
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimization and sustainability assessment of a continuous flow Ru-catalyzed ester hydrogenation for an important precursor of a β2-adrenergic receptor agonist†
Michael
Prieschl
ab,
Jorge
García-Lacuna
b,
Rachel
Munday
c,
Kevin
Leslie
c,
Anne
O'Kearney-McMullan
c,
Christopher A.
Hone
 *ab and
C. Oliver
Kappe
*ab
*ab and
C. Oliver
Kappe
*ab
aCentre for Continuous Synthesis and Processing (CCFLOW), Research Center Pharmaceutical Engineering (RCPE), Inffeldgasse 13, 8010 Graz, Austria
bInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstraße 28, A-8010 Graz, Austria. E-mail: christopher.hone@rcpe.at; oliver.kappe@uni-graz.at
cChemical Development, Pharmaceutical Technology & Development, Operations, AstraZeneca, Macclesfield, UK
First published on 11th August 2020
Abstract
The development of a ruthenium-catalyzed continuous flow ester hydrogenation using hydrogen (H2) gas is reported. The reaction was utilized for the reduction of an important precursor in the synthesis of abediterol, a β2-adrenoceptor agonist that has undergone phase IIa clinical trials for the treatment of asthma and chronic obstructive pulmonary disorder. The reaction was investigated within a batch autoclave by using a design of experiments (DoE) approach to identify important parameter effects. The optimized flow process was successfully operated over 6 h with inline benchtop 19F NMR spectroscopy for reaction monitoring. The protocol is shown to be high yielding (98% yield, 3.7 g h−1) with very low catalyst loading (0.065 mol%). The environmental impact of the Ru-catalyzed hydrogenation was assessed and compared to an existing stoichiometric lithium aluminum hydride (LAH) reduction and sodium borohydride (NaBH4) reduction. The process mass intensity (PMI) for the Ru-catalyzed hydrogenation (14) compared favorably to a LAH reduction (52) and NaBH4 reduction (133).
The reduction of esters into their corresponding alcohols is traditionally performed in batch reactors using stoichiometric metal hydride reagents, such as LiAlH4 and NaBH4.1 The highly reactive hydride species should be carefully handled.2 While these reactions are often high yielding and selective, a stoichiometric reagent is necessary which consequently results in a large amount of waste that can be hazardous and expensive to destroy.3 Moreover, the workup is particularly challenging owing to the highly exothermic hydrolysis step that forms precipitates. Catalytic reductions of esters using hydrogen gas have been demonstrated as an atom economic alternative to using stoichiometric reagents, with minimal waste generated.4 Heterogeneous catalysts are generally limited to the use of harsh conditions and are incompatible with substrates containing sensitive functional groups.4 Heterogeneously catalyzed ester hydrogenations were described as early as 1931 by Adkins and co-workers.5 Adkins-type catalysts (CuO/CuCr2O4) typically utilize very harsh conditions (>200 °C and >200 bar).5 Despite the requirement for elevated temperatures and pressures, these catalysts are still used in modern applications for unselective reduction of fatty acids and their esters.6 Subsequently, milder methods were developed for the reduction of the esters which also conserve olefin functionality, thus facilitating the production of unsaturated fatty alcohols.7 More recent methods have further progressed to the use of less harsh conditions, for instance a bimetallic Ag–Au catalyst was shown to reduce dimethyl oxalate at temperatures as low as 145 °C and 30 bar pressure.8 The heterogeneous hydrogenation of esters was recently achieved at room temperature using ruthenium-based catalysts with phosphorus ligands covalently attached to a polymeric support under 50 bar pressure.9
Homogeneous catalysts have been shown to perform with high turnover numbers under relatively mild conditions and display high functional group tolerance.4,10–15 Ruthenium based catalyst systems have been demonstrated as highly efficient catalysts for ester hydrogenations.4,12–15 In 2006, the group of Milstein introduced a new type of ruthenium catalyst utilizing pincer ligands for the hydrogenation of esters.12 Subsequently, the Takasago International Corporation reported Ru-MACHO (A) as an efficient catalyst system.13 Ru-MACHO is uninhibited by alcohols, thus is catalytically active in alcoholic solvents and is also not deactivated by the product. Gusev and co-workers reported the development of Ru-SNS (B) as an alternative catalyst system.14 The benefit of Ru-SNS is that it does not use phosphine ligands, which can be relatively expensive. The switch from stoichiometric hydride reductions to Ru-catalyzed hydrogenations is estimated to reduce the E-factor (kg waste per kg product) by approximately 3 to 5-fold.15 Currently, Ru-MACHO and Ru-SNS are the most industrially viable homogeneous ruthenium catalysts for ester hydrogenation.
The use of hydrogen gas within batch reactors generally requires the use of high pressure to ensure sufficient dissolution of gas within the liquid phase. Consequently, specialized and expensive equipment is required and scale-up can be challenging. Continuous flow reactors have been demonstrated as a safe and scalable technology for the scale-up of gas–liquid reactions.16–18 In recent years, there has been a greater focus on developing green processes that avoid waste and hazardous compounds.19 Sustainable chemical processes rely not only on effective chemistry but also on the implementation of reactor technologies that enhance reaction performance, reduce energy consumption and improve overall safety. The utilization of continuous flow reactors can significantly contribute towards this endeavor.20 Energy efficient heating enables the sustainable utilization of intensified conditions for maximizing yield and throughput. Precise parameter control, such as mixing, temperature and pressure, can improve product yield and selectivity. The safe use of highly atom efficient routes that would be inaccessible or too dangerous under traditional batch conditions is possible with continuous flow reactors.21 The reduction of different functional groups within continuous flow reactors has been achieved by a number of research groups, and was recently reviewed by Riley and co-worker in 2018.22,23
