
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeparation of precious metals by split-anion extraction using water-saturated ionic liquids†
Viet Tu
Nguyen
 ,
Sofía
Riaño
and
Koen
Binnemans
*
,
Sofía
Riaño
and
Koen
Binnemans
*
KU Leuven, Department of Chemistry, Celestijnenlaan 200F, P.O. box 2404, B-3001 Leuven, Belgium. E-mail: Koen.Binnemans@kuleuven.be
First published on 6th November 2020
Abstract
A split-anion solvent extraction process was developed for the separation of precious metal ions Au(III), Pt(IV), Pd(II) and Rh(III) from aqueous chloride media using water-saturated ionic liquids. The metal extraction and stripping behavior of the chloride form [A336][Cl], bromide form [A336][Br] and the iodide form [A336][I] of the quaternary ammonium ionic liquid Aliquat 336 were compared. The three ionic liquids extracted Au(III), Pd(II) and Pt(IV) quantitatively in most cases, whereas the co-extraction of Rh(III) was strongly dependent on the acidity and the chloride concentration. Among the studied ionic liquids, [A336][I] achieved the highest separation factors between Pd(II)/Rh(III), Pt(IV)/Rh(III), and Au(III)/Rh(III) at 6 mol L−1 Cl−. Additionally, the selective stripping of the individual metal ions Pd(II), Au(III), and Pt(IV) was only possible from loaded [A336][I] using ammonia solution (NH4OH), sodium thiosulfate (Na2S2O3), and thiourea ((NH2)2CS), respectively. A closed-loop flow sheet was designed for the recovery of the precious metals from chloride media using split-anion extraction with [A336][I]. The integrated process was demonstrated to be suitable for the purification of Rh(III), Pt(IV) and Pd(II) from a complex metal feed such as the leachate of spent automotive catalysts. The ionic liquid-based split-anion extraction process is simple, selective and effective for the sustainable separation of the precious metals, using only one green extractant [A336][I], which can be regenerated for consecutive extraction-stripping cycles.
Introduction
Nowadays, the overall supply of the platinum-group metals (PGMs) and gold from primary and secondary sources can hardly meet the global demand, which is forecasted to grow in the coming years. Therefore, the recycling of the precious metals from secondary material streams is an opportunity to reduce the metal deficit and environmental footprint related to primary mining.1 The extraction, separation and purification of precious metals is a challenge due to their poor chemical reactivity, and their complex and similar chemical properties. Traditional refining of precious metals involves a series of precipitation–dissolution steps that are no longer considered efficient due to their low degree of separation, poor yields, complexity of the operations and being not environmentally friendly.2 A considerable amount of research and development has been conducted over the last decades to replace traditional refining practices with solvent extraction processing technology. In general, solvent extraction-based refining processes for precious metals provide more advantages over the traditional precipitation methods: reduced processing time, better yields and purities, larger throughput and the capability for continuous operation and process control.3 In industrial separation processes operated by Vale Acton – Precious Metal Refinery, Anglo American Platinum, and Matthey Rustenburg Refiners, only five extractants are used: methyl isobutyl ketone (MIBK), trioctylamine (TOA), tri-n-butylphosphate (TBP), 2-hydroxy-5-nonylaceto-phenone oxime (LIX 84I) and di-n-octyl sulphide.2,4 Despite their advantages over the classical precipitation route, the solvent extraction processes for the purification of precious metals still have drawbacks such as slow kinetics (i.e. for Pd extraction), poor selectivity, the need to use different types of extractants to achieve full separation and the use of aromatic diluents.4–7Ionic liquids have been considered as greener and safer alternatives to the traditional organic solvents in hydrometallurgy.8–10 Ionic liquids are organic salts that consist entirely of ions and they often have a melting point below 100 °C.11–13 In practice, ionic liquids with hydrophobic cationic moieties associated with simple anions (i.e. Cl− and NO3−) are preferred in solvent extraction, as for instance, the quaternary phosphonium ionic liquid Cyphos IL 101 ([P66614][Cl]) and the quaternary ammonium ionic liquid Aliquat 336 ([A336][Cl]).14–20 Ionic liquids have properties that are advantageous for solvent extraction such as negligible volatility, low flammability, low vapor pressure and they do not accumulate static electricity. Moreover, their ionic structure and metal complex solvation are totally different from apolar aliphatic or aromatic diluents.11,12 Although pure ionic liquids are considered as safer and more environmentally friendly alternatives to traditional organic extractants and diluents in metal extraction, their use remains limited due to their high viscosity. High viscous systems usually have slow mass transfers and their implementation in continuous mode is challenging.15,18,21
The ionic liquid [P66614][Cl] (Cyphos IL 101) has been used in solvent extraction of PGMs from chloride media because it behaves as an anion exchanger, as its chloride counter-ion can be easily exchanged with aqueous anionic species such as [PdCl4]2− and [PtCl6]2−.15,20,22,23 Svecova et al. carried out the selective separation of Pd(II) from Rh(III) from a highly acidic solution using the phosphonium-based ionic liquids [P66614][Cl] and [P66614][Br].24
The term “split-anion extraction” was introduced to describe an ionic-liquid-based solvent extraction in which different anions are present in the aqueous and organic phases. The metal ions are extracted from an aqueous feed solution containing anions (i.e. SO42− and Cl−) that form weakly extracting complexes to an organic phase through complex formation with anions (i.e. Br−, I−, and SCN−) present almost exclusively in the ionic liquid. For instance, Larsson and Binnemans studied the extraction of rare earths ions from chloride solution to a water-immiscible organic phase containing thiocyanate- or nitrate-based ionic liquids (i.e. [P66614][SCN], [P66614][NO3], [A336][SCN], and [A336][NO3]).25 The ionic liquids provide the sources of anions (i.e. NO3− and SCN−) that coordinate strongly and transport the target metals from the aqueous chloride phase into the organic phase. The split-anion extraction can be predicted by the Hofmeister series: SO42− < Cl− < Br− < NO33− < I− < ClO4− < SCN−. Hydrophilic anions (i.e. SO42−, Cl−) with high charge density preferentially distribute to the aqueous where they are better solvated. Whereas, hydrophobic anions (i.e. I−, ClO4−, and SCN−) with low charge density are less hydrated and tend to remain in the organic phase.26 The main advantage of split-anion extraction is that highly selective extraction and/or stripping of metals can be achieved by simply varying the anions present in the ionic liquids, without changing the aqueous phase. During the split-anion extraction, the formation of a complex between the metal ions and the anions in the ionic liquid is prominent reaction. Meanwhile, there is only a limited anion exchange of the different anions from one phase to the other to maintain electrical neutrality.25–28
In this paper, a split-anion extraction process for the recovery of precious metals (i.e. Au(III), Pt(IV), Pd(II) and Rh(III)) from acidic chloride media using water-saturated ionic liquids is presented. The metal extraction and stripping behavior of the chloride form [A336][Cl], bromide form [A336][Br] and the iodide form [A336][I] of the quaternary ammonium ionic liquid Aliquat 336 were compared to select the most suitable extractant. A flow sheet for the recovery of Au(III), Pt(IV), Pd(II) and Rh(III) was designed and applied to the separation of Pt(IV), Pd(II) and Rh(III) from a leachate of spent automotive catalysts. The split-anion extraction using only water-saturated [A336][I] as green extractant exhibits a simple, efficient and selective recovery of individual precious metals with high purity. The developed process avoids the use of water-immiscible organic solvents, many of which are flammable, volatile or toxic (i.e. kerosene, toluene, dodecane, chloroform, dichloromethane, and diethyl ether), minimizes the risk of air pollution, and excludes the risk of a buildup of static electricity in the solvent extraction plant. In addition, the regeneration of the ionic liquid phase was studied to ensure the sustainability of the closed-loop process.
