
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterface tailoring by a versatile functionalization platform for nanostructured wood biocomposites†
Céline
Montanari
 ,
Peter
Olsén
* and
Lars A.
Berglund
,
Peter
Olsén
* and
Lars A.
Berglund
Department of Fiber and Polymer Technology, Wallenberg Wood Science Center, KTH Royal Institute of Technology, Teknikringen 56, 100 44 Stockholm, Sweden. E-mail: polsen@kth.se
First published on 27th October 2020
Abstract
Wood templates are promising biobased substrates that can be functionalized for controlled nanostructure, and the development of novel nanotechnologies. A sustainable, selective and versatile chemical functionalization platform for cellulosic wood templates is developed. Bulk wood templates are delignified using peracetic acid, and the nanoporous delignified wood templates are functionalized with cyclic anhydrides (maleic, itaconic and succinic) from renewable resources. The synthetic pathway uses solvent-free reaction conditions, short reaction times, and has possibilities for chemical recovery. Anhydrides become covalently attached inside the nanoporous wood cell wall and yields wood templates with lower moisture sorption, preserved nano- and ultrastructure, and high degree of esterification with carboxyl content as high as 3.93 mmol g−1. The functionalization platform enables interface tailoring at the molecular scale with possibility for further reaction and covalent modifications. This is demonstrated by the preparation of transparent wood biocomposites. The biocomposites exhibit controlled wood–polymer interfacial adhesion with high optical transmittance. The favorable interaction mechanisms at the cell wall level result in mechanically strong biocomposites.
Introduction
In sustainable societal development, cellulosic materials are of great interest, but can seldom be used in advanced applications without some modification. It is therefore desirable to design chemical functionalization strategies based on environmentally friendly pathways. Wood is from renewable resources, and is designed and optimized by nature to withstand high loads and provide long-term durability to the organism. The high specific strength and stiffness of wood originates from its anisotropic alignment of tubular fibers and the hierarchical structure, ranging from the macroscale down to nanoscale.1,2 The tubular fiber cells have a cell wall of a few micrometers in thickness, surrounding cylindrical pore space (lumen) of about 20 μm in diameter. The cell wall is a nanocomposite of strong, axially aligned cellulose nanofibrils in a hydrated polymer matrix of lignin and hemicelluloses. By chemical treatment and/or suitable drying procedure, this cell wall can become nanoporous, while the strong nanofibrils are preserved. This anisotropic wood structure with hierarchical porosity is suitable as a porous template reinforcement in new and advanced biocomposite materials. Not only can such biocomposites have good structural properties, but the nanoporous cell wall structure can also be modified by novel routes for additional wood functionalities. For example, nanoparticles can diffuse into the cell wall for improved fire retardancy,3,4 epoxy can replace lignin inside the cell wall for oil–water separation,5 and optical dyes can be added to create a wood laser.6For wood templates filled by a polymer matrix, improved molecular interactions between the polymer and the wood cell wall is a remaining challenge. Much effort has been devoted to developing chemical methods for improved interfacial adhesion in wood fiber composites.7,8 Cell wall accessibility can be enhanced by partial removal of cell wall components, which could facilitate monomer impregnation and result in high performance composites.9–13 This includes partial or complete removal of lignin, as well as some of the hemicelluloses. Recently, delignified wood templates with preserved nanostructure have been investigated to develop new wood nanotechnologies.14–21 As lignin is removed, the interior cell wall accessibility is improved, but at the expense of increased sensitivity of physical properties to humid conditions. To address this, chemical functionalization methods of the cell wall components have been developed. Chemical functionalization of the cell wall polysaccharides can reduce the hygroscopic nature of cellulosic fibers and improve stress-transfer at the fiber–polymer interface.22–25 The main strategy is to replace hydrophilic groups, such as hydroxyls, with more hydrophobic functionalities. Esterification, etherification and graft polymerization are common methods for the chemical functionalization of cellulosic materials, where wood-related hydroxyl groups are commonly targeted.26,27 However, etherification and graft-polymerization are hampered by the presence of residual moisture in the fiber structure that initiate polymerization.28,29 Likewise, esterification of wood template with e.g. acetic anhydride requires the use of organic solvents, such as pyridine.30 Thus, most of reported synthetic pathways rely on the use of both organic solvents and toxic reagents. Sustainable pathways for the chemical functionalization of wood would therefore be a significant advance. The ideal chemical functionalization platform would preferably be environmentally friendly, sustainable, based on renewable resources, non-toxic, rapid, selective, atom economic and result in comparable or better material properties compared with traditional pathways.31
In order to address this challenge, we report a sustainable and versatile strategy to functionalize cellulosic wood templates, addressing both the delignification protocol and the functionalization platform. The functionalized wood templates can be used in advanced biocomposite applications. The synthetic strategy was designed in accordance to the principles of green chemistry by using renewable materials, solvent-free reaction conditions, non-toxic reagents, and offering possibilities for chemical recovery.32,33 First, a green and selective peracetic acid delignification protocol was employed on wood veneers, to promote chemical and physical cell wall accessibility. Secondly, the delignified wood template was decorated with new chemical functionalities via ring-opening esterification of three renewable cyclic anhydrides, maleic, itaconic and succinic anhydrides, under neat conditions.
Previous studies of anhydride modification have been focused on plant fibers,34–38 and the effects of anhydride modification on dimensional stability of fibers were assessed.39,40 Sehaqui et al.41 and Huang et al.42 esterified cellulose fibers using succinic anhydride to facilitate the production of nanofibers. The possibility to introduce carboxyl groups in wood has also been explored for applications such as copper remediation and photostability.43–45 However, the majority of the procedures use organic solvents and harmful catalysts. Iwamoto and Endo used solvent-free conditions to modify wood flour with maleic anhydride,46 but the study is a technical approach for plant fiber defibrillation into nanofibrils.
In contrast, the present study creates a green chemical functionalization platform for macroscopic wood templates of interest for multifunctional structural applications. The surface activation of the template via ring-opening esterification of the anhydrides generates new reactive sites, including terminal carboxylic acid groups, di- and mono-substituted alkenes, suitable for further modification.47–50 As a demonstration, the green functionalization platform of wood templates was applied for the preparation of transparent wood biocomposites. The mechanical and optical performance of the final biocomposites transcend state-of-the-art chemical modification strategies, which do not comply with basic principles for environmental sustainability.32
The present functionalization strategy is characterized by short reaction times and very high degrees of esterification. This green functionalization platform is applicable to a wide range of cellulosic materials for controlled cell wall accessibility and functionality at molecular scale. It also provides controlled fiber–polymer interfacial adhesion for advanced biocomposites applications.
The specific engineering science objectives are to explore biobased cyclic anhydrides for wood template functionalization, determine nanoscale porosity and interface interaction effects, investigate nanostructural organization in wood biocomposites, and the applicability of green anhydride treatment for transparent wood materials.
