
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDihydrolevoglucosenone (Cyrene™) as a bio-renewable solvent for Cu(0)wire-mediated reversible deactivation radical polymerization (RDRP) without external deoxygenation†
Arkadios
Marathianos‡
 a,
Evelina
Liarou‡
a,
Ellis
Hancox
abc,
James L.
Grace
bc,
Daniel W.
Lester
a and
David M.
Haddleton
*ab
a,
Evelina
Liarou‡
a,
Ellis
Hancox
abc,
James L.
Grace
bc,
Daniel W.
Lester
a and
David M.
Haddleton
*ab
aDepartment of Chemistry, University of Warwick Library Road, Coventry, CV4 7AL, UK. E-mail: d.m.haddleton@warwick.ac.uk
bMonash Institute of Pharmaceutical Sciences, ARC Centre of Excellence in Convergent Bio-Nano Science and Technology, Monash University, Parkville, Melbourne, Victoria 3052, Australia
cDrug Delivery, Disposition and Dynamics Theme, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia
First published on 17th August 2020
Abstract
Biorenewable dihydrolevoglucosenone (Cyrene™) is used as an effective dipolar aprotic solvent for Cu(0) wire-mediated RDRP of various monomers without external deoxygenation being applied. The solvent is used to give products with a broad range of molar masses (Mn ∼ 700–28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), in situ chain extension and as low as 7.8 × 10−4 eq. of [Cu(II)Br2] relative to initiator.
000), in situ chain extension and as low as 7.8 × 10−4 eq. of [Cu(II)Br2] relative to initiator.
Controlled radical polymerization (CRP) has enabled the synthesis of a large number of well-defined materials,1–6 with well-defined shape, different functionalities and molecular architectures.7,8 Amongst them, Cu-mediated reversible deactivation radical polymerization (Cu-RDRP) has evolved into an efficient technique for the polymerization of many functional vinyl monomers, including hydrophilic and hydrophobic (meth)acrylates, styrenics and (meth)acrylamides using many different dipolar solvents (e.g. water, DMSO, DMF, NMP), verifying the versatility of this technique.10–12 However, many of these solvents have been added to the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) restricted substances list due to their negative environmental effects and are becoming undesirable reagents in many applications. In this context, several solvent selection guides have been published in order to replace many commonly used volatile organic solvents or compounds that could form toxic gases upon incineration (COx, NOx, SOx), with solvents derived from natural or/and renewable resources.13–15
Dihydrolevoglucosenone (Cyrene™) is a bio-based and fully biodegradable (99%, 14d) aprotic dipolar solvent that can be synthesized in a two-step process from biomass, mainly from cellulose.9,16 Cyrene is not mutagenic, has a median lethal dose (LD50) > 2000 mg kg−1 (above the value of high toxicity solvents, 50 mg kg−1) and it is hardly ecotoxic.17 Therefore, it has potential as a greener and safer alternative to widely used dipolar aprotic solvents, such as DMF, NMP, DMAc and DMSO.17–19 Furthermore, Cyrene has similar solvating behavior with the abovementioned solvents, demonstrating similar Hansen solubility parameters.20 A further characteristic of Cyrene is that it is completely miscible with water, since it is in equilibrium with its hydrate (a geminal diol), in contrast to most conventional ketones in water.21 Cyrene™ is currently manufactured on a relatively small scale by Circa Group in Tasmania leading to relatively high prices when compared to DMSO. However, as of 2020 new 1000 and 5000 tonne plants are planned for construction in Europe which should reduce the costs to become within 1.5–2 of DMSO at current pricing.22
Recently, Cyrene has attracted significant interest as a bio-degradable solvent in the synthesis of metal–organic frameworks,17 synthetic transformations,20 metal catalyzed processes,23 in the synthesis of ureas24 and amides.25,26 Moreover, upon further hydrogenation, Cyrene could lead to more renewable chemicals that can be used as precursors for drugs, flavors and polymers.27 In the field of polymer synthesis, the use of Cyrene as solvent has been underexplored, with few examples of its use being as a co-solvent in low toxicity solvent systems for polyamideimide and polyamide amic acid resin manufacture,28 in sustainable membrane preparation29 and in membrane performance tests of interpenetrating polymer networks.30 Conversely, in some cases Cyrene has been used as a monomer precursor. In a recent work, Ray et al. reported the development of bio-acrylic polymers from a methacrylic monomer synthesized from Cyrene.31 This monomer was polymerized under different free radical polymerization conditions (bulk, solution and emulsion) and it was found that polymers obtained from emulsion polymerization had higher yields and molecular weights, in contrast with solution polymerization.