Abediterol (AZD0548) (Fig. 1) is a potent, long-acting inhaled β2-adrenoceptor agonist that was first pharmacologically characterized in 2012.24 It has undergone phase IIa trials for the treatment of asthma and chronic obstructive pulmonary disease (COPD).25 A route for the synthesis of the lipophilic amine tail portion of abediterol was published in 2019.26 An annual demand in the order of kilograms would be expected due to the very high potency of the drug candidate. The first step in the synthesis is a lithium aluminum hydride (LAH) reduction to afford 2,2-difluoro-2-phenylethanol (2) (Scheme 1a). An alternative protocol which uses stoichiometric NaBH4 has also been reported for the transformation.27
 | ||
| Fig. 1 Structure of abediterol (AZD0548), a β2-adrenoceptor agonist. | ||
 | ||
| Scheme 1 Synthesis of 2,2-difluoro-2-phenylethanol (2): (a) a previously reported LAH reduction (ref. 26); and NaBH4 reduction (ref. 27); (b) continuous flow homogeneous Ru-catalyzed hydrogenation (this work). | ||
We were interested in developing a Ru-catalyzed continuous flow protocol with hydrogen gas as a sustainable, safe and scalable alternative for the synthesis of 2 (Scheme 1b). Ikariya and coworkers previously demonstrated the use of Ru-MACHO for the hydrogenation of alpha-fluorinated esters to their corresponding alcohols under batch conditions.28
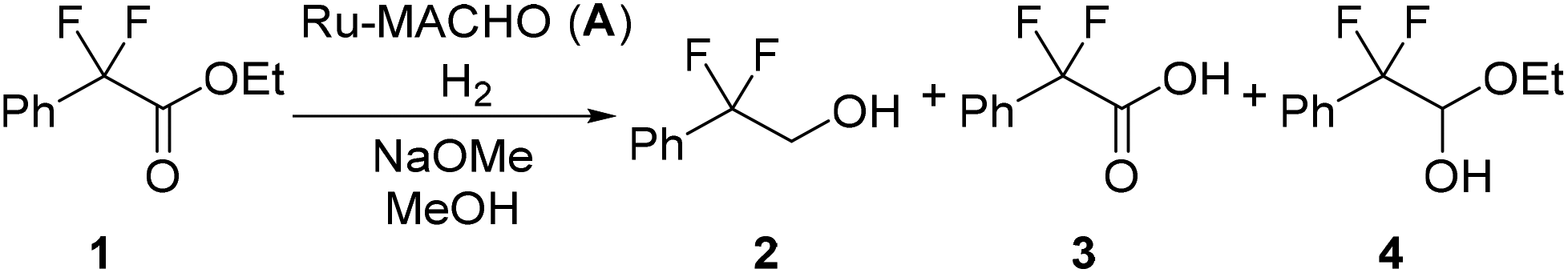
Reaction optimization experiments for the hydrogenation of ethyl 2,2-difluoro-2-phenylacetate (1) were performed on a 5 mmol scale within a batch autoclave. Commercially available Ru-MACHO (A) was used as catalyst (Fig. 2) and sodium methoxide (NaOMe) as base (Scheme 2). A relatively short reaction time of 1 h was used for all batch reactions to facilitate easy transfer from batch to flow. Furthermore, we were also interested in identifying conditions that dissolved all reaction components. The conversion of ester 1 (−103.9 ppm) to alcohol 2 (−106.7 ppm) could be monitored offline by 19F NMR with a low field benchtop spectrometer (Spinsolve Ultra 43 MHz, Magritek). Methanol was used as solvent due to its relatively green credentials.29 Toluene (PhMe), tetrahydrofuran (THF), methyl tetrahydrofuran (MeTHF) and tert-butyl alcohol/PhMe were also screened, but provided inferior results (Table S1†). These poorer results were probably caused by the limited base solubility in the solvent. For the batch optimization, a design of experiments (DoE) approach was selected. A four-parameter, two-level full factorial experimental design was implemented, corresponding to 19 experiments including 3 center point repeats to measure reproducibility (Table 1). Pressure was varied between 10 and 30 bar, temperature between 40 and 60 °C, catalyst loading between 0.03 and 0.10 mol% and base between 0.1 and 0.3 equivalents. During the initial experiments, an acid side product 3 (−101.3 ppm) and a hemiacetal intermediate 4 (−108.7 and −111.0 ppm) could also be identified. The methyl ester 5 derivative (−103.7 ppm) was observed from transesterification, but this did not influence the course of the hydrogenation reaction. The responses for the conversion of 1, alcohol 2, acid side product 3, and hemiacetal intermediate 4 were measured during experiments.
 | ||
| Scheme 2 Ru-catalyzed hydrogenation of ester 1 to alcohol 2. Acid 3 is generated as a side product and hemiacetal 4 as an intermediate. | ||
 | ||
| Fig. 2 Hydrogenation catalysts: Ru-MACHO (A), Ru-SNS (B) and Ru-MACHO-BH (C). | ||
| Entry | p (bar) | T (°C) | Catalyst loading (mol%) | Base (eq.) | Conversion 1 (%) | Alcohol 2 (%) | Side product 3 (%) | Hemiacetal 4 (%) |
|---|---|---|---|---|---|---|---|---|
| a Standard reaction conditions: 1 (5 mmol scale) in MeOH (2.5 mL) with stirring at 600 rpm for 1 h. Conversion and product distribution were determined by integration of 19F NMR. Conversion of 1 was calculated based on the combined integration of the ethyl ester 1 and methyl ester 5. | ||||||||
| 1 | 10 | 40 | 0.03 | 0.1 | 33.0 | 23.3 | 7.3 | 2.3 |
| 2 | 30 | 40 | 0.03 | 0.1 | 72.9 | 66.8 | 5.0 | 1.1 |
| 3 | 10 | 60 | 0.03 | 0.1 | 64.7 | 56.9 | 7.3 | <1 |
| 4 | 30 | 60 | 0.03 | 0.1 | >99 | 94.9 | 5.1 | <1 |
| 5 | 10 | 40 | 0.1 | 0.1 | 47.0 | 38.1 | 6.6 | 2.4 |
| 6 | 30 | 40 | 0.1 | 0.1 | >99 | 96.3 | 3.7 | <1 |
| 7 | 10 | 60 | 0.1 | 0.1 | 99.1 | 93.0 | 5.9 | <1 |
| 8 | 30 | 60 | 0.1 | 0.1 | >99 | 95.2 | 4.8 | <1 |
| 9 | 10 | 40 | 0.03 | 0.3 | 27.7 | 7.7 | 8.0 | 12.1 |
| 10 | 30 | 40 | 0.03 | 0.3 | 71.1 | 57.8 | 7.6 | 5.6 |
| 11 | 10 | 60 | 0.03 | 0.3 | 86.6 | 76.9 | 8.9 | <1 |
| 12 | 30 | 60 | 0.03 | 0.3 | >99 | 94.2 | 5.8 | <1 |
| 13 | 10 | 40 | 0.1 | 0.3 | 75.1 | 64.3 | 6.0 | 4.7 |
| 14 | 30 | 40 | 0.1 | 0.3 | >99 | 94.7 | 5.3 | <1 |
| 15 | 10 | 60 | 0.1 | 0.3 | >99 | 94.1 | 5.9 | <1 |
| 16 | 30 | 60 | 0.1 | 0.3 | >99 | 94.3 | 5.7 | <1 |
| 17 | 20 | 50 | 0.065 | 0.2 | >99 | 93.8 | 6.2 | <1 |
| 18 | 20 | 50 | 0.065 | 0.2 | >99 | 94.7 | 5.3 | <1 |
| 19 | 20 | 50 | 0.065 | 0.2 | >99 | 95.2 | 4.8 | <1 |
Gratifyingly, very high conversion and selectivity towards the desired alcohol 2 were achieved for a number of experiments (Table 1). Under milder conditions, the conversion of ester 1 and yield of alcohol 2 were lower, while intermediate 4 was observed at higher levels for these experiments. Side product 3 was observed at similar levels for all the experiments. The experimental repeats displayed very good reproducibility (entries 17–19). Furthermore, it was shown that by lowering temperature, pressure or catalyst loading (10 bar, 40 °C, 0.03 mol% A) it is possible to shift the selectivity of the reaction towards the hemiacetal intermediate 4.
