
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCleavage of ethers and demethylation of lignin in acidic concentrated lithium bromide (ACLB) solution†
Zheng
Li
 a,
Eka
Sutandar
a,
Thomas
Goihl
a,
Xiao
Zhang
b and
Xuejun
Pan
*a
a,
Eka
Sutandar
a,
Thomas
Goihl
a,
Xiao
Zhang
b and
Xuejun
Pan
*a
aDepartment of Biological Systems Engineering, University of Wisconsin-Madison, 460 Henry Mall, Madison, WI 53706, USA. E-mail: xpan@wisc.edu
bSchool of Chemical Engineering and Bioengineering, Washington State University, 2710 University Drive, Richland, WA 99354, USA
First published on 9th October 2020
Abstract
The methoxyl group is the most abundant functional group of lignin and affects the properties, reactivity, and application of lignin. Efficient demethylation is always of interest in the area of lignin chemistry and application. This study demonstrated a new method for cleaving ether compounds and demethylating lignin in acidic concentrated lithium bromide (ACLB) solution under mild conditions. It was found that the ACLB system could universally cleave ether compounds except for diaryl ethers. The study on lignin model compounds (creosol, syringol, and 1,2,3-trimethoxybenzene) verified that ACLB could demethylate them to corresponding phenols. Four real lignin samples produced from various sources by different methods were also efficiently demethylated by 69–82% in ACLB. The lignin demethylation resulted in more phenolic hydroxyl groups, which benefits some downstream applications of lignin. This study also provided new insights into the cleavage of the ether bonds in lignin. In addition to the methyl–aryl ether bond, ACLB could cleave other ether bonds of lignin in β-O-4, β-5, and β-β structures except for the 4-O-5 bond in the diphenyl structure. The ether bonds were cleaved via the SN2 substitution except for the β-O-4 bond, which was primarily cleaved via the benzyl cation and enol ether intermediates, leading to Hibbert's ketones. Some of the β-O-4 structures were transformed into benzodioxane (BD) structures, which were stable in the ACLB system.
Introduction
The valorization of lignin to high-value products has been challenging due to the complicated structure and unsatisfactory reactivity of lignin, which is built up from the C9 units including coniferyl alcohol (G) with one methoxyl group, sinapyl alcohol (S) units with two methoxyl groups, and p-coumaryl alcohol (H) with no methoxyl substituent.1–4 The content and structure of lignin vary among different taxonomic groups, cell types, and even layers of the plant cell wall and are greatly influenced by developmental and environmental cues.3 With few exceptions, angiosperm (hardwood) lignin typically consists of S and G units with a trace of H units, while gymnosperm (softwood) lignin contains almost exclusively G units along with a small amount of S and H units.5,6 Herbage (grass) lignin usually possesses more H units than wood lignin.3 These units are interlinked mainly via carbon–carbon (C–C) and ether (C–O–C) bonds, including 5–5 (biphenyl), β-5 (phenylcoumaran), β-β (resinol), and β-1 (1,2-diarylpropane), and β-O-4 (β-aryl ether) and 4-O-5 (diphenyl ether), respectively.4 When lignin is isolated from the plant cell wall, its structure changes, dependent on the chemicals and reaction conditions involved in the lignin isolation. For example, milled wood lignin (isolated by finely grinding the plant cell wall followed by solvent extraction) and enzymatic lignin (isolated by enzymatic removal of carbohydrates followed by purification) maintain lignin structures relatively intact, while the lignins isolated by the chemical processes (such as pulping and pretreatment) using alkalis, acids, and organic solvents at high temperature lose most of their β-O-4 bonds and obtain new condensation structures (C–C linkages).7–10Lignin has multiple functional groups including methoxyl, aromatic hydroxyl, aliphatic hydroxyl, carbonyl, and carboxyl groups, which affect the properties and reactivity of lignin. The quantity of the functional groups is determined by the source and isolation method and condition of the lignin. The methoxyl group (OMe) is the most abundant function group in lignin, counting for up to 24% in lignin and 6% in whole wood. Methoxyl groups are critical to the properties, reactivity, and application of lignin. For example, since lignin units without methoxyl groups are easier to couple each other to form interunit C–C bonds,11 the lignin with a high content of methoxyl groups, such as hardwood lignin, usually has a low density of interunit C–C linkages, and such lignin is easier to be depolymerized.12 Fewer C–C linkages lead to the higher mobility of lignin molecules and therefore lower glass transition temperature.13 Methoxyl groups help stabilize phenoxyl radicals, which contributes to the antioxidant capacity of lignin.14,15 On the other hand, demethylation (converting methoxyl to a phenolic hydroxyl group) can improve lignin reactivity toward crosslinking, degradation, and other modifications. For example, demethylated lignin had a higher reactivity in lignin-based phenolic resins, resulting in a stronger binding strength and a lower formaldehyde emission.16–20 Demethylated lignin showed a higher adsorption capacity of heavy metals.21 It was reported that the demethylation favored the opening of the lignin benzene ring during biological degradation22–24 and could promote the Fenton reaction in lignin biodegradation by rot fungi.25 Demethylated lignin was a more reactive precursor for the production of polyphenols.26 The methyl group cleaved from lignin could be selectively converted to acetic acid over RuCl3 in the presence of CO and H2O.27 Besides, demethylation is a key step to produce dimethyl sulfoxide from lignin.28,29
The demethylation of lignin, which cleaves the stable ether bond between the benzene ring and methyl group, usually requires special reagents (mostly toxic, expensive, and unstable) and/or harsh conditions (such as high temperature and strong acidity).30,31 For example, molten pyridine hydrochloride is a classic reagent for cleaving the aryl methyl ether at a temperature as high as 200 °C.32 Concentrated HBr and HI can selectively demethylate lignin, which have been used for quantitating the methoxyl group of lignin.26,33–35 Iodic reagents such as iodocyclohexane and trimethylsilyl iodide are also effective for the demethylation.36–39 Besides, BBr3 and AlI3 can demethylate lignin and lignin model compounds at or below room temperature.40–42
The present work thus aimed at developing a new method for lignin demethylation under mild conditions using an acidic concentrated lithium bromide (ACLB) solution. This system was first introduced in our previous studies for the fractionation and saccharification of raw biomass and quantitation of lignin.43,44 With low-concentration acid (0.04–0.3 mol L−1 or 0.1–1 wt% HCl or HBr) as a catalyst, the ACLB system was able to effectively cleave β-aryl ether (β-O-4), and NMR evidence indicated that the α-O-4 ether bond in phenylcoumaran (β-5) and the α-O-γ ether bond in resinol (β-β) were partially cleaved.44–46 It was confirmed that the cleavage of the β-O-4 ether bond was completed via the Br− and H+ promoted benzyl cation and enol ether intermediates, leading to the formation of Hibbert's ketones, and some of the benzyl cations transformed to a benzodioxane (BD) structure.46 However, the mechanisms of BD formation and the cleavage of the ether bonds in phenylcoumaran and resinol had not been clearly and sufficiently addressed.
