
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-catalytic oxidative depolymerization of lignin in perfluorodecalin to produce phenolic monomers†
Parinaz
Hafezisefat
a,
Jake K.
Lindstrom
a,
Robert C.
Brown
 *a and
Long
Qi
*b
*a and
Long
Qi
*b
aDepartment of Mechanical Engineering, Iowa State University, Ames, Iowa 50011, USA. E-mail: rcbrown3@iastate.edu
bU.S. DOE Ames Laboratory, Iowa State University, Ames, Iowa 50011, USA. E-mail: lqi@iastate.edu
First published on 15th September 2020
Abstract
We demonstrate for the first time non-catalytic, oxidative cracking with molecular oxygen (O2) to depolymerize native lignin into oxygenated phenolic monomers. Maximum monomer yield of 10.5 wt% was achieved at 250 °C after only 10 min of reaction and included vanillin, syringaldehyde, vanillic acid, and syringic acid. High rates of oxidation are attributed to the use of perfluorodecalin as solvent. Perfluorodecalin is a perfluorocarbon (PFC), characterized by their chemical stability and exceptionally high solubility for O2. Monomer yields were typically five-fold higher in perfluorodecalin compared to solvents more commonly employed in lignin conversion, such as methanol, butanol, acetonitrile, and ethyl acetate. Phenolic monomer production in perfluorodecalin favors high temperatures and short reaction times to prevent further oxidation of the produced monomers. Lignin oil obtained under oxidative conditions in perfluorodecalin showed lower molecular weight and smaller polydispersity compared to other solvents. Increasing the reaction time further decreased the molecular weight, while increasing reaction time in an inert atmosphere increased the molecular weight of the lignin oil. High concentrations of O2 in perfluorodecalin not only increased lignin depolymerization but suppressed undesirable condensation reactions. Depolymerization is likely initiated by thermally induced homolytic cleavage of ether linkages in lignin to form phenoxy and carbon-based radicals. These radicals bind with O2 as a radical scavenger and further react to form phenolic monomeric products rather than repolymerizing to large oligomers. The PFC process was scaled from 5 mL to 250 mL without any loss of yield. Because most organic compounds are not soluble in perfluorodecalin, recycling is easily achieved via liquid–liquid separation.
Introduction
Lignin, a heterogeneous aromatic biopolymer that comprises 15 to 30 wt% of lignocellulosic biomass, is mainly composed of p-coumaryl, coniferyl, and sinapyl alcohols connecting by C–O and C–C bonds. Lignin, as the sole component of biomass containing aromatic structures, is a promising feedstock for renewable chemicals.1 However, lignin is produced chiefly as the by-product of industries focused on recovering carbohydrates, with little attention directed toward preserving lignin for chemical synthesis. For instance, in Kraft pulping, responsible for over 90% of all chemical pulps,2 the high alkalinity of the process converts lignin into a condensed and more recalcitrant form known as technical lignin,3 which relegates it to relatively low-value applications such as boiler fuel for heat and power generation. To retain the intrinsic value of virgin lignin, biorefineries will have to employ either mild depolymerization processes, such as ammonia-based fractionation4 and low-temperature organosolv techniques,5 or employ processes that continuously stabilize phenolic products as they are released from biomass. This second approach, sometimes referred to as the lignin-first strategy for deconstructing lignocellulose,3 is receiving increasing attention, with reductive catalytic fractionation (RCF)6 and formaldehyde-assisted fractionation7 as prominent examples.Reductive depolymerization of lignin employs redox catalysts to break β-O-4 and α-O-4 ether linkages2 with molecular hydrogen or hydrogen donor molecules as the reducing agent to stabilize deconstruction products. However, most of these reductive methods have been ineffective in breaking carbon–carbon bonds, and hydrogen is a relatively expensive input to the process. Alternatively, oxidative deconstruction of lignin using molecular oxygen (O2) or even air is potentially much cheaper than reductive depolymerization, although this possibility is just emerging.8 Interestingly, nature has harnessed oxidation as the main pathway to biologically degrade lignin, employing enzymes at ambient temperatures with molecular oxygen from air.9
Lignin oxidation can produce a range of functionalized chemicals of significant economic value such as aromatic acids, aromatic aldehydes, and aliphatic carboxylic acids.10,11 Several challenges stand in the way to its practical implementation. Strong oxidants, including hydrogen peroxide,12 ozone,13 and peroxy acids,14 and expensive and difficult to recycle catalysts15,16 are commonly required for effective oxidation of lignin. Recently, lignin oxidation by O2 has attracted increasing attention. However, O2 is a weak oxidizing agent, requiring either highly acidic17 or alkaline18 reaction conditions to catalyze the oxidation of lignin.10 For example, Beckham and co-workers systematically optimized the alkaline aerobic oxidation of lignin which is the heart of alkaline pulping process. In the presence of 2 M NaOH and 5 bar O2 in 30 min at 150 °C, maximal 14% and 23% yields of monomers were obtained from whole poplar biomass with or without additional copper sulfate respectively.19 The generation of highly alkaline wastewater when processing under basic conditions and the possibility of oxidizing organic solvents under acidic processing conditions are major challenges associated with oxidative depolymerization using O2. Moreover, many organic solvents used in deconstruction of lignin20 are highly flammable and explosive, making their use in the presence of oxygen problematic.
We hypothesize that radicals generated during high temperature homolytic cleavage of lignin ether bonds (phenoxy radicals and carbon-based radicals) can catalyze the oxidation of lignin without the need for acid or base catalysts. This hypothesis is motivated by the observation that thermal depolymerization of lignin under inert conditions shows clear evidence of homolytic cleavage to produce lignin radicals.21,22 Based on the bi-radical nature of oxygen, we further hypothesize that a balance between O2 and freshly generated carbon-based radicals from lignin suppresses undesirable repolymerization reactions to generate carbon–carbon bonds23 which is well established for radical polymerization chemistry.24
To investigate these questions, a solvent with high solubility toward both O2 and lignin is desirable; unfortunately, as described below, these are mutually exclusive criteria. Three major interactions influence the solubility of one substance in another. The most general of these is the nonpolar interaction, which is known as the dispersion interaction (δD). Since this interaction arises from atomic forces, it is a feature of all types of molecules. The second is dipolar interaction (δP) caused by permanent dipole forces between molecules. Intermolecular hydrogen bonding (δH) is the third major interaction.25 From these three so-called Hansen solubility parameters (HSPs), the total solubility parameter, δt, and solubility parameter distance, Ra, are calculated:
| (Ra)2 = 4(δD2 − δD1)2 + (δP2 − δP1)2 + (δH2 − δH1)2 | (1) |
| δt2 = δD2 + δP2 + δH2 | (2) |
To screen solvents for their ability to dissolve O2, Ra was calculated for several perspective solvents including water, acetonitrile, and butanol (Tables S1 and S2†). Solvents frequently used in lignin utilization have large Ra relative to O2 resulting in low O2 solubility. On the other hand, Ra for perfluorodecalin relative to O2 is very small (2.9 MPa0.5), making it an intriguing solvent for oxidative deconstruction of lignin. Perfluorodecalin is a type of perfluorocarbon (PFC) containing only carbon and fluorine atoms. Indeed, as shown in Fig. 1, PFCs dissolve 40–50 vol% oxygen at room temperature and pressure, which is several-fold higher than most other potential solvents.
 | ||
| Fig. 1 Solubility of oxygen (vol%) in PFCs at ambient temperature and atmospheric pressure (estimated by solubility parameter distance of oxygen and solvents). | ||
Perfluorodecalin is a non-toxic, non-flammable, thermally stable, non-bio-accumulating and non-ozone-depleting solvent which is used as artificial blood. Although perfluorocarbons in general are classified as greenhouse gases, perfluorodecalin has a high boiling point (142 °C) in contrast to the very low boiling points of PFCs (typically much below 0 °C) used in refrigeration and semi-conductor manufacturing. Perfluorodecalin's high boiling point mitigates its fugitive emissions. If employed in an industrial process, perfluorodecalin would be recovered in a sealed decantation process to protect against fugitive emissions. Industrial decanting equipment ranges from simple settling tanks, in which the heavy perfluorodecalin would be drained from the bottom, or decanter centrifuges, for continues decanting.