We were interested in comparing the results for the hydrogenation of ethyl 2,2-difluoro-2-phenylacetate (1) with less reactive methyl trifluoroacetate as substrate (Table S2†). Under similar conditions, lower conversion and higher amounts of a hemiacetal intermediate were observed as the kinetic product. The thermodynamic alcohol product could be favored under more aggressive conditions. These results suggest that a less reactive fluorinated hemiacetal is stable enough to resist further conversion to the alcohol under carefully controlled reaction conditions.
The responses from the optimization experiments, shown in Table 1, were fitted to polynomial models by using a statistical experimental design software package (Modde v12). Models were successfully fitted for the alcohol 2 and side product 3 from the 19F NMR data by using multiple linear regression (MLR) (Fig. S6 and S7†). Models were generated by including all main and interaction terms and then non-significant terms were removed. A good fit was achieved for both models with R2 = 0.79 and R2 = 0.72 for the alcohol 2 and side product 3 respectively. The increase of pressure, temperature and catalyst loading were shown to have a positive influence on the formation of alcohol 2. The base loading did not show an influence on the yield of 2. The increase in pressure and catalyst loading resulted in a slight decrease in the yield of side product 3. Temperature displayed no influence over side product 3 formation. Whereas increasing the base loading resulted in a higher yield of 3. The models generated from the DoE were used to explore the experimental design space and to identify promising initial conditions for translation to flow (Fig. 3).
 | ||
| Fig. 3 Model-predicted contour plots showing the influence of different parameters on: (a) alcohol 2 yield; and (b) side product 3 yield. Constant conditions: 1 (5 mmol scale) in MeOH (2.5 mL) with stirring at 600 rpm for 1 h. | ||
The formation of side product 3 was shown to increase with the amount of NaOMe added. Furthermore, significant heat was produced upon exposure of ester 1 to NaOMe, thus we were interested in identifying a strategy to control this exotherm. The reaction resulted in recovered starting material 1 in the absence of base, because base is necessary to form the active catalyst. Ru-MACHO-BH (C) has been reported to work successfully without the addition of base for pre-activation of the catalyst.30 We attempted Ru-MACHO-BH on our system; however, Ru-MACHO-BH under base-free conditions resulted in only recovered starting material 1 (Table S4†). On addition of base, the reaction then worked successfully affording alcohol 2 in 98% yield. These results indicated that the catalyst could be decomposing during reaction preparation. We observed that Ru-MACHO-BH rapidly decomposes, with the hydrolysis of the borane group, in the presence of air (Fig. S8†). Thus, we selected Ru-MACHO (A) as the catalyst of choice for the flow experiments.
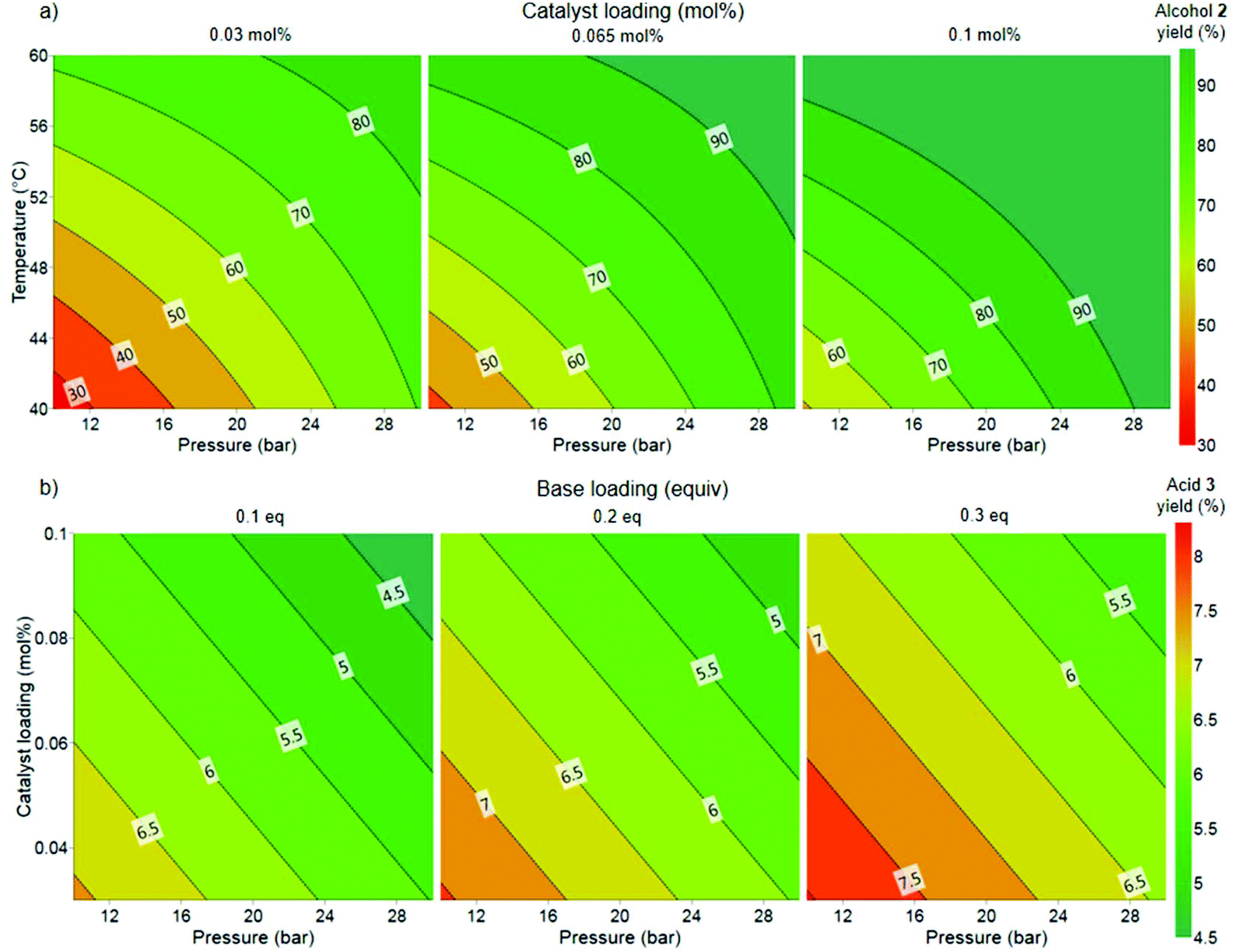
Flow experiments were performed using a Uniqsis FlowSyn system (Fig. 4). The two liquid feeds were introduced with high performance liquid chromatography (HPLC) pumps and hydrogen gas was introduced through a mass flow controller (MFC, Bronkhorst EL-FLOW). The two liquid feeds were mixed within an arrow-shaped mixer. Subsequently, the liquid and gas feed were combined using a Y-shaped mixer. A gas–liquid segmented (Taylor) flow regime was observed for all experiments. The reaction was performed within a heated stainless steel reactor coil (60 mL, 1/8 in. OD, 1/16 in. ID). Pressure was applied by using an adjustable back pressure regulator (BPR). For all flow experiments, fractions were collected every 5 minutes and analyzed offline with 19F NMR. In the initial flow configuration, the base and catalyst were introduced as one feed but this led to irreproducible results (Table 2, entry 1). Interestingly, the catalyst has been previously reported to slowly decompose in the presence of base.31 Furthermore, as stated previously, the presence of a base results in the formation of side product 3. Thus, to avoid any undesired reactions occurring within the feed solutions, the catalyst and substrate in MeOH were introduced as one feed and the NaOMe in MeOH as the second feed.
