
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRAFT thermoplastics from glycerol: a biopolymer for development of sustainable wood adhesives†
Michael
Forrester
 a,
Andrew
Becker
a,
Austin
Hohmann
a,
Nacu
Hernandez
a,
Fang-Yi
Lin
a,
Nicholas
Bloome
a,
Grant
Johnson
a,
Hannah
Dietrich
a,
Joe
Marcinko
b,
R. Chris
Williams
c and
Eric
Cochran
*a
a,
Andrew
Becker
a,
Austin
Hohmann
a,
Nacu
Hernandez
a,
Fang-Yi
Lin
a,
Nicholas
Bloome
a,
Grant
Johnson
a,
Hannah
Dietrich
a,
Joe
Marcinko
b,
R. Chris
Williams
c and
Eric
Cochran
*a
aDepartment of Chemical and Biological Engineering, Iowa State University, Ames, Iowa, USA. E-mail: ecochran@iastate.edu
bPolymer Synergies, Mantua, New Jersey, USA
cCivil, Construction and Environmental Engineering, Iowa State University, Ames, Iowa, USA
First published on 19th August 2020
Abstract
The increasing demand for bioderived plastics and rubbers and the large supply of glycerol makes it an excellent starting chemical for the production of biopolymers. Little success in commercially viable glycerol polymers has yet to be realized. In particular, high molecular weight thermoplastics have been especially elusive due to the multifunctional nature of glycerol. This work details the production of glycerol–acrylic biopolymers. By esterifying glycerol with acrylic acid, and subsequent RAFT polymerization to suppress the gelation, we were able to achieve glycerol thermoplastics with high molecular weights (1 MDa). After studying the thermal/mechanical properties of the polymer, it was found that these glycerol polymers had a high degree of tack. When added to wood as an adhesive, it was found that performance was comparable or exceeded standard wood adhesives such as Poly (Methylene diphenyl diisocyanate) (PMDI) and formaldehyde based adhesives. This yields wood adhesives that have less toxicity, lower environmental impact, and higher renewability.
1. Introduction
The long-term supply prospects, volatile pricing and environmental impacts associated with the use of petroleum-based fuels has placed great pressure on both producers and consumers to find greener alternatives. This has led to concerted efforts in industry and academia to develop new bio-based fuels;1–3 conversion is not 100% efficient and thus the utility of byproducts must be considered in holistic life cycle and technoeconomic assessments. If byproducts are not valorized, the high-volume and low-value nature of fuels markets make the economics of biofuels less appealing. However, if high-value/high-volume materials such as polymers can be developed from these byproducts, the economics improve dramatically. The growth of the biodiesel industry, for example, has led to an abundance of crude glycerol available at prices as low as $0.10 per pound. Crude glycerol, comprised of roughly 20% water, soot, alcohols and other organics requires extensive refining to be suitable for most commercial uses. Unfortunately, refinement quickly erodes any cost advantage and therefore the demand for crude glycerol remains dwarfed by its supply. Thus, crude glycerol is currently one of the cheapest and most readily available biorenewable feedstocks.4–6 Accordingly, there is a significant economic opportunity for high-volume end use applications that can tolerate its low quality. Polymers and adhesives are particularly attractive market segments since high purity materials are often unnecessary for industrial uses. Glycerol has several attractive features that make it an interesting monomer precursor. Its three hydroxyl groups can be differentiated in countless ways through various chemistries to achieve new function and properties. For example, by taking advantage of glycerol's multifunctionality, its solubility can be readily tailored from its native hydrophilicity. Moreover, glycerol has a very low volatility, is non-flammable, and has a very low environmental and personal toxicity. Additionally, the atom economy of reactions of glycerol tend to be high with water being the most common byproduct. Because of these benefits, there have been many recent studies on glycerol derived polymers.Polymers derived from the polycondensation of glycerol have been extensively studied. For example, glycerol can be self-polymerized at elevated temperatures in the presence of a catalyst to produce oligomeric or crosslinked hyperbranched poly(glycerol).7,8 Diacids such as sebacic acid can be used to make low molecular polymers as is demonstrated in the work done by Wang et al.9 Cai et al. describe a poly(glycerol sebacate) that is cured to produce a thermoset shape-memory material.10 Glycerol polycondensates reported to date face several barriers to widespread commercial applicability; they are either oligomeric/low-molecular-weight, crosslinked, and/or produced with resource-intensive techniques like protecting groups. Higher molecular weights are necessary to achieve the requisite thermal and mechanical properties desired for most applications.
Radical polymerization has also been used to produce glycerol derived polymers. Whittaker et al. and Zhang et al. synthesized poly(1-glycerol acrylate) by acrylation of solketal and cleavage of the acetal groups to yield the strictly monofunctional 1-glycerol acrylate monomer; subsequent radical polymerization then readily afforded linear high molecular weight polymers.11,12 However, the monomer synthesis requires refined glycerol, takes multiple steps, uses volatile organic solvents and involves intensive purification. While these requirements may be acceptable for extremely high-value applications like biomedical materials, 1-glycerol acrylate may not be considered “green” and would moreover be too costly for most industrial end-uses.
To meet the challenges of environmental responsibility, cost-effectiveness and utility, we report the discovery of a family of hyperbranched thermoplastic acrylic polymers produced from unpurified mixtures of glycerol multiacrylates formed through a simple non-exothermic, non-pressurized, esterification process (see Scheme 1). By polymerizing these mixtures directly without further purification, the process avoids separations costs and supports a dramatically higher yield of useable end product. The resultant poly(acrylated glycerol) (PAG) polymers range from hydrophobic to hydrophilic, have glass transition temperatures (Tg) ranging from −40 °C to 10 °C, and vary from brittle solids to soft elastomers. We have discovered that the glycerol multiacrylate monomer can be produced directly from crude glycerol, and that for certain high volume end-uses the resultant polymers can be used directly with only a single precipitation/re-dissolution step. This significantly improves the economics and reduces barriers to market entry for any product formulated from this polymer. While the bulk of this work has focused on methanol/isopropanol as our solvent/precipitation media, we have also demonstrated the ability to polymerize in water. Further optimization of this aqueous polymerization method, coupled with the commercial production of bio-based acrylic acid by companies such as ADM and Cargill, could allow PAG to be produced from solely bioderived feedstocks with minimal processing for water-based applications. Herein we detail the synthesis of the glycerol multiacrylate monomers and subsequent reversible addition–fragmentation chain transfer (RAFT) polymerization to high molecular weight (up to 1 MDa) with suppressed macro-gelation. The thermal, mechanical, chemical, and solubility properties of PAG are explored. Finally, an exemplary application of PAG as a wood adhesive is described.