The high gas solubility of PFCs arises from their feeble van der Waals forces. Perfluorocarbons are more stable and chemically more inert than their hydrocarbon counterparts. In contrast to highly flammable hydrocarbons, PFCs are so inert that they can be used as fire extinguishants, and are resistant to thermal decomposition up to 400 °C.26 Their high O2 solubility and low toxicity27,28 make them attractive as artificial blood.29 As a reaction media, the low solubility of most organic compounds in PFCs facilitates product recovery, a fact that has been exploited in a biphasic catalytic system for methane oxidation.30,31
For polymers like lignin, only solvents within a certain range of HSPs can dissolve them. This range is often represented as Hansen sphere and only solvents within this space are likely to dissolve the polymer. In a three-dimensional HSP space, the center point of a solute's Hansen sphere corresponds to its three HSP values and RO (interaction radius) as the radius of the sphere that encompasses all good solvents of the polymer while excluding the poor solvents.25 Hansen solubility sphere of lignin is illustrated in Fig. S1.† The Ra/RO ratio is called the RED number. RED numbers less than 1.0 show the high affinity of solvent and solute and higher RED numbers show lower affinities.25Fig. 2 is a two-dimensional projection of the Hansen sphere for lignin (dashed circle) and several organic solvents (dots) in the δH−δP plane. Methanol and butanol are closer to the center of lignin circle, with Ra ranging between 8 to 16 MPa0.5, indicating that they are more likely to dissolve lignin than the other solvents. Acetonitrile and ethyl acetate, with Ra ranging between 16 to 24 MPa0.5, are better solvents than perfluorodecalin, benzene, and water, but not as good as methanol and ethanol. In general, solvents with high lignin solubility potential have low O2 solubility and vice versa.
 | ||
| Fig. 2 Hansen solubility parameters of lignin and solvents25 (the dashed circle is the maximum circumference of the Hansen solubility sphere in the δH–δP plane for lignin). | ||
To explore our hypotheses about non-catalytic oxidation of lignin and the role of O2 in suppressing undesirable repolymerization reactions, a solvent with high O2 solubility is desired. We have selected perfluorodecalin as our reaction media despite its poor solubilization of lignin. To compare its performance to more traditional solvents, we also performed experiments with two polar protic solvents (methanol and ethanol) and two polar aprotic solvents (acetonitrile and ethyl acetate). In a typical experiment, ball milled red oak powder and a solvent were sealed in a batch reactor, purged and pressurized at ambient temperature with O2 to the desired partial pressure (50–300 psi) and then further pressurized with molecular nitrogen (N2) to reach a total pressure of 300 psi. The reactor was heated to the desired temperature for the desired reaction time followed by cooling and recovery of liquid and solid products. The liquid was extracted with acetone and perfluorodecalin was recycled for other experiments. The acetone-extracted lignin oil was then analyzed by gas chromatography with flame ionization detection or mass spectrometry (GC-FID and GC-MS) and gel permeation chromatography (GPC) and HSQC NMR.
Results and discussion
Solvent effect of perfluorodecalin
The effectiveness of perfluorodecalin in promoting oxidative degradation of lignin was benchmarked against four organic solvents at 250 °C and 300 psi O2 pressure for 10 min of reaction time. Phenolic monomer yields were quantified by calibrating GC for prominent products, which are depicted in Fig. 3. Other products are illustrated in Fig. S2–S6.