 | ||
| Fig. 4 Continuous flow setup for the Ru-catalyzed hydrogenation of ester 1 to alcohol 2. MFC = mass flow controller for the introduction of H2 and BPR = back pressure regulator. The dashed blue box indicates the part of the setup utilized for the inline analysis with NMR which was implemented during the long run. | ||
| Entry | p (bar) | C1.0 [M] | Base [eq.] | t res [min] | Conv. 1 [%] | Yield 2 [%] |
|---|---|---|---|---|---|---|
| a 0.4 mL min−1 total liquid flow rate, both liquid feeds were pumped at equal flow rates, 30 mLn min−1 H2 flow rate, 0.065 mol% catalyst loading, 60 °C temperature. Reagents were introduced for 30 min then switched to carrier solvent. b Pre-stirring of base and catalyst for 15 min. c Anhydrous MeOH and fresh NaOMe solution stored under Ar used. d 0.66 mL min−1 total liquid flow rate, 50 mLn min−1 H2 flow rate. e 1 mL min−1 total liquid flow rate, 50 mLn min−1 H2 flow rate. f Ru-SNS used as catalyst. | ||||||
| 1ab/1bb | 20 | 1.0 | 0.1 | 50 | 80/9 | 73/0 |
| 2a/2b | 20 | 1.0 | 0.1 | 50 | 34/50 | 26/40 |
| 3a/3b | 30 | 1.0 | 0.2 | 55 | >99 | 91/93 |
| 4c | 30 | 1.0 | 0.2 | 55 | >99 | 95 |
| 5c | 30 | 1.5 | 0.2 | 55 | >99 | 96 |
| 6ac | 20 | 1.0 | 0.2 | 55 | >99 | 96 |
| 6b (long run)c | 20 | 1.0 | 0.2 | 70 | >99 | 98 |
| 7c,d | 20 | 1.0 | 0.2 | 30 | >99 | 87 |
| 8c,e | 20 | 1.0 | 0.2 | 25 | >99 | 91 |
| 9f | 20 | 1.0 | 0.2 | 55 | 23 | 1 |
We selected to commence our flow experiments at a lower concentration (1 M) than the batch experiments to ensure the solubility of all the reaction components. A >90% product yield at 0.065 mol% catalyst loading, 20 bar pressure and 60 °C temperature was predicted from the model generated from the batch studies. A reduction in base loading had also been demonstrated in the DoE to reduce the yield of the acid side product 3 (Fig. 3b), but not influence the yield of alcohol 2 (Fig. 3a). The two liquid feeds were each pumped at 0.2 mL min−1 and the hydrogen gas at 30 mLn min−1, corresponding to a residence time of approximately 50 min and 3.3 equiv. of H2. 2 equiv. of H2 is necessary for the transformation, therefore only a relatively small H2 excess (1.3 equiv.) is used. A low base loading of 0.1 equivalents unexpectedly resulted in a lower conversion than expected (entry 1) and provided inconsistent results. This drop in conversion and the irreproducibility can be explained by the nature of the side reaction. The base is consumed in the presence of water by the reaction of esters 1 and 5 to the acid 3. Thus, the reaction is very sensitive to changes in the water content at low base loadings. Full conversion and more than 90% yield of 2 could be achieved by increasing the base loading to 0.2 equivalents and the pressure to 30 bar (entry 3). More importantly, the reaction displayed good reproducibility at these conditions. The use of anhydrous MeOH and fresh NaOMe solution provided an increase in desired alcohol 2 (entry 4), whilst decreasing the formation of the acid side product 3. The throughput could be increased by operating at a higher concentration (1.5 M), without a drop in conversion or yield observed (entry 5). The pressure could also be decreased whilst maintaining full conversion and high yield (entry 6a). The reaction was also performed at a shorter residence time within the reactor setup, corresponding to 0.33 mL min−1 for each liquid pump and 50 mLn min−1 H2 to provide 35 min residence time. These conditions resulted in >99% conversion of 1 and 87% alcohol 2, with the remaining present as the acid 3 side product (entry 7). This result indicates that some formation of the side product 3 from remaining substrate 1 could also be occurring after the reactor within the collection vessel. We conducted control experiments using acid 3 as starting material instead of ester 1. In these experiments only recovered acid 3 and some transesterification to the methyl ester 5 (5–7%) was observed (Table S3†). The reaction provided 91% yield of alcohol 2 even when using 2.2 equiv. of H2 and 25 min residence time (entry 8). A reaction with Gusev's Ru-SNS catalyst (B) was performed as a comparison (entry 9). Under similar conditions, the use of Ru-SNS resulted in low conversion of 1, trace amount of desired product, and high selectivity to hemiacetal 4. A lower activity of Ru-SNS, with higher selectivity towards the hemiacetal intermediate 4 when compared to using Ru-MACHO as catalyst, has been reported by Dub and co-workers.32
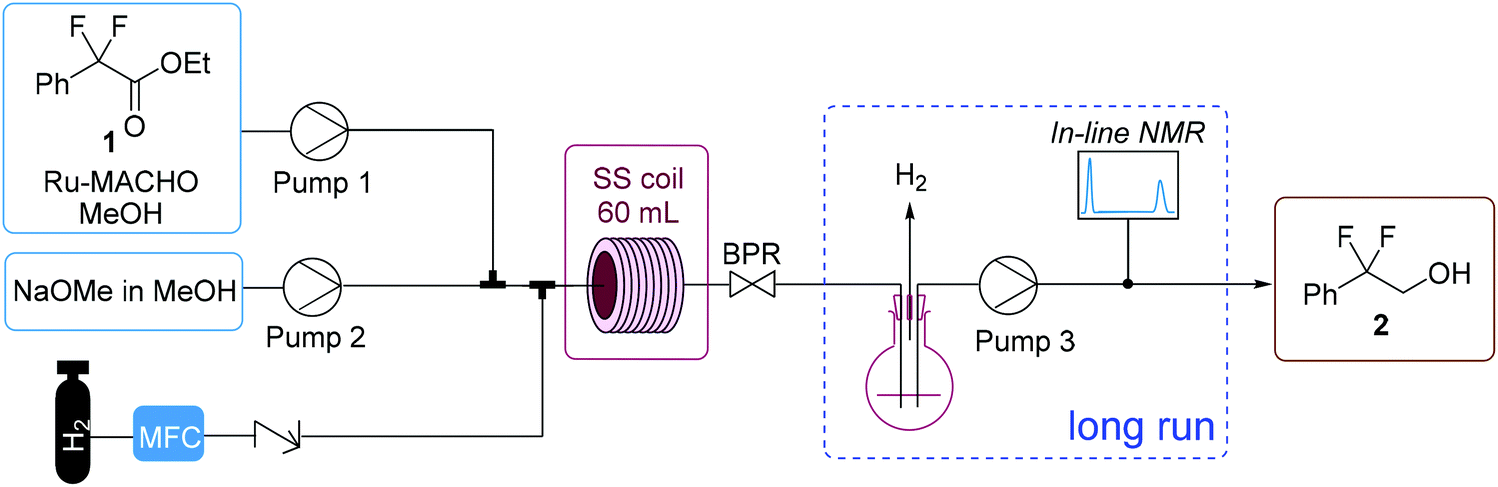