Experimental
Materials and reagents
Aliquat® 336 ([A336][Cl], a commercial mixture of quaternary ammonium chlorides, with 88.2–93.0% trioctylmethyl-ammonium chloride), sodium thiocyanate (≥98%), sodium perchlorate (98%), ammonium chloride (≥99.5%), potassium thiocyanate (≥98.5%), ammonium thiocyanate (≥99%), p-cymene (≥99%), and standard solutions of individual metals (1000 mg L−1) for ICP-OES analysis were purchased from Sigma-Aldrich (Diegem, Belgium). Hydrogen hexachloroplatinate(IV) hydrate (H2PtCl6·5H2O, 99.99%); hydrogen tetrachloro-aurate(III) hydrate (HAuCl4·3H2O, 99.99%); rhodium(III) chloride hydrate (RhCl3·3H2O, 99.98%), and palladium(II) chloride (PdCl2, 99.9%) were obtained from Johnson Matthey (Hertfordshire, UK). Potassium bromide (≥99.5%), potassium iodide (≥99%), thiourea (≥99%), sodium thiosulfate pentahydrate (≥99.5%), and potassium perchlorate (≥99%) were purchased from Acros Organic (Geel, Belgium). Hydrochloric acid (37 wt%) and nitric acid (68 wt%) were purchased from VWR Chemicals (Leuven, Belgium). Ammonia solution (25 wt%) was obtained from Chem-Lab NV (Zedelgem, Belgium). The silicone solution in isopropanol used for TXRF analysis was purchased from SERVA Electrophoresis GmbH (Heidelberg, Germany). All chemicals were used as received without any further purification.Instrumentation
The viscosity of the ionic liquids was measured with a rolling-ball type viscometer (Anton Paar, DMA 4500 M). The densities of both aqueous and organic phases were determined using a densitometer with an oscillating U-tube sensor (Anton Paar, DMA 4500 M). The uncertainty in density measurement was ±10−2 g cm−3. The temperature was automatically controlled with a resolution of 0.01 °C. The refractive index was measured using a refractometer (Abbemat 200, Anton Paar) with a standard deviation of ±0.0002. The water content of the ionic liquids was determined with a volumetric Karl Fischer titrator (Mettler-Toledo DL 38) in combination with the Stromboli oven. The samples were heated at 120 °C in the oven and the evaporated water was transported into the titration vessel by means of a dry inert gas stream. The chloride content in the prepared ionic liquids was quantified using a benchtop total reflection X-ray fluorescence (TXRF) spectrometer (Picofox S2, Bruker). The quartz glass sample carriers were first treated with 20 μL of a silicon solution in isopropanol to avoid spreading of the sample droplet, followed by drying in a hot air oven at 60 °C for 5 min. The samples were prepared by adding 100 μL of a Cu standard solution, 100 μL of a NH3 solution (25 wt%) and 750 μL of ethanol to 40 mg of the ionic liquids [A336][Br] and [A336][I]. A droplet (2.0 μL) of this mixture was placed on the quartz glass carrier. Subsequently, the quartz glass carrier was dried in a hot air oven at 60 °C for 30 min and measured in the TXRF spectrometer for 200 s. 1H NMR spectra were recorded on a Bruker Avance 300 spectrometer, operating at 300 MHz. Tetramethylsilane was used as a reference to determine the chemical shifts, which are noted in parts per million (ppm). All samples were diluted in CDCl3 for NMR measurement. The 1H NMR spectra were analyzed with SpinWorks software. The Fourier Transform Infrared (FTIR) spectra of the ionic liquids were recorded on a Bruker Vertex 70 spectrometer with an ATR module in wavenumber range from 4000–400 cm−1. The data was analyzed using the OPUS software. The metal concentrations in the aqueous phase were determined with inductively coupled plasma optical emission spectrometry (PerkinElmer Optima 8300 ICP-OES Spectrometer, USA) using a Cross-Flow Scott torch module with proper start-up conditions (i.e. RF power 1500 W, plasma 10 L min−1, auxiliary 0.2 L min−1, nebulizer 0.7 L min−1 and pump flow 1.5 mL min−1). The calibration curve was prepared with multi-element solutions of 0.01, 0.1, 1.0, and 10 mg L−1. Samples diluted in 5% v/v HNO3 were measured at different wavelengths (i.e. platinum (214.423 and 265.945 nm); palladium (340.458 and 363.470 nm); rhodium (233.477 and 343.489 nm); and gold (242.795 and 267.595 nm)) using yttrium (5.0 mg L−1) as an internal standard. The concentration of iodide in the aqueous solutions was quantified with ICP-OES at wavelength 178.215 nm. Iodine standard solutions in the range of 1–100 mg L−1 were prepared by dissolving high-purity potassium iodide in 5% v/v HNO3. The analytical quality control was verified with 0.2 and 2.0 mg L−1 samples prepared from the standard solution.Synthesis of [A336][Br] and [A336][I]
[A336][Br] and [A336][I] were synthesized via a metathesis reaction. A sample of 100 g (0.202 mol) [A336][Cl] was equilibrated with approximately 100 mL of an aqueous 3.0 mol L−1 KBr or KI solutions during one hour at 2000 rpm, respectively. Afterwards, the aqueous phase was removed and the organic phase was contacted two more times with the fresh KBr or KI solution at the same experimental conditions. The newly synthesized organic phase was washed twice with 100 mL of Milli-Q water (>18.2 MΩ cm−1 resistivity) to remove chloride and potassium impurities. The chloride content in the ionic liquids analysed with TXRF was less than 20 ppm.[A336][Br]: colourless, yield 95.0%, water content 9.28 wt%, density 0.959 g cm−3 (298 K), viscosity 183 mPa s (298 K). 1H NMR (300 MHz, CDCl3, δ/ppm): 0.88 (t, 9H, J = 4.77 Hz), 1.26–1.36 (m, 32H), 1.63–1.67 (m, 6H), 3.33 (s, 3H), 3.46 (t, 6H, J = 5.16 Hz). FTIR (ν cm−1): 3417, 2926, 2853, 1618, 1461, 1376, 1052, 891, 720.
[A336][I]: yellow-brownish, yield 90.1%, water content 8.46 wt%, density 1.018 g cm−3 (298 K), viscosity 557 mPa s (298 K). 1H NMR (300 MHz, CDCl3, δ/ppm): 0.88 (t, 9H, J = 4.80 Hz), 1.27–1.38 (m, 32H), 1.66–1.70 (m, 6H), 3.32 (s, 3H), 3.44 (t, 6H, J = 6.33 Hz). FTIR (ν cm−1): 3419, 2923, 2854, 1610, 1462, 1378, 1055, 893, 723.
Solvent extraction procedure
Extraction experiments were generally performed by shaking an equal volume of 1.00 mL of two immiscible phases (the metal chloride feed solution and the water-saturated ionic liquid [A336][X] (X = Cl, Br, and I)) in 4.0 mL vials using a Nemus Life Thermo Shaker TMS-200 (Nemus LIFE AB, Lund, Sweden), at 298 K and 2000 rpm, for 60 min to ensure equilibrium, unless stated otherwise. The aqueous feeds containing desired concentration of Pt(IV), Pd(II), Rh(III), and Au(III) were prepared by diluting a certain volume of the metal stock solutions in 0.001–6.0 mol L−1 Cl− (i.e. HCl, NaCl, LiCl, CaCl2, MgCl2). The ionic liquids were saturated with water prior to any extraction to reduce the viscosity and to avoid volume change during solvent extraction. In most experiments, the concentrations of water-saturated ionic liquids were kept constant at 1.75 mol L−1 [A336][I], 1.80 mol L−1 [A336][Br], and 1.70 mol L−1 [A336][Cl], except in case of stoichiometric studies in which the ionic liquids were diluted in p-cymene. After extraction, the phase disengagement was accelerated by centrifuging at 5000 rpm for 5 min with a Heraeus Labofuge 200 centrifuge (Thermo Fisher Scientific, Asse, Belgium). The aqueous phase (raffinate) was then diluted in 5% v/v HNO3 to determine metal content with ICP-OES. The concentration of the metals in the organic phase was calculated from the difference between the metal concentration in the aqueous phase before and after extraction using mass balance. All experiments were performed in triplicate and the data are presented as average values with a standard deviation of less than 3.0%.The distribution ratio (D) is the ratio of the concentration of the metal ion in the ionic liquid phase ([M]IL) and the aqueous phase ([M]aq), at equilibrium:
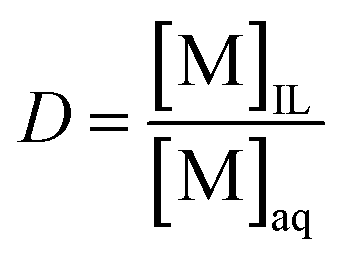 | (1) |
The percentage extraction (%E) is defined as:
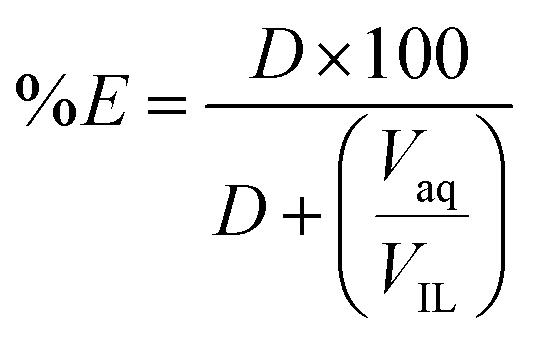 | (2) |
The separation factor αM1,M2 between two metals can be calculated as:
 | (3) |
The stripping experiments were performed by equilibrating individual stripping solutions such as water, CS(NH2)2/HCl, NaSCN, NaClO4, Na2S2O3, NH4Cl, NH4OH, KClO4, KSCN, NH4SCN with the loaded organic phases (LO) at a certain O/A phase ratio in 1.5 mL vials at 298 K and 2000 rpm for 60 min to ensure equilibrium. After centrifugation and phase separation, the aqueous phase was separated and diluted in 5% v/v HNO3 for further analysis with ICP-OES. The percentage stripping (%S) is defined as the amount of metal stripped from the organic phase to the total amount of metal in the organic phase before stripping:
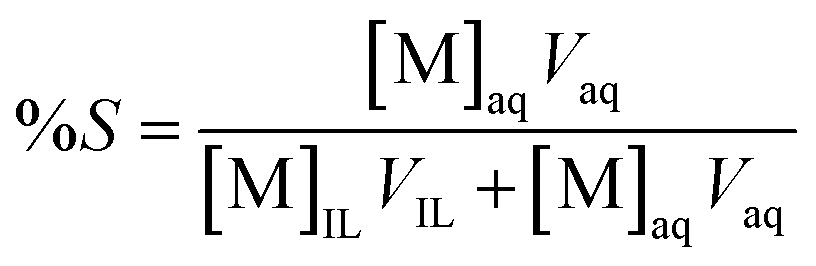 | (4) |
After complete stripping, the ionic liquids were regenerated and reused effectively during three cycles of extraction-stripping.