In this current work, we report the use of Cyrene as a bio-alternative solvent for the polymerization of various monomers using Cu(0) wire-mediated RDRP without applying any external deoxygenation or addition of external additives (Scheme 1). Well-defined polymers with high conversions, narrow molecular weight distributions and high end-group fidelity were obtained, enabling the synthesis of diblock and triblock copolymers. Moreover, a wide range of molar masses from 700 to 28000 g mol−1 (targeted DPn = 5–800) of poly(methyl acrylate) (pMA) were obtained in the absence of deoxygenation and with Cyrene as solvent. The use of a low catalyst concentration was investigated for the Cu(0) wire-RDRP of MA (targeted DPn = 50), to test the limits of this system. Finally, the oxygen tolerance profile of the polymerization was elucidated by employing an oxygen probe.
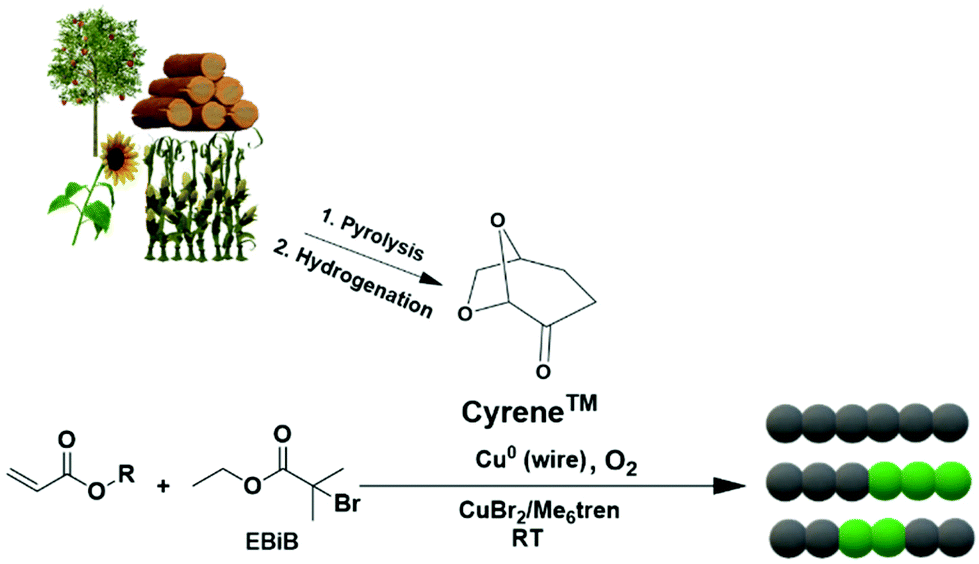 | ||
| Scheme 1 Cu(0) wire-mediated RDRP in Cyrene, a bio-alternative solvent derived from biomass (cellulose) in a two-step process.9 | ||
The choice of solvent is important for a successful Cu-RDRP process due to the effect on the rate of the polymerization,32,33 the solubility of the Cu complexes, monomers and polymers,34–36 as well as potential of solvent coordination to the copper.37 Initially, in order to investigate Cyrene as a solvent for Cu(0) wire-mediated RDRP, polymerizations were conducted under an inert nitrogen atmosphere. For this purpose, MA was used as monomer (targeted DPn = 50), ethyl α-bromoisobutyrate (EBiB) as initiator, 5 cm of Cu(0) wire and Cu(II)Br2 as copper source and the aliphatic multidentate ligand tris(2-(dimethylamino)ethyl)-amine (Me6Tren) as ligand with Cyrene as solvent (Table S1, entry 1, Fig. S1 & S4a†). Polymerization was carried out at ambient temperature with [EBiB]:[Me6Tren]:[Cu(II)Br2] = 1:0.18:0.05 (as previously reported when DMSO was used as solvent38), leading to pMA50 with high monomer conversion (>95% after 18 h), Mn, NMR = 4400 g mol−1 and ĐSEC = 1.09. It is noted that, although it has previously been reported that under basic conditions and in the presence of amines, Cyrene can exhibit sensitivity, we observed no evidence of side reactions in this study (Fig. S2†).23 We think this is as the reactions were conducted at ambient temperature with low concentrations of Me6Tren (18.7 mM for pMA50). Apart from EBiB, we explored the possibility to use the more hydrophobic dodecyl 2-bromoisobutyrate as an initiator for the polymerization of MA (targeted DPn = 50), resulting in pMA33 (74% conversion after 24 h) with Mn, SEC = 4000 g mol−1 and Đ = 1.08 (Table S1, entry 2, Fig. S4b†). Apart from MA, the efficiency of Cyrene as solvent for the synthesis of polymethacrylates was explored. In this context, the polymerization of methyl methacrylate (MMA) was conducted under similar conditions with methyl α-bromophenylacetate as initiator, yielding pMMA50 (95% after 18 h) with Mn, SEC = 6000 g mol−1 and Đ = 1.12 (Table S1, entry 3, Fig. S3 & S4c†). Therefore, based on these initial experiments it was anticipated that Cyrene could be efficiently employed as an alternative, biodegradable solvent for the Cu(0) wire-RDRP of both MA (acrylates) and MMA (methacrylates).