Results and discussion
Green functionalization platform for cellulosic wood templates
A two-step approach was used to prepare functionalized wood templates via a sustainable synthetic platform. The platform is based on ring-opening esterification of three cyclic anhydrides (maleic, itaconic and succinic anhydrides) from renewable resources. Fig. 1 summarizes the processing routes for green modification of balsa wood template. Balsa wood with a density of 304 kg m−3 was selected as raw material. Balsa has aligned vessel elements of ≈280 μm in diameter and fiber cells of ≈40 μm in diameter with microscale cell walls consisting of nanoscale composite layers. The cellular structure of the native wood (NW) is shown in Fig. 2a, where characteristic vessels, fibers and ray cells are apparent. In the cell wall composite layers, oriented cellulose microfibrils of nanoscale diameter are embedded in a molecularly mixed matrix of hemicellulose and lignin. In the living tree, this hemicellulose/lignin matrix is hydrated and the cell wall contains about 30% moisture.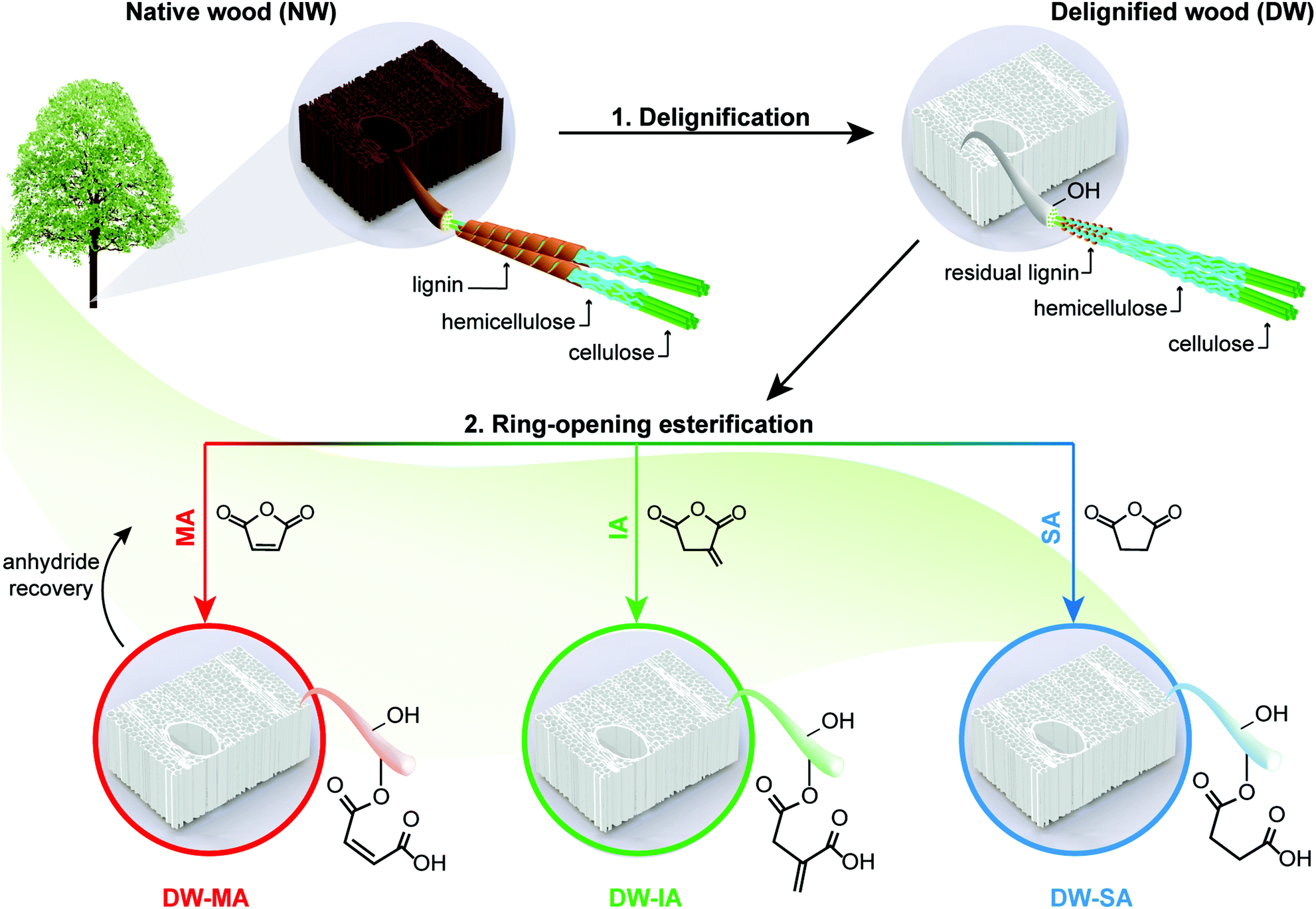 | ||
| Fig. 1 Schematic representation of sustainable functionalization platform for cellulosic wood templates. First, the wood template is delignified and then functionalized via ring-opening esterification of renewable cyclic anhydrides. Maleic anhydride (MA), itaconic anhydride (IA) and succinic anhydride (SA) were left to react under neat conditions, resulting in functionalized wood templates with new chemical functionalities inside the cell wall. | ||
 | ||
| Fig. 2 Cross-sectional SEM images of the (a) native wood (NW), (b) delignified wood (DW) with increasing magnification from left to right, and functionalized templates (c) DW-MA, (d) DW-IA, and (e) DW-SA, where the insets are the respective cell wall micrographs. | ||
The cell wall biopolymers contain abundant hydroxyl groups useful as reactive sites for functionalization. Balsa is chemically composed of 54% cellulose, 22% hemicelluloses, and 24% lignin (Fig. S1†). Lignin is distributed throughout the secondary cell wall and is predominant in the intercellular middle lamella and cell corners. The lignin is rich in chromophores providing wood with its natural dark color.
A mild peracetic acid (PAA) delignification treatment is used to selectively remove lignin and associated chromophores, resulting in a white delignified wood (DW) template with nanoscale porosity. Compared with environmentally hazardous chlorite delignification used in previous studies,19,51 PAA delignification is equally selective at removing lignin, while preserving similar cellulose and hemicellulose contents as chlorite delignification (Fig. S1†). A problem with the latter method, is the formation of chlorinated organic compounds during delignification.52,53 In contrast, PAA is chlorine-free, environmentally friendly, yields non-toxic decomposition products and is directly produced from two renewable chemicals, acetic acid and hydrogen peroxide.54,55 The partial PAA delignification leads to approximately 34% mass loss; and in the present study the resulting DW scaffold is used as biocomposite reinforcement where a thermoplastic polymer matrix is filling the pore space.
Wood morphology and microstructure are preserved after mild delignification (Fig. 2b). The PAA delignification was optimized to have a residual lignin content of about 2% in the DW, which prevents cell wall delamination and stabilizes the template. The native cell wall morphology is well preserved at microscale, enabling exploitation of the hierarchical wood structure for advanced material applications. PAA delignification also preserves cellulose fibril structure, cellulose molar mass as well as hemicellulose distribution in the cell wall, avoiding severe chemical degradation.56 Preserved cellulose molar mass suggests preserved fibril stiffness and strength, while high hemicellulose content prevents cellulose fibril aggregation and largely preserves nanoscale fibril distribution. The removal of lignin is confirmed by the absence of characteristic lignin peaks in the FTIR spectra (Fig. S2†). Interestingly, the nanoscale cell wall porosity increases upon delignification as indicated in the high-resolution SEM image to the right in Fig. 2b. Microscale porosity is apparent in formerly lignin-rich cell corners, and nanoscale pores can be distinguished in the cell wall. Moreover, a significant increase in specific surface area is observed after delignification from 9 m2 g−1 (NW) to 207 m2 g−1 (DW), see SBET in Table 1 and isotherms in Fig. S3.† The large increase in specific surface area is crucial for enhanced chemical accessibility and potential for high degree of chemical functionalization.
| S BET (m2 g−1) | Carboxyl content (mmol g−1) | Weight gain (wt%) | CI (%) | T onset (°C) | Moisture contenta (wt%) | |
|---|---|---|---|---|---|---|
| a Measured at 94% relative humidity. | ||||||
| NW | 9 (2) | |||||
| DW | 207 (22) | 0.28 (0.02) | 70 | 256 | 31 | |
| DW-MA | 87 (2) | 1.23 (0.07) | 17.8 | 71 | 239 | 21 |
| DW-IA | 99 (9) | 2.59 (0.11) | 28.9 | 72 | 237 | 18 |
| DW-SA | 41 (6) | 3.93 (0.34) | 33.2 | 71 | 225 | 17 |
The next step is to provide the wood template with suitable chemical functionalization inside the cell wall. The chemical modification of the wood template was performed under solvent-free conditions with three different renewable cyclic anhydrides: maleic anhydride (MA), itaconic anhydride (IA) and succinic anhydride (SA). MA and SA can be produced from biomass-derived furfural,57–65 while IA is derived from itaconic acid produced by fermentation of biomass.66–68 The reaction time required for functionalizing the DW was dependent on the cyclic anhydride. The SA ring-opening esterification was achieved after only 0.5 h at 130 °C, while DW-MA and DW-IA were obtained after 24 h reaction at 100 °C and 80 °C, respectively. Note that the minimum temperature at which the reaction can be performed corresponds to the melting temperature of the anhydride (Fig. S4†), and the reaction temperature was optimized to result in high yield (Fig. S5†). SEM micrographs of the functionalized wood templates show that the microstructure is preserved after the chemical modification (Fig. 2c and d). The SEM images of DW-MA, DW-IA and DW-SA indicate similar microstructure as the DW. At 100 nm scale, the cell wall nanostructure and porosity are retained.
Mechanistically, the modification proceeds through ring-opening esterification of renewable cyclic anhydrides, reacting accessible hydroxyls in the DW template. The delignified template is swollen by the molten anhydrides, allowing reactions inside the wood cell wall. The esterification can occur from cellulose, hemicellulose or residual lignin hydroxyl groups.36,69–74 The residual water in the wood template (≈4 wt%) hydrolyzes the cyclic anhydrides and creates free dicarboxylic acids, making the reaction self-catalyzed.75 Dependent on the anhydride, different functional groups are added to the DW template (Fig. 1). SA esterification substitutes part of the hydroxyls to carboxylic acids, while MA and IA esterification reactions result in carboxyl groups as well as di-substituted and mono-substituted alkenes, respectively. Therefore, each modification independently leads to novel, functional and nanostructured wood templates with specific active sites available for further modification.