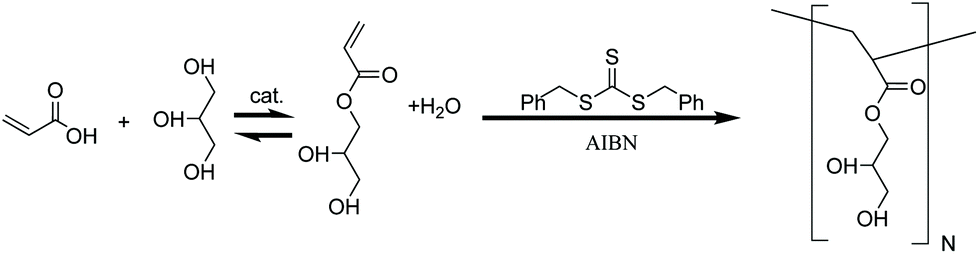 | ||
| Scheme 1 Simplified synthetic route to produce PAG from crude glycerol. Esterification of crude glycerol yields 1-glycerol acrylate in addition to several other polymerizable species as quantified by GCMS (Fig. 1). The mixture of monomers is directly polymerized via RAFT using a chain transfer agent (CTA) and radical initiator (azobisisobutyronitrile, AIBN) to thermoplastic branched chain polymers. | ||
2. Results and discussion
Tables 1 and 2 summarize the monomers and polymers we prepared for this study. Acrylated glycerol monomers are denoted as AGx, where x indicates the stoichiometic ratio of acrylic acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycerol in the esterification step. PAG polymers are designated PAGx-Y, where Y indicates low (A), medium (B), or high (C) molecular weight.
:glycerol in the esterification step. PAG polymers are designated PAGx-Y, where Y indicates low (A), medium (B), or high (C) molecular weight.
| Sample code | AA/HQ/TPPa, eq. | Relative abundance (%) | |||||
|---|---|---|---|---|---|---|---|
| a Equivalents of acrylic acid (AA), hydroquinone (HQ), and triphenylphosphine (TPP) on a basis of 1 eq. of glycerol. | |||||||
| GCMS peak no. | — | 1 | 2–3 | 4 | 5–6 | 7, 9–10, 16, 20, 23–24 | 19, 21–22, 25–28 |
| Identity & abbreviation | — | Glycerol (G) | Glycerol monoacrylate (2GA, 1GA) | Hydroquinone (HQ) | Glycerol diacrylate (1,2DGA, 1,3DGA) | Inactive (I) | Active (A) |
| AG1.3 | 1.3/0.039/0.025 | 38.8 | 16.4 | 4.0 | 1.4 | 20.8 | 18.6 |
| AG1.8 | 1.8/1/0.052/0.025 | 30.1 | 19.4 | 4.0 | 1.7 | 22.3 | 22.6 |
| AG2.2 | 2.2/0.065/0.025 | 20.1 | 17.9 | 8.6 | 3.2 | 21.0 | 22.3 |
| Sample code | AG/AIBNa, eq. | MeOH/AGb, mL g−1 | X |
T
gd, °C |
α |
G
x
f, MPa |
ω
x
g, rad s−1 |
Sh, g L−1 |
M
ni, kDa |
Đj |
Target Mn (kDa) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DSC | DMA | |||||||||||
| a Equivalents of acrylated glycerol (AG) & azobisisobutyronitrile (AIBN) on a 1 eq. basis of chain transfer agent (CTA). b Methanol (MeOH). c Conversion (X). d Glass transition (Tg). e Rheology terminal slope (α). f Crossover modulus (Gx). g Crossover frequency (ωx). h Solubility (S). i Molecular weight (Mn). j Dispersity (Đ). | ||||||||||||
| PAG1.3-A | 70/0.3 | 1.23 | 0.97 | −22.0 | −5.0 | 0.77 | 80 | 34000 |
<LOD | 192 | 3.1 | 10 |
| PAG1.3-B | 700/3 | 4.35 | 0.89 | −10.9 | 16.0 | 0.65 | 87 | 1337 | 20 | 528 | 2.4 | 100 |
| PAG1.3-C | 7000/30 | 4.35 | 0.94 | −15.0 | 4.0 | 0.64 | 94 | 2487 | 30 | 882 | 1.95 | 1000 |
| PAG1.3-CW | 7000/30 | 4.35 | 0.70 | — | — | — | — | — | — | 1563 | 8.5 | 1000 |
| PAG1.8-A | 60/0.3 | 2.17 | 0.97 | −14.8 | 16.0 | 0.77 | 115 | 6700 | <LOD | 200 | 2.76 | 10 |
| PAG1.8-B | 600/3 | 4.47 | 0.94 | −0.3 | 30.0 | 0.67 | 81 | 106 | <LOD | 645 | 2.47 | 100 |
| PAG2.2-A | 50/0.3 | 3.38 | 0.95 | −9.5 | 16.0 | 0.74 | 100 | 3000 | <LOD | 194 | 2.85 | 10 |
Glycerol multiacrylate monomers were prepared through Fisher esterification without solvent simply by combining crude- or anyalytical-grade glycerol and acrylic acid with catalyst and radical inhibitor as illustrated in Scheme 1. Triphenylphosphine (TPP), a base catalyst, and p-toluenesulfonic acid (TSA) were evaluated as catalysts. Table 1 indicates the feed ratios for each monomer entry. Reaction progress (X) was monitored through the change in 1H-NMR chemical shift of the alkene signal from δ = 5.8 ppm for acrylic acid to δ = 5.95 ppm for acrylates (see Fig. S1†). The reaction establishes equilibrium due to the accumulation of water; this was established in about 2 h for TSA catalyzed reactions with a limiting alkene conversion of X = 0.60. Base catalyzed esterification further shifted the equilibrium to about X = 0.70, irrespective of feed ratio, over about 48 h. The base catalyzed route was used to produce AG1.3, AG1.8 and AG2.2. Exemplary reaction kinetics for AG1.3 appear in Fig. S1.†
