm, A. I. Frenkel and G. Li, J. Am. Chem. Soc., 2018, 140, 16042–16047 CrossRef CAS.
- S. Tang, X. Yin, G. Wang, X. Lu and T. Lu, Nano Res., 2019, 12, 457–462 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS.
- J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS.
- J. Zhang, J. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, X. Fu and X. Wang, Energy Environ. Sci., 2011, 4, 675–678 RSC.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS.
- Z. Zhang, J. Huang, Y. Fang, M. Zhang, K. Liu and B. Dong, Adv. Mater., 2017, 29, 1606688 CrossRef.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS.
- Y.-F. Mu, W. Zhang, G.-X. Dong, K. Su, M. Zhang and T.-B. Lu, Small, 2020, 16, 2002140 CrossRef CAS.
- P. Kuang, L. Zhang, B. Cheng and J. Yu, Appl. Catal., B, 2017, 218, 570–580 CrossRef CAS.
- S.-S. Yi, J.-M. Yan and Q. Jiang, J. Mater. Chem. A, 2018, 6, 9839–9845 RSC.
- S. Cao, H. Li, T. Tong, H.-C. Chen, A. Yu, J. Yu and H. M. Chen, Adv. Funct. Mater., 2018, 28, 1802169 CrossRef.
- C. Hu, X. Wang, Z. Qi and C. Li, Infrared Phys. Technol., 2020, 105, 103200 CrossRef CAS.
- P. Huang, M. Cheng, H. Zhang, M. Zuo, C. Xiao and Y. Xie, Nano Energy, 2019, 61, 428–434 CrossRef CAS.
- X. She, J. Wu, H. Xu, J. Zhong, Y. Wang, Y. Song, K. Nie, Y. Liu, Y. Yang, M.-T. F. Rodrigues, R. Vajtai, J. Lou, D. Du, H. Li and P. M. Ajayan, Adv. Energy Mater., 2017, 7, 1700025 CrossRef.
- M. J. Bojdys, J.-O. Müller, M. Antonietti and A. Thomas, Chem. – Eur. J., 2008, 14, 8177–8182 CrossRef CAS.
- M. Shi, G. Li, J. Li, X. Jin, X. Tao, B. Zeng, E. A. Pidko, R. Li and C. Li, Angew. Chem., 2020, 59, 6590–6595 CrossRef CAS.
- Y. Zhou, L. Zhang and W. Wang, Nat. Commun., 2019, 10, 506 CrossRef CAS.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- Y. Li, S. Chen, D. Xi, Y. Bo, R. Long, C. Wang, L. Song and Y. Xiong, Small, 2018, 14, 1702109 CrossRef.
- Y. Yang, F. Li, J. Chen, J. Fan and Q. Xiang, ChemSusChem, 2020, 13, 1979–1985 CrossRef CAS.
- S. Ji, Y. Qu, T. Wang, Y. Chen, G. Wang, X. Li, J. Dong, Q. Chen, W. Zhang, Z. Zhang, S. Liang, R. Yu, Y. Wang, D. Wang and Y. Li, Angew. Chem., 2020, 59, 10651–10657 CrossRef CAS.
- L. Liu, Y. Jiang, H. Zhao, J. Chen, J. Cheng, K. Yang and Y. Li, ACS Catal., 2016, 6, 1097–1108 CrossRef CAS.
- Q.-S. Chen, S.-G. Sun, Z.-Y. Zhou, Y.-X. Chen and S.-B. Deng, Phys. Chem. Chem. Phys., 2008, 10, 3645–3654 RSC.
- J. Jansson, A. E. C. Palmqvist, E. Fridell, M. Skoglundh, L. Österlund, P. Thormählen and V. Langer, J. Catal., 2002, 211, 387–397 CrossRef CAS.
- H.-K. Lin, C.-B. Wang, H.-C. Chiu and S.-H. Chien, Catal. Lett., 2003, 86, 63–68 CrossRef CAS.
- X. Zu, X. Li, W. Liu, Y. Sun, J. Xu, T. Yao, W. Yan, S. Gao, C. Wang, S. Wei and Y. Xie, Adv. Mater., 2019, 31, e1808135 CrossRef.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS.
- Ş. Neaţu, J. A. Maciá-Agulló, P. Concepción and H. Garcia, J. Am. Chem. Soc., 2014, 136, 15969–15976 CrossRef.
- A. Wuttig, M. Yaguchi, K. Motobayashi, M. Osawa and Y. Surendranath, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4585–E4593 CrossRef CAS.
- I. V. Chernyshova, P. Somasundaran and S. Ponnurangam, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9261–E9270 CrossRef CAS.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- M. R. Singh, J. D. Goodpaster, A. Z. Weber, M. Head-Gordon and A. T. Bell, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8812–E8821 CrossRef CAS.
- J. Liu, H. Shi, Q. Shen, C. Guo and G. Zhao, Green Chem., 2017, 19, 5900–5910 RSC.
- Q. Song, J. Li, L. Wang, Y. Qin, L. Pang and H. Liu, J. Catal., 2019, 370, 176–185 CrossRef CAS.
- Q. Xu, B. Zhu, C. Jiang, B. Cheng and J. Yu, Sol. RRL, 2018, 2, 1800006 CrossRef.
- Y.-X. Liu, H.-H. Wang, T.-J. Zhao, B. Zhang, H. Su, Z.-H. Xue, X.-H. Li and J.-S. Chen, J. Am. Chem. Soc., 2018, 141, 38–41 CrossRef.
- B. Jiang, X.-G. Zhang, K. Jiang, D.-Y. Wu and W.-B. Cai, J. Am. Chem. Soc., 2018, 140, 2880–2889 CrossRef CAS.
- A. Salehi-Khojin, H.-R. M. Jhong, B. A. Rosen, W. Zhu, S. Ma, P. J. A. Kenis and R. I. Masel, J. Phys. Chem. C, 2013, 117, 1627–1632 CrossRef CAS.
- F. Lei, W. Liu, Y. Sun, J. Xu, K. Liu, L. Liang, T. Yao, B. Pan, S. Wei and Y. Xie, Nat. Commun., 2016, 7, 12697 CrossRef CAS.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02836c |
| This journal is © The Royal Society of Chemistry 2020 |