Simulation of multi-stage counter-current extraction and stripping
The batch simulation for counter-current extraction of precious metals from aqueous chloride media was conducted using water-saturated [A336][I] at O/A = 1/3 and 2000 rpm for 60 min. The aqueous feed solution was prepared to mimic the leachate from end-of-life autocatalyst containing 10.5 mg L−1 Pt(IV), 24.0 mg L−1 Pd(II), 6.81 mg L−1 Rh(III) and impurities (i.e. 1427 mg L−1 Al, 60.3 mg L−1 Ce, 4.03 mg L−1 Cr, 80.2 mg L−1 Fe, 8.70 mg L−1 La, 310 mg L−1 Mg, 3.25 mg L−1 Mn, 2.28 mg L−1 Mo, 4.74 mg L−1 Ni, 0.96 mg L−1 Pb, 3.28 mg L−1 Si, 28.7 mg L−1 Sn, 60.4 mg L−1 Zn, 419 mg L−1 Zr). A diagram summarizing the different contacts that were carried out in the two-stage counter-current solvent extraction experiment is presented in Fig. 1. The fresh organic phase was mixed with the fresh feed solution in the first stage (E1-1). The raffinate (R1-1) obtained in the first cycle was used as aqueous feed solution in the second stage (E2-1), in which the fresh organic phase was present. Subsequently, the loaded organic phase (LO2-1) was contacted with a fresh feed solution in the first stage (E1-2). The process was repeated up to five complete cycles from (E1-1) to (E1-5) until the steady state was achieved. The final raffinates (R2-2, R2-3, and R2-4) had similar metal concentration and resembled the streams that would exist in an actual continuous countercurrent extraction. The loaded organic phases obtained from the simulation extraction (LO1-3, LO1-4, and LO1-5) were collected and scrubbed with 1 mol L−1 NaCl to remove the co-extracted impurities (i.e. Al, Fe, Mn, Pb, Mg) at O/A = 1/1, during 60 min at 2000 rpm before the stripping experiments. The batch simulation of three counter-current stages for the stripping of Pd(II) from the scrubbed organic phase was investigated using 1.0 mol L−1 NH4OH in 1 mol L−1 NaCl at O/A = 3/1 in 4.0 mL vials during 20 min at 2000 rpm. Finally, the batch simulation of two-stages counter-current stripping of Pt(IV) from Pd-free loaded organic phase was executed using 0.1 mol L−1 thiourea in 0.2 mol L−1 HCl at O/A = 3/1 during 20 min at 2000 rpm.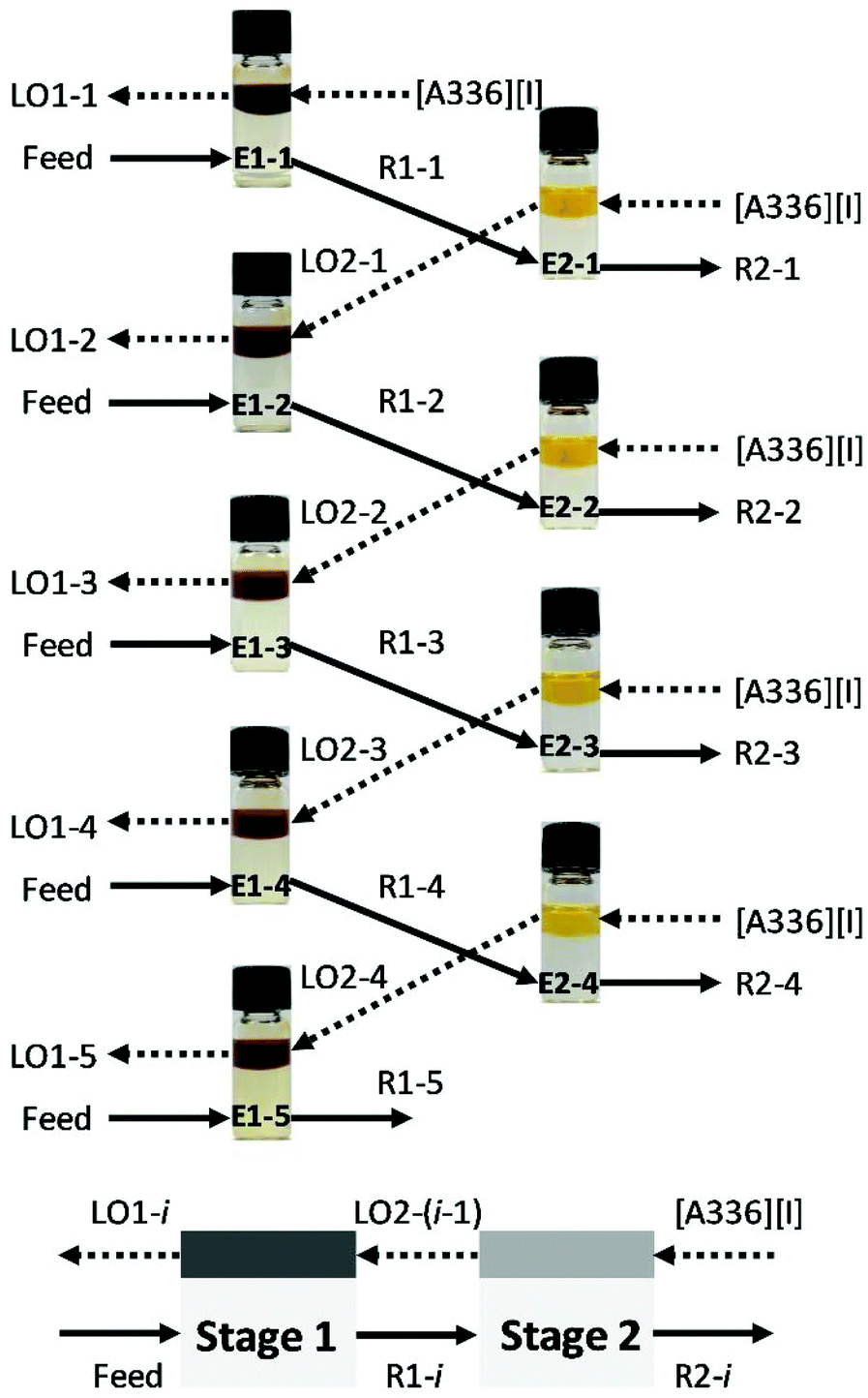 | ||
| Fig. 1 Batch simulation of two-stage counter-current extraction of precious metals with water-saturated [A336][I]. | ||
Reduction of the precious metals into metallic forms
The raffinate obtained after the batch simulation extraction was treated with formic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) to selectively produce metallic Rh at 90 °C in 60 min. The black rhodium was filtered and washed with 0.25 mol L−1 HCl to remove impurities. Similarly, the strip product solution containing Pd was neutralized with an excess amount of formic acid to reduce [Pd(NH3)4]2+ into black Pd. Highly pure Pd (99.8%) was obtained after being washed with 0.25 mol L−1 HCl. The cementation of Pt from the Pt strip product solution was performed using granular Zn (20–30 mesh) in 2.0 mol L−1 HCl at 25 °C in 4 h. The solid was filtered and individually washed with 0.25 mol L−1 HCl and 0.25 mol L−1 HNO3 to get highly pure metallic Pt (99.5%).
:1 v/v) to selectively produce metallic Rh at 90 °C in 60 min. The black rhodium was filtered and washed with 0.25 mol L−1 HCl to remove impurities. Similarly, the strip product solution containing Pd was neutralized with an excess amount of formic acid to reduce [Pd(NH3)4]2+ into black Pd. Highly pure Pd (99.8%) was obtained after being washed with 0.25 mol L−1 HCl. The cementation of Pt from the Pt strip product solution was performed using granular Zn (20–30 mesh) in 2.0 mol L−1 HCl at 25 °C in 4 h. The solid was filtered and individually washed with 0.25 mol L−1 HCl and 0.25 mol L−1 HNO3 to get highly pure metallic Pt (99.5%).
Results and discussion
Split-anion extraction of precious metals
The effect of the hydrochloric acid concentration on the extraction of precious metals was investigated in the range of 0.001 to 6.0 mol L−1 HCl. Fig. 2 shows that Pt(IV), Pd(II), and Au(III) are quantitatively extracted (>99%) by the three water-saturated ionic liquids [A336][X] (X = Cl, Br, I) at any of the acid concentrations investigated. On the other hand, the distribution ratios of Rh(III) are strongly dependent on the HCl concentration. The co-extraction of Rh(III) decreases when increasing the HCl concentration. In general, high concentrations of HCl (6.0 mol L−1 HCl or higher) suppress the Rh(III) extraction. | ||
| Fig. 2 Effect of hydrochloric acid concentration on the percentage extraction of precious metals. Aqueous phase: 680 mg L−1 Pt(IV), 675 mg L−1 Pd(II), 128 mg L−1 Rh(III), and 28.3 mg L−1 Au(III), 0.001–6.0 mol L−1 HCl; organic phase: water-saturated (A) 1.75 mol L−1 [A336][I]; (B) 1.80 mol L−1 [A336][Br]; and (C) 1.70 mol L−1 [A336][Cl]; O/A = 1/1; equilibrium time 60 min; 298 K; 2000 rpm. | ||
The separation factors αPt,Rh, αPd,Rh, and αAu,Rh are all larger than 103 for the three ionic liquids indicating that Pt(IV), Pd(II), and Au(III) are selectively extracted over Rh(III) at 6.0 mol L−1 HCl. This is in agreement with what has been reported by Svecova et al.24 High distribution ratios for the extraction of Rh(III) using the phosphonium ionic liquids [P66614][Cl] and [P66614][Br] were obtained at 1.0 mol L−1 HCl, but the distribution ratios decreased as the HCl concentration above 4.0 mol L−1. The separation of rhodium from other precious metals is one of the most difficult areas in precious metal refining, mainly due to the complex chemistry of rhodium in chloride solutions. Benguerel et al. used available thermodynamic and kinetic data to propose a speciation diagram, in which at least two different rhodium aquo/chloro anionic species (see eqn (5)) are present in relatively high acidic chloride solutions (>4.0 mol L−1 Cl−), i.e. [RhCl5(H2O)]2−, [RhCl6]3−, and to a lesser extent [RhCl4(H2O)2]−.29–31
| [RhCl6]3− + H2O ⇄ [RhCl5(H2O)]2− + Cl− | (5) |
Highly charged octahedral complexes such as [RhCl6]3− and [RhCl5(H2O)]2− with an effective radius of the solvation shell suppress the interaction between extractant and Rh(III) species, and are particularly difficult to extract due to its hydrophilic nature and/or to a lesser degree to steric effects; it is difficult to pack three [A336]+ cations around a single anion, and thus the distribution coefficient is low.
The fact that the percentage extraction of Rh(III) with [A336][I] (25.7%) is lower than with [A336][Cl] (81.5%) and [A336]Br (83.3%) at, for example, 1.0 mol L−1 HCl can be explained by the nature of the split-anion extraction. Anions with a high charge density, such as chloride, are strongly hydrated and tend to remain in the aqueous phase, whereas anions with a low charge density such as iodide, are more hydrophobic and will have a higher affinity for the ionic liquid phase. Furthermore, it has also been reported that Rh(III) forms halide complexes with Cl− and Br− but not with I−, and thus the split-anion extraction with the latter is impossible. However, the co-extraction of Rh(III) with [A336][I] is probably due to distribution of [RhCl5(H2O)]2− into the ionic liquid phase as a result of the salting-out effect. In addition, the conversion of [A336][I] to [A336][Cl], which does extract Rh more efficiently, may also be responsible for co-extraction of Rh(III) (eqn 6 and 7).