Inspired by the observations above from the previous studies that the ACLB system can cleave the ether bonds in lignin, we hypothesized that the ACLB system could be developed into a new method for cleaving regular ether compounds under milder conditions. In particular, we were interested in establishing a new method based on the ACLB system for lignin demethylation to produce phenolic hydroxyl-rich lignin for downstream lignin application. In addition, we wanted to revisit the cleaving mechanisms of the ether bonds of lignin in the ACLB system. Herein, we first tested the ACLB system for cleaving different types of ether compounds and demethylating lignin model compounds with different numbers of methoxyl groups. We then carefully investigated the demethylation of a real lignin, ethanol poplar lignin (EPL), in the ACLB system. Three other real lignin samples including hardwood kraft lignin (HKL), corn stover lignin (CSL), and ethanol lodgepole pine lignin (ELPPL) were also tested to verify the applicability of the ACLB system for lignin demethylation. The mechanisms of the cleavage of the ether bonds in lignin and the formation of BD in the ACLB system were revisited and clarified based on the new findings from the present study.
Experimental
Materials
All chemicals were used as received. Creosol (98%), syringol (99%), pyrogallol (99%), and pyridine (99%) were purchased from Alfa Aesar. 1,2,3-Trimethoxybezene (98%) was purchased from Frontier Scientific. LiBr (99%), Na2SO4 (99%), NaCl (99%) and deuterated DMSO (99%) were purchased from Sigma-Aldrich. Tetrahydrofuran (THF, HPLC grade) was purchased from Fisher Scientific. HBr (48%) was provided by Honeywell. HCl (37%) was provided by Mecron. Acetic anhydride was purchased from MP Biochemicals. 4-Nitrobenzaldehyde (98%) was purchased from Ark Pharm. Deuterated chloroform (99.75%, contained 1 v/v% of TMS) was from Acros Organics. Hardwood Kraft lignin (HKL) was supplied by Westvaco (New York, NY). Corn stover lignin (CSL) was extracted using NaOH from the corn stover residue after furfural production from hemicellulose followed by enzymatic hydrolysis of cellulose to glucose for ethanol. The extracted CSL was precipitated with acid, washed to neutral, and then freeze-dried. The preparation and properties of ethanol lodgepole pine lignin (ELPPL) and ethanol poplar lignin (EPL) were described in our previous studies.14,47Experimental approaches
The theoretical mass loss induced by the demethylation of ArOCH3 to ArOH was calculated using eqn (1).
 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20. Mass spectra ranging from m/z = 50–550 were obtained using electron impact ionization (EI). The concentration of the products was calculated using the external standard method in the software of GC-MS. The yield of the products was calculated using eqn (2).
:20. Mass spectra ranging from m/z = 50–550 were obtained using electron impact ionization (EI). The concentration of the products was calculated using the external standard method in the software of GC-MS. The yield of the products was calculated using eqn (2). | (2) |
:20. The concentration of a product was semi-quantitated by the internal standard method with hexane as an internal standard based on the following eqn (3): | (3) |
The contents of OMe, ArOH, and AlkOH in lignin were quantitated using 1H NMR with 4-nitrobenzaldehyde as an internal standard. In brief, about 5–10 mg 4-nitrobenzaldehyde and 10–15 mg acetylated lignin were fully dissolved in 0.5 mL CDCl3. 1H NMR spectra were recorded on a Bruker AV III 500 MHz spectrometer (Billerica, MA) with an operating frequency of 500 MHz. The data were processed using MestReNova desktop NMR data processing software (version 11.0.4). The phase and baseline in all the spectra were corrected automatically before integration. The contents of OMe, ArOH, and AlkOH were calculated using eqn (4)–(6):14
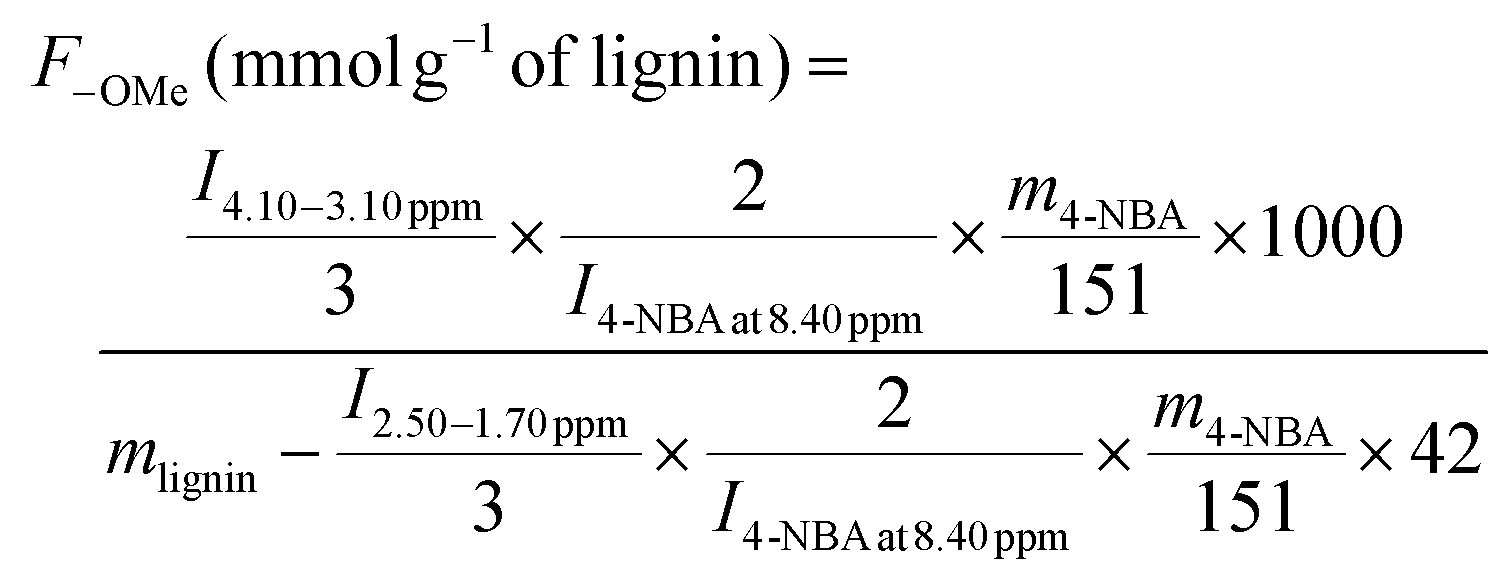 | (4) |
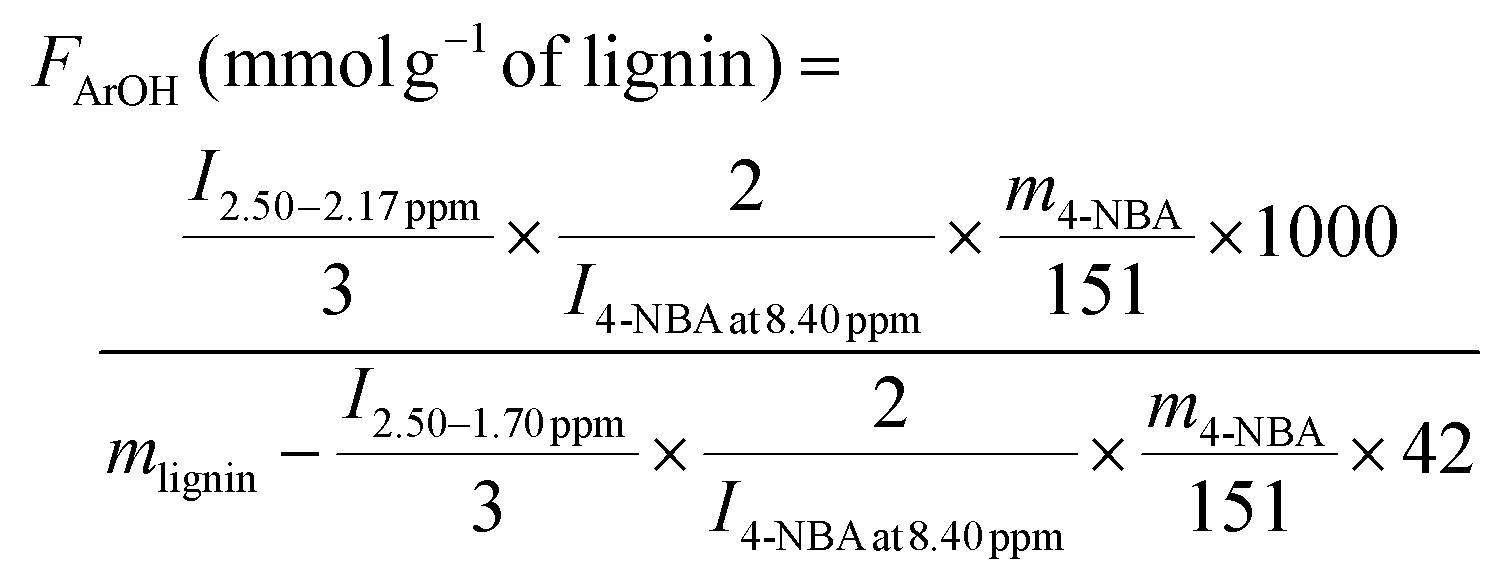 | (5) |
 | (6) |
For the collection of HSQC spectra, about 50 mg unacetylated lignin was fully dissolved in 0.5 mL DMSO-D6. The samples were analyzed on a Bruker AV III 500 MHz spectrometer equipped with a DCH (13C-optimized) cryoprobe (Billerica, MA). The test was conducted at 25 °C with Bruker's standard hsqcetgpsisp 2.2 pulse program (acquisition times 200 ms and 8 ms in 1H and 13C dimensions, inter-scan relaxation delay 1 s). Data were analyzed using Bruker's Topspin 4.0.8 software.
000, 500, and 50 Å) and a 50 mm × 7.8 mm i.d. Phenogel 5U guard column (Phenomenex, Torrance, CA). Lignin acetate (about 3 mg) was dissolved in 5 mL HPLC grade tetrahydrofuran (THF) without a stabilizer, and about 500 μL the solution was injected into the GPC system. THF was used as the eluent at a flow rate of 1 mL min−1. The column temperature was 30 °C. Polystyrene standards were used for calibration. Lignin samples and polystyrene standards were detected using a variable wavelength detector (VWD) at 280 and 254 nm, respectively.
Results and discussion
Cleavage of ether compounds in the ACLB system
Ethers are not commonly reactive toward many reactions, except for highly strained cyclic ethers (oxirane and oxetane). A textbook method for cleaving an ether is to heat it with concentrated HBr or HI because they are sufficiently acidic to protonate the ether, while bromide and iodide are good nucleophiles for the substitution. Specifically, the protonation of the ether oxygen forms an oxonium ion, and then the nucleophile (Br− or I−) attacks the backside of the electrophilic α-carbon of the ether via the SN2 mechanism, leading to the cleavage of the ether bond.To investigate the capability of the ACLB to cleave ethers, different ether compounds including linear and cyclic ethers and aliphatic and aromatic ethers were treated. As summarized in Table 1, all the ether compounds except diphenyl ether could be cleaved in the ACLB. The conversion and product yield depended on the structure of the ethers and reaction conditions. The cleavage of an ether via the SN2 substitution by Br− yields an alcohol and a bromide. The alcohol can further react with Br− to give another bromide, while the bromide intermediate can be further hydrolyzed into another alcohol. Both the alcohol and the bromide were detected in the products. The yield of the products listed in Table 1 (entries 1–7) is the combined yield of the alcohol and the bromide.
| Entry | Substrate | Conversion (%) | Target product | Yield (%) |
|---|---|---|---|---|
| ND – not determined. Reaction conditions (unless indicated separately): 0.1 g substrate, 10 g 53 wt% LiBr with 6 wt% HBr, 100 °C, and 4 h.a Not accurate due to the mass loss of the volatile reactant and products.b Room temperature and 1 h.c 0.18 wt% HBr, room temperature, and 2 h.d 1.3 wt% HBr, room temperature, and 2 h.e 6 wt% HBr, room temperature, and 2 h.f 6 wt% HBr, 120 °C, and 4 h. | ||||
| 1 |