An exemplary 1H-NMR spectra of AG1.3 appears in Fig. S1 (see ESI† for additional spectra). Clearly, the trihydroxy functionality of the glycerol substrate is subject to multiple additions of acrylic acid. As such, the glycerol region of the NMR spectra is complex due to the variety of different addition products such as a distribution of mono-, di-, and tri-glycerol acrylates. Additionally when comparing to the time 0 NMR, the ratio of catalyst protons to alkene protons steadily decrease ending at around half the original alkene concentration (see Fig. S2†). The radical oligomerization of vinyl groups is an obvious culprit due to the high temperature used, although the considerable amount of radical inhibitor used in the reaction should limit this pathway. Acid titration of the reaction products supports this, showing a 50% lower acid number than expected based on the conversion of acid to acrylate. Since the NMR does not show higher conversion of acrylic to acrylic ester, another mechanism must be reducing the acid number. It is known that acrylic acid can oligomerize via carboxylic-alkene Michael addition.13 This is especially prevalent at elevated temperatures in the presence of water and would be a likely explanation for the lower acid number. GC-MS experiments, Fig. 1, further elucidate the composition of the glycerol acrylate reaction product. The complexity of these mixtures is typical of glycerol ester systems.14 To show compatibility of the acrylation process with crude glycerol, AG1.3 was replicated using a crude glycerol feedstock; the GC-MS is similar irrespective of initial purity (Fig. S3†), suggesting that further purification is unnecessary. The major components of the AGx mixtures are shown in Fig. 1 and summarized in Table 1; full details appear in Table S1.† The majority of the eluents, >90 mol%, can be identified as glycerol (G), glycerol acrylates, oligomeric glycerol acrylates, hydroquinone (HQ) and catalyst. 1-Glycerol acrylate (1GA), 2-glycerol acrylate (2GA), 1,2-glycerol diacrylate (12DGA), and 1,3-glycerol diacrylate (13DGA) can be identified unambiguously and are labeled as such in Fig. 1. The mass spectra of several peaks do not permit conclusive identification. However, many of these show significant presence of mass fragment m/z = 55, characteristic of the acrylate group. Accordingly, unidentified compounds are categorized as “active” (A, containing acrylate) or inactive (I) based on the presence of m/z = 55. It does appear that there is some amount of oligomerization through alkene radical polymerization; however, these peaks are below the 0.1% area threshold used in this analysis.
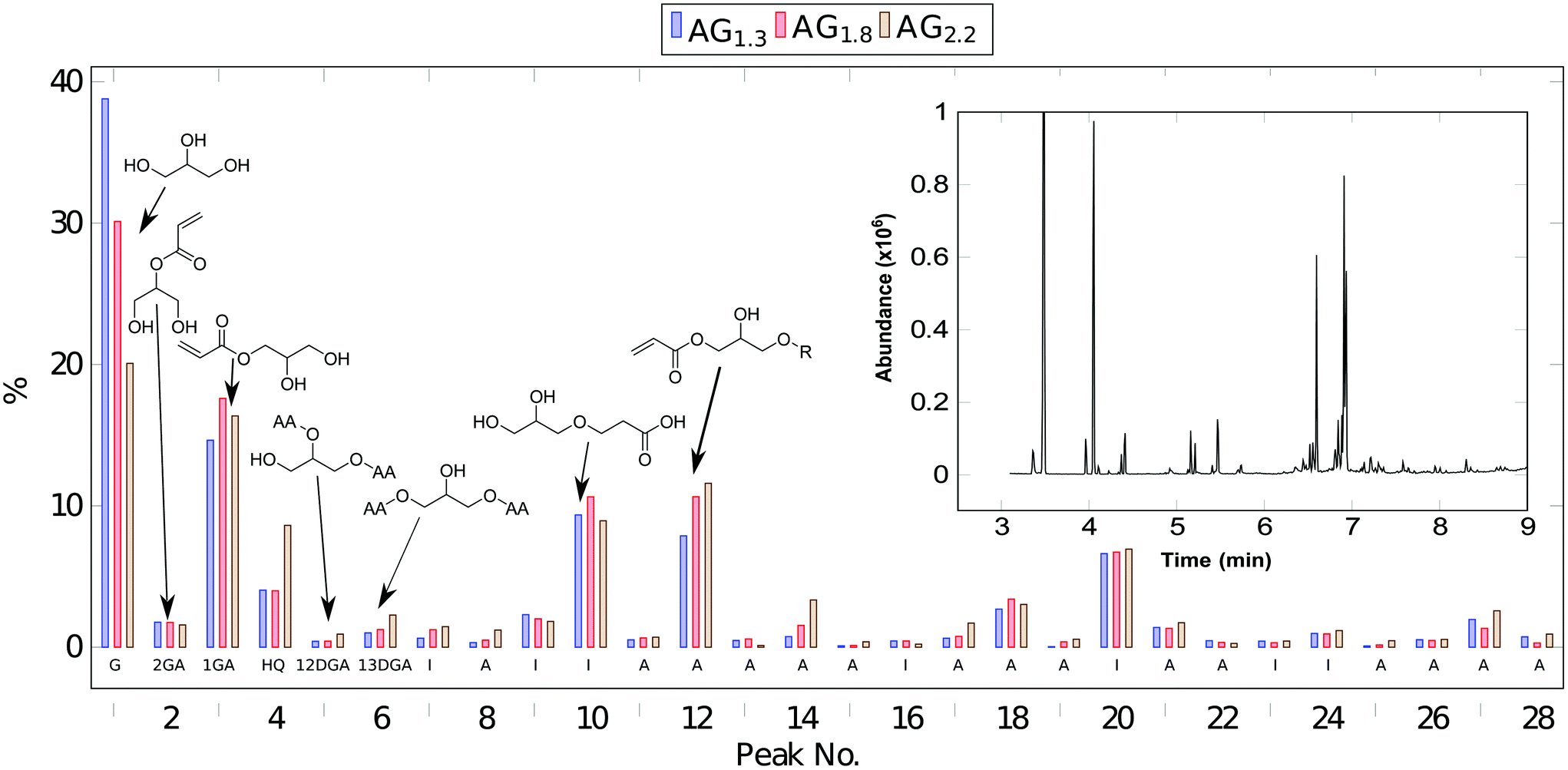 | ||
| Fig. 1 Bar chart showing the relative abundance of compounds identified via GC-MS in AGx specimen, by peak serial number. An exemplary GC-MS chromatogram for AG1.3 is shown in the inset. Note that the elution times have been converted to peak numbers. Chemical structures for key peaks are shown directly on the histogram; labels A, I, G, 1GA, 2GA, 12GA, 13GA further encode peak assignments as described in Table 1. | ||
Given the complexity of the monomer composition, this process provides a unique mixture of monomers that, upon polymerization, yields a polymer that cannot be obtained by other esterification techniques such as Steglich, acid halide, transesterification, and polymer grafting. Thus, as anticipated, the degree of acrylate substitution is dependent on the acrylic acid feed ratio (cf.Table 1). This multifunctional character is inevitable via direct esterification, and the monomer is expected to be highly susceptible to crosslinking and gelation upon polymerization through the vinyl groups. Indeed, AG nearly immediately forms a macrogel if radically polymerized in the absence of a RAFT-active chain transfer agent (CTA).