Molecular scale modification effects of the nanostructured cell wall were then investigated. The FTIR spectrum of the DW reference was compared with the DW-MA, DW-IA, and DW-SA spectra (Fig. 3a). The substitution of the hydroxyl groups with terminal carboxyl functionalities results in increased carboxyl content in the functionalized templates, identified by the high intensity C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration peak at 1725 cm−1 and a decrease in intensity of the O–H stretching peak at 3400 cm−1. The esterification is confirmed by the increase of the absorbance band at 1155 cm−1, attributed to the C–O stretching of the aliphatic ester. For DW-MA and DW-IA, the introduction of reactive vinyl bonds is also indicated by the peak at 1638 cm−1. The SA modification reaction proceeded with an esterification rate about twice that of IA and eight times greater than MA (Fig. 3b and Fig. S6†). The rate difference correlates well with previously observed rates of hydration for SA, IA and MA.76 The carboxyl charge of the template, as determined by conductometric titration, was high for all esterification reactions, with values of 1.23 mmol g−1 for DW-MA, 2.59 mmol g−1 for DW-IA and up to 3.93 mmol g−1 for DW-SA (Table 1). The charge corresponds to a degree of substitution of 0.19 for DW-MA, 0.42 for DW-IA and 0.64 for DW-SA. Obviously, this means that modification is taking place inside the wood cell wall. These values correlate with the intensity increase of the carboxyl peak at 1725 cm−1 in the FTIR spectra, with the following reactivity trend SA > IA > MA. This carboxyl functionality is very interesting, since it has recently been used successfully for strong, wood-based films, where modification is based on a less sustainable TEMPO-oxidation method.77
O stretching vibration peak at 1725 cm−1 and a decrease in intensity of the O–H stretching peak at 3400 cm−1. The esterification is confirmed by the increase of the absorbance band at 1155 cm−1, attributed to the C–O stretching of the aliphatic ester. For DW-MA and DW-IA, the introduction of reactive vinyl bonds is also indicated by the peak at 1638 cm−1. The SA modification reaction proceeded with an esterification rate about twice that of IA and eight times greater than MA (Fig. 3b and Fig. S6†). The rate difference correlates well with previously observed rates of hydration for SA, IA and MA.76 The carboxyl charge of the template, as determined by conductometric titration, was high for all esterification reactions, with values of 1.23 mmol g−1 for DW-MA, 2.59 mmol g−1 for DW-IA and up to 3.93 mmol g−1 for DW-SA (Table 1). The charge corresponds to a degree of substitution of 0.19 for DW-MA, 0.42 for DW-IA and 0.64 for DW-SA. Obviously, this means that modification is taking place inside the wood cell wall. These values correlate with the intensity increase of the carboxyl peak at 1725 cm−1 in the FTIR spectra, with the following reactivity trend SA > IA > MA. This carboxyl functionality is very interesting, since it has recently been used successfully for strong, wood-based films, where modification is based on a less sustainable TEMPO-oxidation method.77
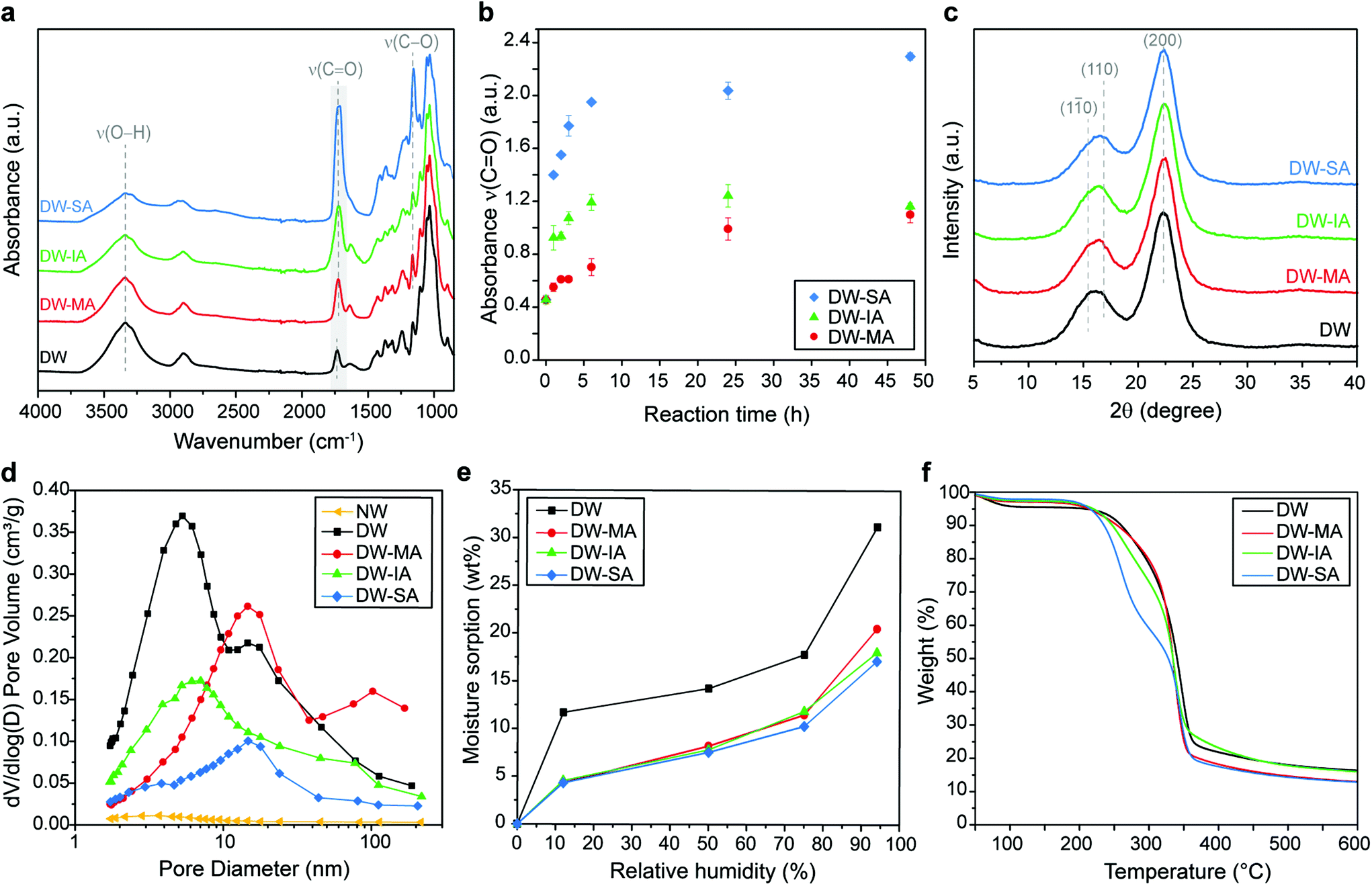 | ||
| Fig. 3 (a) FTIR spectra of native wood (NW), delignified wood (DW), DW-MA, DW-IA and DW-SA. (b) Absorbance of the ν(CO) band of DW-MA, DW-IA and DW-SA as a function of reaction time after functionalization performed at 130 °C. (c) XRD diffractograms of DW and functionalized templates. (d) Pore volume distribution of NW, DW, and functionalized templates. (e) Moisture sorption of at various relative humidities and (f) TG curves of the DW and functionalized templates. | ||
In fully sustainable synthetic pathways, the chemicals should be recoverable and reused for further reactions. Overall, the reaction system is indeed designed so that all reactants can be recovered and reused after chemical reaction, without altering the degree of esterification (Fig. S7†). This results in high yields and good atom economy. Present data show that molten anhydrides subjected to higher reaction temperatures and shorter reaction times, results in a higher degree of esterification and a more eco-friendly process.
The X-ray diffractograms (Fig. 3c) indicate that crystallinity (from cellulose) and ultrastructure of the DW was not altered by chemical functionalization with anhydrides. The characteristic diffraction peaks of cellulose are observed for all functionalized DW samples and are similar to DW. The crystallinity index (CI) of 70% was maintained after modification (Table 1), suggesting that the ring-opening esterification occurs primarily on the more accessible amorphous hemicellulose structure.
The FTIR data and charge values confirmed high degree of modification of the DW template with the different anhydrides. However, whether the anhydride is covalently attached or just absorbed in the substrate remains uncertain. To answer this, we performed carboxyl deprotonation experiments of the functionalized template at pH 9 in a sodium bicarbonate buffer. Deprotonation of the functionalized template results in a hydrogel-like structure with poor mechanical integrity, see ESI Fig. S8.† Fig. S9† shows the absorbance band of the carboxyl groups of the functionalized DW templates in their corresponding carboxylate form. The shift of the carboxyl peak to lower wavenumber is direct evidence of covalent bonding of the anhydrides to the wood template. The presence of chemically bonded moieties within the cell wall is complemented by data showing high weight gain of the functionalized DW templates (Table 1), confirming high anhydride content inside the cell wall.