Some of our previous work has focused on the Cu-RDRP of various monomers without any type of deoxygenation.39–42 Thus, we explored the possibility to polymerize a variety of hydrophobic monomers including MA, MMA, tert-butyl acrylate (tBA), benzyl acrylate (BzA), trifluoroethyl acrylate (TFEA) and styrene (St), via Cu(0)-RDRP by omitting deoxygenation, thus rendering this approach more user-friendly. Cu(0)-RDRP of MA over 3 h resulted in 90% monomer conversion with Mn, SEC = 4800 g mol−1 and Đ = 1.08, the time needed for the other monomers varied from 20 to 45 h all leading to high conversions, good control over the Mn and relatively low dispersities (Table S2, Fig. S5–S11†). These results are similar to the Mn, SEC, dispersity and conversion observed for the non-deoxygenated Cu(0) wire-RDRP of MA in commonly used organic solvents, as described by Liarou et al.39 It is noteworthy that following the polymerization of tBA the formation of a biphasic system was observed, with a top polymer-rich phase and a bottom solvent-rich layer containing the majority of the catalyst and residual monomer (3%).
Subsequently, we were interested in investigating the ability to achieve a range of molecular weights in Cyrene as solvent and in the presence of air/oxygen. By targeting DPn values from 5 to 800, polymers with molecular weights from 700 to 28000 g mol−1 with narrow dispersities were obtained (Table 1, Fig. 1). When low DPs were targeted (higher [EBiB]) (Table 1, entries 1–4) high conversion of MA and very good agreement between theoretical and experimental Mn values was observed. However, moving to higher DPn values with lower [EBiB], and thus lower concentration of propagating chains led to an increase of the polymerization times, while lower yields and loss of initiator efficiency (Ieff) was also seen (Table 1, entries 5–7, Table S4, Fig. S12–S18†). The higher molar masses required prolonged reaction times leading the polymerization to be more susceptible to side reactions. It has also been reported that the initiator participates in the O2-consumption mechanism,39 therefore low [EBiB] could affect the rate of O2-consumption and subsequently result in loss of Ieff and chain termination.