A long run experiment was performed over a total operation time of nearly 6 hours to demonstrate the stability of the process (entry 6b). A gas–liquid separator was incorporated into the flow setup after the BPR to enable inline analysis (Fig. 4). After separating the gas from the liquid stream, the liquid was fed using a HPLC pump through an inline flow cell (0.8 mL internal volume, max. pressure 10 bar) for monitoring by a benchtop low field NMR spectrometer (Spinsolve Ultra 43 MHz, Magritek).33 This enabled the online monitoring of the reaction progress for all the main reaction species by 19F NMR spectroscopy, with a spectrum acquired approximately every 20 s and >800 measurements taken in total. Fig. 5 shows the percentage of starting material 1, alcohol 2 and side product 3 over operation time as determined by integration of the peaks for the spectra generated by the inline 19F NMR measurements. The system performed consistently for the duration of the run. Moreover, the data points for the inline measurements very closely corresponded to the values for the data points of the collected fractions that were measured manually offline by 19F NMR, thus validating the inline method. The residence time was longer for the long run due to the consumption of H2 gas. For the optimization experiments, the feeds were injected via a sample loop (6 mL each) over 30 min. For the long run, feeds were introduced continuously through the pumps. Therefore H2 is not consumed throughout the entire reactor for the optimization experiments, thus accounting for the difference in the observed residence time between the optimization experiments and long run (entry 6b). Overall, the process was operated at “steady-state” conditions for approximately 220 minutes. A >99% conversion of 1 and 98% selectivity towards 2 was observed for the combined fractions from operation at “steady-state” conditions. After removal of MeOH, a simple extraction protocol was performed using ethyl acetate (EtOAc) as solvent to obtain alcohol 2 in 98% isolated yield based on “steady-state” operation. A throughput of 3.7 g h−1 was obtained, corresponding to a space–time yield of 1.0 g/(L min). One could envisage how the throughput could be increased by transfer to a larger scale coil reactor without a drop in performance as described elsewhere.17a
 | ||
Fig. 5 Results of the inline 19F NMR measurements for the long run. Values are calculated by using19 F NMR integrals of the peaks. Ester 1 and 5 ( ), alcohol 2 ( ), alcohol 2 ( ), and side product 3 ( ), and side product 3 ( ). Alcohol 2 ( ). Alcohol 2 ( ) measured offline for validation. Conditions used are given in Table 2, entry 6. ) measured offline for validation. Conditions used are given in Table 2, entry 6. | ||
The flow protocol provides benefits in terms of scalability, safety and product quality over a batch autoclave protocol. The addition of reagents can be carefully manipulated to provide the desired stoichiometry. The exotherm associated with base addition can be minimized, because heat generated can be removed quickly. Typical commercial batch reactors can operate between 2 and 6 bar, therefore higher pressures require more specialized and expensive equipment. There is no headspace filled with gas within a flow reactor and a flow reactor facilitates improved safety due to the small volumes of pressurized equipment needed. Furthermore, a flow protocol operates at steady-state, therefore providing consistent product output as shown by Fig. 5. The in-line NMR also enables improved understanding the performance of the continuous process, thus aiding in waste prevention through an increase in process understanding.
Table 3 shows the results from the green metrics assessment for the Ru-MACHO hydrogenation, the LAH reduction, and the NaBH4 reduction. For the comparison of the environmental impact, a green metrics toolkit developed by Clark and co-workers in 2015 was used.34 The values used for the calculations are shown in Tables S5–S7.† All the reactions reach full conversion and provide good selectivity and yield, however; the Ru-MACHO hydrogenation reaches a higher value for yield. In addition, the Ru-MACHO protocol performs better for atom economy (AE) and reaction mass efficiency (RME), therefore has a higher optimum efficiency (OE). The Green Chemistry Institute Pharmaceutical Roundtable selected process mass intensity (PMI) as their preferred mass-based green metric.35 PMI corresponds to the total mass used in a process divided by the mass of the product. The reaction PMI accounts for all the chemicals in the reaction, whereas the total PMI accounts for the chemicals used in the reaction and the work-up procedure. The total PMI is over 3-fold lower for the Ru-MACHO hydrogenation (14) when compared to the LAH reduction (52), and over 6-fold lower when compared to the NaBH4 protocol (133). These results demonstrate that the Ru-MACHO flow protocol is substantially more sustainable than the LAH and NaBH4 reductions. The E-factor shows the same trend, whereby a higher E factor is observed for the LAH (51) and NaBH4 (132) reductions, largely due to the high amount of solvent necessary for the work-up compared to a Ru-MACHO hydrogenation (13).
| Metric | Ru-MACHO | LAHa | NaBH4a |
|---|---|---|---|
| a Calculations based on LAH and NaBH4 reduction protocols reported in ref. 26 and 27 respectively. | |||
| Conv. [%] | >99 | >99 | >99 |
| Yield [%] | 98 | 93 | 91 |
| AE | 78 | 66 | 66 |
| RME | 75 | 61 | 54 |
| OE | 96 | 91 | 81 |
| PMI reaction | 6 | 10 | 23 |
| PMI work-up | 8 | 41 | 110 |
| PMI total | 14 | 52 | 133 |
| E factor | 13 | 51 | 132 |
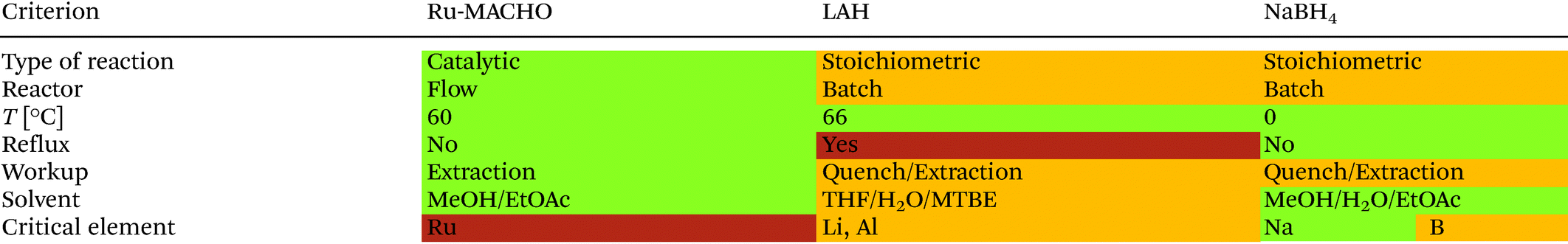
Table 4 shows a comparison for the qualitative green metrics between the LAH and Ru-MACHO reductions. In the green metrics toolkit, colored flags (green, amber, red) are given to each reaction to assess how green they are regarding each criterion. A green flag means “preferred”, amber “is acceptable-some issues” and red is “undesirable”. The Ru-MACHO hydrogenation receives green flags because it is catalytic and performed in flow. On the other hand, the LAH and NaBH4 reductions require stoichiometric reagent and are currently performed in batch, which results in amber flags for these criteria. All reactions are operated within an energy efficient temperature window (0–70 °C) which results in green flags. The LAH reduction uses THF at reflux conditions and is thus less energy efficient than the Ru-MACHO reaction, which is performed below its reflux temperature. Running the LAH reaction at reflux results in an approximately 6-fold increase in energy consumption as opposed to performing the reaction at 5 °C below reflux.34,36 Due to the high amount of energy used when heating to reflux, this results in a red flag for the LAH reduction. The simple extraction for the Ru-MACHO protocol results in a green flag, whereas the LAH and NaBH4 reactions require a number of extractions and an exothermic aqueous quench (amber flag).
|
The Ru-MACHO reaction only uses green solvents (methanol and ethyl acetate), and also the NaBH4 reduction (methanol and ethyl acetate), while the LAH reduction uses THF and MTBE which are considered of medium concern (amber flag). The main drawback of the Ru-MACHO protocol is that ruthenium is considered a critical element, for which the supply could run out in the following 5–50 years (red flag). Although, the ruthenium catalyst is employed at a very low loading (0.065 mol%). An additional amber flag is added because the catalyst is not currently recovered. The supply of sodium is considered to be sustainable into the future, but boron supply is expected for 100–500 years so results in an amber flag. The supply for lithium and aluminum are predicted to be sufficient for 100–500 more years, which results in an amber flag.37 However, the demand of lithium is rising rapidly due to its use in Li-ion batteries.
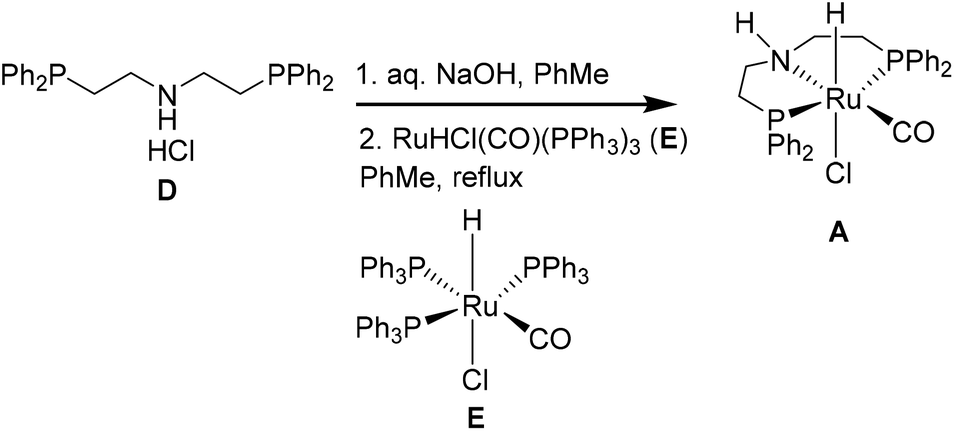
Currently, Ru-MACHO is not recycled as part of the continuous flow protocol, therefore we also evaluated the green metrics associated with its batch synthesis (Table 5 and Table S8†). Ru-MACHO (A) can be prepared from the commercially available bis[(2-diphenylphos-phino)ethyl]ammoniumchloride (D) and carbonylchlorohydridotris(triphenylphosphine)ruthenium(II) (E).13,38 These metrics are very favorable, especially when considering that only 0.065 mol% of Ru-MACHO are used for the hydrogenation.
The hydrogenation is applied to the synthesis of a precursor for a drug candidate, therefore the contamination by Ru metal should be considered. Furthermore, for executing this chemistry on industrial scale, strategies should be considered for the recovery of Ru because it is a critical element. A maximum contamination of 10 ppm is the regulatory limit for Ru metal within a pharmaceutical.39 Hessel and co-workers reviewed strategies for the separation and recycling of homogeneous transition metal catalysts in continuous flow systems.40 Conceivably, one of the simplest strategies is the use of scavenging agents in solution or through the incorporation of a scavenging column inline. A sustainable method for the removal of Ru was published in 2018.41 Here, treatment of the post-reaction mixture with an isocyanide scavenger and then treatment with acid, followed by a simple filtration provided Ru levels below 5 ppm. This type of strategy could be readily incorporated inline for the current process.
Conclusions
A gas–liquid continuous flow Ru-catalyzed hydrogenation protocol was developed for the preparation of an important precursor for the β2-adrenergic receptor agonist. 2,2-Difluoro-2-phenylethanol is a key precursor to abediterol, which has undergone phase IIa clinical trials for the treatment of asthma. The reaction only consumes H2 as a stoichiometric reagent. The flow reaction uses H2 as an inexpensive, atom–economic, and environmentally friendly feedstock to generate gas–liquid segmented flow patterns, which allows the reaction to be completed within 1 h residence time. The flow process operates at 60 °C and 20 bar with a much smaller excess (3.3 equivalents of H2) of gas than required for batch processes. A low catalyst loading (0.065 mol%) afforded the desired alcohol product in 98% isolated yield. In particular, the continuous flow protocol was operated for nearly 6 h run time (total duration) to produce 13.7 g of the API precursor. 19F NMR was successfully incorporated inline for real-time process monitoring of the long run. The environmental impact of the hydrogenation was assessed and compared to an existing stoichiometric lithium aluminum hydride reduction (LAH). The process mass intensity of the hydrogenation represents over a 3-fold reduction when compared to the LAH reduction, and over a six-fold reduction when compared to the NaBH4 reduction. The flow protocol represents an improvement in terms of atom economy, safety and scalability, and also reduces energy consumption and solvent usage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CCFLOW Project (Austrian Research Promotion Agency FFG no. 862766) is funded through the Austrian COMET Program by the Austrian Federal Ministry of Transport, Innovation and Technology (BMVIT), the Austrian Federal Ministry for Digital and Economic Affairs (BMDW), and by the State of Styria (Styrian Funding Agency SFG). The INFRA FLOW project (Zukunftsfonds Steiermark no. 9003) is funded by the State of Styria (Styrian Funding Agency SFG). J. G. thanks the Banco Santander-Fundación San Pablo-CEU for a mobility fellowship.Notes and references
- (a) J. Seyden-Penne, Reductions by the Alumino- and Borohydrides in Organic Synthesis, 1997, 2nd edn, Wiley-VCH, New York Search PubMed; (b) J. Málek, Org. React., 1985, 34, 1–317 Search PubMed; (c) J. Málek, Org. React., 1988, 36, 249–590 Search PubMed.
- L. A. Paquette, T. Ollevier and V. Desyroy, Lithium Aluminum Hydride, in Encyclopedia of Reagents for Organic Synthesis, 2004, DOI:10.1002/047084289X.rl036.
- J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156–1184 CrossRef CAS.
- J. Pritchard, G. A. Filonenko, R. Van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2015, 44, 3808–3833 RSC.
- (a) H. Adkins and K. Folkers, J. Am. Chem. Soc., 1931, 53, 1095–1097 CrossRef CAS; (b) H. Adkins, H. I. Cramer and R. Connor, J. Am. Chem. Soc., 1931, 53, 1402–1405 CrossRef CAS; (c) K. Folkers and H. Adkins, J. Am. Chem. Soc., 1932, 54, 1145–1154 CrossRef CAS; (d) R. Connor, K. Folkers and H. Adkins, J. Am. Chem. Soc., 1932, 54, 1138–1145 CrossRef CAS.
- (a) D. S. Thakur, B. D. Roberts, T. J. Sullivan and A. L. Vichek, Hydrogenation catalyst, process for preparing and process of using said catalyst, US Patent, US5155086, October 13th 1992 Search PubMed; (b) U. R. Kreutzer, J. Am. Oil Chem. Soc., 1984, 61, 343–348 CrossRef CAS.
- Y. Pouilloux, F. Autin and J. Barrault, Catal. Today, 2000, 63, 87–100 CrossRef CAS.
- J. Zheng, H. Lin, Y. N. Wang, X. Zheng, X. Duan and Y. Yuan, J. Catal., 2013, 297, 110–118 CrossRef CAS.
- F. J. L. Heutz, C. Erken, M. J. B. Aguila, L. Lefort and P. C. J. Kamer, ChemCatChem, 2016, 8, 1896–1900 CrossRef CAS.
- S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18(2), 289–302 CrossRef CAS.
- For catalyst systems using metals other than ruthenium, see: (a) T. Zell, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2014, 53, 4685–4689 CrossRef CAS PubMed; (b) S. Chakraborty, H. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause and H. Guan, J. Am. Chem. Soc., 2014, 136, 7869–7872 CrossRef CAS PubMed; (c) K. Junge, B. Wendt, H. Jiao and M. Beller, ChemCatChem, 2014, 6, 2810–2814 CrossRef CAS; (d) S. Elangovan, M. Garbe, H. Jiao, A. Spannenberg, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2016, 128, 15590–15594 CrossRef; (e) N. A. Espinosa-Jalapa, A. Nerush, L. J. W. Shimon, G. Leitus, L. Avram, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2017, 23, 5934–5938 CrossRef CAS PubMed; (f) D. Srimani, A. Mukherjee, A. F. G. Goldberg, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12357–12360 CrossRef CAS PubMed; (g) T. J. Korstanje, J. I. van der Vlugt, C. J. Elsevier and B. de Bruin, Science, 2015, 350, 298–302 CrossRef CAS PubMed.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2006, 45, 1113–1115 CrossRef CAS PubMed.
- W. Kuriyama, T. Matsumoto, O. Ogata, Y. Ino, K. Aoki, S. Tanaka, K. Ishida, T. Kobayashi, N. Sayo and T. Saito, Org. Process Res. Dev., 2012, 16, 166–171 CrossRef CAS.
- D. Spasyuk, S. Smith and D. G. Gusev, Angew. Chem., Int. Ed., 2013, 52, 2538–2542 CrossRef CAS PubMed.
- A. Zanotti-Gerosa, D. Grainger, L. Todd, G. Grasa, L. Milner, E. Boddie, L. Browne, I. Egerton and L. Wong, Chim. Oggi, 2019, 37(4), 8–11 CAS.
- (a) C. J. Mallia and I. R. Baxendale, Org. Process Res. Dev., 2016, 20, 327–360 CrossRef CAS; (b) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed; (c) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed; (d) M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- (a) M. D. Johnson, S. A. May, J. R. Calvin, J. Remacle, J. R. Stout, W. D. Diseroad, N. Zaborenko, B. D. Haeberle, W. M. Sun, M. T. Miller and J. Brennan, Org. Process Res. Dev., 2012, 16, 1017–1038 CrossRef CAS; (b) S. A. May, M. D. Johnson, J. Y. Buser, A. N. Campbell, S. A. Frank, B. D. Haeberle, P. C. Hoffman, G. R. Lambertus, A. D. McFarland, E. D. Moher, T. D. White, D. D. Hurley, A. P. Corrigan, O. Gowran, N. G. Kerrigan, M. G. Kissane, R. R. Lynch, P. Sheehan, R. D. Spencer, S. R. Pulley and J. R. Stout, Org. Process Res. Dev., 2016, 20, 1870–1898 CrossRef CAS.
- For reviews on continuous flow hydrogenation, see: (a) M. Irfan, T. N. Glasnov and C. O. Kappe, ChemSusChem, 2011, 4, 300–316 CrossRef CAS PubMed; (b) P. J. Cossar, L. Hizartzidis, M. I. Simone, A. McCluskey and C. P. Gordon, Org. Biomol. Chem., 2015, 13, 7119–7130 RSC; (c) T. Yu, P. Song, W. Nie, C. Yi, Q. Zhang and P. Li, ChemSusChem, 2020, 13, 300–316 Search PubMed.