Although high-resolution SEM images indicate similar nanoporous microstructure as the DW after anhydride reactions (Fig. 2c–e), BET measurement reveals disappearance of smaller pores after functionalization (Fig. 3d). This is an additional support of ring-opening esterification occurring inside the cell wall, since anhydrides occupy part of the pore space. Indeed, the total pore volume is decreased and the pore size distribution is shifted from 2–7 nm (DW) to higher range of pore size after chemical functionalization, with a predominance around 10–11 nm. Moreover, the specific surface area decreases gradually as the density of carboxyl groups increases (Table 1). This is attributed to stronger inter-fibril interactions and formation of hydrogen bonds that leads to physical crosslinking of the cell wall.78,79 This is further supported by the position of the CO peak at 1725 cm−1 in FTIR spectra of the functionalized templates (Fig. 3a), assigned to carboxyls apt to form interfibril hydrogen bonds.80–83
The esterification of the DW template leads to a large reduction in moisture uptake compared with unmodified DW (Fig. 3e and Fig. S10†). At 94% RH, DW-MA, DW-IA and DW-SA show a decrease in moisture content by 34%, 42% and 45%, respectively, when compared with DW (Table 1). The reduction in moisture sorption is attributed to a bulking effect of the cell wall where anhydride molecules reduce the chemical accessibility for water, thereby helping in limiting hygroscopicity effect. The reduced moisture sensitivity of the esterified DW templates is also observed by contact angle measurements, where the initial contact angle is greater for all the esterified DW compared with the DW reference (Fig. S11†). Note that although the esterified DW templates have reduced moisture sensitivity, they are still hydrophilic in nature and wetting occurs at the same time as the DW reference.
The thermal stability of the functionalized templates was reduced when compared with DW (Fig. 3f). Indeed, the increase in the carboxylic acid content caused a decrease in thermal degradation temperature from 256 °C for DW to 225 °C for DW-SA (Tonset, Table 1), which is attributed to the increased acidity of the template.
Transparent wood biocomposites application
To demonstrate the potential of the chemical functionalization platform, the wood templates were evaluated as reinforcement in transparent wood (TW) biocomposites. TW was prepared by pre-polymerized MMA impregnation, with and without prior functionalization of the DW template, followed by polymerization (Fig. 4a). The type of functionality in the DW template dictates and modulates the physical and chemical bonding interactions at the interface between the template and the polymer matrix.35,48Fig. 4a shows that different molecular scale interactions between wood and PMMA are formed depending on the template functionalization. The interphase tailoring mechanism is dependent on how the functionalized DW templates interplay with MMA during the polymerization step. For DW-MA, MA reacts with MMA monomers during the polymerization and copolymer structures are formed. The macromolecular structure formed during copolymerization of MA and MMA is a statistical copolymer of isolated MA units in blocks of PMMA. This relates to the inability of MA, and its corresponding esters, to homopropagate under these reaction conditions.84,85 Translated to the DW-MA, this leads to the formation of isolated PMMA block structures covalently bound at the interface, see Scheme S1 in the ESI.† In contrast, IA and its corresponding esters homopolymerize to high molecular weight polymers at these conditions.86,87 Thus, during the MMA polymerization step a densely polymerized IA and MMA copolymer interphase is formed (Scheme S2†). This may result in a covalently attached copolymer structure between MMA and IA at the wood cell wall interface.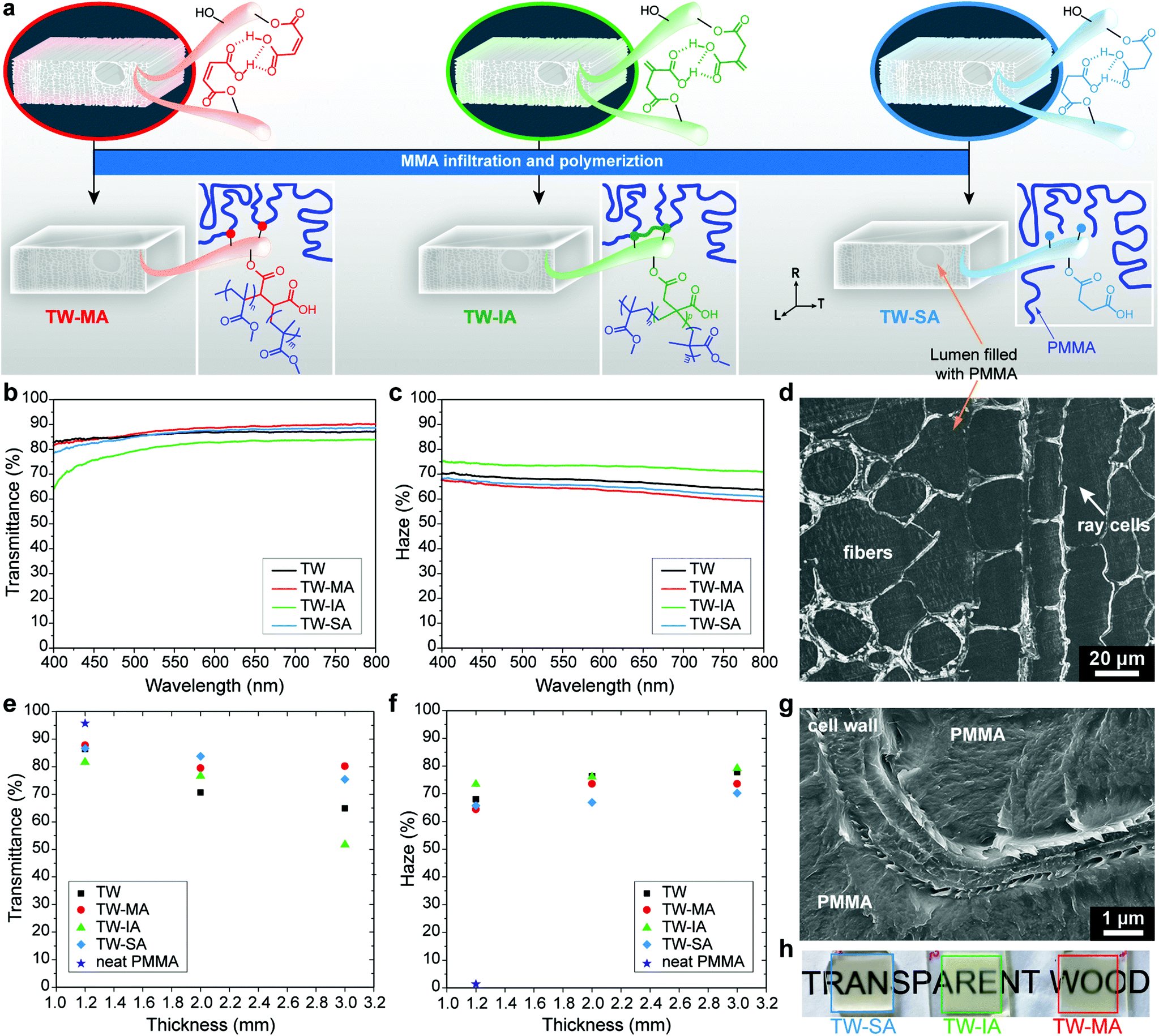 | ||
| Fig. 4 (a) Illustration showing the preparation of transparent wood (TW) from the functionalized templates. Optical transmittance (b) and haze (c) of TW and TW-MA, TW-IA, TW-SA. (d) SEM micrograph of TW-SA infiltrated with PMMA. Optical transmittance (e) and haze (f) as a function of sample thickness. (g) SEM micrograph of TW-SA showing the interface between the wood cell wall and PMMA matrix. (h) Photographs of 2 mm thick TW-MA, TW-IA and TW-SA. | ||
The covalent link between the PMMA matrix and the wood cell wall was confirmed in TW-MA and TW-IA solvent leaching experiments, where non-covalently attached PMMA chains in the wood template were removed by dissolution. Fig. S12† shows that the carbonyl peak associated with PMMA remains high in the FTIR spectra of TW-MA and TW-IA, even after leaching. Not only does this prove that PMMA grafting occurred, it also confirms the presence of PMMA inside the wood cell wall. For TW-SA, only secondary physical interaction takes place between the DW-SA template and PMMA chains, and all the PMMA is removed by solvent leaching. To confirm the removal of PMMA from TW-SA, we performed deprotonation experiment on the leached DW-SA template, and the FTIR spectrum in Fig. S12d† shows the shift of the carboxylic acid peak to lower wavenumber associated with carboxyls in their carboxylate form. This shows that SA moieties are still covalently bonded to the cell wall after PMMA leaching, which is advantageous for recyclability of the leached DW-SA template with possibilities for further modification.