†Fig. 4 illustrates the gas chromatogram of lignin oil obtained using different solvents. The spectra for bio-oil produced in perfluorodecalin contains significantly more and larger peaks in the phenolic region (20–50 min) compared to bio-oil from other solvents. Perfluorodecalin, although a poor solvent for lignin and lignin-derived products, yielded 10.5 wt% of phenolic monomers. In contrast, acetonitrile, ethyl acetate, methanol, and butanol, which are exceedingly more effective in solubilizing lignin and its products, produced only 2.6, 1.0, 2.6, and 2.5 wt% of phenolic monomers, respectively. These counterintuitive results indicate that increasing solubility of oxygen can strongly enhance the rate of oxidative lignin disassembly. The important role of oxygen is evident by comparing these results to those for lignin disassembly in a N2 atmosphere. Yield of phenolic monomers in perfluorodecalin was drastically lower compared to deconstruction in O2 atmosphere, only 1.0 wt%. In the absence of O2, the major monomeric product was cinnamaldehyde, an intermediate in the biosynthesis of lignin.32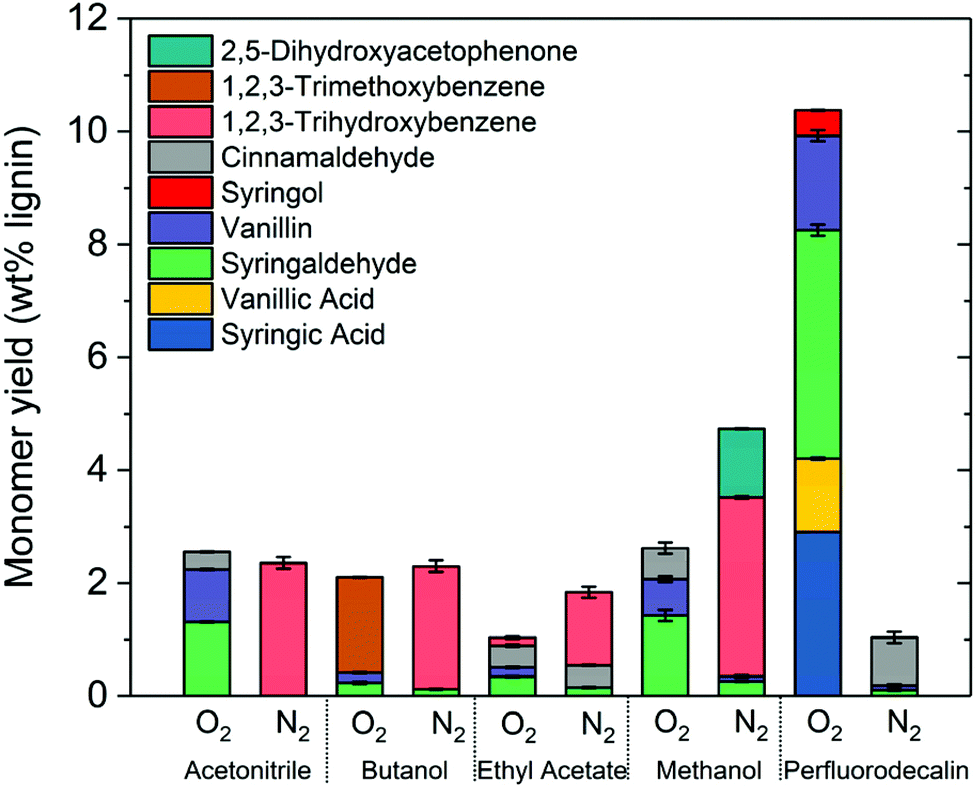 | ||
| Fig. 3 Comparing aromatic monomer yields from lignin depolymerization for different solvents in nitrogen and oxygen environments (250 °C, 300 psi, 10 min reaction time). | ||
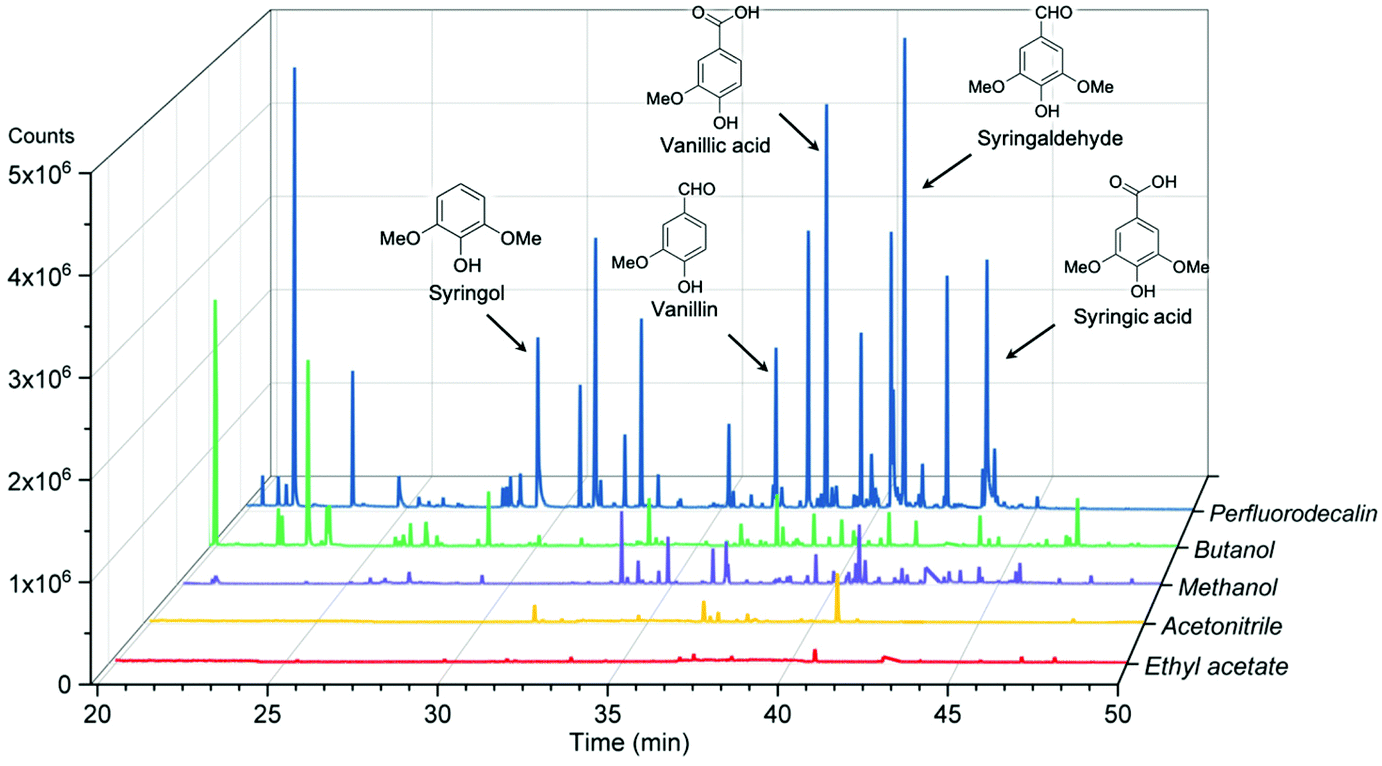 | ||
| Fig. 4 Gas chromatograms of acetone-extracted lignin oils produced from oxidation of red oak in different solvents at 250 °C and 300 psi O2 pressure for 10 min reaction time. | ||
In contrast, the major monomeric products in the presence of O2 included benzaldehydes (1.7 wt% vanillin and 4.1 wt% for syringaldehyde) and benzoic acids (1.3 wt% for vanillic acid and 2.9 wt% for syringic acids). No cinnamaldehyde was detected, suggesting enhanced oxidation of intermediates after the lignin was thermally decomposed. Syringol was also detected, yielding about 0.5 wt%, possibly formed from the thermally induced decarbonylation of benzaldehydes or decarboxylation of benzoic acids.
For all other solvents, the major product for reaction in the absence of oxygen was 1,2,3-trihydroxybenzene. Monomer yields were either not improved or even decreased when O2 was admitted to these other solvent processes. Besides syringaldehyde and vanillin, cinnamaldehyde was found in all cases suggesting insufficient oxygen dissolved in these other solvents.
In contrast to other studies on oxidative depolymerization of lignin, we achieved significant yields of phenolic monomers in only 10 min without catalysts (acidic, basic, or transition metals) by operating at high temperatures, which induced thermal generation of radicals from lignin that further reacted with O2 dissolved in perfluorodecalin. Non-catalytic lignin depolymerization via hydrogenolysis in ethanol/isopropanol mixtures has been reported to produce monomers at high yield.33 However, to the best of our knowledge, effective non-catalytic, reagent-free oxidation of lignin has not been previously reported in the literature. From a safety standpoint, this was achieved without the use of flammable, peroxide-generating solvents. In comparison, Welton et al.34 and Prado et al.35 oxidized lignin in ionic liquids with O2 and hydrogen peroxide, but monomer yield after 5 h at 100 °C was less than 1 wt% and the bio-oil was contaminated with ionic liquid cations. Voitl et al.17 oxidized Kraft lignin in methanol/water (170 °C, 20 min, 5 bar pressure) with phosphomolybdic acid as catalyst to yield 5.0 wt% phenolic monomers. Deng et al.20 oxidized organosolv lignin in methanol (185 °C, 24 h, 1 bar pressure) with Pd/ceria as catalyst to produce 7.6 wt% phenolic monomers.
Thermal degradation of lignin without acid/base catalysts is initiated by lignin-derived radicals. We previously have shown that thermal degradation of lignin can produce radicals.21 The presence of cinnamaldehyde observed under N2 is also an indicator for the thermal decomposition of lignin model compounds and lignin via a radical-based mechanism.36,37 The hydroxyl and methoxy groups in lignin promote the formation of stable radicals. To demonstrate this, we used benzyl phenyl ether (BPE) as a model compound containing an α-O-4 lignin ether linkage. Only 4% conversion for BPE was observed for 10 min of reaction time at 250 °C and 300 psi O2. The absence of substantial oxidative cleavage arises from the failure to efficiently generate radicals from non-hydroxylated aryl ethers. Therefore, cleavage of lignin ether linkages generates radicals stabilized by hydroxylated phenyl rings,22 and phenoxy and carbon-based radicals drive non-catalytic oxidation of lignin.