- (a) J. C. Anastas and P. T. Warner, Green Chemistry: Theory and Practice, 1998, Oxford University Press, New York Search PubMed; (b) H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC; (c) P. Poechlauer, J. Colberg, E. Fisher, M. Jansen, M. D. Johnson, S. G. Koenig, M. Lawler, T. Laporte, J. Manley, B. Martin and A. O'Kearney-McMullan, Org. Process Res. Dev., 2013, 17, 1472–1478 CrossRef CAS.
- (a) D. Dallinger and C. O. Kappe, Curr. Opin. Green Sustain. Chem., 2017, 7, 6–12 CrossRef; (b) L. Rogers and K. F. Jensen, Green Chem., 2019, 21, 3481–3498 RSC; (c) A. J. Blacker, J. R. Breen, R. A. Bourne and C. A. Hone, The Growing Impact of Continuous Flow Methods on the Twelve Principles of Green Chemistry, in Green and Sustainable Medicinal Chemistry: Methods, Tools and Strategies for the 21st Century Pharmaceutical Industry, Royal Society of Chemistry, Cambridge, U.K., 2016, ch. 12, pp. 140–155 Search PubMed.
- B. Gutmann and C. O. Kappe, J. Flow Chem., 2017, 7, 65–71 CrossRef CAS.
- D. L. Riley and N. C. Neyt, Synthesis, 2018, 50, 2707–2720 CrossRef CAS.
- For selected examples of ester reductions in flow using stoichiometric reagents, see: (a) D. Mandala, S. Chada and P. Watts, Org. Biomol. Chem., 2017, 15, 3444–3454 RSC; (b) L. Ducry and D. M. Roberge, Org. Process Res. Dev., 2008, 12, 163–167 CrossRef CAS; (c) D. Webb and T. F. Jamison, Org. Lett., 2012, 14, 568–571 CrossRef CAS PubMed; (d) M. Yoshida, H. Otaka and T. Doi, Eur. J. Org. Chem., 2014, 6010–6016 CrossRef CAS; T. Fukuyama, H. Chiba, H. Kuroda, T. Takigawa, A. Kayano and K. Tagami, Org. Process Res. Dev., 2016, 20, 503–509 Search PubMed; (e) J. M. De Muñoz, J. Alcázar, A. De La Hoz and A. Díaz-Ortiz, Eur. J. Org. Chem., 2012, 260–263 CrossRef; (f) S. B. Ötvös and C. O. Kappe, ChemSusChem, 2020, 13, 1800–1807 CrossRef PubMed.
- M. Aparici, M. Gómez-Angelats, D. Vilella, R. Otal, C. Carcasona, M. Viñals, I. Ramos, A. Gavaldà, J. De Alba, J. Gras, J. Cortijo, E. Morcillo, C. Puig, H. Ryder, J. Beleta and M. Miralpeix, J. Pharmacol. Exp. Ther., 2012, 342, 497–509 CrossRef CAS PubMed.
- (a) W. Timmer, E. Massana, E. Jimenez, B. Seoane, G. de Miquel and S. Ruiz, J. Clin. Pharmacol., 2014, 54, 1347–1353 CrossRef CAS PubMed; (b) J. Beier, R. Fuhr, E. Massana, E. Jiménez, B. Seoane, G. De Miquel and S. Ruiz, Respir. Med., 2014, 108, 1424–1429 CrossRef CAS PubMed.
- R. H. Munday, L. Goodman and G. M. Noonan, Tetrahedron Lett., 2019, 60, 606–609 CrossRef CAS.
- M.-T. Hsieh, K.-H. Lee, S.-C. Kuo and H.-C. Lin, Adv. Synth. Catal., 2018, 360, 1605–1610 CrossRef CAS.
- (a) T. Otsuka, A. Ishii, P. A. Dub and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 9600–9603 CrossRef CAS PubMed; (b) A. Ishii, T. Ootsuka, T. Ishimaru and M. Imamura, Method for producing β-fluoroalcohol, US Patent, US8658840B2, 25th February 2014 Search PubMed.
- (a) D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS; (b) R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binksaand and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- W. Kuriyama, T. Matsumoto, Y. Ino and O. Ogata, Ruthenium carbonyl complex having tridentate ligand, its production method and use, US Patent, US8471048B2, 25th June 2013 Search PubMed.
- A. Anaby, M. Schelwies, J. Schwaben, F. Rominger, A. S. K. Hashmi and T. Schaub, Organometallics, 2018, 37, 2193–2201 CrossRef CAS.
- P. A. Dub, R. J. Batrice, J. C. Gordon, B. L. Scott, Y. Minko, J. G. Schmidt and R. F. Williams, Org. Process Res. Dev., 2020, 24, 415–442 CrossRef CAS.
- For a review on inline monitoring using benchtop NMR, see: P. Giraudeau and F. X. Felpin, React. Chem. Eng., 2018, 3, 399–413 RSC.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- A very similar LAH batch protocol for substrate 1 was reported, but using room temperature and a longer (18 h) reaction time, to afford the product 2 in 66% yield, see: M. A. Higgins, L. R. Marcin, F. C. Zusi, R. Gentles, M. Ding, B. C. Pearce, A. Easton, W. A. Kostich, M. A. Seager, C. Bourin, L. J. Bristow, K. M. Johnson, R. Miller, J. Hogan, V. Whiterock, M. Gulianello, M. Ferrante, Y. Huang, A. Hendricson, A. Alt, J. E. Macor and J. J. Bronson, Bioorg. Med. Chem., 2017, 25, 496–513 CrossRef CAS . Operating at room temperature would result in a green flag for this criterion.
- T. Watari, K. Nansai and K. Nakajima, Resour., Conserv. Recycl., 2020, 155, 104669–104685 CrossRef.
- Q. Yao, Ruthenium, carbonylchloro[2−(diphenylphosphino–κP)–N –[2−(diphenylphosphino–κP)ethyl]ethanamine–κN, ]hydro–in, Encyclopedia of Reagents for Organic Synthesis, 2015 DOI:10.1002/047084289X.rn01800.
- D. R. Abernethy, A. J. DeStefano, T. L. Cecil, K. Zaidi and R. L. Williams, Pharm. Res., 2010, 27, 750–755 CrossRef CAS PubMed.
- I. V. Gürsel, T. Noël, Q. Wang and V. Hessel, Green Chem., 2015, 17, 2012–2026 RSC.
- G. Szczepaniak, A. Ruszczyńska, K. Kosiński, E. Bulska and K. Grela, Green Chem., 2018, 20, 1280–1289 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02225j |
| This journal is © The Royal Society of Chemistry 2020 |