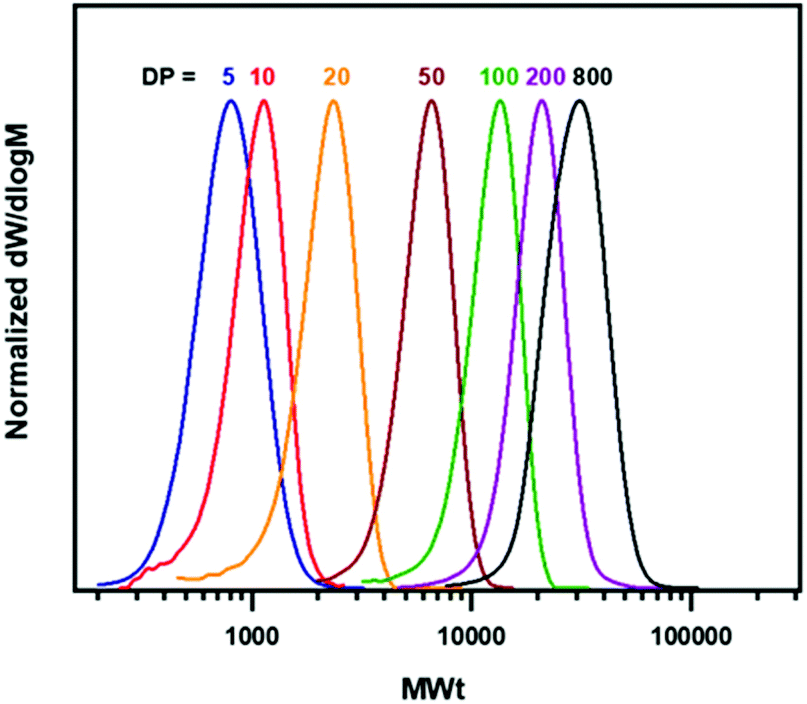 | ||
| Fig. 1 CHCl3-SEC derived molecular weight distributions showing the evolution of MWts for various DPn values (5, 10, 20, 50, 100, 200, 800) of pMA synthesized by Cu(0) wire-mediated RDRP in Cyrene without deoxygenation. Conditions: [EBiB]:[Cu(II)Br2]:[Me6Tren] = 1:0.05:0.18, VCyrene = 50% v/v with respect to monomer. | ||
| Entrya | DPn | Time (h) | Mon. Conv.b (%) | M n, theor. (g mol−1) |
M
n, SECc |
Đ |
|---|---|---|---|---|---|---|
|
a In all polymerizations the volume ratio of monomer to solvent was maintained at 1:1.
b Conversion was calculated via1 H NMR in CDCl3.
c Determined by CHCl3 SEC analysis and expressed as molecular weight relative to pMMA narrow molecular weight standards.
|
||||||
| 1 | 5 | 3 | 99 | 630 | 700 | 1.12 |
| 2 | 10 | 3 | >99 | 1100 | 1000 | 1.11 |
| 3 | 20 | 3 | >99 | 1900 | 2000 | 1.12 |
| 4 | 50 | 3 | 90 | 4100 | 4800 | 1.08 |
| 5 | 100 | 24 | 86 | 7600 | 11600 |
1.09 |
| 6 | 200 | 44 | 88 | 15000 |
19100 |
1.08 |
| 7 | 800 | 96 | 43 | 29800 |
27800 |
1.10 |
To verify the ω-Br functionality of the obtained polymers, matrix assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-ToF-MS) was employed for pMA20.43,44 A single peak distribution corresponding to bromine-capped polymer chains was found, indicating the retention of the ω-end (Fig. 2a & b) with a calculated mass for bromine terminated polymer with DPn = 25 of 2369.9 Da and an observed mass = 2370.2 Da with the associated isotopic pattern expected for incorporation of bromine, confirming the structure as shown in Fig. 2a. This led us to proceed to the synthesis of AB and ABA block copolymers consisting of methyl and ethyl acrylate (A = MA, B = EA) (Table S3,†Fig. 2c) via sequential monomer addition, thus verifying that the end-group fidelity could be attained not only in the case of the pMA10 macroinitiator, but also for the pMA10-b-pEA10 diblock, leading to a well-defined triblock copolymer synthesized in the presence of oxygen.