The effects of the template functionalization on optical and mechanical properties of the TW biocomposites are summarized in Table 2. The PMMA infiltration of functionalized templates results in highly transparent composites even at relatively high thickness. Fig. 4b shows that optical transmittance of 1.2 mm thick TW biocomposites increases after chemical functionalization with MA and SA. In contrast, the transmittance of TW-IA is lowered, because of the slight yellow coloring of DW-IA, which originates from IA polymerization (see photographs in Fig. S13†). As a consequence, light absorbance is higher in this TW biocomposite compared with the others. Moreover, the haze of TW-MA and TW-SA is reduced when compared with the TW reference without the anhydride modification (Fig. 4c). Haze is a measure of the transmitted light that is scattered at wide angles, due to optical defects. The favorable optical properties are related to the non-porous microstructure of wood/PMMA, as exemplified by TW-SA (Fig. 4d). TW-IA exhibits higher haze than the TW reference; one reason could be fibril aggregation due to IA polymerization within the cell wall during functionalization.
| Density (kg m−3) | V f (%) | Transmittance (%) | Haze (%) | Ultimate strength (L) (MPa) | Elastic modulus (L) (GPa) | Ultimate strength (T) (MPa) | Elastic modulus (T) (GPa) | |
|---|---|---|---|---|---|---|---|---|
| TW | 1205 | 6.4 | 70.6 (0.6) | 76.3 (0.1) | 41.4 (8.6) | 4.8 (1.0) | 11.4 (5.8) | 2.9 (0.7) |
| TW-MA | 1200 | 6.0 | 79.5 (2.5) | 73.6 (1.6) | 46.2 (6.0) | 4.3 (0.8) | 18.0 (2.7) | 3.1 (0.2) |
| TW-IA | 1202 | 6.5 | 76.6 (1.4) | 76.2 (1.1) | 54.9 (6.8) | 4.3 (1.1) | 33.2 (3.9) | 3.3 (0.7) |
| TW-SA | 1206 | 6.2 | 83.8 (1.6) | 66.9 (2.3) | 61.7 (3.5) | 4.5 (0.3) | 12.8 (2.3) | 2.9 (0.6) |
The favorable optical properties of functionalized templates are even more apparent in thicker TW composites (Fig. 4e and f). The 2 mm thick TW-MA, TW-IA and TW-SA composites demonstrate higher optical transmittance than unmodified TW, regardless of the anhydride employed during ring-opening esterification (Table 2). Thus, TW-SA results in 18% higher transmittance than unmodified TW, augmenting from approximately 71% (TW) to 84% (TW-SA) at a wavelength of 550 nm. Moreover, the optical haze in 2 mm thick composites decreases by 12% with SA functionalization. The increase in optical transmittance is attributed to the favorable interface between PMMA and functionalized cell wall. This is further supported by cross-sectional SEM micrographs (Fig. 4g and Fig. S14†), showing well-integrated interfaces between the functionalized DW cell wall and the PMMA phase. In contrast, poor interface with debonding gaps (air pockets) responsible for strong light scattering can be observed at the interface between DW cell wall and PMMA in the TW reference (Fig. S15†). In addition, the improvement in optical transmittance correlates with degree of esterification of the DW template. High degree of esterification may improve interactions inside the cell wall at the interface between PMMA and functionalized fibrils. Decreased haze may also be caused by lowered extent of fibril aggregation or other optical defects in the cell wall. The sensitivity of optical properties to wood cell wall functionalization chemistry is strongest at the greatest thickness. Indeed, 3 mm thick TW-MA has slightly higher optical transmittance than TW-SA although haze is lower for TW-SA. The high transparency of TW-MA, TW-IA and TW-SA is illustrated in the photographs in Fig. 4h.
To benchmark the potential of this green functionalization platform, the optical performance of the TW biocomposites was compared with acetylation modification using acetic anhydride, following the method reported by Li et al.30 For similar thickness, our green functionalization treatment results in significantly better optical performance for TW-SA, somewhat better for TW-MA and similar performance to acetylated wood for TW-IA. For 2 mm thick acetylated TW, an optical transmittance of 77 ± 0.5% and a haze of 75 ± 0.2% were obtained. Although acetylation also leads to improved optical properties, the chemical treatment is toxic and environmentally hazardous, and necessitates an extra bleaching step prior to MMA infiltration. The SA and IA functionalizations result in both higher transmittance and lower haze at 2 mm thickness, and the methods are green, solvent-free, and time-efficient.
Strong wood–polymer interfaces can result in high strength composites for structural applications, in particular for high wood content.13 The bulking effect of the cell wall (Fig. 3d) and enhanced interfacial adhesion from esterification (Fig. 4g) may improve ultimate strength. Decreased moisture sorption of the esterified wood templates (Fig. 3e), also means reduced moisture sensitivity during service for the TW biocomposites. The stress–strain curves of the functionalized TW-MA, TW-IA and TW-SA composites loaded in the longitudinal direction are shown in Fig. 5a. The TW-MA, TW-IA and TW-SA composites show higher strength than TW. Moreover, increase in strength correlated with increased anhydride content in the cell wall (Table 1). TW-SA demonstrates an ultimate strength of 61.7 MPa, which is 50% higher than for unmodified TW (Table 2). The increase in ultimate strength of the biocomposites may be related to effects from the anhydrides on cell wall properties.35 Estimation of the effective strength of the DW reinforcement in the biocomposite shows that it reaches 339 MPa for DW-SA, whereas it is 12 MPa for the DW reference (see Table S1†). This demonstrates that improved interfacial adhesion results in more favorable stress transfer in the esterified TW biocomposites. The strong interfacial adhesion of the esterified TW biocomposites is further supported by the relatively flat fracture surfaces and very short fiber pull-out observed in the SEM images in Fig. S16.† Compared with the TW reference, the longitudinal Young's modulus was slightly reduced for esterified templates. Overall, the mechanical properties of the TW biocomposites prepared from functionalized balsa templates are comparable with previous works (Fig. S17†), although the wood volume fraction is low (≈6%).
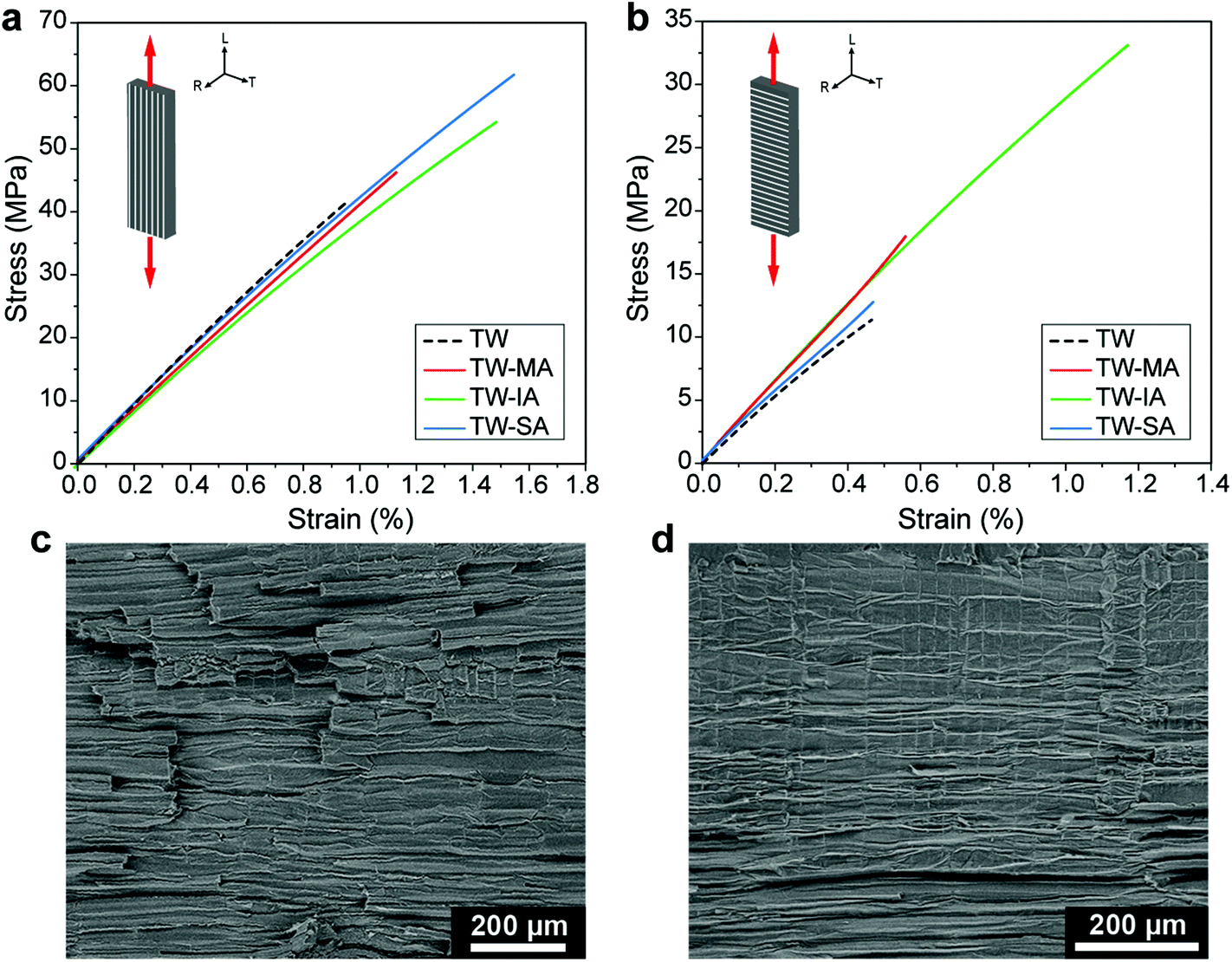 | ||
| Fig. 5 Stress–strain curves in tension for TW, TW-MA, TW-IA and TW-SA with loading along the fibers in the longitudinal direction (a) and in the transverse direction (b). SEM micrographs of transverse fracture surfaces of TW-IA (c) and TW-SA (d). | ||
The effects of covalent interface attachment between the PMMA matrix and the wood cell wall in the case of TW-MA and TW-IA are assessed by transverse direction loading experiments (Fig. 5b). A large fraction of the interface area is loaded in transverse tension in this experiment. TW-MA and TW-IA show much higher strengths than TW-SA and TW. The transverse strength of TW-IA is remarkably high, ≈33 MPa, compared with ≈11 MPa for the TW reference and ≈13 MPa for TW-SA (Table 2). In analogy with transverse loading of polymeric fiber composites, debonding at the wood–polymer interface may initiate failure. The SEM fracture surfaces are much more rough for TW-IA (Fig. 5c) than for TW-SA (Fig. 5d). Possibly, covalently linked wood–polymer interfaces in TW-IA and TW-MA can sustain higher strain before debonding; and other subcritical damage mechanisms are initiated, so that ultimate strength is increased.