The effect of variation of the volume phase ratio on the extraction of precious metals was determined in the range 1/1 to 1/10 at a fixed total volume (1.2 mL) of organic and aqueous phase. Fig. 3 shows a quantitative extraction (>99%) of Pt(IV), Pd(II), and Au(III) was obtained within the investigated range except for the percentage extraction of Pd(II) with [A336][Cl] which drops to 86.2% at O/A = 1/10. On the other hand, increasing in the volume of the aqueous phase results in a gradual decrease in Rh(III) extraction from 18.3% to 1.70% with [A336][Cl], from 26.8% to 1.64% with [A336][Br], and from 9.32% to 0.23% with [A336][I]. The lower the phase ratio O/A was used, the more selective extraction of Pt(IV), Pd(II), and Au(III) over Rh(III) was achieved. In practical, the O/A ratio from 1/1 to 1/3 is preferred for continuous solvent extraction of the metals using mixer-settlers.
 | ||
| Fig. 3 Effect of volume phase ratio O/A on the percentage extraction of precious metals. Aqueous phase: 680 mg L−1 Pt(IV), 675 mg L−1 Pd(II), 128 mg L−1 Rh(III), and 28.3 mg L−1 Au(III), 6.0 mol L−1 HCl; organic phase: water-saturated (A) 1.75 mol L−1 [A336][I]; (B) 1.80 mol L−1 [A336][Br]; and (C) 1.70 mol L−1 [A336][Cl]; O/A = 1/1 to 1/10; equilibrium time 60 min; 298 K; 2000 rpm. | ||
The influence of the initial metal concentration on the extraction the precious metals was determined. As shown in Fig. 4, Pt(IV), Pd(II), and Au(III) was completely extracted regardless of the metal concentration. Meanwhile, a slight decrease trend was observed for the extraction of Rh(III) with increasing metal concentration in the feed. At a compared experimental conditions, the water-saturated [A336][I] exhibits the most selective extraction of Pt(IV), Pd(II), and Au(III) over Rh(III).
 | ||
| Fig. 4 Effect of initial metal concentration on the percentage extraction of precious metals. Aqueous phase: 0.68–17.0 g L−1 Pt(IV), 0.68–17.6 g L−1 Pd(II), 0.13–3.20 g L−1 Rh(III), and 0.03–0.71 g L−1 Au(III), 6.0 mol L−1 HCl; organic phase: water-saturated (A) 1.75 mol L−1 [A336][I]; (B) 1.80 mol L−1 [A336][Br]; and (C) 1.70 mol L−1 [A336][Cl]; O/A = 1/1; equilibrium time 60 min; 298 K; 2000 rpm. | ||
So far, the water-saturated ionic liquids have been used for extraction of precious metals. Therefore, the concentrations of ionic liquids are constant and cannot be varied. However, in order to investigate the extraction stoichiometry via slope analysis and Job's method, the ionic liquids were diluted in p-cymene, which is derived from bio-mass and is presented as a greener substitute of toluene. In addition, p-cymene whose flash point is equal to 47 °C is a model of commercial diluents dedicated to solvent extraction, for example, SOLVESSO 150 (flash point 64 °C). The dependence of precious metals extraction on different concentrations of ionic liquids was studied. As shown in Fig. 5, the extraction of Pd(II) and Pt(IV) linearly increased with increasing the ionic liquid concentration in the range 1.0–14 mmol L−1 [A336][X] (X = Cl−, Br−, and I−).
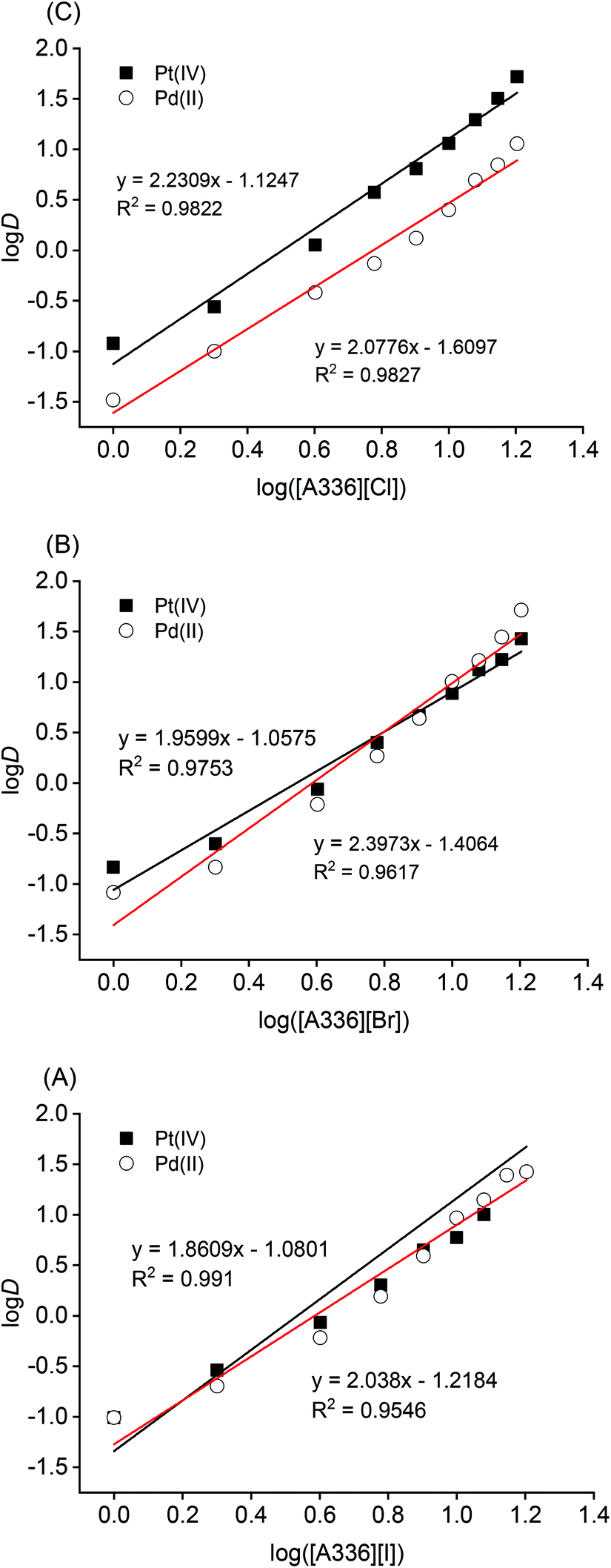 | ||
| Fig. 5 Effect of ionic liquid concentration on the extraction of precious metals. Aqueous phase: single-element solution 680 mg L−1 Pt(IV) or 675 mg L−1 Pd(II) in 6.0 mol L−1 HCl; organic phase: 1.0–14 mmol L−1 [A336][X]/p-cymene; O/A = 1/1; equilibrium time 60 min; 298 K; 2000 rpm. | ||
The extraction mechanism of Au(III), Pt(IV), and Pd(II) using basic extractant [A336][Cl] is generally described as an anion exchange process, in which negatively charged metal complexes [MCly]n− present in the aqueous chloride media are exchanged for anions Cl− in the ionic liquid phase (eqn (6)):
 | (6) |
On the other hand, the extraction of Au(III), Pt(IV), and Pd(II) from chloride media using water-saturated ionic liquids [A336][X] (X = Br− or I−) was proposed to proceed via the split-anion process (eqn (7)). According to Hofmeister series, Br− or I− anions are more hydrophobic than Cl− and stabilize in the organic phase.25–28 Therefore, the ionic liquids play essential roles of providing the anions Br− or I− that form complexes with the precious metals ions. The extraction mechanism involves two concomitant reactions. First, PdCl42− and PtCl62− reacts with X− (i.e. Br− and I−) from the ionic liquids to form PdX42− or PtX62−. Second, PdX42− or PtX62− are extracted towards the ionic liquids.10,32,33
 | (7) |
Eqn (7) is not valid for Rh(III) extraction with [A336][I] because Rh(III) does not form iodo-complexes. Noted that at least (y − n)X− was exchanged for (y − n)Cl− when the water-saturated [A336][X] was contacted with a 6 mol L−1 HCl feed solution. As a result, the mixture of [A336][I] and [A336][Cl] was obtained during the extraction (eqn (7)). The limited anion exchange reaction between X− and Cl− is necessary to maintain the electrical neutrality.
Assuming ideal behavior in the organic phase and constant activity coefficient in the aqueous phase, the equilibrium constant for reaction (7) can be written as:
 | (8) |
Taking into account the definition of distribution ratio D in eqn (1), the following expression is obtained:
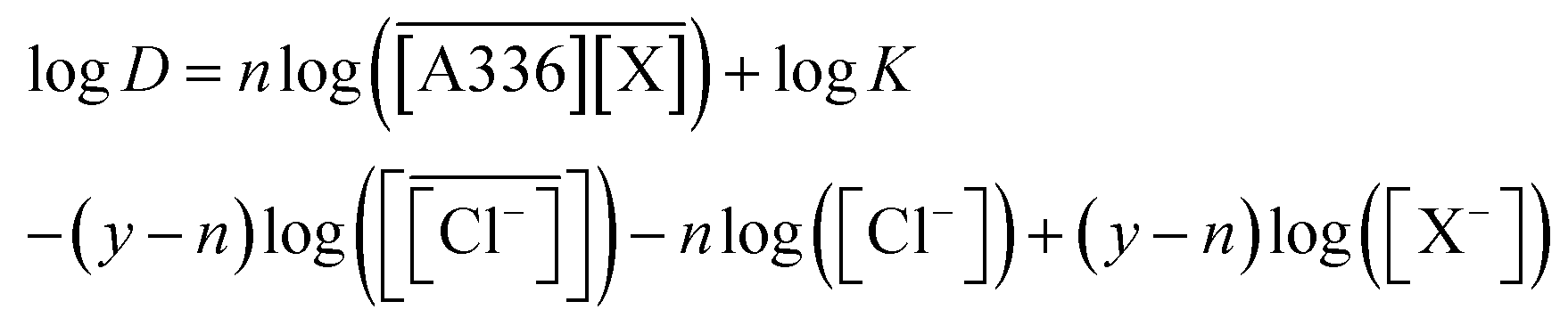 | (9) |
 | (10) |
Thus, a plot of logD against log([A336][X]) (X = Cl−, Br−, I−) should give a straight line with a slope of n and an intercept equal to (logK′).