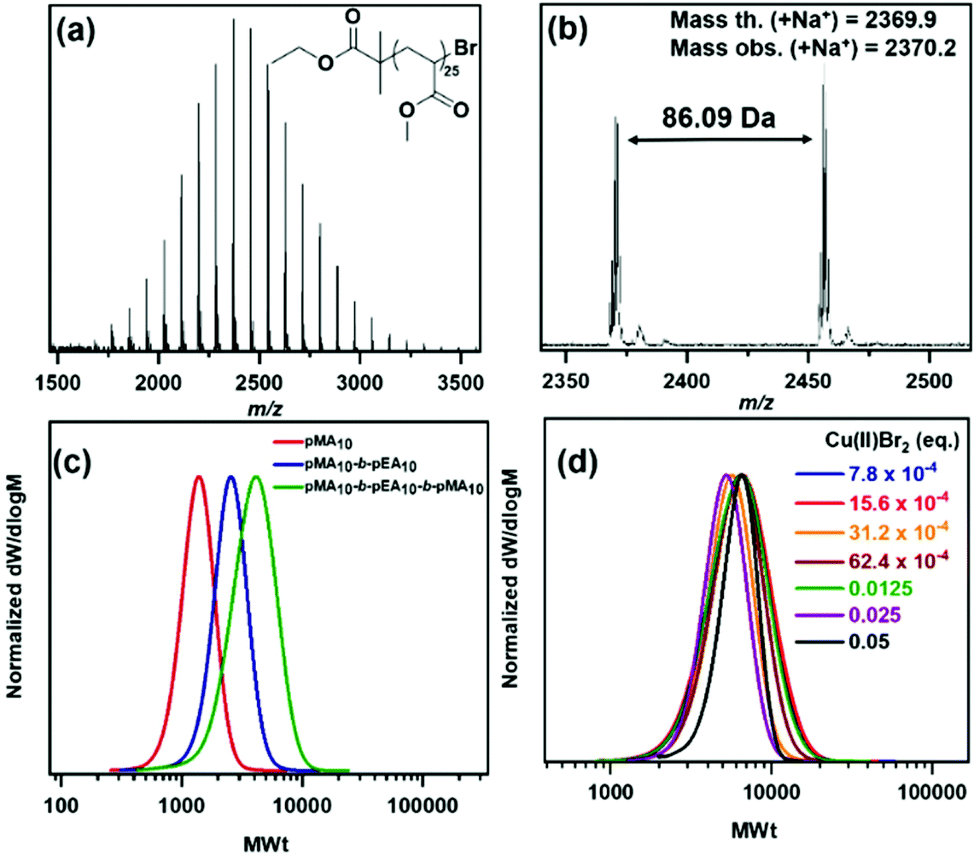 | ||
| Fig. 2 (a) and (b) MALDI-ToF-MS spectra for the –Br terminated pMA25. CHCl3-SEC derived molecular weight distributions showing the evolution of MWts of (c) pMA10 (red), pMA10-b-pEA10 (blue) and pMA10-b-pEA10-b-pMA10 (green) and (d) pMA50 synthesized by Cu(0) wire-mediated RDRP in Cyrene without any type of deoxygenation with various [Cu(II)Br2]. | ||
Apart from the facile removal of Cu(0) via simply removing the stirrer bar with the wrapped copper wire, we were also interested in exploring the limits of this chemistry by lowering the [Cu(II)Br2]. Consequently, polymerization of MA (targeted DPn = 50) was conducted by varying the amount of Cu(II)Br2. Starting from 0.05 equivalents of Cu(II)Br2 and reducing its amount by half, we were able to limit the catalyst content to 7.8 × 10−4 eq., which is 64-fold lower than the 0.05 eq. of Cu(II)Br2 previously used. All polymerizations were carried out at ambient temperature for 3 h, achieving monomer conversions from 92 to 97% (Table 2). By comparing the SEC results from these polymerizations (Table 2, Fig. 2d), slightly higher molecular weights and dispersities were observed with decreasing [Cu(II)Br2]. It might have been expected that by changing the [Me6Tren]: [Cu(II)Br2] ratio, the equilibrium between dormant and active species is affected and this is depicted in the molecular characteristics i.e. Mn and Đ.
| Cu(II)Br2a (eq.) |
Time (h) | Mon. Conv.b (%) | M n, theor. (g mol−1) |
M
n, NMRb |
M
n, SECc |
Đ |
|---|---|---|---|---|---|---|
|
a In all polymerizations, the volume ratio of monomer to solvent was maintained at 1:1.
b Conversion was calculated via1NMR in CDCl3.
c Determined by CHCl3 SEC analysis and expressed as molecular weight relative to pMMA narrow molecular weight standards.
|
||||||
| 0.00078 | 3 | 97 | 4400 | 4900 | 5600 | 1.20 |
| 0.00156 | 3 | 94 | 4200 | 5500 | 5600 | 1.20 |
| 0.00312 | 3 | 92 | 4200 | 4800 | 5500 | 1.20 |
| 0.00625 | 3 | 94 | 4200 | 4800 | 5600 | 1.15 |
| 0.0125 | 3 | 92 | 4200 | 4500 | 5100 | 1.12 |
| 0.025 | 3 | 92 | 4200 | 4600 | 4800 | 1.10 |
| 0.05 | 3 | 97 | 4400 | 4800 | 4800 | 1.08 |
Finally, the oxygen consumption profile for polymerization to pMA in Cyrene was monitored by use of an oxygen concentration probe. As shown in Fig. S19,† in the absence of headspace, full oxygen consumption takes place within the first 5 minutes of the reaction (2 mL total reaction volume), while even when 0.5 mL of headspace is present, the oxygen consumption lasts for 24 minutes. This observation comes in agreement with previous work by Liarou et al., and verifies the importance of headspace, which is highlighted in smaller scale reactions (2 mL total reaction volume). As has previously been reported,39 all the reagents of the polymerization (Cu(0)-wire, initiator, Cu(II)Br2/Me6Tren) contribute to oxygen consumption, and synergistically lead to full consumption of dissolved oxygen, which occurs within the first minutes of the reaction (in the absence of headspace) (Fig. S19†). It is noted that the [O2] at t = 0 in the reaction mixture containing Cyrene was < 6 mg L−1, while in the case of DMSO it was >8 mg L−1. However, the effect of solvent on the evolution of oxygen consumption will be further discussed in a forthcoming publication.