The gelation suppression effect in controlled radical polymerizations (CRP) such as RAFT has been well-studied in systems with dilute multivinyl monomers, and more recently for systems comprised entirely of multivinyl monomers.15–17 When a monomer with x vinyl groups is added to a propagating radical chain, (x − 1) vinyl groups remain that could further participate in initiation or propagation reactions, leading to intermolecular branches and intramolecular loop formation. Rather than immediately forming a pervasive polymer network at the early stages of polymerization, CRP promotes the formation of branched structures that only overlap to form a macrogel at monomer conversions far higher than classical gelation theory predicts. The architecture of the resultant hyperbranched chains is thus statistically defined and sensitive to several kinetic parameters. As such, these materials have some conceptual similarities to the hyperbranched polymers (HBPs) formed through ABx step growth monomers as originally anticipated by Flory.18,19 Namely, a multivinyl monomer with a functionality of x can potentially branch in a similar fashion to an ABx. Branching probabilities in both systems are governed by reactivity ratios and steric constraints, and the distribution of linear segments and the degree of branching are impacted by both x and kinetic considerations. However, ABx-based HBPs can have at most 1 loop and are incapable of gelation. In contrast, looping is highly prevalent multivinyl HBPs and is a critical feature of the gel point suppression phenomenon.20
To better quantify CRP-mediated gel point suppression, Lin et al. recently introduced a phenomenological parameter termed the “crosslinking tendency” (CT), which is simply the ratio of the excess vinyl content to the number of primary chains per multivinyl monomer.15
In a retrospective analysis of several CRP studies, Lin found that for CT < 100 the intermolecular crosslinking mechanism that leads to macrogelation is suppressed. We exploited this feature of CRP to design RAFT polymerizations with CT ≤ 20 to form high molecular weight hyperbranched thermoplastics from the multivinyl AGx mixtures (see ESI† for CT calculation details).
Table 2 enumerates the synthetic conditions we employed to synthesize PAGx-Y at “ideal” number average molecular weights of 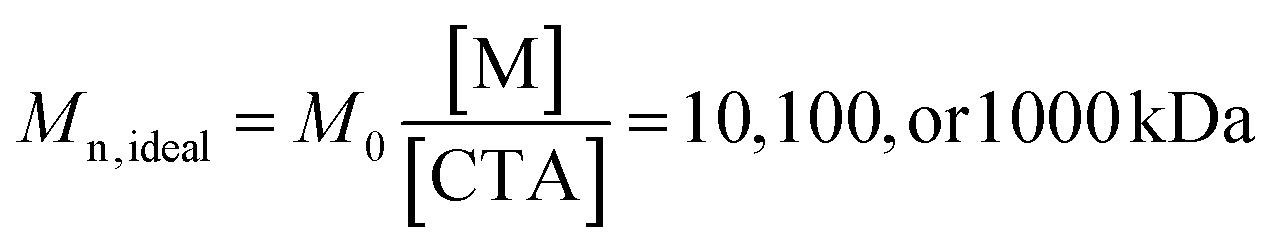 .
.
Due to the high polarity of the AGx monomer series, the polymerization can easily be conducted in green solvents like water or ethanol. We have evaluated the polymerization kinetics and degree of gel-point suppression in water, methanol and ethanol. Gel permeation chromatography (GPC) data appears in Fig. 3. The molecular weight distribution is multimodal with dispersity values Đ ∈ 1.95–3.1; these features are common to hyperbranched polymers.21 The CT value increases with the degree of acrylate substitution;15 accordingly, the gel-point conversion Xg decreases significantly and limits the achievable molecular weight. The Xg values for the PAGx-Y polymers are plotted vs. CT value in the manner previously shown by Lin et al.15 in Fig. 2. 1H-NMR analysis on the solution after polymerization reveals that Xg > 88% can be achieved when methanol is the solvent (see Table 2). Similarly, aqueous polymerizations reach alkene conversions of at least 70% without gelation. Accordingly, the process can be completed without the use of organic solvents, and could easily be reduced to a 1-pot synthesis beginning with raw materials and ending with PAG in aqueous solution. Interestingly, in both aqueous and alcoholic systems, the higher molecular weight targets (1 MDa) seem to have a significantly higher conversion than is expected given the high CT value. One probable explanation is that the high amount of hydrogen bonding present in this system may be limiting the accessibility of double bonds already very near a backbone chain.