Fig. 5 shows a straight line of slope 2, which is an indication of the number of molecules of extractant involved per metal ion in the extraction. In other words, experimental data suggests that two molecules of ionic liquid [A336][X] are associated in the extraction of Pd(II) and Pt(IV). Accordingly, the extracted species in the organic phase could be  and
and  .
.
To confirm the stoichiometry (eqn (7)), Job's plot was studied by varying the molar fractions of precious metals from 0 to 1, while keeping the total molar concentration of metal and ionic liquid constant at 5 mmol L−1. As shown in Fig. 6, the extrapolated maximum of Pd(II) and Pt(IV) extraction occurs at molar fractions close to 0.31–0.33, which suggests the metal/ionic liquid stoichiometry is 1/2. Two moles of ionic liquid [A336][X] are needed to extract 1 mol of Pd(II) or Pt(IV). The findings are in line with those obtained from slope analysis.
 | ||
| Fig. 6 Job's plot for the extraction of precious metals with [A336][I]/p-cymene. Aqueous phase: single-element solution 100–900 mg L−1 Pt or 54–489 mg L−1 Pd in 6 mol L−1 HCl; organic phase: 0.38–4.49 mmol L−1 [A336][I]/p-cymene; O/A = 1/1; equilibrium time 60 min; 298 K; 2000 rpm. | ||
Stripping of precious metals from loaded ionic liquids
So far the proposed method allows the extraction of Au(III), Pd(II) and Pt(IV) from Rh(III) using only one extractant. The separation of Au(III), Pd(II) and Pt(IV) into individual elements can be achieved by means of selective stripping. Aqueous solutions that may form stable complexes with precious metals, such as CS(NH2)2/HCl, NaSCN, NaClO4, Na2S2O3, NH4Cl, NH4OH, NH4Cl + NH4OH (1:1 v/v), KClO4, KSCN, and NH4SCN were individually used as stripping reagents. Fig. 7 shows the percentage stripping of the precious metals from loaded ionic liquids as a function of different stripping agents. Although after the extraction most of the Rh(III) is left in the raffinate, its stripping behavior was also studied to determine its partitioning. The stripping of Rh(III) from loaded [A336][Cl] could be achieved from 18% to 38% using aqueous solution (i.e. H2O, NH4OH, NH4Cl, or mixture of NH4OH and NH4Cl solutions). The reason is that the highly negatively charged chloro-complexes RhCl63− less associate with the organic cation [A336+]. In other words, the RhCl63− is more water stabilized species and tends to stay in the aqueous phase. Note that the Rh(III) stripping efficiency decreased from [A333][Cl] > [A336][Br] > [A336][I]. The observation in stripping behavior of Rh(III) is in accordance with Hofmeister series for anions.25–28 Whereas, the stripping of precious metals from [A336][Br] and [A336][I] with deionized water is inefficient due to the strong ionic bonds between [A336]+ cations and the halide complexes ([PtX6]2−, [PdX4]2−, and [AuX4]− (X = Br, and I) in the organic phase.
 | ||
| Fig. 7 Stripping behavior of precious metals from the loaded ionic liquids. Aqueous phase: 1.0 mol L−1 stripping reagent; loaded organic phase: (A) [A336][I], 678 mg L−1 Pt(IV), 672 mg L−1 Pd(II), 11.8 mg L−1 Rh(III), and 27.7 mg L−1 Au(III); (B) [A336][Br], 676 mg L−1 Pt(IV), 675 mg L−1 Pd(II), 31.9 mg L−1 Rh(III), and 28.9 mg L−1 Au(III); (C) [A336][Cl], 680 mg L−1 Pt(IV), 674 mg L−1 Pd(II), 25.5 mg L−1 Rh(III), and 28.8 mg L−1 Au(III); O/A = 1/1; equilibrium time 60 min; 298 K; 2000 rpm. | ||
The use of stripping agents containing low charge density anions such as SCN− and ClO4− is problematic due to their hydrophobic nature and the anion exchange that will take place with the ionic liquid phase when the strip solution is mixed with the halide ionic liquids. This will form a new ionic liquid and it will not be possible to directly reuse the ionic liquid in a new cycle of extraction. Furthermore, these stripping agents were not efficient. Fig. 7 shows that Pd(II) can be selectively stripped with NH4OH from [A336][I] and not from [A336][Cl] and [A336][Br], under the same experimental conditions. Not only NH4OH but also Na2S2O3 is promising for selective recovery of individual precious metals from the loaded [A336][I]. For instance, selective stripping of 89.8% Pd(II) was achieved with an NH3 solution, which results in co-stripping of only 0.97% Pt(IV), 4.25% Rh(III), and 0.04% Au(III) in a single contact. On the other hand, a Na2S2O3 solution yielded quantitative and selective stripping of Au(III) from [A336][I].
The limited stripping efficiency of Pd(II) with NH4OH from the loaded organic ionic liquids [A336][Cl] and [A336][Br] is attributed to the co-extraction of HCl at high acidity of the aqueous feed (Table S1 in the ESI†). The water content of the ionic liquids after equilibration with the aqueous phase is 18.4%, 9.28%, and 8.46% for [A336][Cl], [A336][Br] and [A336][I], respectively. The initial and equilibrium acid concentrations in the aqueous phases were calculated based on the interpolation table for refractive index (n20)-normality relationship for solutions of hydrochloric acid proposed by Olsen et al.34 The concentration of HCl in the organic phase was estimated using the mass balance with the assumption that the phase ratio O/A remained constant. Note that the ionic liquid [A336][Cl] extracts more HCl than [A336][Br] and [A336][I] at a given concentration of 5.77 M HCl (Table S1 in the ESI†).
Therefore, different concentrations of the stripping agent NH4OH were studied to ensure that a sufficient amount of NH4OH was used for not only the neutralization of the HCl already present in the organic phase, but also for the stripping of Pd(II). Table 1 shows that higher concentrations of NH4OH are required to successfully strip Pd(II) from [A336][Cl] and [A336][Br]. Furthermore, higher selectivity for stripping is obtained when stripping from the ionic liquid [A336][I], even when using high concentrations of NH4OH. However, the use of high concentrations of NH4OH for stripping is not recommended because the ionic liquid can be decomposed.35 In order to avoid the need of high concentrations of NH4OH for stripping, NaCl can be used as a chloride source in the feed, instead of HCl. Advantages of the use of NaCl are less aggressive media, its low price and the fact that it improves and accelerates the phase disengagement, a critical factor when carrying out solvent extraction in continuous mode.
| Loaded ionic liquid | NH4OH (mol L−1) | Percentage stripping (%S) | |||
|---|---|---|---|---|---|
| Pt | Pd | Rh | Au | ||
| a Aqueous phase: 1.0–6.0 mol L−1 NH4OH. Loaded organic phase from 6.0 M HCl feed: (A) [A336][Cl], 680 mg L−1 Pt(IV), 674 mg L−1 Pd(II), 25.5 mg L−1 Rh(III), and 28.8 mg L−1 Au(III); (B) [A336][Br], 676 mg L−1 Pt(IV), 675 mg L−1 Pd(II), 31.9 mg L−1 Rh(III), and 28.9 mg L−1 Au(III); (C) [A336][I], 678 mg L−1 Pt(IV), 672 mg L−1 Pd(II), 11.8 mg L−1 Rh(III), and 27.7 mg L−1 Au(III); O/A = 1/1; 60 min; 298 K; 2000 rpm. | |||||
| [A336][Cl] | 1.0 | 0.16 | 1.95 | 24.5 | 0.61 |
| 2.0 | 29.8 | 97.1 | 71.3 | 72.6 | |
| 6.0 | 39.5 | 94.0 | 88.7 | 85.5 | |
| [A336][Br] | 1.0 | 0.11 | 2.30 | 9.23 | 0.07 |
| 2.0 | 49.5 | 87.1 | 49.2 | 0.49 | |
| 6.0 | 39.4 | 80.0 | 68.1 | 36.9 | |
| [A336][I] | 1.0 | 0.97 | 89.8 | 4.25 | 0.04 |
| 2.0 | 3.74 | 90.3 | 5.76 | 0.08 | |
| 6.0 | 25.9 | 94.7 | 13.9 | 0.12 | |
The effect of chloride concentration on the extraction of precious metals with [A336][Cl], [A336][Br], and [A336][I] was studied in the range of 0.001 to 6.0 mol L−1 by introducing calculated amounts of different chloride salts at a given proton concentration of 0.001 mol L−1. The results are shown for Rh(III) in Fig. 8. As expected, the complete extraction (>99%) of Pt(IV), Pd(II), and Au(III) with three ionic liquids is independent of the chloride ion concentration. However, high chloride concentrations strongly suppress the extraction of Rh(III) as it was also seen when HCl was used (Fig. 2). Furthermore, the influence of the source of the chloride on the extraction of precious metals with three ionic liquids was also investigated by using different chloride sources such as LiCl, NaCl, NH4Cl, MgCl2, and CaCl2. Fig. 8 shows no significant salting-out effect on the extraction of precious metals, particularly Rh(III). A change of the cation of the chloride salt has a negligible effect on the extraction of precious metals. Thus, NaCl can be used as a replacement for HCl for selective extraction of Pt(IV), Pd(II), and Au(III) over Rh(III) from the aqueous feed solution.