Promoting lignin deconstruction while slowing monomer condensation
Experiments on the effect of reaction time (5, 10, and 20 min) on molecular weight distribution of the bio-oil produced in perfluorodecalin under N2 and O2 atmospheres provides insight into the role of oxygen in promoting lignin deconstruction while also slowing monomer condensation. In Fig. 5, the GPC of lignin oil produced in perfluorodecalin under an O2 atmosphere show two major signals in the low MW region, suggesting successful oxidative degradation of lignin ether linkages by O2. The signals at around 200 Da and 250 Da correspond to benzaldehydes and benzoic acids, respectively, as confirmed by authentic samples. Under N2, the bio-oil was mostly composed of oligomers with only small amounts of monomers produced by anaerobic thermal decomposition.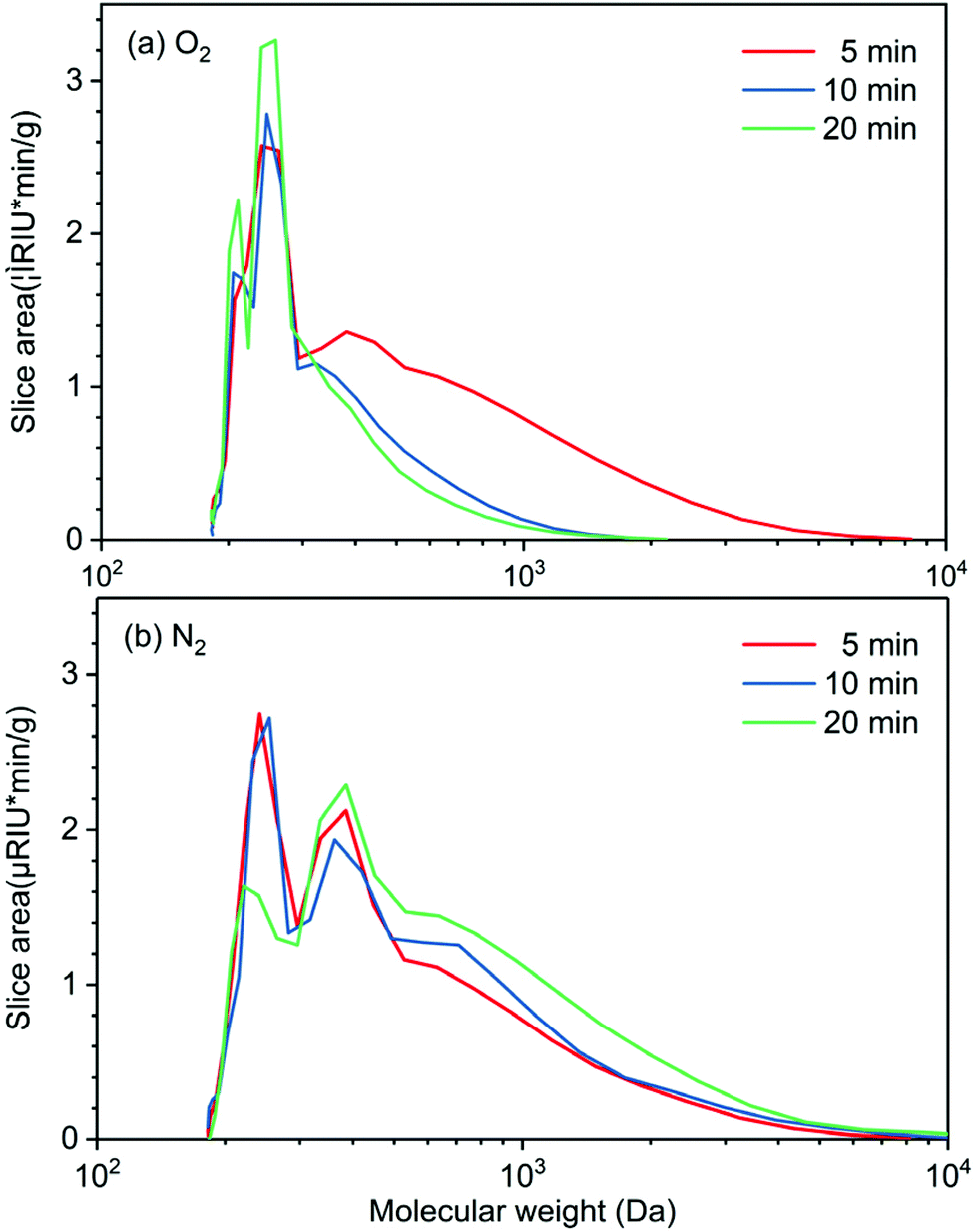 | ||
| Fig. 5 Molecular weight distribution of lignin oil produced from lignin depolymerization under (a) O2 atmosphere and (b) N2 atmosphere for several reaction times. Reaction conditions: 100 mg red oak, 5 ml perfluorodecalin, 250 °C, 300 psi. | ||
In the presence of O2, increasing reaction time further decreased the intensity of high MW regions and increased monomer peaks, as shown in Fig. 5a and Table 1. In contrast, the progression with time under N2 shifts the MW of lignin oil toward larger molecules, suggesting a condensation process in the absence of O2 arising from the irreversible formation of C–C bonds.
| Gas | Time (min) | M n | M w | PD |
|---|---|---|---|---|
| O2 | 5 | 323 | 511 | 1.58 |
| 10 | 272 | 314 | 1.16 | |
| 20 | 256 | 287 | 1.12 | |
| N2 | 5 | 334 | 505 | 1.52 |
| 10 | 347 | 557 | 1.65 | |
| 20 | 383 | 661 | 1.73 | |
GPC profiles of lignin oils produced in various solvents in the presence of O2 are depicted in Fig. 6. Lignin oils produced in alcohols, methanol, and butanol are primarily composed of high molecular-weight species centered at ∼3000 Da, reflecting the fact that short-chain alcohols are good solvents for lignin dissolution and possible participation in solvolysis. Ethyl acetate and acetonitrile are polar aprotic solvents, delivering bio-oils of similar distribution, centered at ∼400 Da. None of the solvents produced appreciable quantities of monomeric products, in agreement with the GC analysis in Fig. 4. Mn, Mw, and PD of these lignin oils are listed in Table S3.† These results indicate that O2 solubility plays a crucial and dominant role in oxidative depolymerization of lignin. The time-dependent GPC results suggest that perfluorodecalin is an outstanding carrier of O2 and boosts thermal oxidation of lignin via kinetic control. As a result of enhanced oxidation, condensation of the intermediate products of depolymerization was greatly reduced.
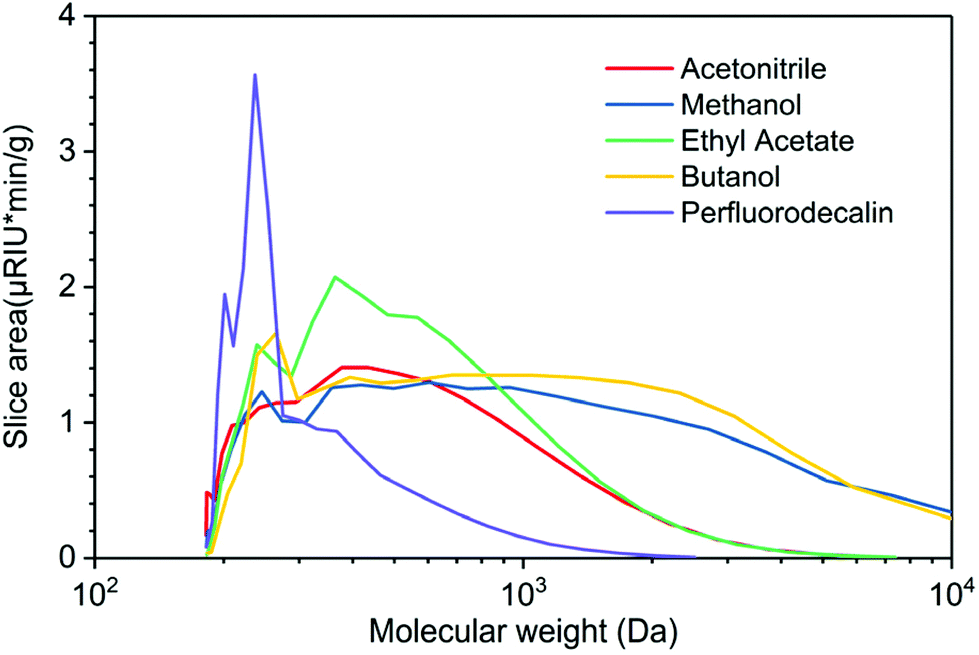 | ||
| Fig. 6 Molecular weight distribution of lignin oil produced from lignin oxidation in perfluorodecalin, acetonitrile, ethyl acetate, methanol, and butanol. Reaction conditions: 100 mg red oak, 5 ml solvent, 250 °C, 300 psi O2, for 10 min reaction time. | ||
Oxidation pathway in perfluorocarbons
To further understand the oxidation mechanism, experiments were performed at various combinations of reaction temperatures and reaction times. At 200 °C and very short reaction times, phenolic monomer yield was very low (Fig. 7). For example, monomer yield was only 1.2 wt% after 10 min. Increasing the reaction time to 60 min enhanced monomer yield to around 4.5 wt%. By increasing the temperature from 200 to 250 °C, total monomer yield significantly increased after 10 min of reaction, increasing from 1.2 wt% to 10.5 wt%. However, phenolic monomer yield at 250 °C actually decreased for reaction time greater than 10 min. We suspect that phenolic monomers like vanillic acid and syringic acids experienced deeper oxidation to light gases such as H2O, CO2, and CO. However, we were not able to confirm this suspicion since we could not collect gaseous products from the experiments. | ||
| Fig. 7 Phenolic monomer yields from lignin oxidation in perfluorodecalin at various temperatures and reaction times. Reaction conditions: 100 mg red oak, 5 ml perfluorodecalin, 300 psi oxygen pressure. | ||
Monomer yield suffered as temperature increased to 300 °C, suggesting that the rate of deep oxidation of monomeric products was increasing faster than the rate of lignin depolymerization. Notably, PFC reaches the supercritical state above 293 °C.38 Under supercritical conditions, rates of diffusion and reaction could be significantly altered, which could be another reason monomer yield decreased at 300 °C.
To further study the role of O2 in mitigating condensation reactions, the effect of O2 pressure on the yield of monomeric products was investigated. Oxidation was performed under O2 partial pressures between 50 and 300 psi at 250 °C. Under N2, the monomeric yield was very low, and almost no oxidation products were produced (vide supra, Fig. 3). However, under the same conditions of temperature and time, increasing oxygen partial pressure increased monomer yield from 6.0 wt% at 50 psi to 10.5 wt% at 300 psi (Fig. 8). Under increasing O2 pressure, vanillin and syringaldehyde yields remained almost constant, but vanillic acid and syringic acid yields increased. Because vanillic acid and syringic acid are products of the oxidation of vanillin and syringaldehyde, respectively, their increases show that vanillin and syringaldehyde are being oxidized rather than condensed as O2 partial pressure is increased.
 | ||
| Fig. 8 Phenolic monomer yield from oxidation of red oak in perfluorodecalin at various starting oxygen partial pressures. Reaction conditions: 250 °C, 300 psi total pressure, for 10 min reaction time. | ||
The positive effect of O2 on monomer yield can be explained by the specific interaction of carbon-based radicals with O2. Free radical polymerization (FRP) is the process by which radicals catalyze polymerization. FRP is initiated from radicals and propagates by the addition of monomers (such as olefins) to the radicals to form C–C bonds, thus growing the polymer chain. Termination of radical polymerization requires an inhibitor. Molecular oxygen, a bi-radical, is a common inhibitor in FRP.24 The active end of the growing polymer chain can react with O2, possibly producing a peroxy radical, which is much less susceptible to further reactions with monomers to form O–C bonds.24
Radicals generated from thermal depolymerization of lignin can directly react with O2 to form oxidation products instead of condensing as occurs in inert environments (Fig. 9a). However, under N2 (Fig. 9b), the carbon-based radicals will be more easily coupled via repolymerization reactions, which eventually reduces monomer yield and increases the MW of the lignin oil (vide ante). If O2 can be transferred from the gas phase to the liquid phase at sufficient rates, it can serve as both depolymerization promoter and capping agent and enhance the formation of phenolic monomers.
 | ||
| Fig. 9 Proposed mechanism of non-catalytic oxidation of lignin. (a) Monomers stabilized by O2 and (b) undesirable lignin condensation products in absence of O2. | ||
The lignin oil was further characterized by HSQC NMR (Fig. 10). As one of the few techniques to deliver structural insights for oligomers, HSQC NMR can be applied to directly identify chemical changes to the interunit linkages in lignin,39 while GPC only provides information on molecular weight. The lignin oil obtained under N2 mainly showed guiacyl units (G5/6) and syringyl (S2/6) units. Lignin oil obtained under O2 contained less guaicyl and syringyl units. Instead, oxidized guiacyl units (G′2) and oxidized syringyl units (S′2/6) increased significantly with the emergence of α-ketone structures.
 | ||
| Fig. 10 HSQC NMR spectra of lignin oils obtained under 300 psi of (a) N2 and (b) O2. Reaction conditions: 100 mg red oak, 5 ml perfluorodecalin, 250 °C, 10 min reaction time. | ||
Fig. 10 displays the aldehyde region for δH/δC in the ranges of 9–10.2 ppm and 188–195 ppm. Lignin oil generated under N2 showed only small areas of benzaldehyde and cinnamaldehyde end groups, while lignin oil obtained under oxygen showed a significant region for benzaldehyde due to syringaldehyde and vanillin production. Cinnamaldehyde radicals are known to initiate condensation reactions.40 As the NMR data illustrates, the oligomer end group with cinnamaldehyde moiety disappeared in the presence of O2. As shown in Fig. 10, carbohydrate-derived products including levoglucosan are also produced under oxidative conditions (δH/δC in the ranges of 3–5 ppm and 50–80 ppm). Some of them have been also identified by GC-MS (Fig. S2–S6†).
Kinetic control of lignin oxidation in perfluorocarbons
Oxidative depolymerization in perfluorocarbons is a transformative approach to lignin utilization. The high oxygen solubility of perfluorodecalin enhances the reaction of lignin-derived radicals with O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 which encourage lignin depolymerization and phenolic monomer stabilization. Perfluorodecalin accelerates O2 diffusion from the head space of the reactor into the liquid medium, enhancing selectivity of lignin depolymerization toward oxidation products.41
23 which encourage lignin depolymerization and phenolic monomer stabilization. Perfluorodecalin accelerates O2 diffusion from the head space of the reactor into the liquid medium, enhancing selectivity of lignin depolymerization toward oxidation products.41
In a mass transfer limited system, illustrated in Fig. 11a, the gas–liquid interface is rich in O2, while the bulk of the liquid remains O2-deprived because of low O2 solubility and high consumption. In this circumstance highly reactive radicals from lignin are expected to readily repolymerize and produce condensed oligomeric lignin oil of high MW. However, in a kinetically controlled system, illustrated in Fig. 11b, enough O2 diffuses into the bulk liquid to react with lignin radicals and produce less reactive radicals. These oxygenated radicals further transform to produce oxidation products like benzaldehydes and benzoic acids and suppress repolymerization reactions.
 | ||
| Fig. 11 The influence of mass transfer on the selectivity of lignin oxidation by O2. (a) Mass transfer-controlled system and (b) kinetically controlled system. | ||
Design of PFC-enabled lignin oxidation process
Most of the experiments described in this paper were performed in small reactors (total volume: 7 ml) loaded with 100 mg biomass and 5 mL perfluorodecalin. The test with the highest monomer yield (100 mg red oak, 5 ml perfluorodecalin, 250 °C, 300 psi O2, 10 min) was repeated in a 500 mL autoclave stirred reactor with 4 g biomass and 250 mL perfluorodecalin to evaluate the feasibility of scaling up the process. In this 50-fold scale-up (based on solvent volume), lignin oil obtained in two replicated trials produced the same monomer yields as smaller-scale tests. The results show that the PFC-enabled non-catalytic lignin oxidation can be scaled without mass transfer limitation for O2, supporting the prospect for industrial applications.The scheme of the proposed oxidative depolymerization process is illustrated in Fig. 12. Perfluorodecalin is stable at reaction conditions (≤300 °C) in this study and PFCs are not miscible with most organic compounds, which allows simple, facile liquid–liquid separation for recovering and recycling PFCs without emulsion formation (Fig. S7†). Recovered perfluorodecalin was extremely pure: no contaminants or decomposition of perfluorodecalin were identified by GC or NMR in the recovered PFC (Fig. S8–S10†). Recycled perfluorodecalin was used for the second runs performed for the purpose of statistical analysis.
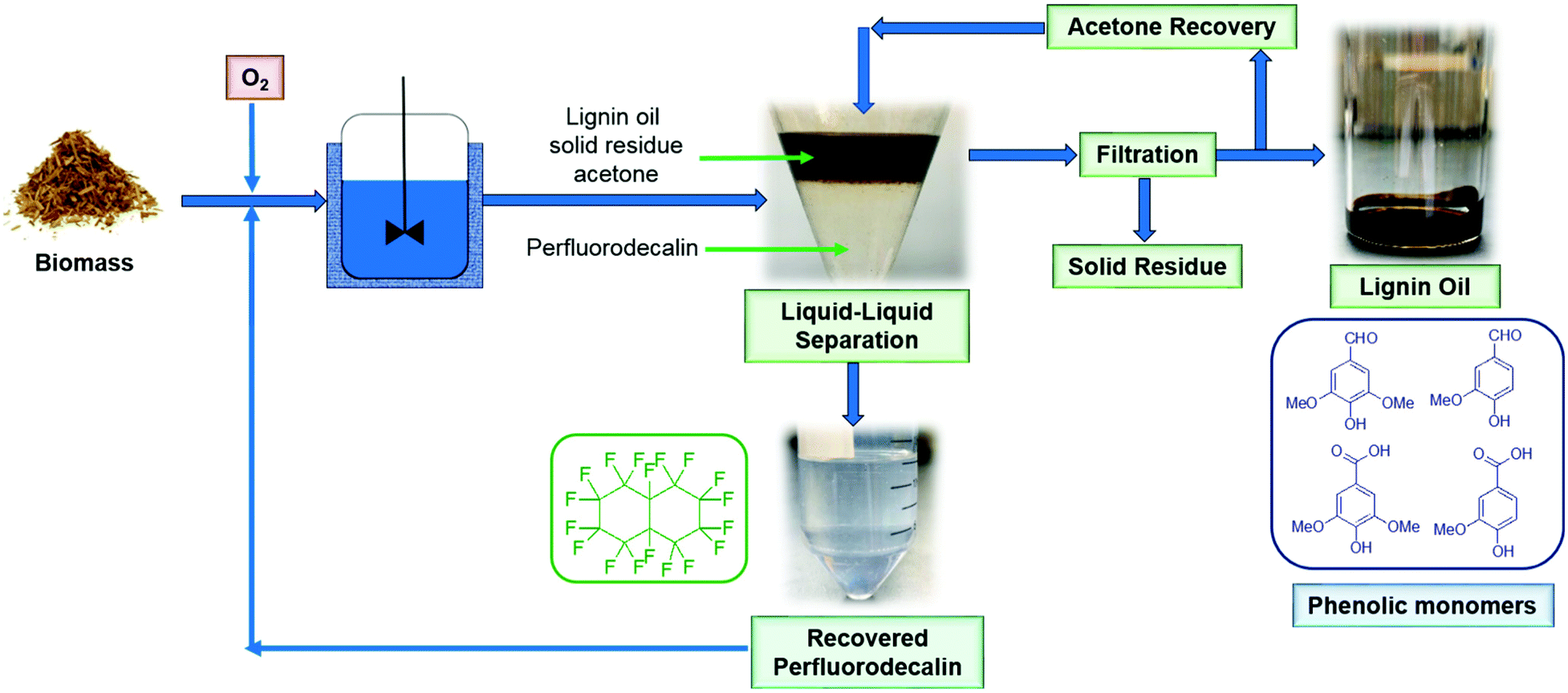 | ||
| Fig. 12 Schematic diagram of the proposed process for oxidative depolymerization of lignin in perfluorodecalin with solvent recycle to produce valuable phenolic monomers at high selectivity. | ||
Furthermore, GC analysis of the lignin oil revealed no residual perfluorodecalin. In contrast, other solvents are likely to decompose or be oxidized if employed in high-temperature oxidation reactions. For instance, butanol is not inert under oxidative conditions (Fig. S5†), and side products, including 1,4-butanediole, 2-methyl butane, 2-methyl butanoic acid, and 1,3-butanediol, were identified by GC.
Conclusions
We have demonstrated for the first time non-catalytic oxidative depolymerization of lignin in perfluorodecalin for the facile and rapid production of valuable phenolic monomers, including vanillin, syringaldehyde, syringic acid, and vanillic acid. Perfluorodecalin achieved 10.5 wt% yield of identifiable phenolic monomers at 250 °C under 300 psi O2 in 10 min, much higher than achieved using conventional organic solvents at comparable reaction conditions. Phenolic monomer yields in the alternative solvents of acetonitrile, ethyl acetate, methanol, and butanol were only 2.6, 1.0, 2.6, and 2.5 wt%, respectively. The superior performance of perfluorodecalin compared to the other solvents despite its poor solubility for lignin or phenolic products demonstrates that O2 solubility plays a crucial role in boosting the rate of oxidative lignin disassembly. Short reaction times, high O2 pressure, and high temperature are desirable for the highest phenolic monomer yield from non-catalytic deconstruction of lignin. Increasing the temperature not only increased the rate of lignin depolymerization but increased the rate of oxidation to phenolic aldehydes and carboxylic acids.In these experiments, molecular oxygen was not only effective in promoting depolymerization but also blocked condensation to recalcitrant phenolic oligomers. Increasing reaction time in perfluorodecalin shifted the lignin oils to lower molecular weight and lower polydispersity. GC analysis revealed that oxygen increased yields of oxygenated phenolic monomers while HSQC NMR showed that phenolic oligomers were also oxygenated. Lignin depolymerization in perfluorodecalin under N2 only produced 1.3 wt% of monomers. GPC analysis of the lignin oil obtained under N2 suggests mostly oligomeric products, and average molecular weight increased with increasing reaction time, suggesting condensation predominated under non-oxidative conditions. This is consistent with the role of O2 as an inhibitor in free radical polymerization reactions. Under oxidative condition, phenoxy and carbon-based radicals, produced by homolytic cleavage of lignin ether linkages, reacted with O2 to produce less reactive species. However, under inert conditions, these radicals were easily repolymerized, causing an increase in the molecular weight of lignin oil. GPC characterization of lignin oils from other solvents indicated products were primarily phenolic oligomers even under O2 atmospheres, suggesting that mass transfer was rate limiting for these solvents.
Non-catalytic lignin oxidation in perfluorocarbons shows promise as a safe, efficient, and economical method to convert lignin to value-added products. The process is readily scalable, and perfluorocarbons are thermally stable. Since PFCs do not dissolve the products of lignin depolymerization, they can be readily recovered by simple liquid phase separation.
Material and method
Chemicals and materials
Perfluorodecalin, N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) with 1% TMCS, pyridine (anhydrous, 99%), vanillin (99%), syringaldehyde (+98%), vanillic acid (+97%), syringic acid (+95%), syringol (+99%), benzyl phenyl ether (98%), pyridine-d5 (99.5 atom% D), and dimethyl sulfoxide-d6 (99.5 atom% D) were all purchased from Sigma-Aldrich. Acetone (+99%), tetrahydrofuran (HPLC grade), methanol (+99%), ethanol (+99%), acetonitrile (+99%), and ethyl acetate (+99%) were all purchased from Fischer Scientific. Red oak wood with particle size of less than 300 μm was obtained from Iowa State University's BioCentury Research Farm (BCRF). The total lignin content of the extractive-free red oak was 23 wt%.Microreactors
Most of the experiments were performed in 7 ml microreactors made of 316 stainless steel tubing (i.d. 0.5′′) and Swagelok fittings. As illustrated in Fig. 13, a microreactor was connected to an oxygen or nitrogen tank by 1/8′′ tubing and also a pressure relief valve to maintain desired reaction pressure during an experiment. A fluidized sand bath (Techne Industrial Bed 51) was used to heat the microreactor to reaction temperature.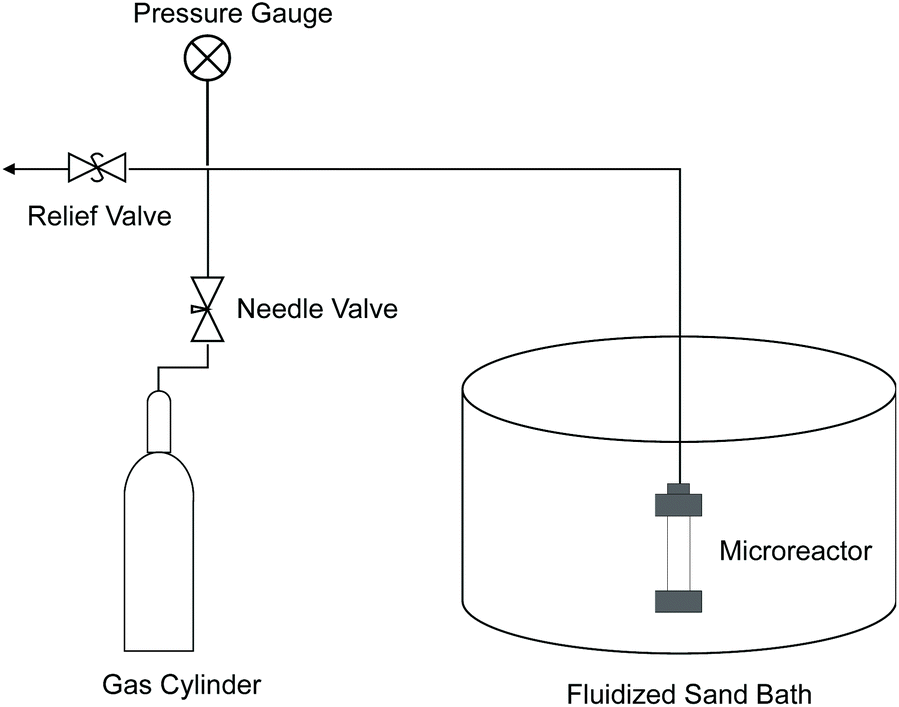 | ||
| Fig. 13 Schematic diagram of microreactor system. | ||
Red oak (100 mg) and perfluorodecalin (5 ml) were added to the reactor. In order to assure mass transfer of products to solvent was not rate limiting in the microreactors, we used a high solvent to substrate mass ratio, which was based on our previous research.42 The reactors were closed and purged five times with nitrogen gas, pressurized with oxygen to the desired oxygen partial pressure, and then nitrogen added to the desired total operating pressure of 300 psi. The reactors were then connected to a custom-built shaker and submerged into a preheated sand bath, which heated the reactor at a rate of 3.5 °C s−1. Reaction time was defined as the time interval between reaching the desired reactor temperature and the conclusion of the test when the reactor was removed from the sand bath and quenched in water.
Autoclave stirred reactor
The microreactor experiment with the highest monomer yield was repeated in a 500 mL Parker Autoclave Engineers EZE-Seal Stirred Reactor. The reactor was modified by addition of a batch feeder and a liquid product cooling vessel (bottoms quench) for rapid heating and cooling of the reactants and products (Fig. 14). In this system, the reactants and solvent are mixed in a small autoclave that serves as feeder. It is connected via a ball valve to a larger autoclave that serves as the reactor. Desired operating pressure is maintained with a back-pressure regulator through which volatile products pass to an “overheads condenser”. At the end of an experiment, the slurry of solvent and products is rapidly cooled by injecting it into the bottoms quench. Provisions are included for pressurizing both feeder and reactor autoclaves to desired pressures with mixtures of nitrogen and oxygen. | ||
| Fig. 14 Block flow diagram of autoclave reactor system used for scaled experiments. | ||
For each experiment, the empty reactor was preheated to the desired temperature using a 1000 W electrical heater and fast-response temperature controller. The feeder autoclave was filled with 250 mL solvent, 4 g red oak, and stirred for 30 min. At this point, the feeder and reactor were purged and pressurized with nitrogen and nitrogen/oxygen mixtures, respectively, to desired partial and total pressures. The valve between feeder and reactor was then opened to rapidly transfer the mixture of reactants and solvent from the feeder to the reactor. This system made possible the heating of the reactants and solvent to desired reaction temperature in less than 3 min, much faster than a conventionally operated autoclave reactor. At the completion of an experiment, the valve between the reactor and bottoms condenser was opened and the pressurized contents were rapidly expelled into the unpressurized bottom condenser, cooling the products below 100 °C within a few seconds.
Product separation and sample preparation
After reaction, liquid products were separated from residual solids by filtration. For oxidation reactions in perfluorodecalin, lignin oil was extracted from perfluorodecalin by acetone (4 × 5 mL). The recovered perfluorodecalin was separated from the remaining acetone for use in future experiments. Other solvents used in experiments were separated from lignin oil by a rotary evaporator.GC analysis
Prior to GC analysis, lignin oil was derivatized via silylation to improve volatility of products like syringic acid and vanillic acid. 10 mg of lignin oil was dissolved in 1 ml pyridine in a small reaction vial to which 100 μl BSTFA (with 1% TMCS) was added. The vial was well mixed, capped tightly, and heated to 60 °C for 15 min. After cooling, the sample was filtered and transferred to a GC vial. A gas chromatograph with mass spectrometer and flame ionization detector (Agilent 7890A GC-MS/FID) was used to analyze samples. GC was equipped with two identical Agilent J&W (VF-1701 ms, 60 m × 0.250 mm and 0.250 μm film thickness) capillary columns for separation of the products. One of these was connected to the MS while the other was connected to the FID. The injection port and FID back detector in the GC were held at 280 °C. The GC oven temperature was ramped from 35 °C (3 min hold time) to 280 °C (4 min hold time) at a heating rate of 5 °C min−1. Response factors of the products were obtained by calibration with commercial standards. The product yields were calculated by the following equation: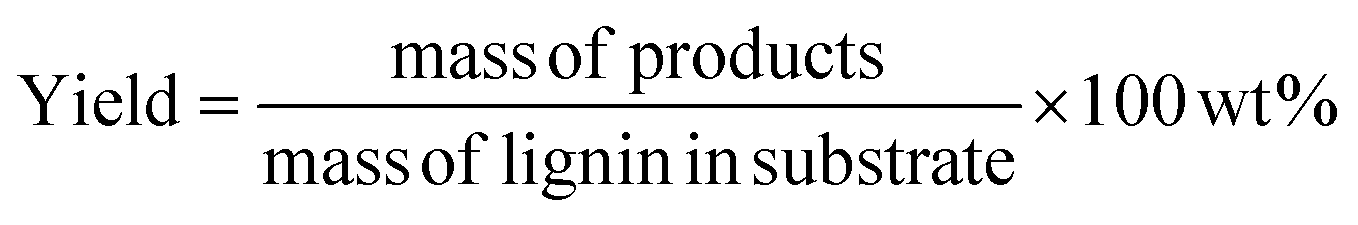 | (3) |
GPC analysis
GPC analysis was performed to assess the molecular weight distribution of lignin oil. 10 mg of lignin oil was dissolved in 5 ml tetrahydrofuran and filtered with a glass microfiber syringe filter (0.45 μm) before transferring to the GPC vial. GPC analysis was performed using a Dionex Ultimate 3000 (Sunnyvale, CA) HPLC system, equipped with a Shodex Refractive Index (RI). Two Agilent columns (PLgel 3 μm 100 Å 300 × 7.5 mm (p/n PL1110-6320) and one Mesopore 300 × 7.5 mm (p/n PL1113-6325) column were used. Tetrahydrofuran was used as eluent, and the instrument was calibrated from 162–45120 g mol−1. The software used to control the instrument and evaluate the samples was Dionex Chromeleon version 6.8.