Conclusions
A green, selective and versatile chemical functionalization platform is presented for renewable cellulosic materials. This chemical functionalization strategy, based on green PAA delignification and ring-opening esterification of renewable cyclic anhydrides, represents a promising sustainable approach with short reaction time and high degree of esterification. Not only are wood surfaces functionalized, but also the nanoporous wood cell wall is modified internally with covalently linked anhydrides. The functionalized templates show high carboxyl content, lowered moisture sorption, and preserved nano- and microstructure. Carboxyls and alkenes can, for instance, interact with inorganic nanoparticles, dyes, electropositive ions and vinyl monomers. The chemical function of the wood template, including reactive double bonds, can be tuned for specific interface tailoring in advanced applications, such as optically transparent wood (TW) biocomposites. These biocomposites are mechanically strong and show improved optical transmittance and reduced haze at greater thickness.Maleic anhydride (MA) and itaconic anhydride (IA) modification provides covalent wood–polymer interface bonding, which translates into substantially increased transverse strength. The thermoplastic polymer matrix is also distributed inside the wood cell wall, at nanoscale, for these two biocomposites (TW-MA, TW-IA), and this is probably also true for TW-SA. The excellent optical and mechanical properties, obtained by sustainable chemical functionalization of nanostructured wood cell walls, are inspiring also for other functional cellulosic materials where eco-friendly preparation methods are favored.
Experimental
Materials
Balsa (Ochroma pyramidale) with an oven-dried density of 304 kg m−3 was purchased from Material AB (Sweden). The reagents and reactants, peracetic acid (PAA, 40%), maleic anhydride (MA, 99%), itaconic anhydride (IA, 95%), succinic anhydride (SA, ≥99%), and 2,2′-azobis(2-methylpropionitrile) (AIBN, 98%) were purchased from Sigma Aldrich (Sweden) and used as received. Acetone (≥99.5%) was purchased from VWR (Sweden). Methyl methacrylate (MMA, 99%, Sigma Aldrich), was passed through a column of aluminum oxide (Sigma Aldrich) prior to use to remove the inhibitor.Chemical functionalization of the wood template
The starting balsa wood veneers have a thickness of 1.2 mm, 2 mm and 3 mm. Delignification was performed using an aqueous PAA solution (4 wt%) adjusted at a pH of 4.8 with sodium hydroxide. The delignification was left to proceed for 4–6 h at 80 °C until the samples became white. After the delignification, the delignified wood (DW) samples were washed three times with deionized water under vacuum condition and finally solvent exchanged to acetone. The samples were left in acetone until further use.Ring-opening esterification was performed on the DW templates using MA, IA, and SA. The reaction was performed on DW under neat conditions without solvent, and left to react without stirring. The optimum reactions conditions at which the DW samples were modified while remaining uncolored were as follow: 24 h at 100 °C for MA, 24 h at 80 °C for IA, and 0.5 h at 130 °C for SA. After the reaction, the DW samples were thoroughly washed with acetone in low-vacuum condition five times to remove any non-bonded reagent. The samples were left in acetone until further use. The functionalized templates modified with MA, IA and SA are abbreviated as DW-MA, DW-IA and DW-SA, respectively.
Preparation of transparent wood biocomposites
The wood templates were infiltrated with a pre-polymerized solution of MMA (≈20% conversion) and 0.3 wt% AIBN under vacuum at room temperature for 24 h. The infiltrated samples were then placed between two glass slides, packaged in aluminum foil, and polymerized at 40 °C for 24 h and then at 70 °C for another 24 h. The wood volume fraction (Vf) was estimated from the DW template weight fraction (Wf) after deduction of the anhydride mass, following the equation:where ρf is the cell wall density (1500 kg m−3) and ρc is the density of the biocomposite. The density of the biocomposite was measured with a pycnometer (Micromeritics AccuPyc 1330, USA).
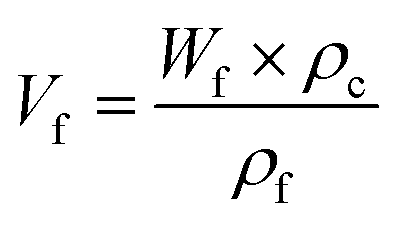
Chemical composition
The lignin content of the native balsa and delignified balsa was determined by the Klason lignin TAPPI T 222 om-2 method.88 Carbohydrate analysis was performed using a Dionex ICS-3000 ion chromatography system (Thermo Fisher Scientific, USA). The main monosaccharides determined are arabinose, galactose, xylose, mannose and glucose. The cellulose content was assumed equal to the glucose content. The effect of the modification on the wood template was characterized using a Spectrum 100 Fourier transform infrared (FTIR) spectrometer (PerkinElmer, USA) equipped with a Golden Gate diamond ATR (Gaseby Specac Ltd, UK). The spectra were recorded at room temperature with a resolution of 4 cm−1 and were normalized based on the highest absorbance band of the fingerprint region at around 1030 cm−1. The carboxyl content of the wood templates was determined by conductometric titration using a Metrohm 702SM Titrino titrator (Switzerland) according to the SCAN-CM 65:02 standard. Each measurement was performed in triplicates. The degree of substitution was calculated from the carboxyl charge data, assuming that wood consists of 100% cellulose and that one –OH group from an anhydroglucose unit (Mw = 162 g mol−1) reacts.Morphology
High-resolution images were captured using a Hitachi S-4800 (Japan) field-emission scanning electron microscope (FE-SEM). Prior to imaging, cross-sections of the wood samples in the wet state were obtained with a microtome (Leica SM2010 R Sliding Microtome, Germany), exchanged to ethanol absolute and placed in a critical point dryer (Autosamdri-815, Tousimis, USA) using CO2 to preserve the wood structure. Cross-sections of the transparent wood composite were prepared using an ultramicrotome (MT-XL, RMC, UK). All samples were Pt/Pd sputtered under vacuum for 10 s (Cressington 208HR, UK). SEM images of fracture surfaces were captured using a TM-1000 tabletop SEM (Hitachi, Japan).Nitrogen physisorption
The analysis was carried out at 77 K using a MicroActive 3Flex 3500 surface area and pore size analyzer (Micromeritics, USA). Before measurements, the samples were dried using supercritical CO2 and degassed at 70 °C for 48 h on a SmartVacPrep setup. Specific surface area (SSA) was calculated from the obtained isotherms according to a multipoint Brunauer–Emmett–Teller (BET) method and the total pore volume was calculated using Barrett–Joyner–Halenda (BJH) model.X-ray diffraction (XRD)
XRD patterns were acquired using a PANalytical X'Pert alpha1 powder diffractometer (Netherlands) with Cu Kα radiation generated at 45 kV and 40 mA. The cellulose crystallinity index (CI) was calculated by peak deconvolution, fitting all individual crystalline peaks and the broad amorphous peak located at around 21.5°, until an iteration with an R2 value of 0.999 was reached.89 The samples were supercritical CO2 dried and cold-pressed before measurements.Moisture sorption
Moisture sorption experiments were carried out in a controlled-humidity chamber at 22 °C. The relative humidity (RH) was sequentially set to 94%, 75%, 50%, 12% and 0%, where 0% RH corresponds to the oven-dry weight. The samples were left at each RH for 2 weeks before weighing. The measurements were performed in quintuplicate. The moisture uptake was calculated according to the following equation:where mRH is the sample weight equilibrated at a specific RH and mdry is the oven-dry weight of the sample.