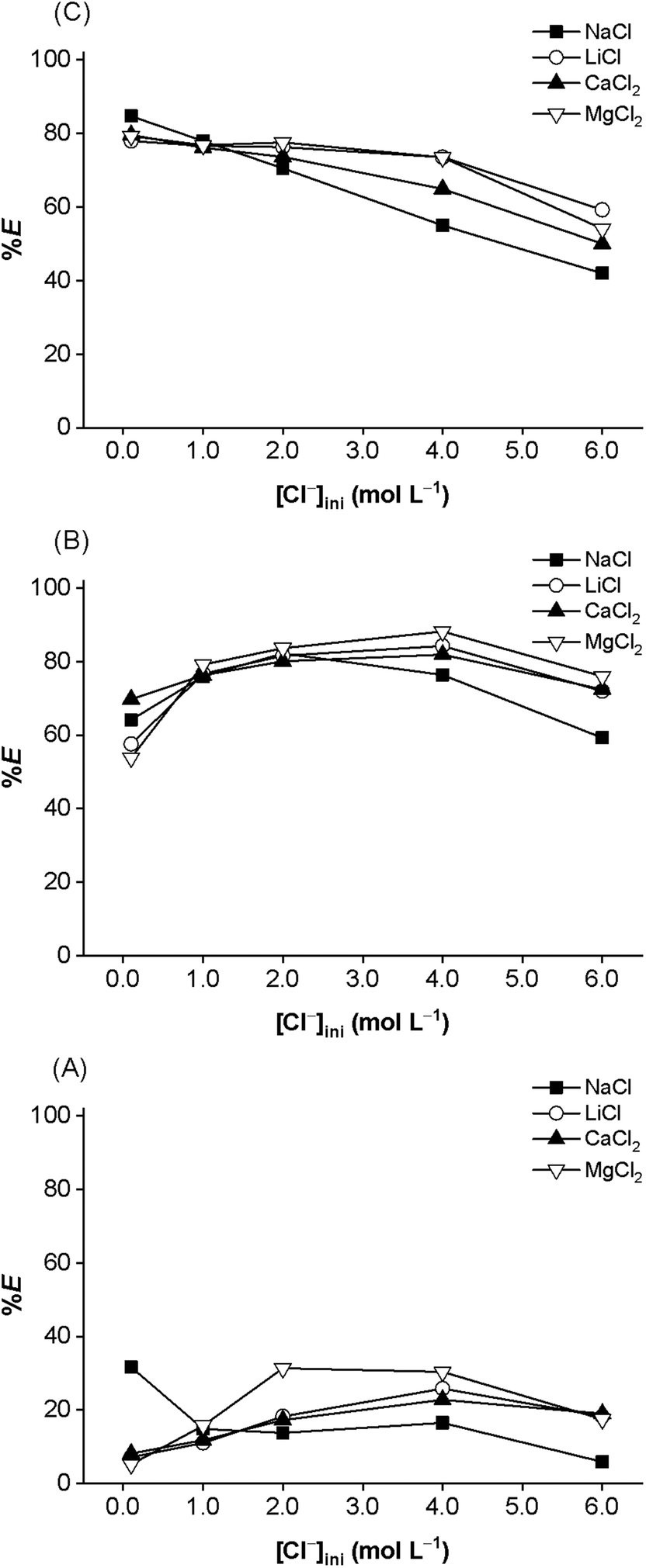 | ||
| Fig. 8 Effect of salting-out on the extraction of Rh(III). Aqueous phase: 680 mg L−1 Pt(IV), 675 mg L−1 Pd(II), 128 mg L−1 Rh(III), and 28.3 mg L−1 Au(III) in 0.001 mol L−1 HCl and total Cl− concentration of 0.001–6.0 mol L−1 Cl−; organic phase: water-saturated (A) 1.75 mol L−1 [A336][I]; (B) 1.80 mol L−1 [A336][Br]; and (C) 1.70 mol L−1 [A336][Cl]; O/A = 1/1; equilibrium time 60 min; 298 K; 2000 rpm. | ||
The stripping behavior of the precious metals in the loaded organic phases prepared from NaCl feed solution was studied. Lower concentrations of NH4OH (i.e. 0.2–0.5 mol L−1) can be used for the selective stripping of 66.8–90.1% Pd(II) from loaded [A336][I] (Table S2 in the ESI†). Meanwhile, the selective stripping of Pd(II) in loaded [A336][I] from 6 mol L−1 HCl feed requires at least 1.0 mol L−1 NH4OH. Using NaCl in the feed solution also allows to quantitatively strip Pd(II) from loaded [A336][Cl] and [A336][Br] with 1.0 mol L−1 NH4OH. However, a significant amount of Pt(IV), Rh(III), and Au(III) are co-stripped with Pd(II). The use of NaCl feed rather than HCl feed also led to higher stripping efficiency of the precious metals with 1.0 mol L−1 CS(NH2)2/HCl under comparable conditions. Among the investigated ionic liquids, the loaded [A336][I] yielded the highest stripping percentage of Pt(IV), Pd(II), Au(III), and Rh(III) using 1.0 mol L−1 CS(NH2)2/HCl as a result of its lowest loading capacity of HCl (Table S1 in the ESI†). Therefore, [A336][I] was chosen for further studies because it allows not only the selective but also the high recovery of the precious metals.
Selective stripping of precious metals from loaded [A336][I]
The sequence of selective stripping of precious metals from loaded [A336][I] is of importance. Depending on the stripping reagent used in the first stripping step, the stripping behavior of the metals can change in the subsequent one. Fig. 7 shows that Na2S2O3 and NH3 solutions have potential for the selective stripping of Au(III) and Pd(II), respectively. After the first stripping step, the Au(III)-free loaded organic and Pd(II)-free loaded organic phase were scrubbed twice with water (O/A = 1/2). Afterwards, stripping behavior of the precious metals from the scrubbed organic phases was examined using individual stripping reagents (i.e. CS(NH2)2/HCl, NH4OH, and Na2S2O3).Table 2 shows that the percentage stripping of precious metals with NH4OH is very low (<2.0%) from the organic phase [A336][I] previously stripped with Na2S2O3. In particular, the second stripping with 1.0 mol L−1 NH3 solution yielded only 0.96% Pt(IV), 0.30% Pd(II), 1.71% Rh(III), and 2.73% Au(III). This finding is not consistent with previous data (Fig. 7), in which selective stripping of Pd(II) was achieved with the same NH3 solution. The first stripping of Au(III) with Na2S2O3 prohibits Pd(II) stripping in a subsequent step. The presence of S2O32− anion, which forms strong complexes with Pt(IV) and Pd(II) in the organic phase, is probably the reason why Pd(II) and Pt(IV) cannot be stripped later on. Furthermore, no selective stripping of precious metals was observed using CS(NH2)2/HCl in the second stripping.
| Sequence | Stripping agents | Percentage stripping (%S) | |||
|---|---|---|---|---|---|
| Pt | Pd | Rh | Au | ||
| a Loaded [A336][I]: 678 mg L−1 Pt(IV), 672 mg L−1 Pd(II), 11.8 mg L−1 Rh(III), and 27.7 mg L−1 Au(III). Aqueous phase: 1.0 mol L−1 Na2S2O3, CH4N2S/HCl, or NH4OH; O/A = 1/1; 60 min; 298 K; 2000 rpm. b Metal concentration in the aqueous phase. | |||||
| 1st stripping | Na2S2O3 | 2.16 (14.6)b | 0.09 (0.61) | 1.63 (0.19) | 98.4 (27.3) |
| 2nd stripping | CS(NH2)2/HCl | 25.4 (172) | 62.6 (421) | 6.85 (0.81) | 2.91 (0.80) |
| NH4OH | 0.96 (6.51) | 0.30 (2.62) | 1.71 (0.20) | 2.73 (0.76) | |
On the other hand, a single contact for selective stripping of Pd(II) with NH3 solution from the loaded organic phase [A336][I] allows the selective recovery of other precious metals during the second stripping (Table 3). For example, complete stripping of Au(III) was achieved with 1.0 mol L−1 Na2S2O3 with co-stripping of only 4.09% Pt(IV), 0.73% Pd(II) and 6.18% Rh(III). The selectivity on the stripping of Au(III) in the second stage can be significantly enhanced by varying the Na2S2O3 concentration and the O/A ratio. Furthermore, 72.1% Pt(IV) was stripped in the second stripping with 1.0 mol L−1 CS(NH2)2/HCl. Co-stripping of other precious metals was 1.32% Pd(II), 11.0% Rh(III), 18.4% Au(III). Despite the fact that stripping with CS(NH2)2/HCl was not fully selective for the recovery of individual PGMs, the result suggests that CS(NH2)2/HCl could be used in the last stripping step, in which Pt(IV) can be recovered from the Pd(II)-free and Au(III)-free organic phases. Therefore, the sequence for stripping is critical to selectively recover precious metals from the loaded organic phase. Pd(II) is preferred to be recovered first using NH3 solution, followed by Au(III) stripping with Na2S2O3 solution, and finally Pt(IV) stripping with CS(NH2)2/HCl.