NMR analysis
The NMR analysis was acquired on a Bruker Biospin NEO 400 MHz spectrometer equipped with liquid-nitrogen cooled 5 mm Prodigy Probe with normal geometry (broadband coil closest to the sample). Bruker's Topspin 3.5 software was used to process spectra. The central solvent peak was used as the internal reference (δH/δC: DMSO-d6, 2.49/39.50). For the HSQC NMR experiment, 100 mg lignin oil was dissolved in a 500 mL solution of DMSO-d6 and pyridine-d5 (4:1, v/v). The Bruker standard pulse sequence ‘hsqcedetgpsisp2.2’ was used with the following parameters: 12 ppm sweep width in F2 (1H), centered at 5.5 ppm, acquiring 3366 data points, 220 ppm sweep width centered at 105 ppm in F1 (13C) acquiring 620 increments, 20 scans per increment, a 1.0 s relaxation delay, and with the evolution time set for a 1-bond 1H–13C coupling constant of 145 Hz, with a total acquisition time of ∼5 h. Peak assignment was performed according to the literature.43 NMR analysis of recovered perfluorodecalin was acquired on the same instrument. Due to the insolubility of perfluorodecalin in all common deuterated solvents, solution 1H, 13C, and 19F NMR spectra of fresh and recycled perfluorodecalin (0.4 mL) were collected using a co-axial insert containing both D2O and DMSO-d6.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to acknowledge the contributions of Preston Gable, Lysle Whitmer, Andrew Friend, Jessica L. Brown, Martin Haverly, and Zachary Paskach in designing, constructing, and operating the novel autoclave reactor system used in some of this research. We also would like to acknowledge Marge Rover and Patrick Johnston who provided valuable advice on analyzing products. R. C. B. and P. H. were supported by the Bioeconomy Institute at Iowa State University. L. Q. was supported by the US Department of Energy (DOE), Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences. The Ames Laboratory is operated for the U.S. DOE by Iowa State University under Contract No. DE-AC02-07CH11358.References
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS.
- W. Schutyser, T. Renders, S. Van Den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- T. Renders, S. Van Den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- A. Mittal, R. Katahira, B. S. Donohoe, S. Pattathil, S. Kandemkavil, M. L. Reed, M. J. Biddy and G. T. Beckham, ACS Sustainable Chem. Eng., 2017, 5, 2544–2561 CrossRef CAS.
- C. S. Lancefield, I. Panovic, P. J. Deuss, K. Barta and N. J. Westwood, Green Chem., 2016, 19, 202 RSC.
- S. Van Den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 4, 6940–6950 Search PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS.
- R. Ten Have and P. J. M. Teunissen, Chem. Rev., 2001, 101, 3397–3413 CrossRef CAS.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2015, 4, 35–46 CrossRef.
- I. Hasegawa, Y. Inoue, Y. Muranaka, T. Yasukawa and K. Mae, Energy Fuels, 2011, 25, 791–796 CrossRef CAS.
- J. R. Silverman, A. M. Danby and B. Subramaniam, React. Chem. Eng., 2019, 4, 1421–1430 RSC.
- R. Ma, M. Guo, K. T. Lin, V. R. Hebert, J. Zhang, M. P. Wolcott, M. Quintero, K. K. Ramasamy, X. Chen and X. Zhang, Chem. – Eur. J., 2016, 22, 10884–10891 CrossRef CAS.
- S. K. Hanson, R. Wu and L. A. P. Silks, Angew. Chem., Int. Ed., 2012, 51, 3410–3413 CrossRef CAS.
- B. Sedai, C. Díaz-Urrutia, R. T. Baker, R. Wu, L. A. P. Silks and S. K. Hanson, ACS Catal., 2013, 3, 3111–3122 CrossRef CAS.
- T. Voitl and P. R. Von Rohr, ChemSusChem, 2008, 1, 763–769 CrossRef CAS.
- P. Cristina, R. Pinto and C. E. Costa, Ind. Eng. Chem. Res., 2013, 52, 4421–4428 CrossRef.
- W. Schutyser, J. S. Kruger, A. M. Robinson, R. Katahira, D. G. Brandner, N. S. Cleveland, A. Mittal, D. J. Peterson, R. Meilan, Y. Román-Leshkov and G. T. Beckham, Green Chem., 2018, 20, 3828–3844 RSC.
- W. Deng, H. Zhang, X. Wu, R. Li, Q. Zhang and Y. Wang, Green Chem., 2015, 17, 5009–5018 RSC.
- K. H. Kim, X. Bai, S. Cady, P. Gable and R. C. Brown, ChemSusChem, 2015, 8, 894–900 CrossRef CAS.
- K. H. Kim, X. Bai and R. C. Brown, J. Anal. Appl. Pyrolysis, 2014, 110, 254–263 CrossRef CAS.
- N. M. Emanuel and G. E. Zaikov, Z. K. Maizus, Oxidation of Organic Compounds: Medium Effects in Radical Reactions, Pergamon Press, Oxford, 1984 Search PubMed.
- V. A. Bhanu and K. Kishore, Chem. Rev., 1991, 91, 99–117 CrossRef CAS.
- C. M. Hansen, Hansen solubility parameters: a user's handbook, CRC press, 2002 Search PubMed.
- J. G. Riess, Artif. Cells, Blood Substitutes, Immobilization Biotechnol., 2005, 33, 47–63 CrossRef CAS.
- B. D. Spiess, J. Appl. Physiol., 2009, 106, 1444–1452 CrossRef CAS.
- J. G. Riess, Chem. Rev., 2001, 101, 2797–2919 CrossRef CAS.
- R. M. Winslow, Br. J. Haematol., 2000, 111, 387–396 CAS.
- J. J. J. Juliette, D. Rutherford, I. T. Horváth and J. A. Gladysz, J. Am. Chem. Soc., 1999, 34, 2696–2704 CrossRef.
- I. T. Horváth and J. Rábai, Science, 1994, 266, 72–75 CrossRef.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS.
- C. Cheng, J. Truong, J. A. Barrett, D. Shen, M. M. Abu-Omar and P. C. Ford, ACS Sustainable Chem. Eng., 2020, 8, 1023–1030 CrossRef CAS.
- G. F. De Gregorio, R. Prado, C. Vriamont, X. Erdocia, J. Labidi, J. P. Hallett and T. Welton, ACS Sustainable Chem. Eng., 2016, 4, 6031–6036 CrossRef.
- R. Prado, A. Brandt, X. Erdocia, J. Hallet, T. Welton and J. Labidi, Green Chem., 2016, 18, 834–841 RSC.
- M. Davaritouchaee, W. C. Hiscox, E. Terrell, R. J. Mancini and S. Chen, Green Chem., 2020, 22, 1182–1197 RSC.
- L. Khachatryan, M. X. Xu, A. J. Wu, M. Pechagin and R. Asatryan, J. Anal. Appl. Pyrolysis, 2016, 121, 75–83 CrossRef CAS.
- S. K. Ray and G. Moss, Adv. Energy Convers., 1966, 6, 89–102 CrossRef CAS.
- J. L. Wen, S. L. Sun, B. L. Xue and R. C. Sun, Materials, 2013, 6, 359–391 CrossRef CAS.
- E. Nagel and C. Zhang, Ind. Eng. Chem. Res., 2019, 58, 18866–18880 CrossRef CAS.
- V. E. Tarabanko and N. Tarabanko, Int. J. Mol. Sci., 2017, 18 Search PubMed.
- A. Ghosh, R. C. Brown and X. Bai, Green Chem., 2016, 18, 1023–1031 RSC.
- D. J. McClelland, A. H. Motagamwala, Y. Li, M. R. Rover, A. M. Wittrig, C. Wu, J. S. Buchanan, R. C. Brown, J. Ralph, J. A. Dumesic and G. W. Huber, Green Chem., 2017, 19, 1378–1389 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02505d |
| This journal is © The Royal Society of Chemistry 2020 |