In summary, we report the use of Cyrene, which derives from renewable resources, as an alternative dipolar aprotic solvent for the Cu(0) wire-mediated RDRP of various hydrophobic monomers. Polymerization proceeded successfully under both deoxygenated and non-deoxygenated conditions, allowing production of polymers with high end-group fidelity at high conversions, allowing for sequential monomer addition for the synthesis of di- and tri- block copolymers. Even with very low [Cu(II)Br2] (as low as 7.8 × 10−4 eq.) the obtained pMAs exhibited controlled macromolecular characteristic. Cyrene offers a valuable biorenewable alternative to harsh aprotic polar solvents which are increasingly seen as unattractive.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the University of Warwick (A. M., E. L.), Syngenta (A. M.) and the Warwick-Monash Alliance (E. H.). We also thank the Polymer Characterization RTP for access to SEC equipment. This collaborative research was conducted with the Australian Research Council (ARC) Centre of Excellence in Convergent Bio-Nano Science and Technology (project number CE140100036). J. L. G acknowledges support from the Australian Research Council (DP200102829). We also thank Professor Matt Gibson for suggesting the use of Cyrene. We also thank Tony Duncan from Circa group for valuable discussion about Cyrene.Notes and references
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed.
- S. Aksakal, R. Liu, R. Aksakal and C. R. Becer, Polym. Chem., 2020, 11, 982–989 RSC.
- A. Theodorou, E. Liarou, D. M. Haddleton, I. G. Stavrakaki, P. Skordalidis, R. Whitfield, A. Anastasaki and K. Velonia, Nat. Commun., 2020, 11, 1486 CrossRef CAS PubMed.
- E. Hancox, E. Liarou, J. S. Town, G. R. Jones, S. A. Layton, S. Huband, M. J. Greenall, P. D. Topham and D. M. Haddleton, Polym. Chem., 2019, 10, 6254–6259 RSC.
- J. E. Camp, ChemSusChem, 2018, 11, 3048–3055 CrossRef CAS PubMed.
- N. H. Nguyen, X. Leng, H.-J. Sun and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3110–3122 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1752–1763 CrossRef CAS.
- Q. Zhang, Z. Li, P. Wilson and D. M. Haddleton, Chem. Commun., 2013, 49, 6608–6610 RSC.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- F. Pena-Pereira, A. Kloskowski and J. Namieśnik, Green Chem., 2015, 17, 3687–3705 RSC.
- S. Kudo, Z. Zhou, K. Norinaga and J.-I. Hayashi, Green Chem., 2011, 13, 3306–3311 RSC.
- J. Zhang, G. B. White, M. D. Ryan, A. J. Hunt and M. J. Katz, ACS Sustainable Chem. Eng., 2016, 4, 7186–7192 CrossRef CAS.
- J. E. Camp, S. B. Nyamini and F. J. Scott, RSC Med. Chem., 2020, 11, 111–117 RSC.
- H. J. Salavagione, J. Sherwood, M. De bruyn, V. L. Budarin, G. J. Ellis, J. H. Clark and P. S. Shuttleworth, Green Chem., 2017, 19, 2550–2560 RSC.
- J. Sherwood, M. De bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650–9652 RSC.
- F. Shafizadeh, R. H. Furneaux and T. T. Stevenson, Carbohydr. Res., 1979, 71, 169–191 CrossRef CAS.
- Private communication with Circa Group on 22nd July 2020.