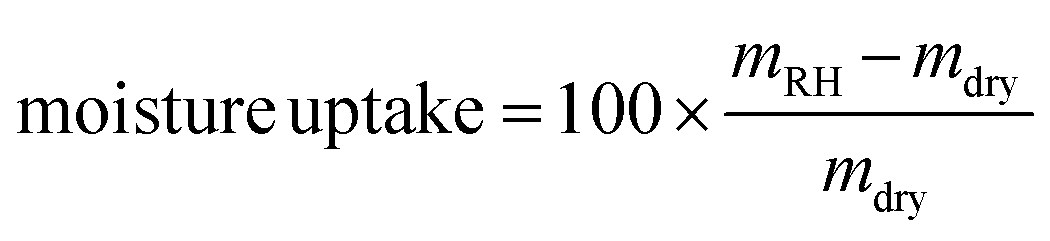
Contact angle
Contact angle measurements where performed using a PGX+ contact angle goniometer (Pocket goniometer, Sweden) with automatic dispenser. MilliQ water was pumped into a droplet placed on the substrate and the dynamic contact angle was recorded.Thermal properties
The thermal stability of the wood template was studied by thermogravimetric analysis (TGA, Mettler Toledo TGA/DSC1, Switzerland) with a heating rate of 10 °C min−1 under nitrogen atmosphere. The temperature at a weight loss of 5% in the temperature range of 100–600 °C was taken as the onset degradation temperature (Tonset). Differential scanning calorimetry (DSC) curves of MA, IA and SA were acquired with a Mettler Toledo DSC1 instrument (Switzerland) at a heating rate of 10 °C min−1 under a nitrogen flow of 50 mL min−1.Optical properties
The transparent wood samples were prepared with dimensions of 1.5 cm × 1.5 cm for optical measurement. Optical transmittance and haze of the samples were acquired with an integrating sphere.90 The sample was set in front of an input port of the integrating sphere, a quartz tungsten halogen light source (model 66181, Oriel Instruments) with strong, stable output mainly in the visible and NIR region was applied as the incident beam. The output light was collected through an optical fiber connected to the output port of integrating sphere. Each measurement was performed in triplicates.Mechanical properties
For mechanical testing, the samples were cut in 5 mm × 5 cm strips and preconditioned for 48 h at 50% RH before measurement. The tests were carried out at a temperature of 22 °C and 50% RH. The tensile tests were performed on a universal testing machine (Instron 5944, USA) equipped with a 2 kN load cell and a video extensometer. All samples were loaded in the longitudinal axis along the fiber direction. The tests were carried out with a 10% min−1 strain rate and span of 20 mm.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 742733). We acknowledge funding from KTH and Knut and Alice Wallenberg foundation through the Wallenberg Wood Science Center and KAW Biocomposites program at KTH Royal Institute of Technology.References
- R. Lakes, Nature, 1993, 361, 511–515 CrossRef.
- U. G. K. Wegst and M. F. Ashby, Philos. Mag., 2004, 84, 2167–2186 CrossRef CAS.
- V. Merk, M. Chanana, T. Keplinger, S. Gaan and I. Burgert, Green Chem., 2015, 17, 1423–1428 RSC.
- Q. Fu, L. Medina, Y. Li, F. Carosio, A. Hajian and L. A. Berglund, ACS Appl. Mater. Interfaces, 2017, 9, 36154–36163 CrossRef CAS.
- Q. Fu, F. Ansari, Q. Zhou and L. A. Berglund, ACS Nano, 2018, 12, 2222–2230 CrossRef CAS.
- E. Vasileva, Y. Li, I. Sychugov, M. Mensi, L. Berglund and S. Popov, Adv. Opt. Mater., 2017, 5, 1700057 CrossRef.
- K. Oksman, Y. Aitomäki, A. P. Mathew, G. Siqueira, Q. Zhou, S. Butylina, S. Tanpichai, X. Zhou and S. Hooshmand, Composites, Part A, 2016, 83, 2–18 CrossRef CAS.
- A. K. Mohanty, M. Misra and L. T. Drzal, Compos. Interfaces, 2001, 8, 313–343 CrossRef CAS.
- P. Grönquist, M. Frey, T. Keplinger and I. Burgert, ACS Omega, 2019, 4, 12425–12431 CrossRef.
- H. Yano, A. Hirose, P. J. Collins and Y. Yazaki, J. Mater. Sci. Lett., 2001, 20, 1125–1126 CrossRef CAS.
- H. Yano, A. Hirose and S. Inaba, J. Mater. Sci. Lett., 1997, 16, 1906–1909 CrossRef CAS.
- M. Frey, L. Schneider, K. Masania, T. Keplinger and I. Burgert, ACS Appl. Mater. Interfaces, 2019, 11, 35305–35311 CrossRef CAS.
- E. Jungstedt, C. Montanari, S. Östlund and L. Berglund, Composites, Part A, 2020, 133, 105853 CrossRef CAS.
- Q. Fu, Y. Chen and M. Sorieul, ACS Nano, 2020, 14, 3528–3538 CrossRef CAS.
- L. A. Berglund and I. Burgert, Adv. Mater., 2018, 30, 1704285 CrossRef.
- F. Jiang, T. Li, Y. Li, Y. Zhang, A. Gong, J. Dai, E. Hitz, W. Luo and L. Hu, Adv. Mater., 2018, 30, 1703453 CrossRef.
- H. Guan, Z. Cheng and X. Wang, ACS Nano, 2018, 12, 10365–10373 CrossRef CAS.
- M. Frey, D. Widner, J. S. Segmehl, K. Casdorff, T. Keplinger and I. Burgert, ACS Appl. Mater. Interfaces, 2018, 10, 5030–5037 CrossRef CAS.
- Y. Li, E. Vasileva, I. Sychugov, S. Popov and L. Berglund, Adv. Opt. Mater., 2018, 6, 1800059 CrossRef.
- M. Frey, G. Biffi, M. Adobes-Vidal, M. Zirkelbach, Y. Wang, K. Tu, A. M. Hirt, K. Masania, I. Burgert and T. Keplinger, Adv. Sci., 2019, 6, 1802190 CrossRef.
- C. Montanari, Y. Li, H. Chen, M. Yan and L. A. Berglund, ACS Appl. Mater. Interfaces, 2019, 11, 20465–20472 CrossRef CAS.
- A. J. Stamm, in Wood and cellulose science, 1964, pp. 248–263 Search PubMed.
- L. T. Drzal and M. Madhukar, J. Mater. Sci., 1993, 28, 569–610 CrossRef CAS.
- P. Olsén, N. Herrera and L. A. Berglund, Biomacromolecules, 2020, 21, 1795–1801 CrossRef.
- A. Sangregorio, A. Muralidhara, N. Guigo, L. G. Thygesen, G. Marlair, C. Angelici, E. de Jong and N. Sbirrazzuoli, Green Chem., 2020, 22, 2786–2798 RSC.
- S. C. Fox, B. Li, D. Xu and K. J. Edgar, Biomacromolecules, 2011, 12, 1956–1972 CrossRef CAS.
- P. Olsén, N. Herrera and L. A. Berglund, Biomacromolecules, 2020, 21, 597–603 CrossRef.
- A. Carlmark, E. Larsson and E. Malmström, Eur. Polym. J., 2012, 48, 1646–1659 CrossRef CAS.
- N. Herrera, P. Olsén and L. A. Berglund, ACS Sustainable Chem. Eng., 2020, 8, 11977–11985 CrossRef CAS.
- Y. Li, X. Yang, Q. Fu, R. Rojas, M. Yan and L. Berglund, J. Mater. Chem. A, 2018, 6, 1094–1101 RSC.