| Sequence | Stripping agents | Percentage stripping (%S) | |||
|---|---|---|---|---|---|
| Pt | Pd | Rh | Au | ||
| a Loaded [A336][I]: 678 mg L−1 Pt(IV), 672 mg L−1 Pd(II), 11.8 mg L−1 Rh(III), and 27.7 mg L−1 Au(III). Aqueous phase: 1.0 mol L−1 NH4OH, CH4N2S/HCl, or Na2S2O3; O/A = 1/1; 60 min; 298 K; 2000 rpm. b Metal concentration in the aqueous phase. | |||||
| 1st stripping | NH4OH | 0.98 (6.64)b | 89.8 (603) | 4.25 (0.50) | 0.04 (0.01) |
| 2nd stripping | CS(NH2)2/HCl | 72.1 (489) | 1.32 (8.87) | 11.0 (1.30) | 18.4 (5.10) |
| Na2S2O3 | 4.09 (27.7) | 0.73 (4.91) | 6.18 (0.73) | 100 (27.7) | |
Taking into account the most stable species that can be formed during the split-anion extraction of precious metals (eqn (7)), the mechanism for stripping of the metals is proposed as:
 | (11) |
 | (12) |
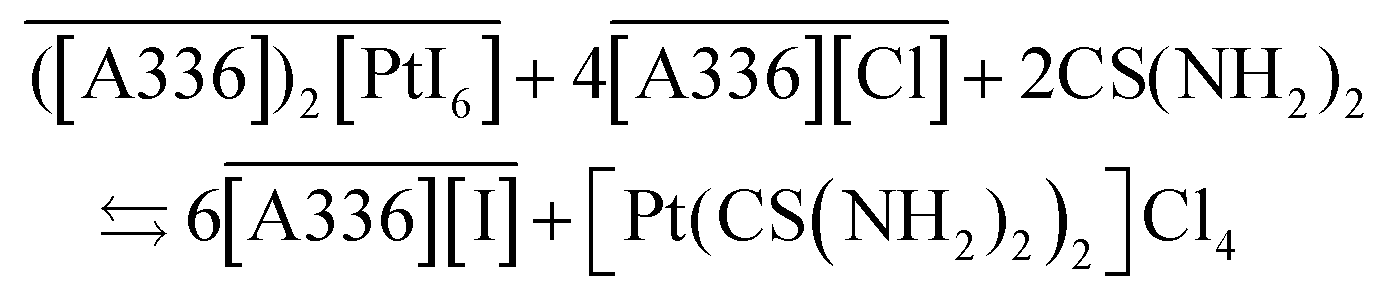 | (13) |
Regeneration of the ionic liquid [A336][I]
The ionic liquids need to be recovered and reused in several extraction-stripping cycles to ensure a sustainable process (Fig. 9). As shown in (eqn 11–13), a great advantage of the process is that [A336][I] automatically regenerated itself after the stripping. The presence of a trace amount of thiourea in the organic phase, however, can affect the extraction-stripping behavior of the precious metals in the subsequent cycles. Therefore, the ionic liquid phase obtained after Pt stripping was scrubbed with 0.1 mol L−1 HCl.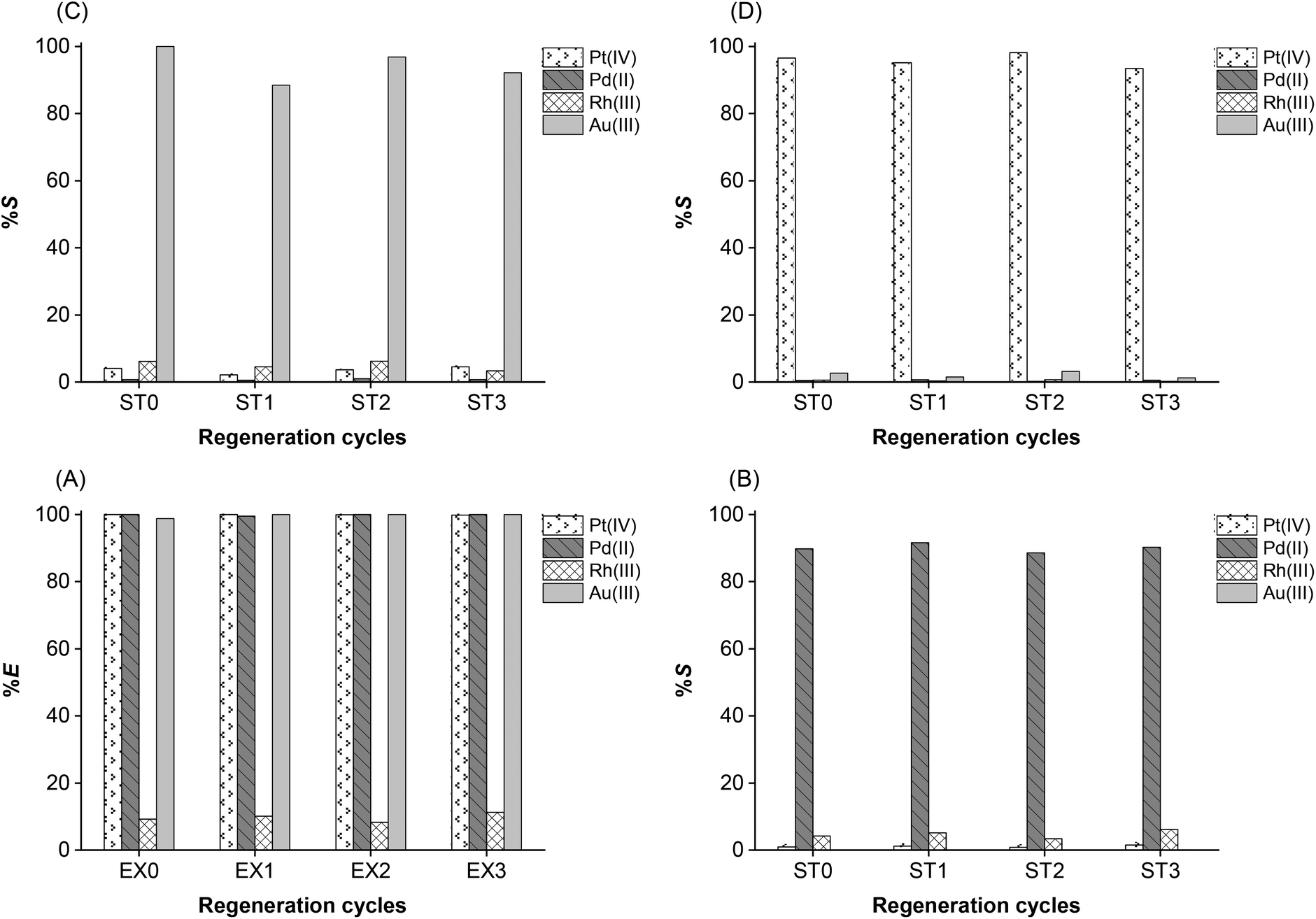 | ||
| Fig. 9 Extraction and stripping of PGMs using fresh and regenerated ionic liquids [A336][I]. (A) Extraction; (B) Pd stripping with 1.0 mol L−1 NH4OH; (C) Au stripping with 1.0 mol L−1 Na2S2O3; (D) Pt stripping with 1.0 mol L−1 CS(NH2)2/HCl; O/A = 1/1; equilibrium time 60 min; 298 K; 2000 rpm. | ||
The stability of the ionic liquids was determined by 1H NMR and FTIR (Fig. S3–S10 in the ESI†). As compared to the fresh ionic liquid, the regenerated [A336][I] shows NMR spectra with corresponding peaks, which indicates that the structural decomposition of the ionic liquid after the extraction-stripping processes is negligible. In addition, the 1H NMR spectra confirm the absence of CS(NH2)2 in the regenerated ionic liquid.
The reusability of the ionic liquid was evaluated within three extraction-stripping cycles under the same experimental conditions. Each cycle was completed by scrubbing of the ionic liquid with diluted HCl solution. As shown in Fig. 9, the percentage extraction of PGMs remains the same in the three regenerated cycles. Likewise, the selective stripping of Pd, Au, and Pt with NH4OH, Na2S2O3, and CS(NH2)2, respectively, is comparable using either fresh or regenerated ionic liquid. These results demonstrate the possibility to reuse the ionic liquid without loss of performance.
Conceptual flow sheet
A conceptual flow sheet for the separation of Rh(III), Pd(II), Pt(IV) and Au(III) from chloride media using the ionic liquid [A336][I] is presented in Fig. 10. First, Pd(II), Au(III) and Pt(IV) are extracted from chloride media using [A336][I], while leaving the majority of Rh in the raffinate. Afterwards, Pd(II), Au(III) and Pt(IV) are stripped from the loaded organic phases (LO) using aqueous solutions of NH4OH, NaS2O3 and CS(NH2)2/HCl, respectively.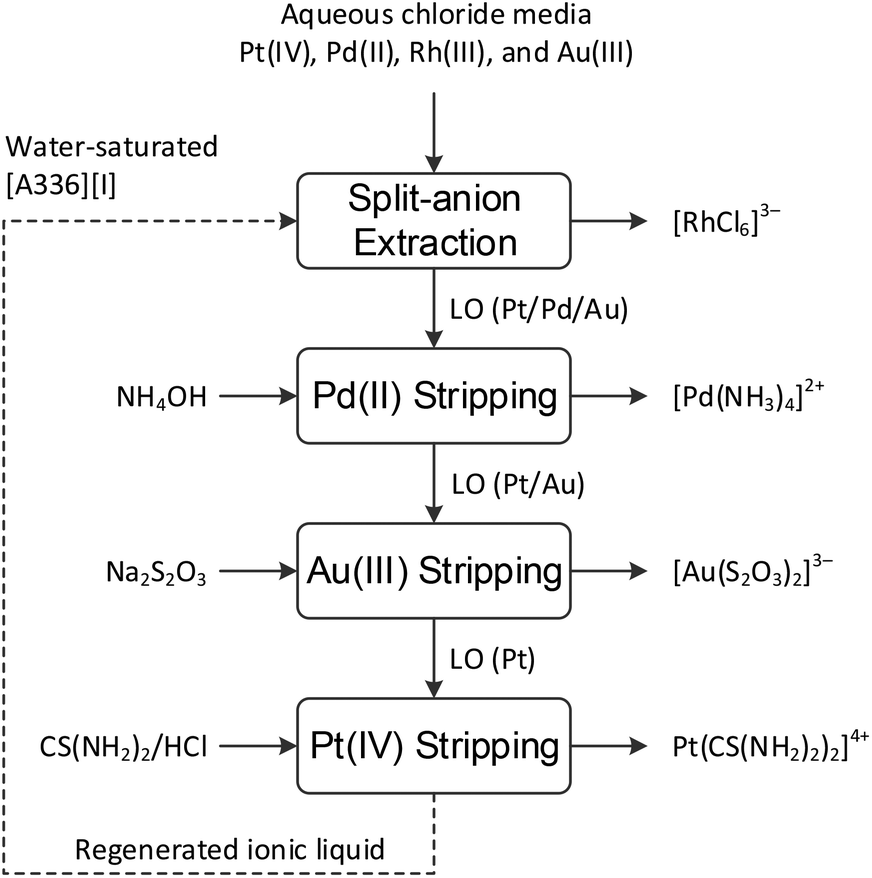 | ||
| Fig. 10 Conceptual flow sheet for separation of rhodium, palladium, platinum and gold from chloride feeds by split-anion extraction using water-saturated [A336][I]. | ||
The developed split-anion extraction makes use of only water-saturated ionic liquid [A336][I] as an environmentally friendlier alternative that allows selective and efficient separation of precious metals from chloride media. Furthermore, the regeneration of the ionic liquid phase without loss of performance up to three extraction-stripping cycles ensures the sustainability of the process.