|
NDa |

|
45a |
| 2 |

|
15 |

|
30 |
| 3b |
|
100 |

|
>95 |
| 4c |

|
19 |

|
3.4 |
| 5d |

|
39 |

|
18 |
| 6e |

|
75 |
|
65 |
| 7 |
|
83 |

|
70 |

|
25 | |||
| 8 |

|
47 |

|
38 |
| 9 |
|
35 |

|
17 |
| 10f |

|
<5 |

|
ND |
Diethyl ether and dibutyl ether were both cleaved to give ethanol/bromoethane and butanol/bromobutane as products, respectively (entries 1 and 2 in Table 1). Because of a larger steric hindrance at the α-carbon for the SN2 substitution, the debutylation of dibutyl ether was slower than the deethylation of diethyl ether.
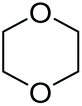
Cyclic ethers are easier to be cleaved than linear ethers because of the high strain of the former. For example, tetrahydrofuran (THF) was ring-opened to 1,4-butanediol and its bromides very rapidly even at room temperature (Table 1, entry 3), which was in agreement with the previous observation that THF was very reactive to undergo polymerization in the presence of Brønsted or Lewis acid.48,49 Dioxane was much more stable than THF in ACLB (entry 7 in Table 1) because the six-membered ring of dioxane is less strained than the five-membered ring of THF. A similar result was observed in other acidic systems as well.49,50 The cleavage of the first ether bond of dioxane occurred faster than the second one, and the yield of the product from the first step (70%) was much higher than that from the second step (25%). However, the cleavage of dioxane was easier than diethyl and dibutyl ethers, since the conversion and product yield of dioxane were higher than those of the linear ethers under the same conditions, as shown in Table 1 (entry 7 vs. entries 1 and 2).
When the steric hindrance at the α-carbon is large, the ether cleavage becomes slower and more difficult. For the SN2 substitution reactions, the relative rates are in the order of methyl (CH3−) > primary carbon (RCH2−) > secondary carbon (R2CH−) ≫ tertiary carbon (R3C−). For example, 2-methyl THF (entry 4 in Table 1) was cleaved in the ACLB at room temperature, but the relative rate was much lower, compared with THF (entry 3 in Table 1), because one of the α-carbon (C2) of 2-methyl THF is a secondary carbon. Increasing the severity of reaction conditions, such as acidity, can promote the reaction. For example, with increasing HBr concentration from 0.18 wt% to 1.3 wt% and 6 wt%, the conversion of 2-methyl THF at room temperature increased from 19% to 39% and 75%, and the yield of the products from 3.4% to 18% and 65%, respectively.
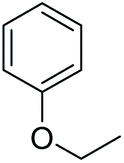
Alkyl aryl ethers were cleaved as well in the ACLB, generating a phenol and an alkyl bromide. For example, anisole was cleaved to phenol and bromomethane under the tested conditions (entry 8 in Table 1). The cleavage of phenetole to phenol and bromoethane was slower than anisole because of the larger steric hindrance of the primary α-carbon in the ethyl side of phenetole (entry 9 in Table 1). The alkyl aryl ether can only be cleaved from the alkyl side via dealkylation because the sp2 hybridized carbon atom bonded to ether oxygen on the benzene ring cannot undergo the SN2 substitution.
Diphenyl ether was stable in ACLB (entry 10 in Table 1) even at a higher temperature due to its high bond dissociation energy (314 kJ mol−1).51,52 Generally, the cleavage of the diphenyl ether is very hard without the assistance of hydrogenation. For the same reason (sp2 hybridized carbon on benzene ring), the ether bond in diphenyl ether cannot be cleaved via the SN2 mechanism. This result suggests that the ether bond of the 4-O-5 linkage in lignin should be stable in the ACLB system.
Demethylation of lignin model compounds in the ACLB system
With the results of cleaving the ether compounds above, lignin model compounds with methoxyl group(s) were studied for the demethylation in ACLB. 2-Methoxy-4-methylphenol (creosol) was chosen as a G type lignin model compound. HBr was used as an acid catalyst, so the ACLB system can be compared with the concentrated HBr system, which is a classic method for ether cleavage. The demethylation mechanism is shown in Scheme 1 (top).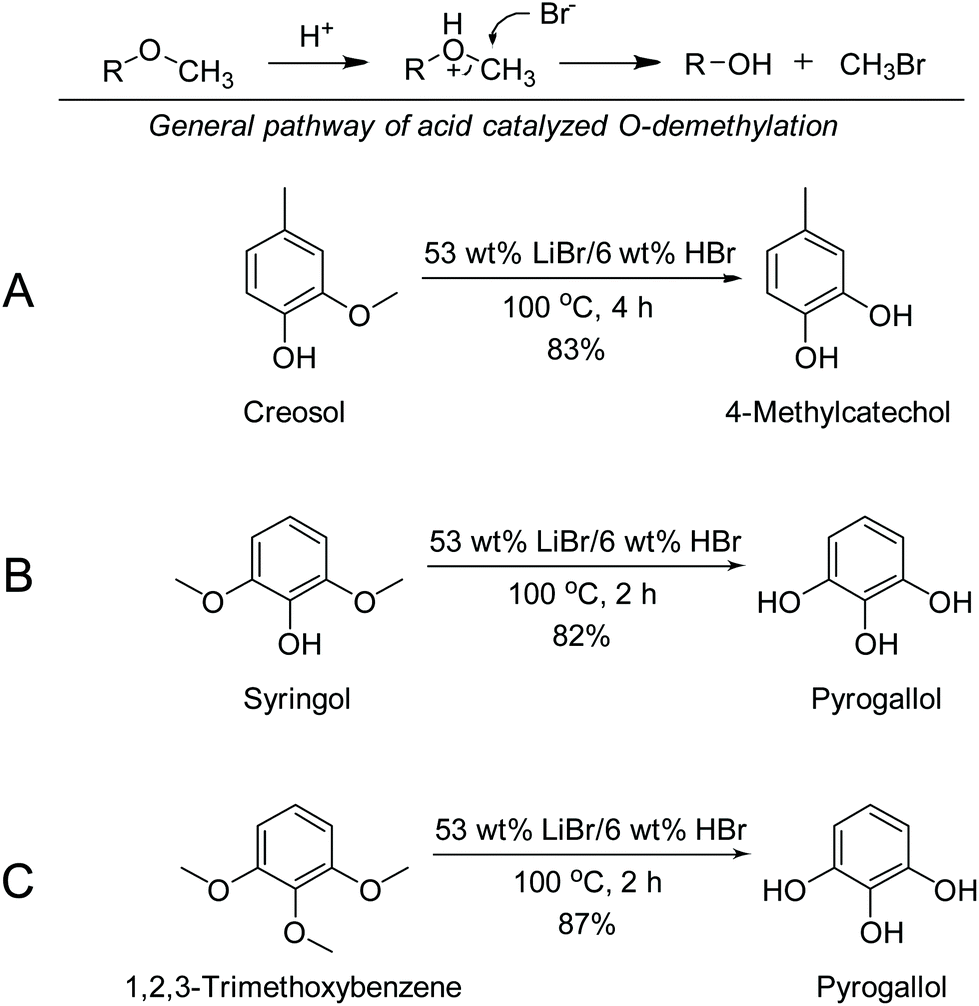 | ||
| Scheme 1 Demethylation of creosol, 2,6-dimethoxyophenol, and 1,2,3-trimethoxybenzene in the ACLB system. | ||
The concentrated HBr system (48 wt%) was first examined for the demethylation of creosol. The concentrated HBr was able to demethylate creosol but required a higher temperature and longer time than the ACLB, as shown in Table 2 (entry 1 vs. entries 6 and 7). Besides, the concentrated HBr had a lower yield of 4-methylcatechol and more condensation compounds (black humin-like precipitates), suggesting that ACLB had better selectivity than the concentrated HBr. Differently, 6% HBr in water was unable to demethylate creosol because the acidity and Br− concentration were too low to effectively protonate and cleave the ether bond, respectively (entry 2 in Table 2). Similarly, creosol was not demethylated in 61 wt% LiBr in the absence of acid after 20 h treatment (entry 3 in Table 2) because protonation of the ether oxygen by H+ is a prerequisite to cleave the ether bond by bromide as a nucleophile via SN2 substitution.
| Entry | Reaction system | Temperature (°C) | Time (h) | Creosol conversion (%) | 4-Methylcatechol yield (%) |
|---|---|---|---|---|---|
| a 0.5 g creosol in 20 mL 48 wt% HBr. b 0.1 g creosol in 10 g water with 1 mL 48 wt% HBr. c 0.1 g creosol in 6.1 g LiBr and 3.9 g water. d 0.1 g creosol in 10 g aqueous LiBr solution at different concentrations with 1 mL 48 wt% HBr. | |||||
| 1a | 48 wt% HBr | 120 | 20 | 100 | 68 |
| 2b | 6 wt% HBr | 100 | 4 | <5 | 0 |
| 3c | 61 wt% LiBr | 100 | 20 | 33 | 0 |
| 4d | 40 wt% LiBr/6 wt% HBr | 100 | 4 | 25 | 0 |
| 5d | 47 wt% LiBr/6 wt% HBr | 100 | 4 | 31 | 28 |
| 6d | 53 wt% LiBr/6 wt% HBr | 100 | 4 | 100 | 83 |
| 7d | 61 wt% LiBr/6 wt% HBr | 100 | 4 | 100 | 80 |
| 8 | 48 wt% NaBr/6 wt% HBr | 100 | 4 | 11 | <5 |
| 9 | 40 wt% KBr/6 wt% HBr | 100 | 4 | 0 | 0 |
| 10 | 61% LiBr/6 wt% HCl | 100 | 20 | 99 | 87 |
| 11 | 46% LiCl/6 wt% HCl | 100 | 4 | 0 | 0 |
As expected, creosol was effectively demethylated in ACLB. For example, creosol was completely converted in 53% and 61% LiBr solutions with 6 wt% HBr at 100 °C, yielding >80% 4-methylcatechol (Scheme 1A and entries 6, 7 in Table 2). The results under other conditions (temperature, acid concentration, and acid type) are available in Table S1.† Why low-concentration acid (e.g., 6% HBr) can protonate the ether bond is because the acidity of HBr is enhanced in the concentrated LiBr solution. When highly oxophilic Li+ coordinates tightly with H2O molecules, the H+ is less hydrated and has high freedom in the solution, which enhances the acidity of the acid, compared with that in the water at the same acid concentration.53 Meanwhile, without coordination with water, high-concentration Br− is naked in the solution and freely available for the SN2 substitution to cleave the ether bond.43,54 These are the reasons why creosol was rapidly and effectively demethylated in the concentrated LiBr solutions at a low acid concentration.
The results in Table 2 (entries 4–7) indicate that LiBr concentration is a crucial factor affecting the demethylation. No demethylation occurred at 40% LiBr concentration (entry 4), apparently due to the insufficient acidity and Br− concentration in the dilute LiBr solution, while 28% demethylation was completed at 47% LiBr concentration (entry 5). Increasing the LiBr concentration to 53% yielded 83% 4-methylcatechol (entry 6). However, further increasing LiBr concentration to 61% did not show a positive effect on the demethylation (entry 7).
When LiBr was replaced by NaBr or KBr, a much lower demethylation rate was observed (entries 8 and 9 in Table 2). Since the solubility of NaBr and KBr was lower and the molecular weight was higher than that of LiBr, the concentration of Br− (the nucleophile for the SN2 substitution) is lower in NaBr and KBr solutions than in LiBr solution. Another more important reason is that the lower charge density of Na+ and K+ because of their larger size makes them have a lower oxophilicity than Li+. Therefore, the coordination of Na+ and K+ with water is weaker than that of Li+. As a result, Br− has more chances to enter the coordination positions of Na+ and K+ than those of Li+, which reduces the availability of Br− in the NaBr and KBr solutions for the SN2 substitution. For the same reason, the freedom (activity) of H+ is reduced in the NaBr and KBr solutions, compared to that in LiBr solution. In other words, NaBr and KBr solutions have a lower acidity and a lower concentration of free Br− than LiBr solution at the same concentration. This is why their performance in the demethylation of creosol follows the order of LiBr > NaBr > KBr, as shown in Table 2.
HCl as a catalyst gave a comparable yield of 4-methylcatechol in the ACLB with HBr (entry 10 vs. entries 6, 7 in Table 2), indicating that the ether cleaving capability of the ACLB is the unique property of the system because of the enhanced acidity and Br− concentration, independent of the proton source (HBr or HCl). However, the LiCl/HCl system was unable to demethylate creosol (entry 11 in Table 2) due to the lower solubility of LiCl (lower Cl− concentration) and more importantly the much lower nucleophilicity of Cl− than Br− for the SN2 substitution.
It is worth mentioning that the demethylation of creosol was much faster than that of anisole in ACLB because the hydroxyl group in creosol increases the electron density of the conjugated system, which eases the protonation of methoxyl oxygen for the following SN2 substitution by Br−.
2,6-Dimethoxyphenol (syringol) and 1,2,3-trimethoxybenzene as S type lignin model compounds with more methoxyl groups were also investigated for demethylation in the ACLB system (Scheme 1B and C). Both model compounds could be completely demethylated to pyrogallol via the intermediates with one or two OMe groups. Besides, it was noted that the hydroxyl-rich intermediates and pyrogallol would condense and form black precipitates with extended reaction.
Demethylation of ethanol poplar lignin (EPL) in the ACLB system
After proving that ACLB is capable of cleaving aryl methyl ether in the three lignin model compounds above, real lignin (EPL) was tested for demethylation under the same conditions. It was observed that the lignin was well dispersed in the system, and the lignin became darker after the reaction. During the reaction, a thin layer of fine bubbles was observed on the surface of the reaction mixture, which was proved by GC-MS to be bromomethane (b.p. = 3.56 °C) produced from the demethylation. The contents of methoxyl (OMe), aromatic hydroxyl (ArOH), and alkyl hydroxyl groups (AlkOH) of the lignin were quantitated by 1H NMR, as described in the Experimental section.14,55 As shown in Fig. 1 (full data available in Table S2†), OMe in EPL was efficiently cleaved, resulting in ArOH, which occurred even at 50 °C. Meanwhile, GPC results revealed that the average molecular weights Mn and Mw of the lignin decreased from 1080 to 980 and 2500 to 1450, respectively (Fig. 1D), suggesting that lignin depolymerization occurred at a mild temperature in ACLB, which was attributed to the cleavage of the residual β-O-4 bonds. The PDI value decreased as well, indicating that the lignin was homogenized in size due to the depolymerization. Increasing the reaction temperature from 50 °C to 100 °C substantially enhanced the demethylation efficiency, leading to 72% OMe cleavage (to 2.19 mmol g−1) with an 81% selectivity to ArOH (8.27 mmol g−1 in total). However, the increased temperature resulted in the repolymerization of lignin, such as the condensation between aromatic C5 or C6 and benzyl cations induced by acid (Scheme S1†),56 and Mn, Mw, and PDI increased from 1050, 1450, and 1.38 to 1620, 2840, and 1.76, respectively. Further elevating temperature to 120 °C brought the cleavage rate of OMe up to 79%. However, the selectivity to ArOH dropped to 78%, and the average molecular weights of the lignin became higher because of promoted repolymerization. To avoid undesired excessive lignin condensation, 100 °C was selected as the working temperature. | ||
| Fig. 1 Demethylation of EPL. (A), (B), and (C) show the quantity of OMe, ArOH, and AlkOH in original and demethylated EPL at different temperatures and reaction times, and in different LiBr concentrations; (D), (E), and (F) show the average molecular weights and polydispersity index (PDI) of original and demethylated EPL at different temperatures, reaction times, and in different LiBr concentrations. Reaction conditions: 0.5 g EPL, 53 wt% LiBr, 6 wt% HBr, 100 °C, and 4 h. All lignin samples were acetylated for NMR and GPC analysis. All reactions were conducted in duplicate except the 30%, 40%, 47%, and 61% LiBr tests (single). | ||
A way to redeem the low OMe cleavage at 100 °C is to extend the reaction time. As shown in Fig. 1B, prolonging reaction time from 4 h to 8 h and 24 h increased the cleavage rate of OMe from 72% to 86% and 93%, respectively. However, the increased demethylation did not proportionally turn out as more ArOH but led to reduced ArOH, which was probably attributed to the formation of the benzodioxane (BD) structure, as reported previously.46 As further discussed in Scheme 2, one ArOH at C3 or C5 is consumed to form the BD structure. The average molecular weights did not change much, suggesting that the repolymerization was not significantly affected by reaction time, although it was very sensitive to reaction temperature (Fig. 1D and E). It was also noted that lignin mass recovery was 79 wt%, which was lower than the reported mass recovery of 83–94 wt% when lignin was treated under mild conditions (low acid concentration) in ACLB.45 The additional mass loss was attributed to the demethylation since the elimination of CH2via the demethylation could lead to an 8.0 wt% mass loss (calculated from OMe content in EPL).
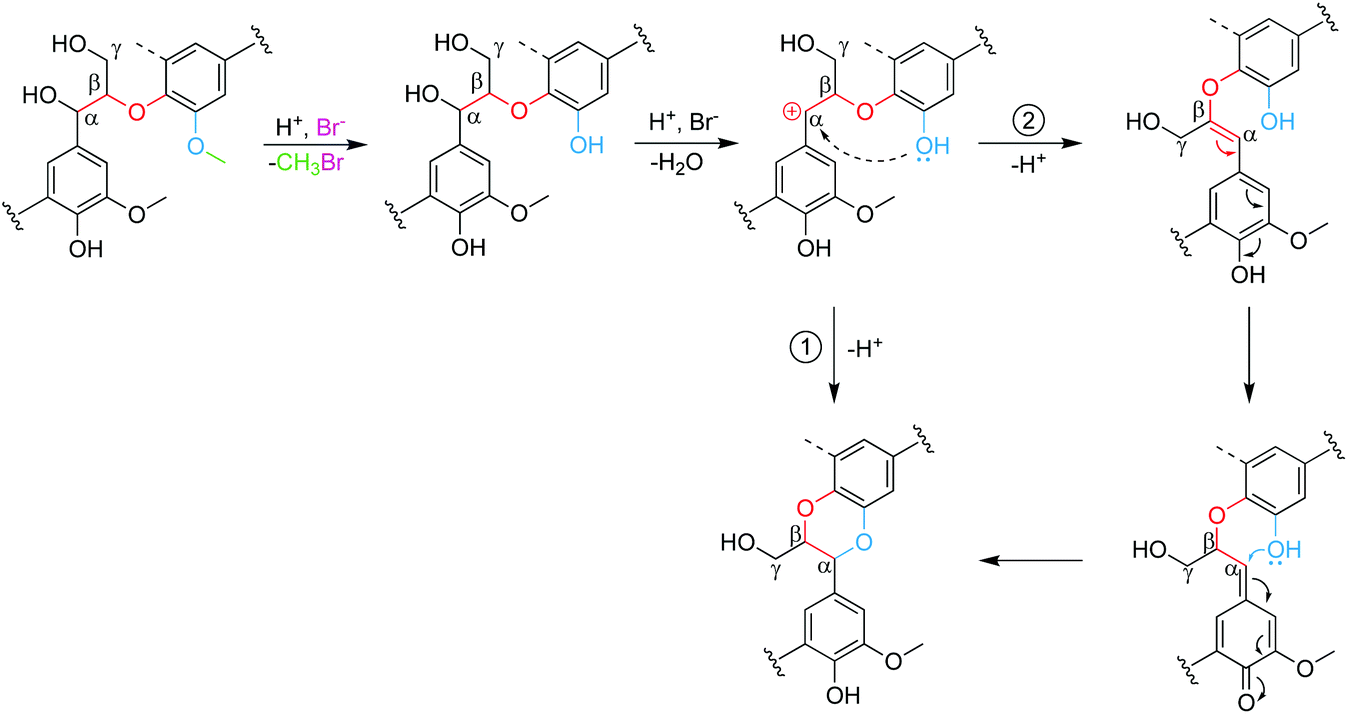 | ||
| Scheme 2 Proposed formation mechanism of the BD structure. | ||
Since the concentration of LiBr played an important role in the conversion of creosol, it is plausible that the demethylation of lignin is also greatly impacted by LiBr concentration. Therefore, different LiBr concentrations from 30% to 61% were applied for EPL demethylation (Fig. 1C and F). The cleavage degree of OMe and ArOH yield increased when LiBr concentration was increased from 30% to 61%. The average molecular weight also became higher with increased LiBr concentration, suggesting that the acidity of ACLB was likewise enhanced, which promoted the repolymerization of EPL.
To compare the lignin demethylation performance in the ACLB and the traditional HBr system, EPL was also treated in concentrated HBr at 100 °C (Fig. 1C and F). The results indicated that concentrated HBr was able to demethylate EPL as well, and the average molecular weights were nearly identical to those of ACLB treated EPL. The cleavage rate of OMe was 84%, but the selectivity to ArOH was only 22%, much lower than that in the ACLB system, which was probably related to the enhanced lignin condensation in the concentrated HBr. As observed above, creosol and 4-methylcatechol underwent severe condensation in concentrated HBr and generated humin-like precipitates.
To investigate the structural changes to EPL during the demethylation in ACLB, the original and demethylated EPL samples were analyzed by HSQC, as shown in Fig. 2 (aliphatic region), Fig. S3† (aromatic region), and Fig. S4† (lower LiBr concentrations). The original EPL contained abundant OMe groups, β-aryl ether A (H/G and S units), phenylcumaran B (β-5), resinol C (β-β), and characteristic p-hydroxybenzoate (also see the aromatic region in Fig. S3G†). After the demethylation in ACLB, residual OMe groups were still visible, while the characteristic signals of A, B, and C subunits were no longer visible (probably chemically shifted) in the aliphatic region due to the cleavage of the ether bonds in these structures (Fig. 2). The signal of the characteristic p-hydroxybenzoate unit, however, did not change after the demethylation (Fig. S3H†). Benzodioxane (BD) was also observed in the ACLB-treated EPL with 53% LiBr (Fig. 2B), as previously observed when wood was treated in a concentrated LiBr solution.46BD-α and BD-β were assigned at δC/δH 76.0/4.81 ppm and 78.4/4.05 ppm, respectively.57 The BD structure was observed in the EPL treated with 40% and 47% LiBr solutions as well, but not that with 30% LiBr (Fig. S4†), suggesting that the demethylation of OMe is the precondition of BD formation because 30% LiBr cannot efficiently demethylate lignin. The formation of the BD structure during the acid-catalyzed lignin depolymerization in the ACLB was reported in our previous study, but the formation mechanism of BD was not clearly addressed.46 Based on the evidence and observation from both the previous46 and the present studies, the BD formation mechanism is elucidated in Scheme 2. The OMe on the benzene ring involved in a β-O-4 linkage is demethylated into ArOH, and meanwhile, the α-carbon of another lignin unit in the same β-O-4 linkage is cationized by H+ and/or Br−.46 The ArOH then attacks the electropositive α-carbon to form the dioxane ring in BD.46,58,59 The ArOH can directly attack the α-cation to form the dioxane ring (Path 1). Alternatively, the α-cation is transformed first to enol ether and then to the quinone intermediate, which couples with the ArOH to form the BD (Path 2).
 | ||
| Fig. 2 Aliphatic regions of the HSQC spectra of original and demethylated EPL in DMSO-d6. (A) Original EPL; (B) demethylated EPL. Reaction conditions: 0.5 g lignin, 53 wt% LiBr, 6% HBr, 100 °C, and 4 h. | ||
BD was detected in the EPL treated in the ACLB at a high HBr concentration (6 wt% or about 1.3 mol L−1), while the previous observation of BD was in the lignins treated at a low acid concentration in the previous study (10–40 mmol L−1 HCl),46 indicating that the ether bonds in the BD structure were stable and could survive under the severe conditions tested, although dioxane was easily cleaved under the same conditions (entry 7 in Table 1). This can be attributed to the following reasons: (a) the ether bonds in BD are not cleavable from the aryl side, as discussed above; and (b) the Cα and Cβ of the alkyl side of the ether bonds in BD are both secondary carbons, which have higher steric hindrance, in particular, the Cα bonded to a benzene ring, to the SN2 substitution for cleaving the ether bonds, compared to the primary carbons in dioxane. By this means, BD can be treated as a “sealed” β-O-4 structure. Apparently, the formation of BD consumed some of the ArOH generated by the demethylation, which explained why ArOH did not increase proportionally with the demethylation degree, as discussed above. Hibbert's ketone (HK) was not detected in the ACLB-treated EPL as in the previous studies,43,45,46 possibly due to the higher acid concentration used in the present study, which promoted the aldol condensation of the ketone.46,60
Although EPL was very well dispersed in the ACLB system, the reaction was still heterogeneous (lignin retained undissolved), so the OMe groups inside lignin particles might be less accessible than those exposed on the surface. To dissolve EPL for a homogeneous reaction, acetic acid, ethanol, and acetone were tested as co-solvents for in the demethylation of EPL (Table S3†). All three co-solvents were miscible with the ACLB to give clear solutions at room temperature, and EPL was soluble in the three co-solvent-ACLB systems. It was found that acetic acid and acetone did not significantly affect the demethylation performance, suggesting that lignin could be demethylated efficiently in the ACLB even without dissolving the lignin. However, the lowest ArOH was detected when ethanol was used as a co-solvent (Table S3†). This is probably due to the etherification between ethanol and ArOH formed from the demethylation, which was supported by the detection of ethoxy groups in the treated lignin by the HSQC spectrum (Fig. S1D†) at δC/δH 64.6/3.44 ppm and 66.3/3.44 ppm. As discussed above, the ethyl-O-aryl bond was more difficult to be cleaved than the methyl-O-aryl bond due to the steric hindrance (Table 1). The re-etherification was probably a reason why both ArOH and AlkOH decreased when the reaction was extended to 24 h in Fig. 1B. Besides, the introduction of ethanol led to much lower average molecular weights of demethylated EPL probably because ethanol prevented the HK moieties generated from the cleavage of the β-O-4 bond from condensation. HSQC spectrum revealed that HK moieties (δC/δH 45.0/3.61 ppm and 67.6/4.17 ppm in Fig. S1D†) were only visible in the demethylated EPL in the ethanol-ACLB. HK was stabilized by ethanol probably via the acetalization of HK or the etherification of the enol form of HK. The signal of O-CH2 was observed in HSQC spectra, which suggested the existence of ethyl ether units (Fig. S1D†). The signals of HK were also exclusively observed in the aromatic region (Fig. S2D†) of the demethylated EPL in the ethanol-ACLB.
Demethylation of other lignins in the ACLB system
To evaluate the compatibility of the ACLB system, three other lignin samples (HKL, CSL, and ELPPL) were treated under the chosen conditions mentioned above for EPL. All three lignin samples were dispersed well in the system and gave homogeneous suspensions. 1H NMR results confirmed that the OMe content in three original lignins followed the order of HKL > ELPPL > CSL since hardwood lignin has more S units, while softwood and corn stover lignin contains more G and/or H units (Fig. S3†). As summarized in Fig. 3, after the treatment in the ACLB, the OMe groups in the lignin samples were effectively cleaved, and the ArOH groups increased correspondingly (Fig. 2A). Similar to EPL, Mn, and Mw of HKL, CSL, and ELPPL all increased after demethylation as a result of repolymerization, which was inevitable in the strongly acidic ACLB system. Different from EPL whose DPI decreased from 2.31 to 1.76, the PDI of HKL, CSL, and ELPPL increased from 1.35, 1.72, and 2.73 to 1.73, 2.25, and 3.00, respectively, after the demethylation.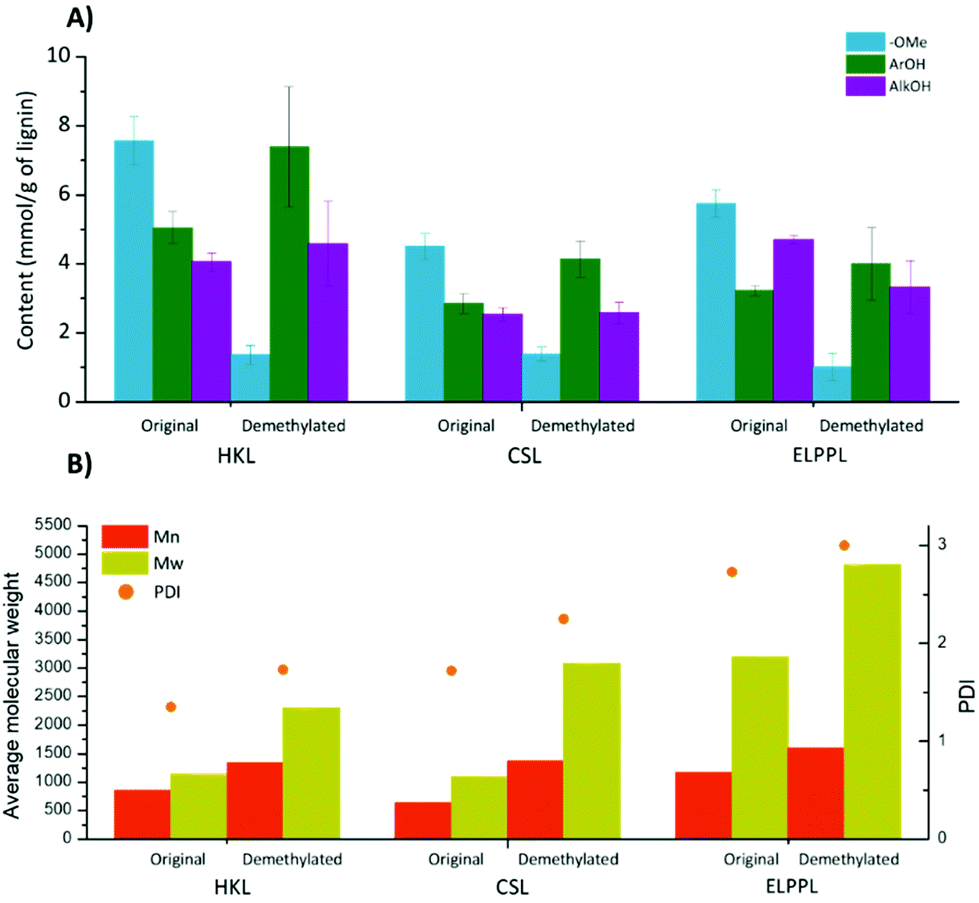 | ||
| Fig. 3 Demethylation of HKL, CSL, and ELPPL in ACLB. (A) OMe, ArOH, and AlkOH in the lignins quantitated by 1H NMR. (B) Mn, Mw, and PDI of the lignins estimated by GPC. Reaction conditions: 0.5 g lignin, 53 wt% LiBr, 6% HBr, 100 °C, and 4 h. All values are the average of triplicated tests (see Table S4† for details). | ||
To gain insights into the structural changes to HKL, CSL, and ELPPL during the demethylation, their HSQC spectra were recorded, as shown in Fig. 4 (aliphatic region) and Fig. S3† (aromatic region). Almost all the ether bonds in A, B, and C structures were cleaved (signals became invisible). Like EPL, the demethylation of HKL also led to the formation of BD structure (Fig. 4B) as HKL is hardwood lignin with more S units (Fig. S3B†). However, BD was not detected in the demethylated CSL and ELPPL. As shown in Scheme 2, the formation of BD requires the presence of the ArOH generated from demethylation and the rotation of the benzene ring for the ArOH to attack the α-carbon of another lignin unit. Hardwood lignin has more S units, which have more OMe and fewer interunit C–C linkages and thereby better mobility for the rotation to facilitate the formation of BD formation, than softwood and grass lignin. This is why BD was detected in the demethylated HKL but not CSL and ELPPL. HK (δC/δH at about 44.5 /3.67 ppm and 67.1/4.19 ppm) was not detected either in the demethylated HKL, CSL, and ELPPL, which further confirmed the instability of HK under the demethylation conditions used in this study.
 | ||
| Fig. 4 Aliphatic region of the HSQC spectra of original and demethylated lignin samples in DMSO-d6. (A) Original HKL; (B) demethylated HKL; (C) original CSL; (D) demethylated CSL; (E) original ELPPL; (F) demethylated ELPPL. Reaction conditions: 0.5 g lignin, 6.1 g LiBr (53 wt% in ACLB), 3.9 g water, 1 mL HBr, 100 °C, and 4 h. | ||
It is worth mentioning that the chemical shifts of S and G units greatly changed after the demethylation (Fig. S3†), probably due to the cleavage of the ether bonds in the A, B, and C structures. Notably, the signals of G2 in all four lignins were completely invisible probably due to the demethylation of ortho-methoxyl groups (Fig. S3†). Meanwhile, the characteristic signals of ferulate also shifted away, likely due to the removal of the unit during the demethylation (Fig. S3D†).
Insights into the cleavage of the ether bonds of lignin in the ACLB system
There are different types of ether bonds in lignin, including β-O-4, 4-O-5, α-O-4 in phenylcoumaran, α-O-γ in resinol, and methyl-O-aryl involving methoxyl groups. The results from the present and previous studies43–46 clearly indicate that the ether bonds of lignin except for 4-O-5 can be cleaved in the ACLB system. However, the cleavage mechanisms and the reaction conditions required are dependent on the ether bonds.Based on the reaction mechanism discussed above, the cleavage of these ether bonds would introduce bromine into lignin side chains. Also, some aliphatic hydroxyl groups in lignin could be substituted by bromide in this system. Indeed, a trace amount of bromine was detected in the ACLB-treated lignin. First, the demethylated lignin was analyzed by SEM-EDS, and the Lα signal of Br at 1.481 keV was observed (Fig. S5†). This is direct evidence of Br existence in the lignin. A negligible amount of Br was also detected in the lignin treated in the LiBr system under milder conditions in a previous study.43 Second, when the demethylated lignin was oxidized by H2O2 with H2SO4 as a catalyst, bromo- and dibromo-acetic acids were detected in the oxidation products by GC-MS (Fig. S6†), which were probably from the oxidation of the lignin sidechains. This provided additional evidence of the existence of Br in the treated lignin.
Can the β-O-4 bond be cleaved via the SN2 mechanism? Chemically, it is not impossible but not favorable because the steric hindrance at Cβ (secondary carbon in the middle of the lignin side chain) is very high for the SN2 substitution. If β-O-4 was primarily cleaved via the SN2 mechanism, BD and HK would not have been produced, which is certainly not the case and contradicts the results from this and the previous studies. Besides, the β-O-4 cleavage via the SN2 mechanism would have introduced a significant amount of bromine to the Cβ position because β-O-4 is the most abundant ether bonds in lignin, but this was not observed.
Conclusions
This work demonstrated the ether cleavage and lignin demethylation in acidic concentrated lithium bromide (ACLB) solution. The results revealed that the ACLB could universally cleave (dealkylate) ether compounds except for diaryl ethers. The system was also capable of demethylating lignin model compounds with methoxyl groups (methyl aryl ethers), leading to corresponding phenols. The cleavage of the ether bonds was completed via the mechanism of ether oxygen protonation followed by the SN2 substitution with bromide.The ACLB was also effective at demethylating real lignin and converting the methoxyl groups into phenolic hydroxyl groups. Under the conditions investigated, 69–82% of methoxyl groups in four lignins from different sources were demethylated. In addition to demethylation, the ACLB was able to cleave other ether bonds of lignin in β-O-4, β-5, and β-β structures except for the 4-O-5 bond in the diphenyl structure. The ether bonds were cleaved via the SN2 mechanism except for the β-O-4 bond, which was primarily cleaved via the benzyl cation and enol ether intermediates, leading to Hibbert's ketones. Some of the β-O-4 structures in hardwood lignin (S-type) were transformed into benzodioxane (BD) structures, which were stable in the ACLB system.
Compared with existing lignin demethylation methods, such as the concentrated HBr method, the ACLB method developed in this study required milder conditions (lower temperature and acid concentration), which would reduce the acid-induced lignin condensation. Besides, ACLB had a better selectivity in converting methoxyl to phenolic hydroxyl groups. A less condensed structure and more phenolic hydroxyl groups would certainly benefit the downstream applications of the demethylated lignin, such as heavy metal adsorbents, antioxidants, lignin-based resin adhesive, and feedstock for dicarboxylic acid production from lignin via oxidation.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project was supported by USDA NIFA (2018-67009-27902).References
- Y. Shao, Q. Xia, L. Dong, X. Liu, X. Han, S. F. Parker, Y. Cheng, L. L. Daemen, A. J. Ramirez-Cuesta, S. Yang and Y. Wang, Nat. Commun., 2017, 8, 1–9 CrossRef CAS.
- X. Zhou, L. J. Broadbelt and R. Vinu, in Adv. Chem. Eng., Elsevier, 2016, vol. 49, pp. 95–198 Search PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS.
- M. Jablonsky, M. Botkova and J. Kosinkova, Cellul. Chem. Technol., 2015, 49, 165–168 CAS.
- H. H. Nimz, D. Robert, O. Faix and M. Nemr, Holzforschung, 1981, 35, 16–26 CrossRef CAS.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS.
- K. M. Holtman, H. Chang and J. F. Kadla, J. Agric. Food Chem., 2004, 52, 720–726 CrossRef CAS.
- A. Guerra, I. Filpponen, L. A. Lucia and D. S. Argyropoulos, J. Agric. Food Chem., 2006, 54, 9696–9705 CrossRef CAS.
- K. M. Holtman, H. Chang, H. Jameel and J. F. Kadla, J. Wood Chem. Technol., 2006, 26, 21–34 CrossRef CAS.
- S. Wang, B. Ru, H. Lin, W. Sun and Z. Luo, Bioresour. Technol., 2015, 182, 120–127 CrossRef CAS.
- D. E. Bland and A. F. Logan, Biochem. J., 1965, 95, 515–520 CrossRef CAS.
- A. E. Kazzaz, Z. H. Feizi and P. Fatehi, Green Chem., 2019, 21, 5714–5752 RSC.
- A. M. Olsson and L. Salmén, Nord. Pulp Pap. Res. J., 1997, 12, 140–144 CAS.
- X. Pan, J. F. Kadla, K. Ehara, N. Gilkes and J. N. Saddler, J. Agric. Food Chem., 2006, 54, 5806–5813 CrossRef CAS.
- I. Sumerskii, T. Zweckmair, H. Hettegger, G. Zinovyev, M. Bacher, T. Rosenau and A. Potthast, RSC Adv., 2017, 7, 22974–22982 RSC.
- J. Li, J. Zhang, S. Zhang, Q. Gao, J. Li and W. Zhang, Polymers, 2017, 9, 428–445 CrossRef.
- H. Schroeder, Method For Recovering And Using Lignin In Adhesive Resins By Extracting Demethylated Lignin, US Pat., US5026808A, 1991 Search PubMed.
- H. Wang, T. L. Eberhardt, C. Wang, S. Gao and H. Pan, Polymers, 2019, 11, 1771–1787 CrossRef CAS.
- S. Wu and H. Zhan, Cellul. Chem. Technol., 2001, 35, 253–262 Search PubMed.
- Y. Song, Z. Wang, N. Yan, R. Zhang and J. Li, Polymers, 2016, 8, 209 CrossRef.
- B. Wang, J. Wen, S. Sun, H. Wang, S. Wang, Q. Liu, A. Charlton and R.-C. Sun, Ind. Crops Prod., 2017, 108, 72–80 CrossRef CAS.
- A. L. Pometto III, J. B. Sutherland and D. L. Crawford, Can. J. Microbiol., 1981, 27, 636–638 CrossRef.
- M. L. Álvarez-Rodríguez, C. Belloch, M. Villa, F. Uruburu, G. Larriba and J. R. Coque, FEMS Microbiol. Lett., 2003, 220, 49–55 CrossRef.
- S. H. Campion and I. D. Suckling, Appita J., 1998, 51, 209–212 CAS.
- T. R. Filley, G. D. Cody, B. Goodell, J. Jellison, C. Noser and A. Ostrofsky, Org. Geochem., 2002, 33, 111–124 CrossRef CAS.
- S. Zhao and M. M. Abu-Omar, Macromolecules, 2017, 50, 3573–3581 CrossRef CAS.
- Q. Mei, H. Liu, X. Shen, Q. Meng, H. Liu, J. Xiang and B. Han, Angew. Chem., Int. Ed., 2017, 56, 14868–14872 CrossRef CAS.
- N. A. David, Annu. Rev. Pharmacol., 1972, 12, 353–374 CrossRef CAS.
- J. Gierer, Wood Sci. Technol., 1980, 14, 241–266 CrossRef CAS.
- TAPPI, T 209 su-72 Methoxyl Content of Pulp and WoodTAPPI Test Methods, TAPPI Press: Atlanta, Georgia, 1972 Search PubMed.
- F. Kačík, D. Kačíková, T. Bubenikova and V. Veľková, Drewno, 2004, 47, 113–119 Search PubMed.
- V. Viswanatha and G. S. K. Rao, J. Chem. Soc., Perkin Trans. 1, 1974, 450–453 RSC.
- H. Lee, X. Feng, M. Mastalerz and S. J. Feakins, Org. Geochem., 2019, 136, 103894 CrossRef CAS.
- K. Sawamura, Y. Tobimatsu, H. Kamitakahara and T. Takano, ACS Sustainable Chem. Eng., 2017, 5, 5424–5431 CrossRef CAS.
- S. Zhao and M. M. Abu-Omar, Biomacromolecules, 2015, 16, 2025–2031 CrossRef CAS.
- P. P. Kulkarni, R. B. Mane, U. V. Desai and P. P. Wadgaonkar, J. Chem. Res., Synop., 1999, 394–395 RSC.
- M. E. Jung and M. A. Lyster, J. Org. Chem., 1977, 42, 3761–3764 CrossRef CAS.
- Y. Song, Z. Wang, N. Yan, R. Zhang and J. Li, Polymers, 2016, 8, 209–223 CrossRef.
- M. Ferhan, N. Yan and M. Sain, J. Chem. Eng. Process Technol., 2013, 4, 1000160 Search PubMed.
- X. Li, J. He and Y. Zhang, J. Org. Chem., 2018, 83, 11019–11027 CrossRef CAS.
- B. G. Harvey, A. J. Guenthner, W. W. Lai, H. A. Meylemans, M. C. Davis, L. R. Cambrea, J. T. Reams and K. R. Lamison, Macromolecules, 2015, 48, 3173–3179 CrossRef CAS.
- J. Tian, C. Yi, H. Fang, D. Sang, Z. He, J. Wang, Y. Gan and Q. An, Tetrahedron Lett., 2017, 58, 3522–3524 CrossRef CAS.
- N. Li, X. Pan and J. Alexander, Green Chem., 2016, 18, 5367–5376 RSC.
- C. G. Yoo, S. Zhang and X. Pan, RSC Adv., 2017, 7, 300–308 RSC.
- X. Yang, N. Li, X. Lin, X. Pan and Y. Zhou, J. Agric. Food Chem., 2016, 64, 8379–8387 CrossRef CAS.
- N. Li, Y. Li, C. G. Yoo, X. Yang, X. Lin, J. Ralph and X. Pan, Green Chem., 2018, 20, 4224–4235 RSC.
- X. Pan, D. Xie, R. W. Yu, D. Lam and J. N. Saddler, Ind. Eng. Chem. Res., 2007, 46, 2609–2617 CrossRef CAS.
- G. Pruckmayr and T. K. Wu, Macromolecules, 1978, 11, 662–668 CrossRef CAS.
- P. A. Delaney, R. A. W. Johnstone and I. D. Entwistle, J. Chem. Soc., Perkin Trans. 1, 1986, 1855–1860 RSC.
- B. H. Kwant, J. Labelled Compd. Radiopharm., 1980, 17, 841–859 CrossRef CAS.
- J. He, C. Zhao and J. A. Lercher, J. Am. Chem. Soc., 2012, 134, 20768–20775 CrossRef CAS.
- C. Zhu, J. Cao, X. Zhao, T. Xie, J. Ren and X. Wei, J. Energy Inst., 2019, 92, 74–81 CrossRef CAS.
- J. A. Duffy and M. D. Ingram, Inorg. Chem., 1978, 17, 2798–2802 CrossRef CAS.
- S. Sen, J. D. Martin and D. S. Argyropoulos, ACS Sustainable Chem. Eng., 2013, 1, 858–870 CrossRef CAS.
- X. Pan and Y. Sano, Holzforschung, 1999, 53, 590–596 CAS.
- J. Li, G. Henriksson and G. Gellerstedt, Bioresour. Technol., 2007, 98, 3061–3068 CrossRef CAS.
- F. Chen, Y. Tobimatsu, D. Havkin-Frenkel, R. A. Dixon and J. Ralph, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1772–1777 CrossRef CAS.
- J. Ralph, C. Lapierre, J. M. Marita, H. Kim, F. Lu, R. D. Hatfield, S. Ralph, C. Chapple, R. Franke and M. R. Hemm, Phytochemistry, 2001, 57, 993–1003 CrossRef CAS.
- J. Ralph, C. Lapierre, F. Lu, J. M. Marita, G. Pilate, J. Van Doorsselaere, W. Boerjan and L. Jouanin, J. Agric. Food Chem., 2001, 49, 86–91 CrossRef CAS.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02581j |
| This journal is © The Royal Society of Chemistry 2020 |