- R. M. Rowell, in Handbook of Wood Chemistry and Wood Composites, CRC Press, 2nd edn, 2005, pp. 381–420 Search PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- A. Llevot, P.-K. Dannecker, M. von Czapiewski, L. C. Over, Z. Söyler and M. A. R. Meier, Chem. – Eur. J., 2016, 22, 11510–11521 CrossRef CAS.
- Y. Hirabayashi and G. G. Allan, Makromol. Chem., 1984, 185, 2371–2376 CrossRef CAS.
- H. Matsuda, Wood Sci. Technol., 1987, 21, 75–88 CAS.
- F. Gellerstedt and P. Gatenholm, Cellulose, 1999, 6, 103–121 CrossRef CAS.
- C. A. S. Hill, in Wood Modification, John Wiley & Sons, Ltd, Chichester, UK, 2006, vol. 61, pp. 77–97 Search PubMed.
- Z. Söyler, K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, Green Chem., 2018, 20, 214–224 RSC.
- C. Clemons, R. A. Young and R. M. Rowell, Wood Fiber Sci., 1992, 34, 353–363 Search PubMed.
- T. Chartier and A. Badev, in Handbook of Advanced Ceramics: Materials, Applications, Processing, and Properties: Second Edition, Elsevier Inc., 2nd edn, 2013, pp. 489–524 Search PubMed.
- H. Sehaqui, K. Kulasinski, N. Pfenninger, T. Zimmermann and P. Tingaut, Biomacromolecules, 2017, 18, 242–248 CrossRef CAS.
- P. Huang, Y. Zhao, S. Kuga, M. Wu and Y. Huang, Nanoscale, 2016, 8, 3753–3759 RSC.
- S. Vitas, T. Keplinger, N. Reichholf, R. Figi and E. Cabane, J. Hazard. Mater., 2018, 355, 119–127 CrossRef CAS.
- S. T. Chang and H. T. Chang, Polym. Degrad. Stab., 2001, 71, 261–266 CrossRef CAS.
- D. Rosu, C. A. Teaca, R. Bodirlau and L. Rosu, J. Photochem. Photobiol., B, 2010, 99, 144–149 CrossRef CAS.
- S. Iwamoto and T. Endo, ACS Macro Lett., 2015, 4, 80–83 CrossRef CAS.
- H. D. Rozman, W. B. Banks and M. L. Lawther, J. Appl. Polym. Sci., 1994, 54, 191–200 CrossRef CAS.
- W. B. Banks, R. H. Din and N. L. Owen, Holzforschung, 1995, 49, 104–108 CrossRef CAS.
- Y. Li, Q. Wu, J. Li, Y. Liu, X.-M. Wang and Z. Liu, Holzforschung, 2012, 66, 59–66 CAS.
- S. Mallon and C. A. S. Hill, Int. J. Adhes. Adhes., 2002, 22, 465–469 CrossRef CAS.
- D. Pokhrel and T. Viraraghavan, Sci. Total Environ., 2004, 333, 37–58 CrossRef CAS.
- M. C. Taylor, J. F. Whitte, G. P. Vincent and G. I. Cunnigham, Ind. Eng. Chem., 1940, 32, 899–903 CrossRef CAS.
- H. Hefti, Text. Res. J., 1960, 30, 861–867 CrossRef CAS.
- Z. Yuan, Y. Ni and A. R. P. Van Heiningen, Can. J. Chem. Eng., 1997, 75, 37–41 CrossRef CAS.
- S. P. Teong, X. Li and Y. Zhang, Green Chem., 2019, 21, 5753–5780 RSC.
- X. Yang, F. Berthold and L. A. Berglund, ACS Appl. Mater. Interfaces, 2019, 11, 10310–10319 CrossRef CAS.
- C. Chatterjee, F. Pong and A. Sen, Green Chem., 2015, 17, 40–71 RSC.
- G. Pavarelli, J. Velasquez Ochoa, A. Caldarelli, F. Puzzo, F. Cavani and J.-L. Dubois, ChemSusChem, 2015, 8, 2250–2259 CrossRef CAS.
- J. Lan, Z. Chen, J. Lin and G. Yin, Green Chem., 2014, 16, 4351–4358 RSC.
- R. Wojcieszak, F. Santarelli, S. Paul, F. Dumeignil, F. Cavani and R. V. Gonçalves, Sustainable Chem. Processes, 2015, 3, 9 CrossRef.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- I. Bechthold, K. Bretz, S. Kabasci, R. Kopitzky and A. Springer, Chem. Eng. Technol., 2008, 31, 647–654 CrossRef CAS.
- J. S. Luterbacher, D. Martin Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816–4838 RSC.
- Z. Du, J. Ma, F. Wang, J. Liu and J. Xu, Green Chem., 2011, 13, 554–557 RSC.
- P. L. Arias, J. A. Cecilia, I. Gandarias, J. Iglesias, M. López Granados, R. Mariscal, G. Morales, R. Moreno-Tost and P. Maireles-Torres, Catal. Sci. Technol., 2020, 10, 2721–2757 RSC.
- T. Willke and K.-D. Vorlop, Appl. Microbiol. Biotechnol., 2001, 56, 289–295 CrossRef CAS.
- J. Cunha da Cruz, A. Machado de Castro and E. F. Camporese Sérvulo, 3 Biotech, 2018, 8, 138 CrossRef.
- M. Okabe, D. Lies, S. Kanamasa and E. Y. Park, Appl. Microbiol. Biotechnol., 2009, 84, 597–606 CrossRef CAS.
- Y. Chen, N. M. Stark, Z. Cai, C. R. Frihart, L. F. Lorenz and R. E. Ibach, BioResources, 2014, 9, 5488–5500 Search PubMed.
- X. W. Peng, J. L. Ren and R. C. Sun, Biomacromolecules, 2010, 11, 3519–3524 CrossRef CAS.
- S. Iwamoto, Y. Saito, T. Yagishita, A. Kumagai and T. Endo, Cellulose, 2019, 26, 4721–4729 CrossRef CAS.
- C. Cai, K. Hirth, R. Gleisner, H. Lou, X. Qiu and J. Y. Zhu, Green Chem., 2020, 22, 1605–1617 RSC.
- P. Olsén, M. Jawerth, M. Lawoko, M. Johansson and L. A. Berglund, Green Chem., 2019, 21, 2478–2486 RSC.
- A. Eraghi Kazzaz, Z. Hosseinpour Feizi and P. Fatehi, Green Chem., 2019, 21, 5714–5752 RSC.
- L. Mezzasalma, J. De Winter, D. Taton and O. Coulembier, Green Chem., 2018, 20, 5385–5396 RSC.
- A. C. D. Rivett and N. V. Sidgwick, J. Chem. Soc., Trans., 1910, 97, 1677–1686 RSC.
- K. Li, S. Wang, H. Chen, X. Yang, L. A. Berglund and Q. Zhou, Adv. Mater., 2020, 2003653 CrossRef CAS.
- R. M. Rowell, B. A. Cleary, J. S. Rowell, C. Clemons and R. A. Young, in Wood–fiber/polymer composites: Fundamental concepts, processes and material options, Forest Products Society, Madison, WI, 1993, pp. 121–127 Search PubMed.
- S. Fujisawa, Y. Okita, H. Fukuzumi, T. Saito and A. Isogai, Carbohydr. Polym., 2011, 84, 579–583 CrossRef CAS.
- S. E. Creager and C. M. Steiger, Langmuir, 1995, 11, 1852–1854 CrossRef CAS.
- E. L. Smith, C. A. Alves, J. W. Anderegg, M. D. Porter and L. M. Siperko, Langmuir, 1992, 8, 2707–2714 CrossRef CAS.
- R. G. Nuzzo, L. H. Dubois and D. L. Allara, J. Am. Chem. Soc., 1990, 112, 558–569 CrossRef CAS.
- L. Sun, R. M. Crooks and A. J. Ricco, Langmuir, 1993, 9, 1775–1780 CrossRef CAS.
- F. Xi, W. Bassett and O. Vogl, J. Polym. Sci., Polym. Chem. Ed., 1983, 21, 891–911 CrossRef CAS.
- A. S. Brown, K. Fujimori and I. E. Craven, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 3315–3325 CrossRef CAS.
- T. Otsu and H. Watanabe, Eur. Polym. J., 1993, 29, 167–174 CrossRef CAS.
- S. Bednarz, A. Wesołowska-Piętak, R. Konefał and T. Świergosz, Eur. Polym. J., 2018, 106, 63–71 CrossRef CAS.
- T 222 om-02 Acid-insoluble lignin in wood and pulp, Tappi Press, 2002 Search PubMed.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 10 CrossRef.
- ASTM D1003-13, Standard Test Method for Haze and Luminous Transmittance of Transparent Plastics, ASTM International, West Conshohocken, PA, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02768e |
| This journal is © The Royal Society of Chemistry 2020 |