Simulation of multistage counter-current solvent extraction of Pt(IV), Pd(II) and Rh(III) in the presence of other impurities
The refining of highly pure precious metals is critical for sustainable use in the catalyst industry. Therefore, the feasibility of the conceptual process was evaluated on the purification of precious metals from a complex metal feed such as the leachate of spent automotive catalysts using water-saturated [A336][I]. The feed solution mimicked the composition of a leachate of spent-auto catalyst. Different parameters were optimized for both extraction and stripping. The results are summarized in Table 4. The water-saturated ionic liquid [A336][I] was used as a green solvent, which decreases the viscosity, accelerates the phase disengagement and therefore facilitates the counter-current experiment. Different O/A ratios were chosen to concentrate the final solutions of Pt and Pd.| Step | Reagent | Concentration | O/A |
|---|---|---|---|
| a Equilibrium time 60 min, 298 K, 2000 rpm. | |||
| Extraction | [A336][I] | Water-saturated | 1/3 |
| Scrubbing of impurities | NaCl | 1.0 mol L−1 | 1/1 |
| Stripping Pd(II) | NH4OH | 1.0 mol L−1 | 3/1 |
| Stripping Pt(IV) | CS(NH2)2/HCl | 0.1 mol L−1 | 3/1 |
To purify precious metals from the leachate of spent automotive catalysts, the conceptual process (Fig. 10) was slightly modified, as shown in Fig. 11. According to the results obtained from the counter-current experiments, one stage was necessary to achieve the quantitative extraction of Pd(II) and Pt(IV) leaving Rh(III) in the raffinate. Subsequently, an extra scrubbing was needed for complete removal of the impurities prior to stripping of Pd(II) and Pt(IV). Three stages were needed for the stripping of Pd(II) with 1.0 mol L−1 NH3 solution and two stages for stripping Pt(IV) with 0.1 mol L−1 acidic thiourea solution at the same O/A phase ratio of 3/1. The stripping of Au(III) was not mentioned because of the negligible amount of Au(III) (<0.1 mg L−1) in the feed solution.
 | ||
| Fig. 11 Proposed flow sheet for separation of precious metals from the leachate of spent automotive catalysts by split-anion extraction using water-saturated [A336][I]. | ||
The mass balance of each metals in counter-current extraction and stripping is presented in Table S3 in the ESI.† Note that most of the impurities are left behind in the raffinate or are removed in the scrubbing step with 1.0 mol L−1 NaCl in a single stage. The raffinate, which contains Rh(III), is highly contaminated with other metals such as Al(III), Ce(III), Fe(III) and Na(I) and therefore a final purification step is needed. The raffinate was further processed with formic acid to produce metallic rhodium. Palladium was also reduced with formic acid, while platinum was reduced using cementation with zinc powder. The metallic forms of the precious metals were obtained with the following purities: 99.8% Pd, 99.5% Pt, and 99.9% Rh. The metallic forms of the PGMs can be used as starting material for producing the nitrate salts that are usually employed for producing the auto-catalysts.
Conclusions
The separation of precious metals (i.e. Pt(IV), Pd(II), Rh(III), and Au(III)) can be achieved by split-anion extraction with water-saturated ionic liquids [A336][X] (X− = Br− and I−). The stripping of the precious metals from loaded [A336][I] is significantly different from the stripping from [A336][Cl] and [A336][Br]. The water-saturated ionic liquid [A336][I] is a good extractant for Au(III), Pt(IV) and Pd(II) and allows the selective stripping of these elements into individual solutions. The process is versatile since it can be applied to complex feeds containing different metals in different ranges of compositions. Counter-current solvent extraction experiments demonstrated the feasibility of carrying out this process in continuous mode. The developed split-anion extraction is an environmentally friendly, simple, and effective alternative for the separation of Rh(III), Au(III), Pd(II) and Pt(IV) using only one extractant. In addition, the closed-loop process with a possible regeneration of the ionic liquid up to several extraction-stripping cycles contributes to the sustainable recycling and refining of the precious metals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research leading to these results received funding from the European Union's Horizon 2020 Research and Innovation program under Grant Agreement no. 730224 (PLATIRUS). This publication reflects only the authors’ view, exempting the Community from any liability. The authors acknowledge Monolithos (Athens, Greece) for providing the spent automotive catalysts.Notes and references
- R. M. Izatt, S. R. Izatt, N. E. Izatt, K. E. Krakowiak, R. L. Bruening and L. Navarro, Green Chem., 2015, 17, 2236–2245 RSC.
- F. Crundwell, M. Moats, V. Ramachandran, T. Robinson and W. Davenport, Extractive Metallurgy of Nickel, Cobalt and Platinum-Group Metals, Elsevier B.V., Oxford, UK, 2011 Search PubMed.
- G. P. Demopoulos, J. Met., 1986, 38, 13–17 CAS.
- F. L. Bernardis, R. A. Grant and D. C. Sherrington, React. Funct. Polym., 2005, 65, 205–217 CrossRef CAS.
- V. T. Nguyen, J. C. Lee, A. Chagnes, M. S. Kim, J. Jeong and G. Cote, RSC Adv., 2016, 6, 62717–62728 RSC.
- M. K. Jha, J. C. Lee, M. S. Kim, J. Jeong, B. S. Kim and V. Kumar, Hydrometallurgy, 2013, 133, 23–32 CrossRef CAS.
- V. T. Nguyen, J. C. Lee, J. Jeong, B. S. Kim, G. Cote and A. Chagnes, Ind. Eng. Chem. Res., 2015, 54, 1350–1358 CrossRef CAS.
- J. G. Huddleston, H. D. Willauer, R. P. Swatloski, A. E. Visser and R. D. Rogers, Chem. Commun., 1998, 1765–1766 RSC.
- U. Domańska, A. Pobudkowska and F. Eckert, Green Chem., 2006, 8, 268–276 RSC.
- N. Papaiconomou, S. Génand-Pinaz, J. M. Leveque and S. Guittonneau, Dalton Trans., 2013, 42, 1979–1982 RSC.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS.
- X. Sun, H. Luo and S. Dai, Chem. Rev., 2012, 112, 2100–2128 CrossRef CAS.
- H. Niedermeyer, J. P. Hallett, I. J. Villar-Garcia, P. A. Hunt and T. Welton, Chem. Soc. Rev., 2012, 41, 7780–7802 RSC.
- K. Larsson and K. Binnemans, Green Chem., 2014, 16, 4595–4603 RSC.
- T. Vander Hoogerstraete, S. Wellens, K. Verachtert and K. Binnemans, Green Chem., 2013, 15, 919–927 RSC.
- S. Wellens, B. Thijs and K. Binnemans, Green Chem., 2012, 14, 1657–1665 RSC.
- S. Riaño and K. Binnemans, Green Chem., 2015, 17, 2931–2942 RSC.
- T. Vander Hoogerstraete and K. Binnemans, Green Chem., 2014, 16, 1594–1606 RSC.
- M. Zhu, J. Zhao, Y. Li, N. Mehio, Y. Qi, H. Liu and S. Dai, Green Chem., 2015, 17, 2981–2993 RSC.
- M. L. Firmansyah, F. Kubota and M. Goto, J. Chem. Technol. Biotechnol., 2018, 93, 1714–1721 CrossRef CAS.
- S. Wellens, R. Goovaerts, C. Möller, J. Luyten, B. Thijs and K. Binnemans, Green Chem., 2013, 15, 3160–3164 RSC.
- A. Cieszynska and M. Wiśniewski, Hydrometallurgy, 2012, 113–114, 79–85 CrossRef CAS.
- A. Cieszynska and M. Wisniewski, Sep. Purif. Technol., 2011, 80, 385–389 CrossRef CAS.
- L. Svecova, N. Papaiconomou and I. Billard, Dalton Trans., 2016, 45, 15162–15169 RSC.
- K. Larsson and K. Binnemans, Hydrometallurgy, 2015, 156, 206–214 CrossRef CAS.
- D. Dupont, D. Depuydt and K. Binnemans, J. Phys. Chem. B, 2015, 119, 6747–6757 CrossRef CAS.
- M. Regadío, T. Vander Hoogerstraete, D. Banerjee and K. Binnemans, RSC Adv., 2018, 8, 34754–34763 RSC.
- S. Riaño, S. Sobekova Foltova and K. Binnemans, RSC Adv., 2019, 10, 307–316 RSC.
- E. Benguerel, G. P. Demopoulos and G. B. Harris, Hydrometallurgy, 1996, 40, 135–152 CrossRef CAS.
- D. Cozzi and F. Pantani, J. Inorg. Nucl. Chem., 1958, 8, 385–398 CrossRef CAS.
- M. H. Mihailov, V. T. Mihailova and V. A. Khalkin, J. Inorg. Nucl. Chem., 1974, 36, 115–120 CrossRef CAS.
- R. Lommelen, T. Vander Hoogerstraete, B. Onghena, I. Billard and K. Binnemans, Inorg. Chem., 2019, 58, 12289–12301 CrossRef CAS.
- W. Vereycken, S. Riano, T. Van Gerven and K. Binnemans, ACS Sustainable Chem. Eng., 2020, 8, 8223–8234 CrossRef CAS.
- A. L. Olsen and E. R. Washburn, Trans. Kans. Acad. Sci., 1937, 40, 117–126 CrossRef CAS.
- D. Landini, A. Maia and A. Rampoldi, J. Org. Chem., 1986, 51, 3187–3191 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Viscosity and density of ionic liquids; FTIR and 1H NMR spectra of the organic phases; extraction of HCl with the water-saturated ionic liquids; stripping of precious metals from loaded organic phase; mass balance of multi-stage counter-current solvent extraction. See DOI: 10.1039/d0gc02356f |
| This journal is © The Royal Society of Chemistry 2020 |