- K. L. Wilson, A. R. Kennedy, J. Murray, B. Greatrex, C. Jamieson and A. J. B. Watson, Beilstein J. Org. Chem., 2016, 12, 2005–2011 CrossRef CAS PubMed.
- L. Mistry, K. Mapesa, T. W. Bousfield and J. E. Camp, Green Chem., 2017, 19, 2123–2128 RSC.
- M. C. Bryan, L. Diorazio, Z. Fei, K. Fraunhoffer, J. Hayler, M. Hickey, S. Hughes, M. McLaws, P. Richardson, G.-D. Roiban, M. Schober, A. G. Smith, A. Steven, T. White, S. Wuyts and J. Yin, Org. Process Res. Dev., 2018, 22, 667–680 CrossRef CAS.
- K. L. Wilson, J. Murray, C. Jamieson and A. J. B. Watson, Org. Biomol. Chem., 2018, 16, 2851–2854 RSC.
- S. H. Krishna, D. J. McClelland, Q. A. Rashke, J. A. Dumesic and G. W. Huber, Green Chem., 2017, 19, 1278–1285 RSC.
- C. M. Kelly, D. E. Noga, J. E. Sidenstick, L. Ben-Asher and A. Kondo, US Pat., 20150299393A1, 2015 Search PubMed.
- T. Marino, F. Galiano, A. Molino and A. Figoli, J. Membr. Sci., 2019, 580, 224–234 CrossRef CAS.
- D. Zhao, J. F. Kim, G. Ignacz, P. Pogany, Y. M. Lee and G. Szekely, ACS Nano, 2019, 13, 125–133 CrossRef CAS PubMed.
- P. Ray, T. Hughes, C. Smith, M. Hibbert, K. Saito and G. P. Simon, Polym. Chem., 2019, 10, 3334–3341 RSC.
- M. Horn and K. Matyjaszewski, Macromolecules, 2013, 46, 3350–3357 CrossRef CAS.
- S. Harrisson, S. R. Mackenzie and D. M. Haddleton, Macromolecules, 2003, 36, 5072–5075 CrossRef CAS.
- E. Liarou, M. Staniforth, J. S. Town, A. Marathianos, M. Grypioti, Y. Li, Y. Chang, S. Efstathiou, E. Hancox, A. M. Wemyss, P. Wilson, B. A. Jones, M. Aljuaid, V. G. Stavros and D. M. Haddleton, Eur. Polym. J., 2020, 123, 109388 CrossRef.
- O. Bertrand, P. Wilson, J. A. Burns, G. A. Bell and D. M. Haddleton, Polym. Chem., 2015, 6, 8319–8324 RSC.
- A. J. Carmichael, D. M. Haddleton, S. A. F. Bon and K. R. Seddon, Chem. Commun., 2000, 14, 1237–1238 RSC.
- T. J. Zerk and P. V. Bernhardt, Dalton Trans., 2013, 42, 11683–11694 RSC.
- R. Whitfield, A. Anastasaki, V. Nikolaou, G. R. Jones, N. G. Engelis, E. H. Discekici, C. Fleischmann, J. Willenbacher, C. J. Hawker and D. M. Haddleton, J. Am. Chem. Soc., 2017, 139, 1003–1010 CrossRef CAS PubMed.
- E. Liarou, R. Whitfield, A. Anastasaki, N. G. Engelis, G. R. Jones, K. Velonia and D. M. Haddleton, Angew. Chem., Int. Ed., 2018, 57, 8998–9002 CrossRef CAS.
- E. Liarou, A. Anastasaki, R. Whitfield, C. E. Iacono, G. Patias, N. G. Engelis, A. Marathianos, G. R. Jones and D. M. Haddleton, Polym. Chem., 2019, 10, 963–971 RSC.
- A. Marathianos, E. Liarou, A. Anastasaki, R. Whitfield, M. Laurel, A. M. Wemyss and D. M. Haddleton, Polym. Chem., 2019, 10, 4402–4406 RSC.
- E. Liarou, Y. Han, A. M. Sanchez, M. Walker and D. M. Haddleton, Chem. Sci., 2020, 11, 5257–5266 RSC.
- J. S. Town, G. R. Jones and D. M. Haddleton, Polym. Chem., 2018, 9, 4631–4641 RSC.
- J. S. Town, G. R. Jones, E. Hancox, A. Shegiwal and D. M. Haddleton, Macromol. Rapid Commun., 2019, 40, 1900088 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02184a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |