
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsocyanide 2.0†
Pravin
Patil
 ,
Maryam
Ahmadian-Moghaddam
and
Alexander
Dömling
*
,
Maryam
Ahmadian-Moghaddam
and
Alexander
Dömling
*
Department of Drug Design, University of Groningen, A. Deusinglaan 1, 9700 AD Groningen, The Netherlands. E-mail: a.s.s.domling@rug.nl; Web: http://www.drugdesign.nl
First published on 29th September 2020
Abstract
The isocyanide functionality due to its dichotomy between carbenoid and triple bond characters, with a nucleophilic and electrophilic terminal carbon, exhibits unusual reactivity in organic chemistry exemplified for example in the Ugi reaction. Unfortunately, the over proportional use of only a few isocyanides hampers novel discoveries about the fascinating reactivity of this functional group. The synthesis of a broad range of isocyanides with multiple functional groups is lengthy, inefficient, and exposes the chemist to hazardous fumes. Here we present an innovative isocyanide synthesis overcoming these problems by avoiding the aqueous workup which we exemplify by parallel synthesis from a 0.2 mmol scale performed in 96-well microtiter plates up to a 0.5 mol multigram scale. The advantages of our methodology include an increased synthesis speed, very mild conditions giving access to hitherto unknown or highly reactive classes of isocyanides, rapid access to large numbers of functionalized isocyanides, increased yields, high purity, proven scalability over 5 orders of magnitude, increased safety and less reaction waste resulting in a highly reduced environmental footprint. For example, the hitherto believed to be unstable 2-isocyanopyrimidine, 2-acylphenylisocyanides and even o-isocyanobenzaldehyde could be accessed on a preparative scale and their chemistry was explored. Our new isocyanide synthesis will enable easy access to uncharted isocyanide space and will result in many discoveries about the unusual reactivity of this functional group.
Introduction
Isocyanides are hubs in chemistry, having reaction plasticity yet conserved reactivities, and can function as integral members of large multimolecular assemblies, or just contribute a few atoms. The isocyanide functional group is of high relevance to different fields of science,1 including astronomy,2 biology,3 information technology,4,5 materials science,6 and most importantly chemistry.7 Isocyanides are isolobal to carbon monoxide and therefore there is a rich metalorganic chemistry.8 In organic chemistry isocyanides are the basis of many heterocycle syntheses and different multicomponent reactions (IMCRs). The fascinating aspects of isocyanide chemistry were reviewed extensively.9–17Although >26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 isocyanides have been described and >3000 are listed to be commercially available, only a very small number accounts for the overwhelming use in chemistry (Fig. 1A). Most isocyanide research focuses on those already known. A recent bibliometric analysis using Sci-Finder revealed that isocyanides are mentioned in >21000 publications, excluding patents. However, just 9 isocyanides (1–9) account for >19000 citations (>90%) of all isocyanide research papers. These reoccurring isocyanides are the commercially available tert-butyl 1, cyclohexyl isocyanide 2, TOSMIC 3, isocyanoaceticacid esters 4 and 9, 2,6-dimethyl phenyl 5, benzyl 6, methyl 7 and phenyl isocyanide 8. Despite this fundamental imbalance, new chemical space is charted by the synthesis and exploration of novel, less commonly used isocyanides (Fig. 1B). Schöllkopf's isocyanide 10 is useful in a plethora of unprecedented heterocycle syntheses, for example, a multicomponent thiazole synthesis (11).18,19 Similarly, N-isocyanoiminotriphenylphosphorane also called Fehlhammer isocyanide 12 after its inventor is a highly versatile example.20–22 Ramazani, for example, introduced 12 in the elegant oxadiazole synthesis (13), which was also probed in complex head-to-tail cyclic peptide syntheses.23,24 Another example is Boc protected o-aminophenyl isocyanide 14, which has been used in the elegant work of Hulme to form hybrid pyridinone-3-yl-benzimidazol-2-ones 15 in just one synthetic step.25 On the other hand, TOSMIC 3, a representative of the well-used isocyanides is a multi-ton technical product which was introduced by van Leusen and has made a major impact on heterocyclic chemistry with important industrial applications, for example, the fungicide Fludioxonil 16.26
000 isocyanides have been described and >3000 are listed to be commercially available, only a very small number accounts for the overwhelming use in chemistry (Fig. 1A). Most isocyanide research focuses on those already known. A recent bibliometric analysis using Sci-Finder revealed that isocyanides are mentioned in >21000 publications, excluding patents. However, just 9 isocyanides (1–9) account for >19000 citations (>90%) of all isocyanide research papers. These reoccurring isocyanides are the commercially available tert-butyl 1, cyclohexyl isocyanide 2, TOSMIC 3, isocyanoaceticacid esters 4 and 9, 2,6-dimethyl phenyl 5, benzyl 6, methyl 7 and phenyl isocyanide 8. Despite this fundamental imbalance, new chemical space is charted by the synthesis and exploration of novel, less commonly used isocyanides (Fig. 1B). Schöllkopf's isocyanide 10 is useful in a plethora of unprecedented heterocycle syntheses, for example, a multicomponent thiazole synthesis (11).18,19 Similarly, N-isocyanoiminotriphenylphosphorane also called Fehlhammer isocyanide 12 after its inventor is a highly versatile example.20–22 Ramazani, for example, introduced 12 in the elegant oxadiazole synthesis (13), which was also probed in complex head-to-tail cyclic peptide syntheses.23,24 Another example is Boc protected o-aminophenyl isocyanide 14, which has been used in the elegant work of Hulme to form hybrid pyridinone-3-yl-benzimidazol-2-ones 15 in just one synthetic step.25 On the other hand, TOSMIC 3, a representative of the well-used isocyanides is a multi-ton technical product which was introduced by van Leusen and has made a major impact on heterocyclic chemistry with important industrial applications, for example, the fungicide Fludioxonil 16.26
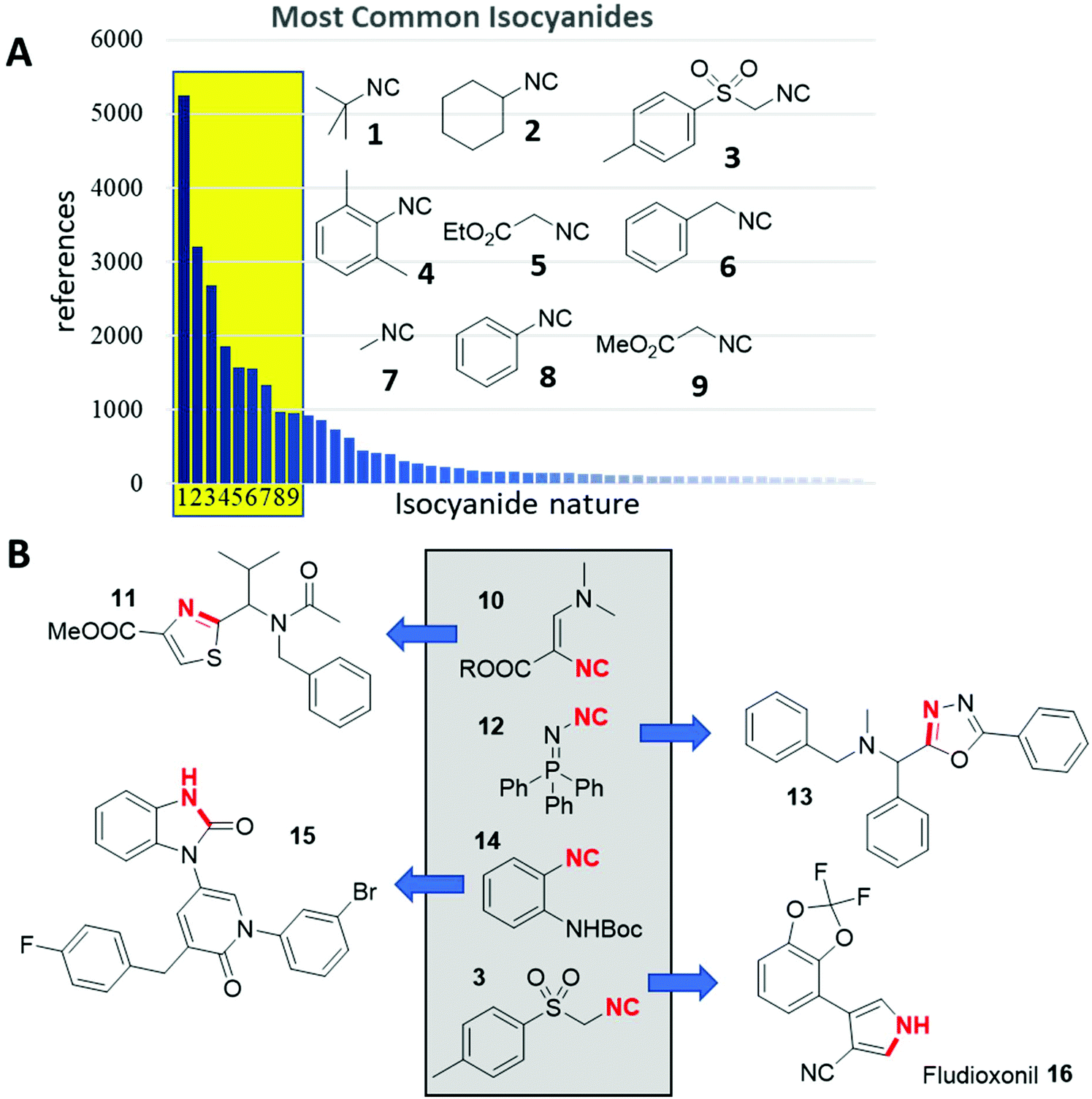 | ||
| Fig. 1 Isocyanides in the past and present. (A) Only a very few isocyanides account for the majority of citations in the chemical literature (Sci-Finder). (B) Some polyfunctional isocyanides leading to interesting scaffolds and products. | ||
Isocyanides were discovered 200 years ago and made initially available by the nucleophilic substitution reaction of AgCN with alkyl halides. Shortly after that, Hoffman invented the direct conversion of primary amines to isocyanides using dichloro carbene reagents. The chemistry of isocyanides got a boost with the discovery of the first isocyanide-based multicomponent reaction (IMCR) by Passerini (P-3CR). A milestone development by Ugi was the reliable synthesis of isocyanides by a two-step procedure, formylation of a primary amine followed by dehydration. Although many other alternative isocyanide procedures exist, such as the improved Hoffman procedure using phase transfer catalysis (PTK),27 the Ugi method accounts for the vast majority of isocyanide syntheses performed today. Isocyanides are highly reactive and many volatile liquid isocyanides stand out by a terrifying odor. The toxicity of isocyanides is less investigated but they don't seem to have acute toxicity.28 Therefore, special measures have to be taken during their preparation. The bench stability of isocyanides depends on their structure and substitution pattern. For example, phenyl isocyanide and some derivatives as well as vinyl isocyanides29 polymerize and 2-(isocyanomethyl)pyridine cyclizes fast. Multiple attempts have been made aiming to improve isocyanide syntheses, such as in situ generation of isocyanides.30–32 In our opinion, during formamide dehydration, phosgene would be the most preferable dehydrating agent due to the byproduct formation of CO2 and chloride salts. However, its use is largely prohibited due to the high toxicity of the gas. Organic chlorophosphate derivatives,33 XtalFluor-E,34 and other fancy dehydrating agents were used to synthesize isocyanides from their formamides. Strong reaction conditions such as use of MW at 100 °C and cyanurchlorid (TCT) are useful only for a few isocyanides, while labile functionalities will not survive such conditions.35 Recently, the purification of isocyanides on reversed-phase silica was introduced as an innovation.36 However, all improvements still suffer from one or another issue such as lengthy laborious procedures, exotic and expensive reagents, massive waste generation, and long exposure of the chemist to potentially hazardous fumes and often show a narrow scope of functionalization. Phosphorus oxychloride (POCl3) is the most used dehydrating agent and yields inorganic phosphate as a side product and is therefore preferable over p-TsCl yielding additional organic waste.37 The most used POCl3 procedure is performed from low temperatures to 50 °C depending on the reactivity of the formamide. Keeping the reaction always under basic conditions is essential to avoid the reduced yields of the product. The workup involves careful hydrolysis to destroy the excess of the dehydrating agent while pH has to be in the basic region to avoid hydrolysis of the isocyanide. Finally, the crude isocyanide needs further purification by distillation, chromatography, or recrystallisation. This step can also be the source of major yield reductions for example through the presence of small amounts of acidic matter. Thus, the classical POCl3-enabled isocyanide synthesis not only needs careful control of the reaction, workup and purification conditions, but is also lengthy and exposes the chemist to the often-smelly isocyanide fumes over a rather long period.
In our laboratory, the access to a large number of isocyanides with high structural diversity is crucial for example to explore the structure–activity relationship (SAR) and to prepare large libraries of IMCRs on a nanoscale or in the DEL format.38–42 Thus, we revisited the formamide to isocyanide dehydration and came up with a completely revised procedure avoiding any type of aqueous workup. The advantages of our very general isocyanide syntheses are increased synthesis speeds and thus decreased exposure of the chemist to badly smelling fumes, very mild conditions giving access to hitherto unknown classes of isocyanides, rapid access to large numbers of functionalized isocyanides, increased yields, high purity, exemplified scalability over 5 orders of magnitude, and less reaction waste resulting in a highly reduced environmental footprint.
Experimental
Having realized that the aqueous work-up is the major problem in the isocyanide synthesis we investigated non-aqueous workup procedures using the formation of benzyl isocyanide as a model reaction (Scheme 1). Inspired by Jamison's idea of in-line isocyanide synthesis and to avoid aqueous work-up we decided to use a reaction set-up that would allow for both the synthesis and purification of the isocyanide.43,44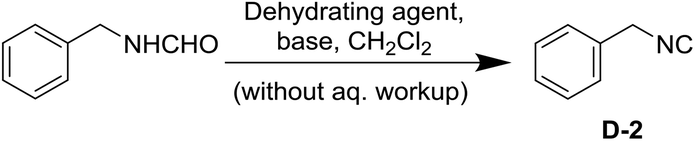 | ||
| Scheme 1 Synthesis of benzyl isocyanide. | ||
An appropriate amount of silica (2 g) (230–400 mesh size) was first coated with phosphorus oxychloride (1 mmol) and triethylamine (3 mmol).45 The column was then packed with the reagent-coated silica (column length and diameter being 6 cm and 0.75 cm, respectively). Subsequently, the formamide (1 mmol) was dry-loaded onto the column. Using dichloromethane (DCM) as the mobile phase, the formamide was passed through the column (Fig. 2, set-up: I). The result, however, was discouraging and we failed to obtain the isocyanide. Increasing the amounts of triethylamine was not helpful, neither.
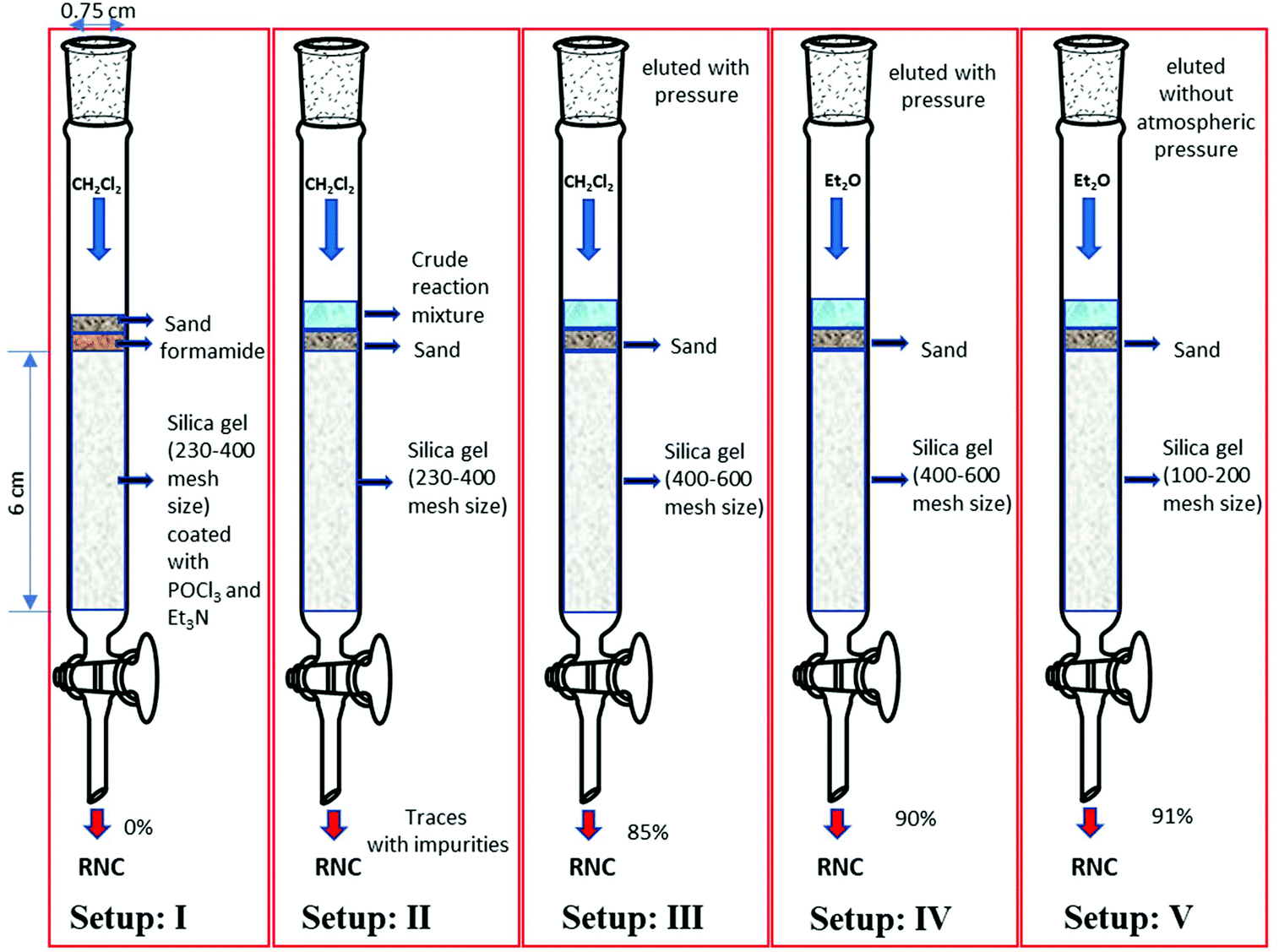 | ||
| Fig. 2 Different set-ups for N-benzyl isocyanide synthesis without aqueous workup and instant purification. | ||
We tested various reaction solvents like dichloromethane, THF, toluene, acetonitrile, isopropyl ether, DCM, and diethyl ether. Among them, only DCM as the reaction solvent gave high yields. This can be explained by the poor solubility of the formamides in most solvents.
Next, we investigated a different dry workup. To a solution of N-benzyl formamide (1 mmol) in DCM (0.1 M, 10 ml), triethylamine (5 mmol) and subsequently phosphorus oxychloride (1 mmol) were added at 0 °C. The reaction mixture was stirred for 1 h. Subsequently, the reaction mixture was poured onto a dry column loaded with silica (230–400 mesh size, 6 cm × 0.75 cm). DCM was used as the mobile phase. A mixture of triethylamine, triethylamine hydrochloride, and the desired isocyanide was obtained, with the isocyanide being the main hydrophobic component of the mixture (Fig. 2, set-up: 2). Encouraged by this result, we experimented with the particle size of the silica. However, smaller particles of the stationary phase (400–600 mesh size) made the mobile phase move through the column much slower. Due to the acid-sensitive nature of isocyanides, prolonged contact of the isocyanide with silica should be avoided as it consequently reduces the yield.46,47
To let the product elute faster, pressure was applied to the column. In this way, we were able to isolate benzyl isocyanide in 85% yield (Fig. 2, set-up: 3). Performing the reaction on 5 and 10 mmol scales afforded the compound in 86% yield along with small amounts of impurities.
To further improve our methodology, we screened different solvent systems as the mobile phase. DCM and diethyl ether in different ratios were utilized. It was observed that flash filtration with diethyl ether alone as the mobile phase and 400–600 mesh size silica afforded benzyl isocyanide in very high yields (90%) and purity, without the impurities usually present in isocyanide syntheses, namely triethylamine, triethylamine hydrochloride, and colored impurities. Next, we investigated the use of 100–200 and 230–400 mesh size silica and diethyl ether as the mobile phase. Surprisingly, the product was isolated with unreacted triethylamine, but without triethylamine hydrochloride and colored impurities. The excess triethylamine was removed during the vacuum evaporation of the solvent, leaving behind the pure isocyanide. Finally, we observed that a column with 100–200 mesh-size silica (Fig. 2, set-up: 5) afforded the product in a slightly higher yield (91%) than with 230–400 mesh size silica and without any air pressure.
Synthesis of isocyanides on a 100 mmol scale
To further validate the method and investigate its scalability, we performed the benzyl isocyanide synthesis on a 100 mmol scale at increased concentration. Triethylamine (5.0 equiv.) was added to a solution of the formamide in DCM (2 M) at room temperature and phosphorus oxychloride (1.0 equiv.) was subsequently slowly added to the reaction mixture at 0 °C. The reaction mixture was stirred at 0 °C for 5 minutes. Reaction monitoring by TLC revealed a considerable increase in speed. Indeed, carrying out the reaction at higher 2 M concentration dramatically reduced the reaction time to less than 10 minutes. Purification was performed using filtration over silica. The column (15 × 5 cm) was dry-packed with 100–200 mesh size silica (140 g) and a layer of sand (1 cm) was put on top of the silica (Fig. 3). | ||
| Fig. 3 Benzylisocyanide on a 10 g scale in less than 1 h. Reaction (A) and purification (B and C) on a 100 mmol scale. | ||
The reaction mixture was poured directly into the column. Initially, 100 ml diethyl ether was used as the mobile phase. Then, the mobile phase was made more polar by adding DCM (10% up to 100% dichloromethane). Fractions of 25 ml were collected. The product eluted in the first four fractions with only 100–150 ml of diethyl ether as the mobile phase. In this way, we were able to obtain the product with minimal 250 ml solvent consumption and in 97% yield.
Next, we applied the methodology in the synthesis of more complex phenylalanine methyl ester isocyanide (G-10) on a scale of 100 mmol (Scheme 2). Synthesis and purification were performed as above, by column chromatography using 140 g 100–200 mesh size silica and diethyl ether and dichloromethane as the mobile phase. The product was obtained in 87% yield in high purity.
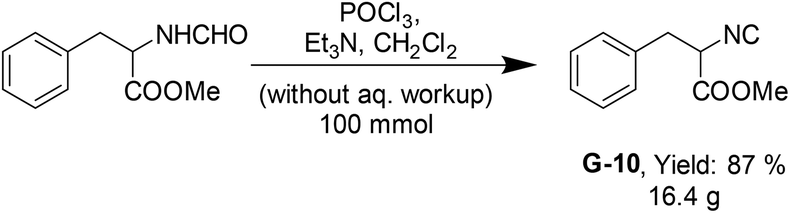 | ||
| Scheme 2 Synthesis of phenylalanine methyl ester isocyanide on a 100 mmol scale. | ||
Synthesis of isocyanides on a 500 mmol scale
The approach also proved successful in the synthesis of isocyanides on even larger scales. 1-Adamantyl isocyanide (H-2) (Fig. 4) was prepared using this methodology on a 0.5 mol scale in 97% yield (Fig. 4). Shortly, to a solution of 1-adamantyl formamide (0.5 mol) in dichloromethane (2 M), triethylamine (5.0 equiv.) was added at room temperature (Fig. 4A). Subsequently, phosphorus oxychloride (1.0 equiv.) was added at 0 °C. The reaction mixture was stirred for 10 min (Fig. 4B). After completion of the reaction as indicated by TLC, the compound was purified using column chromatography. The crude reaction mixture was loaded directly on a column (20 × 9 cm) dry-packed with 600 g 100–200 mesh size silica. Diethyl ether was used as the mobile phase and fractions of 250 ml were collected. The compound was eluted within the first four fractions (Fig. 4C). The solvent was evaporated under reduced pressure to afford the pure product as a yellow solid (Fig. 4D).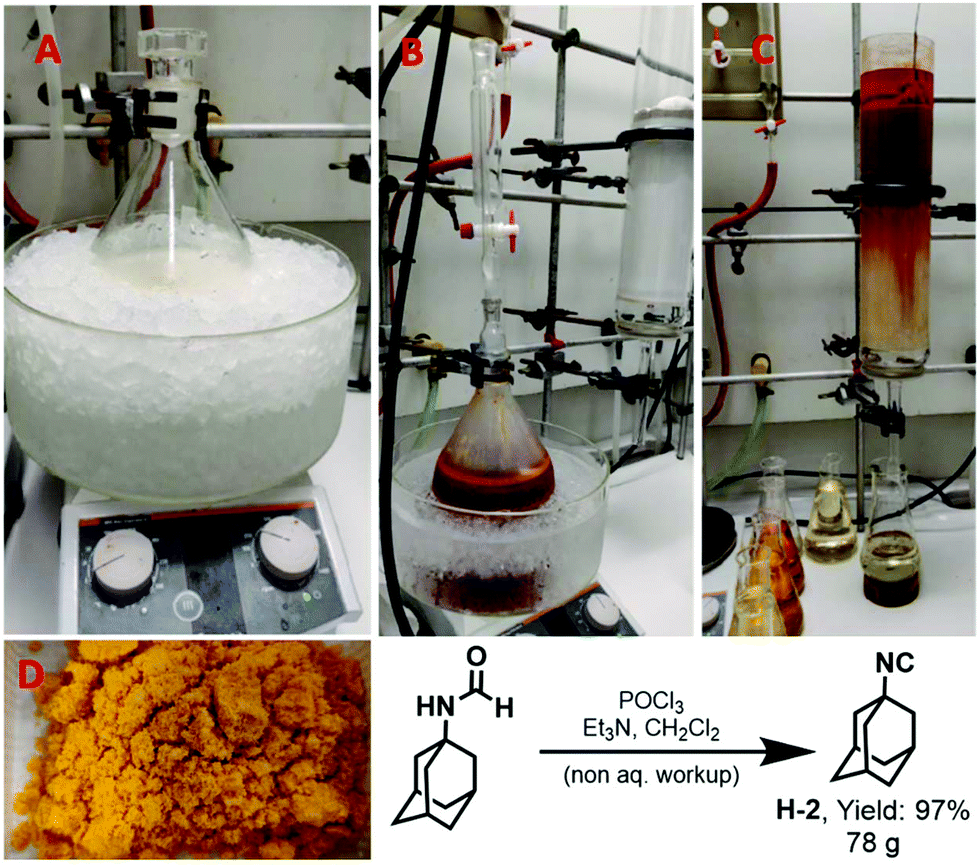 | ||
| Fig. 4 Synthesis of 1-adamantyl isocyanide (H-2) on a 500 mmol scale. A: Reaction mixture before POCl3 addition, B: reaction mixture after POCl3 addition, C: column purification, and D: isolated isocyanide. | ||
The amount of reaction solvent dramatically reduced the reaction time from several hours, mostly reported in the literature, to less than 15 min. Elimination of the aqueous workup during the purification made the process of isocyanide synthesis not only more efficient and quicker but also less laborious. Ease of synthesis and purification led to a dramatic increase in the preparation speed of isocyanides. Moreover, by reducing the amount of time and resources required to make a given isocyanide, the new methodology offers excellent economic and environmental benefits over the conventional approach. Another significant added value of the new methodology is that it considerably reduces exposure to the highly unpleasant odors of many isocyanides during the reaction, work-up and purification, making isocyanide synthesis much less of an unpleasant experience for the organic chemist.
Parallel synthesis of isocyanides in a 96-well plate format on a 0.2 mmol scale
Having established an innovative and easy process of synthesis, work-up and purification, we next investigated parallel synthesis in a 96-well format (Fig. 5). The reactions were performed on a scale of 0.2 mmol per well. For this purpose, a commercial 96-well plate with removable 1.5 ml glass vials was used (ESI†). To the pre-weighed formamides in the wells was added 0.1 ml dichloromethane (2 M), followed by the addition of triethylamine (5 eq.) at room temperature (Fig. 5A). The well plate was cooled to −10 °C in an ice/acetone bath and 0.2 mmol of POCl3 in 1 M solution of dichloromethane was added to the reaction mixture inside the wells using an 8-channel pipette as shown in Fig. 5B. After 5 minutes, the reaction mixture was diluted with 0.5 ml diethyl ether and transferred to a 96-well polypropylene filtration plate using a multi-channel pipette (Fig. 5C). The 96-well filtration plate was modified for this experiment by replacing the original filter paper with small balls of cotton at the bottom of the wells (Fig. 5D). Each well served as a mini-column and was packed with 400 mg 100–200 mesh size silica (Fig. 5E).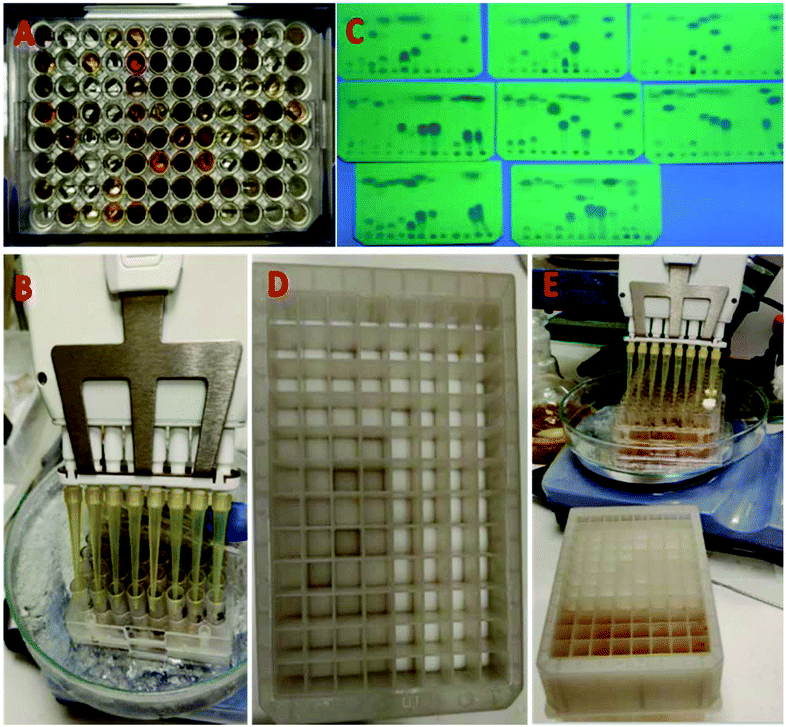 | ||
| Fig. 5 Set-up for the parallel synthesis of isocyanides in a 96-well format on a 0.2 mmol scale per isocyanide. (A) Top view of the 96-well glass reaction plate with multiple colored reaction mixtures. (B) Parallel addition of POCl3 to the wells; (C) TLC of the 96-well plate within 2 minutes after the addition of POCl3; (D) filtration plate with silica gel for purification; (E) efficient transfer of reaction mixtures to the filtration plate using 8- and 12-channel pipets. | ||
The isocyanide-containing solution was conveniently transferred from the 96-well glass reaction plate onto the 96-well filtration plate using an 8-channel pipette (Fig. 6A). The mobile phase in each well was allowed to flow through the columns by gravity and collected in a 96-deep-well (1.5 ml per well) collection plate (Fig. 6B). The remaining isocyanides in the columns were washed first with diethyl ether and afterwards with 0.5 ml solutions of each 25%, 50%, 75%, and 100% DCM in diethyl ether. The fractions were collected in other 96-well collection plates (Fig. 6C).
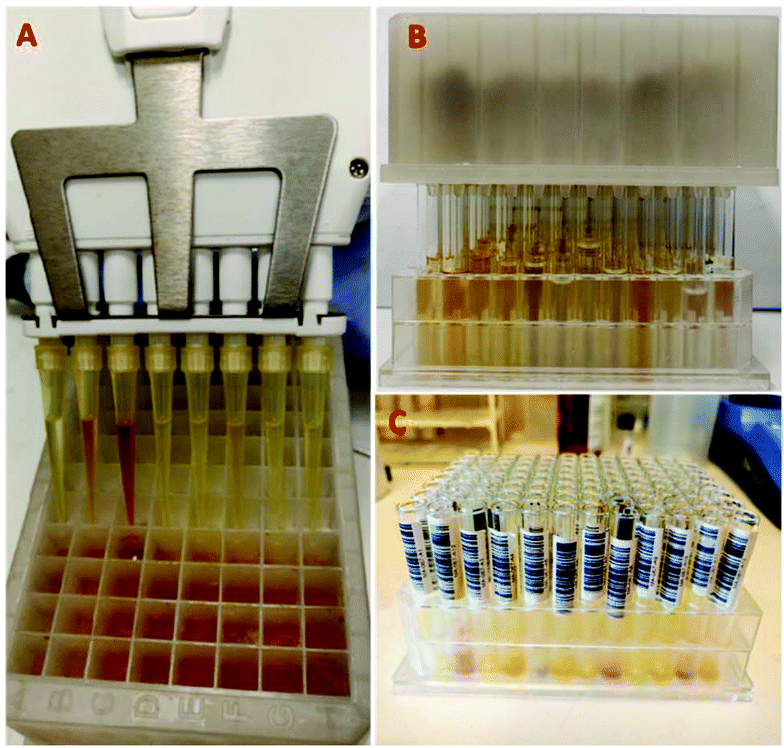 | ||
| Fig. 6 Parallel purification of isocyanides using a 96-well filtration plate. A: Transferring the reaction mixture to the filtration plate using an 8-channel pipette. B: Filtration plate on top of a 96-well collection plate. C: Pure isocyanides in a 96-well plate in pre-weighed barcoded glass vials after solvent evaporation to facilitate automation. | ||
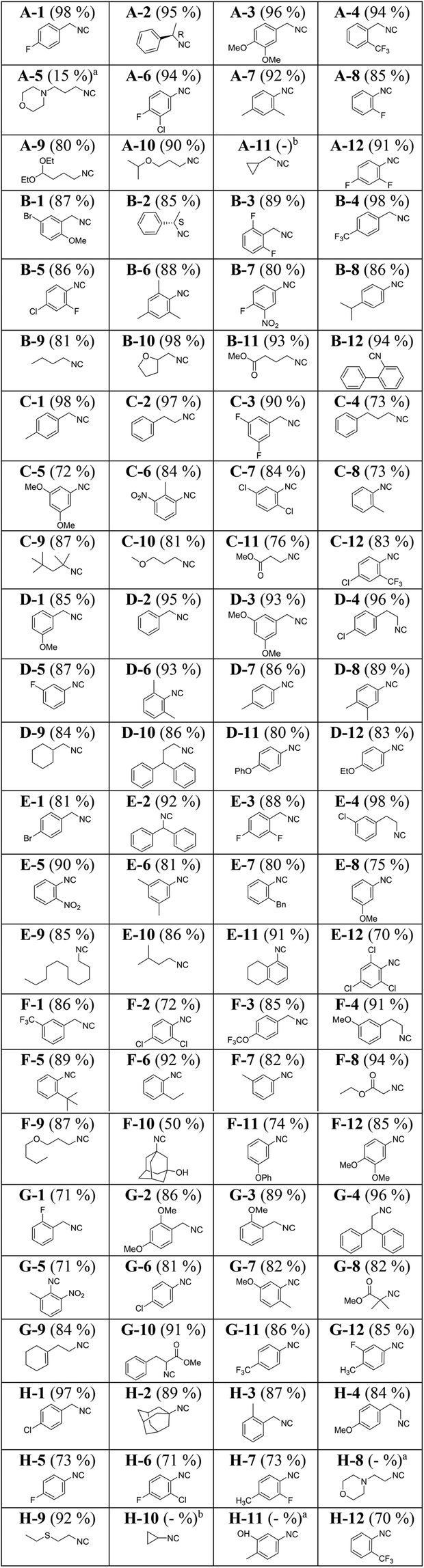
The purity of the fractions was investigated by TLC. It was observed that the first two collection plates contained the highest amount of isocyanides. As expected, the eluted solutions in the first two collection plates contained, in addition to the desired isocyanides, an appreciable amount of triethylamine. The later fractions eluted with 100% dichloromethane contained mostly the hydrochloride salt of triethylamine as well as inorganic impurities but only trace amounts of the isocyanides. Subsequently, the isocyanide-containing fractions were transferred to pre-weighed and barcoded 3 ml vials and the solvent and triethylamine were evaporated under reduced pressure (Genevac). The use of pre-weighed and barcoded vials facilitates the automation of yield determination and storage (Fig. 6C). All wells underwent strict analytical control by NMR and SFC MS (ESI†) and the structures and yields of the isocyanides are depicted in Table 1.
| % yields showed in this table are the isolated yields of isocyanides. aQuantitative product formation was observed by TLC, but the compound didn't elute effectively from the column due to its polar nature. bQuantitative product formation was observed by TLC, but the compound could not be effectively isolated due to its volatile nature. |
|---|
|
Parallel synthesis of isocyanides on a 1.0 mmol scale
Next, we resynthesized 56 randomly selected isocyanides from the 96-well plate along with a few new isocyanides on a larger 1 mmol scale (Table 2, for the synthesis details see in the ESI†).| a Quantitative conversion and product formation were observed by TLC, but the compound was difficult to isolate by this method due to its high volatility. |
|---|

|
Generally, 1 mmol scale synthesis afforded the isocyanides in higher yield and purity. For instance comparing entries B-9 and I-36, E-10 and I-47, D-9 and I-38, C-10 and I-45, B-10 and I-44, D-12 and I-34, G-9 and I-41, and H-2 and I-49 reveals that scaling up the experiments from 0.2 mmol to 1 mmol has been associated with an average increase of 8% yield. Intriguingly, a great structural diversity of isocyanides was obtained in high yields: cyclic, acyclic, aliphatic, bulky and amino acid-derived isocyanides with a lot of different functional groups, including halogens, boronic acid esters, hydroxyl, ester, ether, nitro, or morpholino. Using enantiopure formamides yielded enantiopure isocyanides (A-2 and B-2). In the case of aryl isocyanides, both electron-donating and -withdrawing substituents work similarly good (I-24: 93%, I-29: 93%, D-3: 93%, I-25: 98%, and I-33: 86%). Constitutionally isomeric isocyanides were isolated with comparable yields (B-3: 89%, C-3: 90%, E-3: 88% and E-6: 81%, D-8: 89%, D-6: 93%, and A-7: 92%). Volatile isocyanides such as A-11 and H-10 were synthesized on a 1 mmol scale in good yields. It should be added though that due to the volatile nature of these isocyanides, triethylamine and diethyl ether could not be effectively removed under reduced pressure. Such isocyanides can be best isolated by distillation. Bulky isocyanides such as tert-octylisocyanide (C-9, 87%), adamantyl isocyanide (H-2, 89%), and 3-hydroxy adamantyl isocyanide (F-10, 50%) were isolated in high or good yields. We failed to isolate the aromatic isocyanide containing a free hydroxyl group on the phenyl ring (H-11). Likely due to its high polarity, it could not be eluted from the column using diethyl ether and dichloromethane as the mobile phase. It is noteworthy that we were also able to isolate 2-formylphenyl isocyanide (I-51) in 85% yield. It has been reported to be highly unstable and reactive.9 Previously, it has been reported by a lengthy 5-step procedure.48
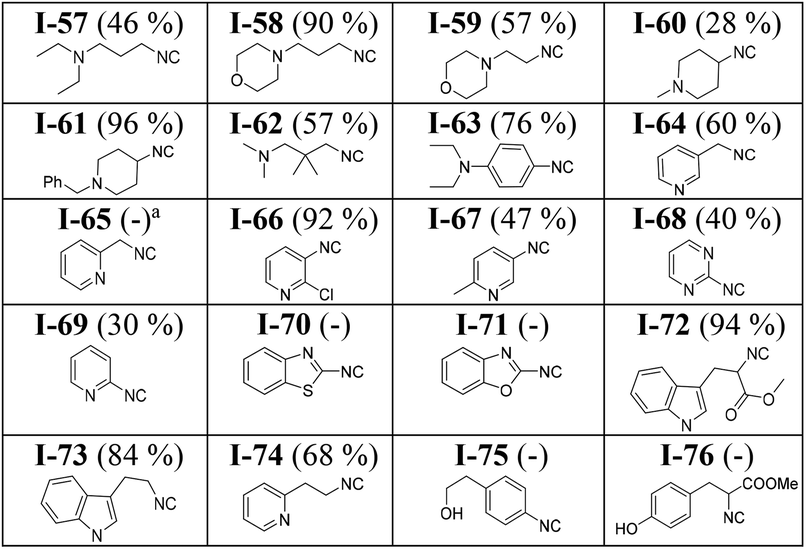
Modification of our approach allowed the synthesis of more polar isocyanides. A slight increase of the polarity of the mobile phase during column chromatography led to the clean isolation of several polar isocyanides in good yields (Table 3). For example, isocyanides containing a tertiary amine (I-57 to I-63) were synthesized and isolated successfully. In general, isocyanides bearing a morpholine ring (I-58 and I-59) are water-soluble and therefore, difficult to isolate using the conventional approach which involving aqueous work-up. By eliminating the aqueous work-up, we were able to obtain these isocyanides in excellent and good yields (I-58, 90% and I-59, 57%).
| a Quantitative product formation was observed by TLC, but the product decomposes upon exposure to silica while performing column chromatography. |
|---|
|
Generally, heterocyclic isocyanides, particularly those having the isocyano group ortho to the heteroatom (I-68 to I-71) are not stable at room temperature.9,49 Such isocyanides are prone to undergo further reaction such as polymerization or cyclization. Synthesis of pyridine- and pyrimidine-containing isocyanides such as I-65 to I-69 at very low temperatures (−78 to −40 °C) has been reported in the literature.50 Due to the reported instability, the isolated isocyanides were immediately used in further reactions.50 In contrast, our methodology enabled the synthesis of these isocyanides with regular ice bath cooling and isolation at room temperature and NMR analysis (ESI†). 2-Isocyanopyridine (I-69, 30%) and 3-(isocyanomethyl)pyridine (I-64, 60%) were isolated in good yields. Despite being stable enough to be isolated, these isocyanides undergo decomposition at room temperature and cannot be stored for a long time. In the cases of I-65, I-70, and I-71 we did not succeed in isolating the desired product in a highly pure form. However, the characteristic isocyanide smell of the reaction mixture and the formation of a new spot on the TLC plate clearly indicated their formation.
The inherent instability of heterocycle-containing isocyanides posed problems in their isolation. A noteworthy exception is 2-bromo-6-isocyanopyridine which was reported recently as a stable universal convertible isocyanide amongst many similar heterocyclic but unstable relatives.50 Thus, we decided to proceed to the synthesis of some MCR-derived compounds with the in situ formation of such isocyanides. By participating in a classical Ugi 4-component reaction and an Ugi-tetrazole reaction, respectively, 2-pyrimidine isocyanide was trapped within the structures of U-1 and UT-1, suggesting that the lifetime of the isocyanide is long enough to take part in a reaction (Scheme 3).
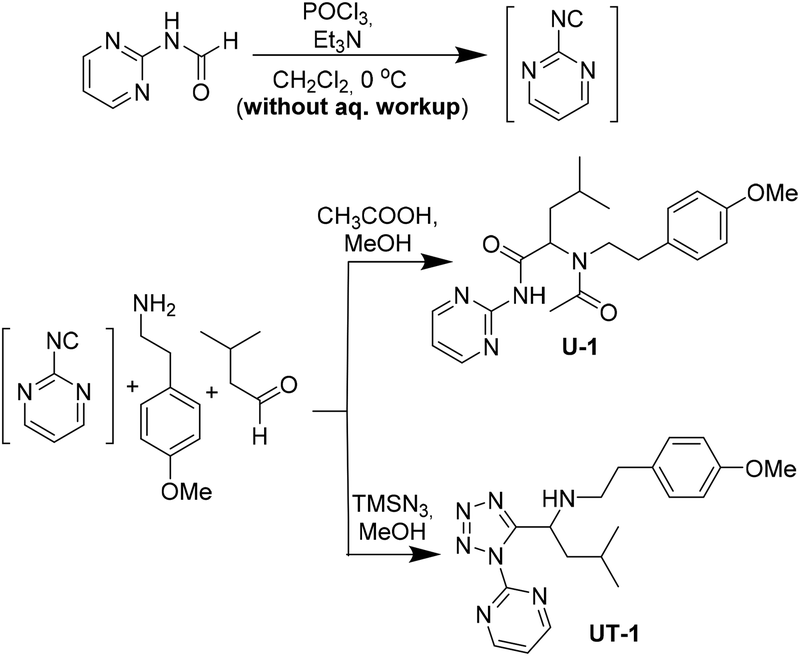 | ||
| Scheme 3 Synthesis of Ugi products using 2-pyrimidine isocyanide. | ||
To further underscore the superiority of our new isocyanide synthesis method, we prepared phenylethyl isocyanide on a 100 mmol scale which can be converted according to our recently published Ugi–Pictet–Spengler procedure to essential medicines including the schistosomiasis-drug Praziquantel (Fig. 7).51–53 For this, we compared our method of phenylethyl isocyanide synthesis with literature methods using POCl3 on a 100 mmol scale (Fig. 7, ESI†). Our method allows the synthesis of the target compounds in less than 1 h on a 100 mmol scale in almost quantitative yields with minimal use of halogenated solvents and no wastewater production. Reported reactions, however, lasted between 7 and 10 h and used a lot of the chlorinated solvent DCM (250–400 ml) and waste water (300–500 ml) and reported mixed yields were from 45% to 99%. Additionally, the reported methods expose the chemist for a considerably longer time to the hazardous fumes of the isocyanide during work-up (∼4 h). The number of workup steps in the reported procedures is between 7 and 9, while our workup comprises a simple silica filtration step followed by solvent evaporation.
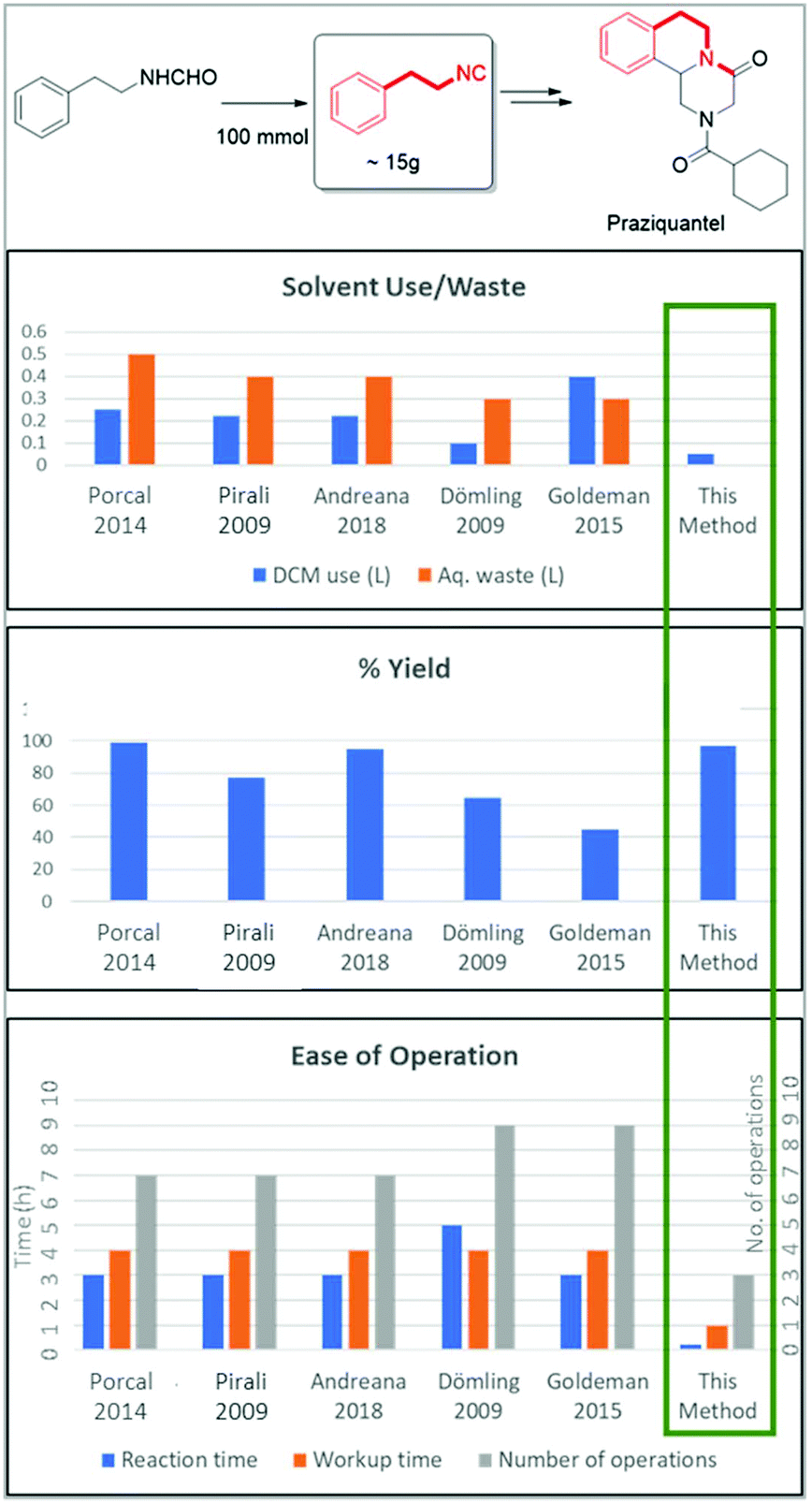 | ||
| Fig. 7 Comparison of different reports of the 100 mmol synthesis of phenylethyl isocyanide (C-2) useful for the production of the schistosomiasis-drug praziquantel. We compared 6 methods for solvent use and waste water production, yield and ease of operation, such as the number of workup steps, time to target and chemist's contact time with the chemical. | ||
Despite the description of thousands of isocyanides, currently their use in chemistry is reduced mostly to a small number of commercially available or easy to synthesize derivatives. The extensive use of isocyanides in chemistry, for example, in medicinal chemistry, still hampers the rapid and efficient access to a large number of functionalized isocyanides. To overcome this limitation, we proposed an innovative approach to quickly and easily synthesize isocyanides. We found that a non-aqueous dry workup by a fast filtration through a silica pad could considerably improve yields. Apart from being time-efficient and reducing resource consumption, the approach enables rapid access to structurally diverse isocyanides in good to very high yields. We describe for the first time a parallel synthesis approach to isocyanides in a 96-well format, which can be used to produce hundreds of novel high-quality isocyanides per week per chemist. An emerging area where large numbers of different isocyanides are of need is high-density information storage.4
To show the superiority of our process over the old processes, a comparison of the environmental factor (E-factor) is an appropriate method.54 The lower the value of E-factor the better the performance of the overall process. We calculated the environmental factor (E-factor) (see the ESI†) for all the above five literature processes for the phenylethyl isocyanide synthesis using the POCl3 method with our process (Table 4).55–59
In all reported literature processes, a high E-factor is displayed due to the excessive organic solvent use for the aqueous workups of a small quantity of phenylethyl isocyanide. On the other hand, in our process, we achieved the lowest E-factor by avoiding aqueous workup and also by running the reaction at higher concentration. In our process, the lower E-factor also indicates the lowest waste generation. Thus, our process is greener than all the known literature processes of synthesis of isocyanide, by a factor of 6–10. Most isocyanides are generally produced by similar procedures with minor changes in the details. To generalize, the current method of dehydrative isocyanide synthesis from formamides is a far greener method than previously described methods.
Conclusions
Our methodology affords isocyanides in high purity and in addition to that, offers a way to get around the unpleasant smell of these chemicals. By virtue of the short reaction time, this methodology allows for the in situ synthesis of unstable isocyanides, making it possible for unstable isocyanides to be used as reactants in a variety of reactions. We have shown that compared to traditional isocyanide synthesis approaches our method is much faster, higher yielding, produces only a fraction of the organic and aqueous waste, and also largely reduces the contact time of the chemist operator with the hazardous materials and fumes, thus considerably reducing the overall footprint of this chemistry. Also, there is no ‘one-size-fits-all’ method for the synthesis of isocyanides; we believe that our protocol is superior to currently used methods with respect to a very large number of workable formamide precursors concerning yields, time, simplicity, and safety of the procedure and sustainability.What are the reasons for the overuse of only a handful of isocyanides? The nature of many isocyanides being hazardous and smelly chemicals prevents chemists from synthesizing less explored derivatives, while only a few isocyanides are commercially affordable. We believe that ensuring that high-quality isocyanide building blocks are developed with a high scaffold-, shape- and functional group-diversity may be all that is needed to drive research into the unstudied parts of isocyanide reaction chemical space. What discoveries are we missing by focusing only on a handful of isocyanide building blocks? We have shown that even isocyanides known as very unstable can be synthesized by our method and reacted further. Much of the work that has emerged from exploring the chemical space of isocyanides over the past years lies fallow. Challenges notwithstanding, making isocyanide-based research tools readily available must be a major objective in the time to come and our protocol will help to do so.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been supported (to AD) by the National Institute of Health (NIH) (2R01GM097082-05), the European Lead Factory (IMI) under grant agreement number 115489, and the Qatar National Research Fund (NPRP6-065-3-012). Moreover, funding was received through ITN “Accelerated Early stage drug discovery” (AEGIS, grant agreement no. 675555), COFUND ALERT (grant agreement no. 665250), Hartstichting (ESCAPE-HF, 2018B012) and KWF Kankerbestrijding grant (grant agreement no. 10504).References
- J. Zhang, P. Yu, S.-Y. Li, H. Sun, S.-H. Xiang, J. Wang, K. N. Houk and B. Tan, Science, 2018, 361, eaas8707 Search PubMed.
- Chemical & Engineering News Archive, 1996, 74, 8.
- F. Y. Lim, T. H. Won, N. Raffa, J. A. Baccile, J. Wisecaver, A. Rokas, F. C. Schroeder and N. P. Keller, mBio, 2018, 9, e00785-18 Search PubMed.
- C. E. Arcadia, E. Kennedy, J. Geiser, A. Dombroski, K. Oakley, S.-L. Chen, L. Sprague, M. Ozmen, J. Sello, P. M. Weber, S. Reda, C. Rose, E. Kim, B. M. Rubenstein and J. K. Rosenstein, Nat. Commun., 2020, 11, 691 Search PubMed.
- A. C. Boukis, K. Reiter, M. Frolich, D. Hofheinz and M. A. R. Meier, Nat. Commun., 2018, 9, 1439 Search PubMed.
- S. Javanbakht and A. Shaabani, ACS Appl. Bio Mater., 2020, 3, 156–174 Search PubMed.
- U. Ivar, Isonitrile Chemistry, Academic Press, New York, 1971 Search PubMed.
- E. Singleton and H. E. Oosthuizen, in Adv. Organomet. Chem, ed. F. G. A. Stone and R. West, Academic Press, 1983, vol. 22, pp. 209–310 Search PubMed.
- M. Giustiniano, A. Basso, V. Mercalli, A. Massarotti, E. Novellino, G. C. Tron and J. Zhu, Chem. Soc. Rev., 2017, 46, 1295–1357 Search PubMed.
- C. Hulme and J. Dietrich, Mol. Divers., 2009, 13, 195–207 Search PubMed.
- T. Zarganes-Tzitzikas, A. L. Chandgude and A. Domling, Chem. Rec., 2015, 15, 981–996 Search PubMed.
- A. Dömling, Chem. Rev., 2006, 106, 17–89 Search PubMed.
- B. Song and B. Xu, Chem. Soc. Rev., 2017, 46, 1103–1123 Search PubMed.
- B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505–3521 Search PubMed.
- V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698–2779 Search PubMed.
- T. Vlaar, E. Ruijter, B. U. Maes and R. V. Orru, Angew. Chem., Int. Ed., 2013, 52, 7084–7097 Search PubMed.
- J. W. Collet, T. R. Roose, E. Ruijter, B. U. Maes and R. V. Orru, Angew. Chem., 2020, 132, 548–566 Search PubMed.
- J. Kolb, B. Beck, M. Almstetter, S. Heck, E. Herdtweck and A. Dömling, Mol. Divers., 2003, 6, 297–313 Search PubMed.
- A. Dömling and K. Illgen, Synthesis, 2005, 662–667 Search PubMed.
- B. Weinberger and W. P. Fehlhammer, Angew. Chem., 1980, 92, 478–479 Search PubMed.
- Y. Wang and C. Zhang, ChemistrySelect, 2020, 5, 171–177 Search PubMed.
- G. M. Ojeda-Carralero, L. G. Ceballos, J. Coro and D. G. Rivera, ACS Comb. Sci., 2020 DOI:10.1021/acscombsci.0c00111.
- A. Ramazani and A. Rezaei, Org. Lett., 2010, 12, 2852–2855 Search PubMed.
- J. R. Frost, C. C. Scully and A. K. Yudin, Nat. Chem., 2016, 8, 1105–1111 Search PubMed.
- Z. Xu, F. De Moliner, A. P. Cappelli and C. Hulme, Angew. Chem., Int. Ed., 2012, 51, 8037–8040 Search PubMed.
- D. V. Leusen and A. M. V. Leusen, Org. React., 2004, 417–666, DOI:10.1002/0471264180.or057.03.
- W. P. Weber and G. W. Gokel, Tetrahedron Lett., 1972, 13, 1637–1640 Search PubMed.
- U. Galli, G. C. Tron, B. Purghè, G. Grosa and S. Aprile, Chem. Res. Toxicol., 2020, 33, 955–966 Search PubMed.
- M. Spallarossa, Q. Wang, R. Riva and J. Zhu, Org. Lett., 2016, 18, 1622–1625 Search PubMed.
- C. G. Neochoritis, S. Stotani, B. Mishra and A. Dömling, Org. Lett., 2015, 17, 2002–2005 Search PubMed.
- P. Golubev, A. Pankova and M. Krasavin, Tetrahedron Lett., 2019, 60, 1578–1581 Search PubMed.
- M.-G. Liu, N. Liu, W.-H. Xu and L. Wang, Tetrahedron, 2019, 75, 2748–2754 Search PubMed.
- G. Kobayashi, T. Saito and Y. Kitano, Synthesis, 2011, 3225–3234 Search PubMed.
- M. Keita, M. Vandamme, O. Mahé and J.-F. Paquin, Tetrahedron Lett., 2015, 56, 461–464 Search PubMed.
- A. Porcheddu, G. Giacomelli and M. Salaris, J. Org. Chem., 2005, 70, 2361–2363 Search PubMed.
- A. Chao, E. Alwedi and F. F. Fleming, Synthesis, 2019, 51, 2122–2127 Search PubMed.
- K. A. Waibel, R. Nickisch, N. Möhl, R. Seim and M. A. R. Meier, Green Chem., 2020, 22, 933–941 Search PubMed.
- C. G. Neochoritis, S. Shaabani, M. Ahmadianmoghaddam, T. Zarganes-Tzitzikas, L. Gao, M. Novotná, T. Mitríková, A. R. Romero, M. I. Irianti, R. Xu, J. Olechno, R. Ellson, V. Helan, M. Kossenjans, M. R. Groves and A. Dömling, Sci. Adv., 2019, 5, eaaw4607 Search PubMed.
- S. Shaabani, R. Xu, M. Ahmadianmoghaddam, L. Gao, M. Stahorsky, J. Olechno, R. Ellson, M. Kossenjans, V. Helan and A. Dömling, Green Chem., 2019, 21, 225–232 Search PubMed.
- Y. Wang, S. Shaabani, M. Ahmadianmoghaddam, L. Gao, R. Xu, K. Kurpiewska, J. Kalinowska-Tluscik, J. Olechno, R. Ellson, M. Kossenjans, V. Helan, M. Groves and A. Dömling, ACS Cent. Sci., 2019, 5, 451–457 Search PubMed.
- M. Hadian, S. Shaabani, P. Patil, S. V. Shishkina, H. Böltz and A. Dömling, Green Chem., 2020, 22, 2459–2467 Search PubMed.
- A. Osipyan, S. Shaabani, R. Warmerdam, S. V. Shishkina, H. Boltz and A. Dömling, Angew. Chem., Int. Ed., 2020, 59, 12423–12427 Search PubMed.
- Z. He, M. Bae, J. Wu and T. F. Jamison, Angew. Chem., Int. Ed., 2014, 53, 14451–14455 Search PubMed.
- M. Movsisyan, E. Delbeke, J. Berton, C. Battilocchio, S. Ley and C. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 Search PubMed.
- N. R. Impens and E. F. Vansant, Interface Sci., 1997, 5, 95–101 Search PubMed.
- A. Van Beijnen, R. Nolte, A. Naaktgeboren, J. Zwikker, W. Drenth and A. Hezemans, Macromolecules, 1983, 16, 1679–1689 Search PubMed.
- A. Van Leusen, B. Hoogenboom and H. Siderius, Tetrahedron Lett., 1972, 13, 2369–2372 Search PubMed.
- A. V. Lygin and A. d. Meijere, Org. Lett., 2009, 11, 389–392 Search PubMed.
- M. A. Mironov and V. S. Mokrushin, Russ. J. Org. Chem., 1999, 35, 693–697 Search PubMed.
- G. van der Heijden, J. A. W. Jong, E. Ruijter and R. V. A. Orru, Org. Lett., 2016, 18, 984–987 Search PubMed.
- H. Cao, H. Liu and A. Dömling, Chem. – Eur. J., 2010, 16, 12296–12298 Search PubMed.
- H. Liu, S. William, E. Herdtweck, S. Botros and A. Dömling, Chem. Biol. Drug Des., 2012, 79, 470–477 Search PubMed.
- A. Boltjes, H. Liu and A. Dömling, Org. Synth., 2017, 94, 54–64 Search PubMed.
- C. H. Lam, V. Escande, K. E. Mellor, J. B. Zimmerman and P. T. Anastas, J. Chem. Educ., 2019, 96, 761–765 Search PubMed.
- M. Ingold, G. V. López and W. Porcal, ACS Sustainable Chem. Eng., 2014, 2, 1093–1097 Search PubMed.
- A. A. Grolla, V. Podesta, M. G. Chini, S. Di Micco, A. Vallario, A. A. Genazzani, P. L. Canonico, G. Bifulco, G. C. Tron, G. Sorba and T. Pirali, J. Med. Chem., 2009, 52, 2776–2785 Search PubMed.
- N. Esmati, A. Maddirala, N. Hussein, H. Amawi, A. Tiwari and P. Andreana, Org. Biomol. Chem., 2018, 16, 5332–5342 Search PubMed.
- A. Dömling, PCT Int. Appl., pat., WO2009115333, 2009.
- W. Goldeman and A. Nasulewicz-Goldeman, Tetrahedron, 2015, 71, 3282–3289 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02722g |
| This journal is © The Royal Society of Chemistry 2020 |