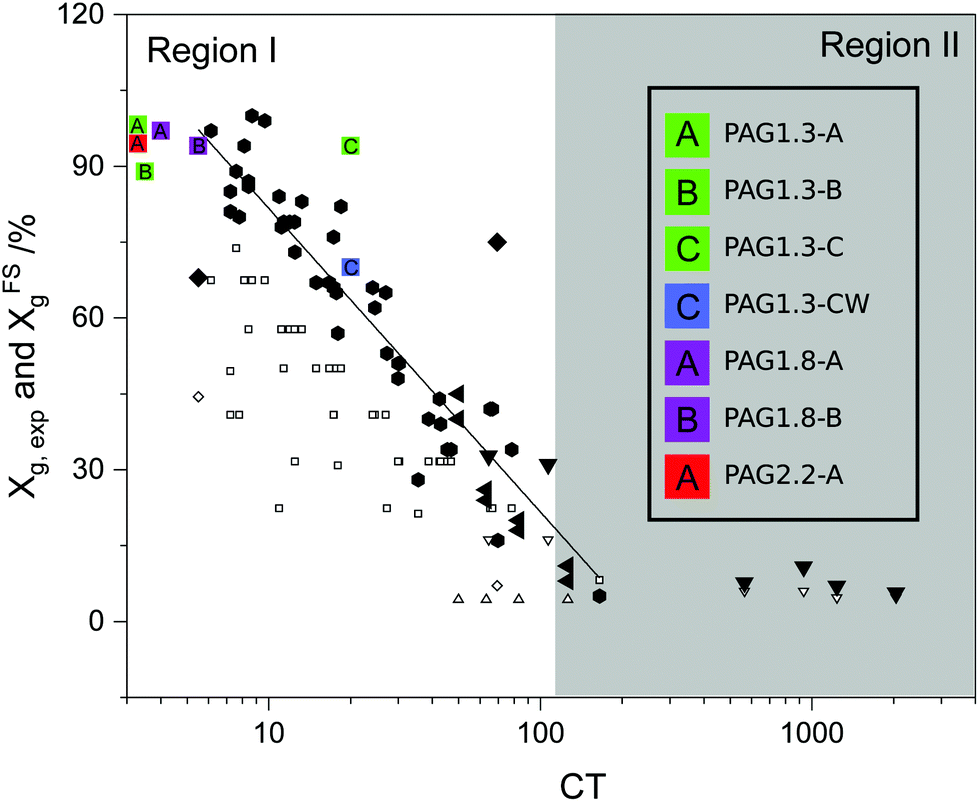 | ||
| Fig. 2 Conversion as a function of crosslinking tendency based on the work of Lin, et al. with data from this work added (colored squares). Other points are the collection of work of Lin and examples in the literature.15 | ||
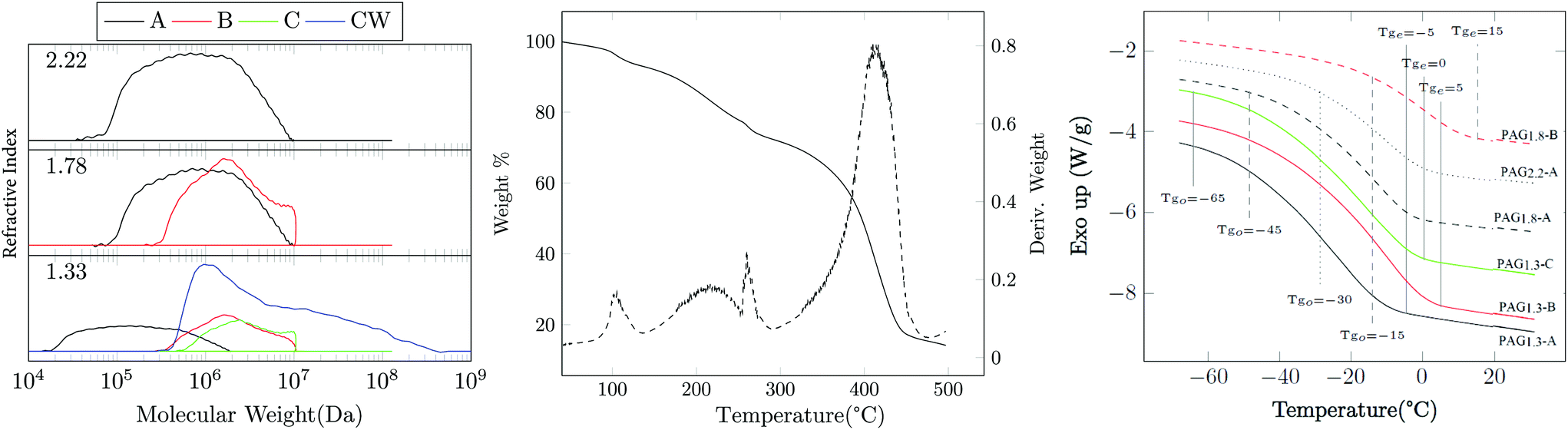 | ||
| Fig. 3 (Left) GPC showing the distribution for each sample. (Middle) TGA of PAG1.3-B showing weight % and derivative weight % curves. (Right) DSC that shows the onset of the glass transition and the end point of the glass transition for each PAG sample. Traces have been shifted vertically for clarity. | ||
GPC analysis shown in Fig. 3 and Table 2 show that Mn, ideal and Mn are substantially different and the Đ values are also quite high. However, molecular weight clearly increases with increasing Mn, ideal, indicating that the CTA is mediating the polymerization as anticipated. The evaluation of Mn, ideal assumes that all monomers are distributed equally amongst each chain transfer agent, which occurs in well controlled non-branching RAFT polymerizations. Intermolecular branching reactions cause the merger of existing primary chains, yielding topologies with two or more CTA units; this has been observed in the RAFT polymerization of other systems such as acrylated epoxidized soybean oil.20,22 Likewise, the multimodal distributions seen in these samples is typical in branched controlled radical systems and is a result of the combination of macrochains in the later stages of polymerization prior to gelation.20,23,24 The choice of solvent slightly alters the several kinetic parameters that govern the evolution of the polymerization. As such, the PAG1.3-C and PAG1.3-CW show significant differences in molecular weight distribution as shown Fig. 3; the aqueous system shows a greater tendency to crosslink, resulting in an attending increase in dispersity and molecular weight compared to the methanol-processed PAG1.3-C (see Table 2). Use of the RAFT process has allowed, as has been demonstrated by the GPC, the ability to produce thermoplastics from a highly functional branching system. While there have been numerous other works that have studied branched structures from bioderived sources, they have largely focused on thermosetting materials.25,26 This work has demonstrated a unique ability to apply gel-point suppression to yield high molecular weight crude glycerol-based polymers.
TGA was performed to determine thermal stability. Fig. 3 shows the TGA of PAG1.3-B which is representative of the results for the other PAG samples. There are four distinct peaks observed in the differential curve. The first mass loss event is the physical de-watering of the material. Despite having been dried prior to TGA analysis, PAG is extraordinarily hygroscopic; either it absorbed more water between transfer from the vacuum ovens to the TGA, or it holds onto water strongly enough to not become fully dry. The secondary transition would be related to the chemical de-watering of PAG. Secondary alcohols are readily eliminated at elevated temperatures. The sharper third transition at 260 °C is most likely oxidative losses due to alkenes formed by water elimination. The final degradation at 400 °C is due to traditional oxidative loss.
DSC and isothermal rheology reveal relationships (DMA) between the degree of acrylation, the molecular weight, and the glass transition temperature Tg (see Fig. 3 and 4 and Table 2). Tg increases with the functionality x, which we attribute to the reduction of segmental mobility as the branch density increases; this is similar to the well-known influence of crosslink density on Tg in thermosets.27 As is ordinarily expected, Tg also increases with Mn, ideal comparing 10 kDa and 100 kDa PAG. However, comparison of PAG at Mn, ideal = 100 kDa and 1 MDa shows a decrease in Tg. The most likely explanation is that the Mn, ideal = 10 kDa polymers are lowly branched materials that behave fairly like oligomeric compounds. As molecular weight increases to 100 kDa the material behaves like a high molecular weight branched material. The molecular weight increasing to 1 MDa shows a decrease in Tg and is likely an effect of chain topology and hydrogen bonding; however, further study is required to fully explain. It is also interesting to note that the breadth of the glass transition as indicated by DSC (Fig. 3) is quite large. For example, PAG1.3-A exhibits onset at Tgo − 65 °C ending at Tge − 5 °C. This is explained by the branching of the polymer, as different branch lengths can cause changes in amount of mobility and thus glass transition.28
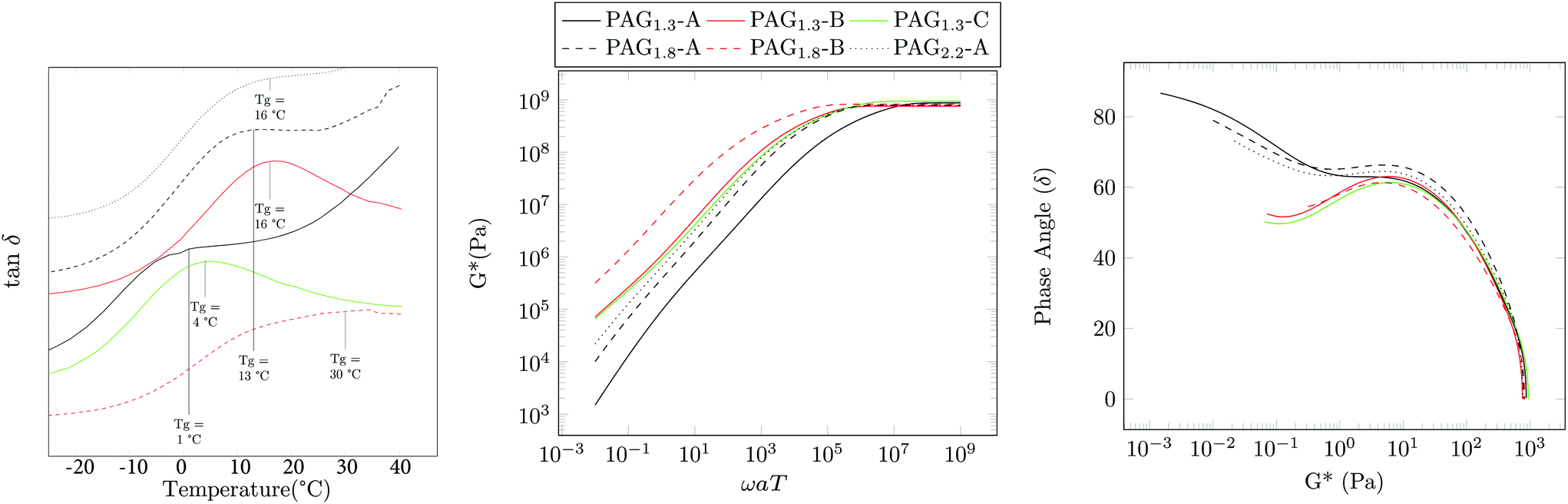 | ||
| Fig. 4 (Left) Isochronal rheology that shows the peaks in tan δ for each PAG sample. Master curve showing complex modulus G* (Middle) and Van Gurp-Palmen plot (Right). | ||
Water solubility (S) was also examined. Table 2 shows the solubility of the various materials. As acrylic functionality is increased there is a corresponding decrease in water solubility. Somewhat unexpectedly, the solubility of the PAG1.3 series improves with increasing molecular weight. This is somewhat counter-intuitive; however, it is likely the use of the hydrophobic CTA seems to be strongly effecting the ability of the polymer to dissolve in water. Finally, master curves of these materials were assembled with dynamic shear rheology in the linear viscoelastic regime through the application of the time temperature superposition principle at a reference temperature Tref = 20 °C. Fig. 4 shows the overlayed results of the complex modulus and the corresponding van Gurp-Palmen plot.29 A trend similar to that of the isochronal rheology and DSC was observed: as functionality increases, the crossover frequency ωx decreases, corresponding to the persistence of solid-like behavior for longer relaxation times. Likewise, ωx decreases on comparing Mn, ideal = 10 kDa with 100 kDa samples, and then increases at Mn, ideal = 1 MDa. These measurements are consistent with our qualitative observations noting that PAG1.33-A is incredibly soft and tacky, whereas PAG1.33-B,C,CW are solid and rubbery. Given the similar Đ values it is expected that the cross-over modulus (Gx) for these materials would be similar and this is observed. The relaxation dynamics of PAG in the terminal regime differ from the classical behavior of linear polymers in a manner consistent with other HBPs. The slope of the viscous modulus with respect to frequency α = dlogG′′/dlogωaT is constant in this regime and assumes a value of unity for linear polymers. PAG shows α < 1, where α decreases with increasing molecular weight. This is typical for hyperbranched systems with more branching resulting in a lower α.20 This coupled with the DSC indicates that the 10 kDa targets have significantly less branching than the 100 kDa or 1 MDa samples. The van Gurp-Palmen plot indicates that the development of the rubbery plateau may begin to be forming, indicative of an onset of entanglement dynamics; however, given the materials’ susceptibility for crosslinking at elevated temperature, further elucidation of the plateau is not possible.
The tack, elasticity, water solubility, and residual reactivity of PAG is suggestive of its potential utility in adhesive applications. PAG1.33-C was evaluated as a water-based sprayable adhesive for wood composites. Both oriented strand board (OSB) and plywood samples were constructed. The OSB internal bond strength was tested and is shown in Fig. 5. It is evident that PAG, when the water content is kept low, can perform as well or significantly better to the commercially available OSB boards.30,31
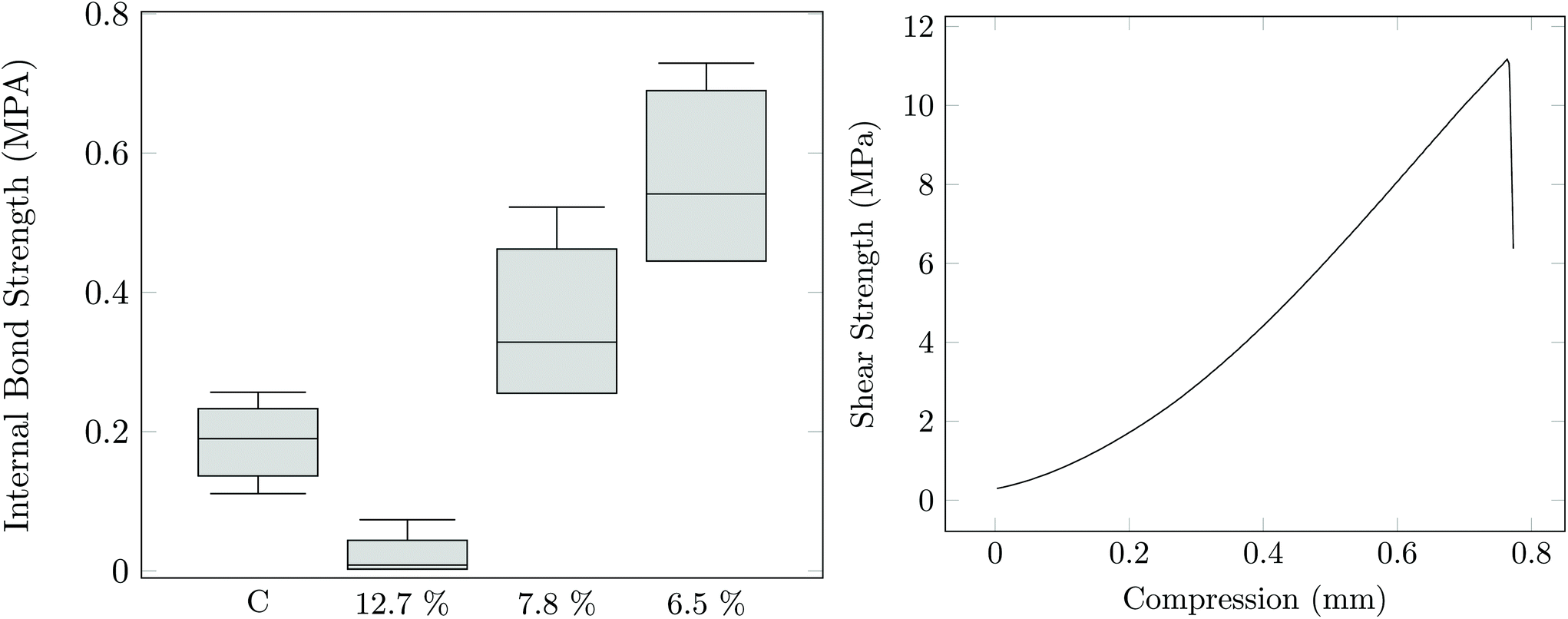 | ||
| Fig. 5 (Left) Bond strength of a control Georgia Pacific OSB board, and the PAG1.3-C sample at various different water contents. (Right) Overlap shear test of plywood samples using PAG1.3-C adhesive. | ||
Plywood was also produced using both pure PAG1.33-C as well as PAG1.33-C/poly (methylene diphenyl diisocyanate, PMDI) mixtures as an adhesive. In Fig. 5 the results of the overlap shear test for a plywood constructed from only PAG are shown. The results show that PAG (10 MPa) has nearly the same shear strength as the pure maple wood (13 MPa). In order to show whether this material is suitable for use in wet environment applications, both a soak and boiling water experiment were performed. Fig. 6 show the results of these experiments. It appears that pure PAG as an adhesive for plywood is unacceptable for application in wet environments; however, with the addition of 10–20% PMDI the adhesive can provide similar, if not better, wet and boiled water strength when compared to both literature values and a 100% PMDI control.32,33 Evidently, sufficient urethane linkages form between the PAG and PMDI to both strengthen the cured material and reduce water affinity through construction of the residual hydroxyl functionality.
 | ||
| Fig. 6 (Left) Wet strength of plywood samples that have been soaked in water at various loadings of PAG1.3-C/PMDI. (Middle) Image of scale-up reaction of PAG1.3-C. This represents 1 kg of material. (Right) Plot showing the strength of samples that have been boiled in water. 100% PMDI control shown in both plots. | ||
3. Conclusions
In this work, a hyperbranched thermoplastic poly(glycerol acrylate) was developed using the RAFT process. This polymer was discovered to have excellent tack, low glass transitions (<30 °C), tunable water solubility, molecular weight, and physical properties. Additionally, there is excellent opportunity to manipulate functionality/properties by modifying the residual hydroxyl groups. When applied to wood as an adhesive, PAG was found to perform similarly or better than traditional PMDI adhesives as well as commercial boards. The properties, lower cost than that of PMDI adhesives (<$1.00 per lb vs. $2.00 per lb) and renewable origins could lead PAG to displacing some of the most common, and toxic/environmentally damaging, chemicals used in the adhesives market. Additionally, given the properties and available functionality, additional study of PAG could be done to develop materials such as pressure-sensitive adhesives, thermoplastic elastomers and viscosity modifiers. This can lead to a variety of products that are inexpensive, less toxic, biorenewable, biodegradable, and environmentally responsible displacing traditional petrochemical products and providing more economic value to high-volume biofuels by utilizing waste products to provide high-volume high-value applications.4. Experimental section
Analytical grade glycerol, triphenylphosphine, p-toluenesulfonic acid, hydroquinone, and acrylic acid were all purchased from Sigma Aldrich with purity of 99% or higher. Crude glycerol was obtained through REG Inc. in Ames, IA. Benzyl bromide, potassium phosphate tribasic, benzyl mercaptan, carbon disulfide, and azobisisobutyronitrile (AIBN), and 4,4′-azobis(4-cyanovaleric acid) were purchased from Sigma Aldrich with purities of 98% or higher. Methanol was purchased from Fischer scientific with a purity of 99.8%.4.1. Acrylation of glycerol
Crude or technical grade glycerol, acrylic acid, hydroquinone, and catalyst were added together in a round bottom flask equipped with a stir bar and reflux condenser. The ratios of reagents used are shown in Table 1. This reaction mixture was heated at 125 °C or 100 °C for 36 or 2 hours at 500 rpm stir rate for TPP/TSA catalyzed reactions respectively. The monomer was then used without purification.4.2. RAFT agent-dibenzyl carbonotrithioate
The synthesis of the used CTA, dibenzyl carbonotrithioate (DBCTT), was well documented in literature, and there are multiple pathways available for this synthesis. We followed the route used by Gooch et al.34 Briefly, acetone, potassium phosphate (1.1 eq.), and benzyl mercaptan (1 eq.) are mixed together and allowed to stir for 10 minutes. 1.7 eq. of carbon disulfide is added and allowed to stir for 10 minutes. Finally 1.05 eq. of benzyl bromide is added and allowed to stir for 10 minutes. The reaction mixture was then filtered and dried under reduced pressure to yield the product.4.3. RAFT polymerization of acrylated glycerol
The conditions of the reaction are shown in Table 2. The appropriate amount of each component were added to a flask with a magnetic stir bar. Then the material was sparged with argon for a period of 15 minutes to an hour depending on the quantity of solution. The solution was then placed in an oil bath at 71 °C for 8 hours at 500 rpm. The poly(acrylated glycerol), or PAG, was then precipitated in isopropanol and dried under reduced pressure at room temperature.This polymerization has also been conducted using water as the solvent and 4,4′-azobis(4-cyanovaleric acid) as the initiator. The conditions were the same as above other than substitution of the solvent and the initiator.
4.4. Overlap shear test, internal bond strength and wet/boiled strength
PAG was tested as an adhesive for the production of OSB and plywood. OSB was made by mixing the polymer with water to obtain a 33 wt% solids in water. 0.1 wt% 4,4′-azobis(4-cynaovaleric acid) was added to the PAG solution. This was then sprayed with a HVLP sprayer onto wood strands tumbling in a rotating mixer. The PAG was applied at 10 wt% relative to the wood. The strands were then dried in the vacuum oven to drive off some of the water. Finally the wood was pressed into a 7′′ × 7′′ form using a Carver press at 400 psi and 204 °C.Internal Bond Strength was determined using an Instron universal test frame. Test samples were prepared by cutting samples into 2′′ × 2′′ sections and adhering testing blocks to the top and bottom surfaces of the testing block with hot melt adhesive. The samples were then strained at a 1 mm min−1 rate until failure. Internal bond strength was determined according to ASTM D1037.
In order to determine the suitability of this adhesive for plywood, wet vacuum and boil exposure tests from PS1–95 Construction and Industrial Plywood were used to determine the water resistance of different methods of manufacturing plywood.
7′′ × 10′′ samples of plywood were made from 1/20′′ Douglas Fir. Samples were cut into 15 1′′ × 3′′ coupons cut so that 5 coupons from each sample can be tested dry, after a wet vacuum test, and after a boiling water exposure test. In the wet vacuum test, samples were immersed in tap water, a vacuum of −12 psig is applied for 30 minutes. They were then further soaked for 4.5 hours at atmospheric pressure.
For the boiling water stability test, samples were boiled for 4 hours in tap water, dried at 65 °C for 15 hours in a convection oven, boiled for 4 additional hours, and then tested wet.
Overlap sheer testing was done by applying a 41 wt% solids mixture to the top surface of a 12.5′′ × 2.5′′ × 1′′ maple board. The adhesive was applied at a 10 mg cm−2 loading. A second 12.5′′ × 2.5′′ × 1′′ board was set on this and then the samples were pressed at 100 psi for 3 hours at 120 °C in a Carver press.
4.5. Analytics
The gel permeation chromatography (GPC) system used was a Malvern Viscotek high temperature GPC run in dimethylformamide. It is equipped with a refractive index detector, a UV-vis detector, a viscometer, and a light scattering detector. The equipment was equipped with Malvern CLM6210 high temperature columns that have a 10 MDa cutoff. Samples were analyzed using the refractive index detector compared to a polymethylmethacrylate sample. Samples were run for 40 minutes through three columns in series.The GCMS used was an Agilent 6890 GC coupled with an Agilent 5975C MS detector. Column used was an Agilent DB-1. The NMR was a Bruker 600 MHz. Samples were run for 32 scans with a 1 second delay. Samples were dissolved in deuterated DMSO. The DSC used is a TA instrument Q2000 DSC. DSC was run for three heating and cooling cycles from −70 °C to 30 °C at a rate of 10 °C min−1. Samples were between 5–15 mg. The data reported was the heating on the third cycle with the exotherm up.
TGA was run using a TA TGA 550. Samples were run using platinum pans with a ramp rate of 10 °C min−1 in an air environment.
Rheology was run on an Ares G2 rheometery using 8 mm parallel plate geometry. The glass transition was determined by doing constant strain temperature sweeps at 10 rad s−1 and the master curve was generated by varying the frequency from 100 rad s−1 to 1 rad s−1. The temperature range was from 40 °C to −80 °C with 10 °C increments.
We defined the solubility of the polymer to be a solution that had an average size less than 1 μm. This was done by use of a Malvern Zetasizer Nano ZS.
Acid titration was done by dissolving 1 mL of the monomer in 10 mL of water and then titrating with 0.1 M NaOH until pH 7. A Hanna HI 208 pH probe was utilized.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We wish to thank ISU Chemical Instrumentation Facility staff member Sarah Cady for training and assistance pertaining to the NMR Bruker 600 MHz NMR. We would also like to thank ISU Keck Lab staff member Lucas Showman for training and assistance on the Agilent 6890 GC. Finally, acknowledge USDA NIFA 2014-38202-22318 and the State of Iowa Board of Regents for financial support.References
- J. L. Williams, WTRG Economics, May, 2008 Search PubMed.
- P. F. Kingston, Spill Sci. Technol. Bull., 2002, 7, 53–61 CrossRef CAS.
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893–1909 RSC.
- M. Ayoub and A. Z. Abdullah, Renewable Sustainable Energy Rev., 2012, 16, 2671–2686 CrossRef CAS.
- U.S. Department of Agriculture, World and U.S. Sugar and Corn Sweetener Prices, 2014, http://www.ers.usda.gov/data-products/sugar-and-sweeteners-yearbook-tables.aspx Search PubMed.
- R. W. M. Pott, C. J. Howe and J. S. Dennis, Bioresour. Technol., 2014, 152, 464–470 CrossRef CAS PubMed.
- H. Zhang and M. W. Grinstaff, Macromol. Rapid Commun., 2014, 35, 1906–1924 CrossRef CAS PubMed.
- P. J. Flory, J. Am. Chem. Soc., 1941, 63, 3083–3090 CrossRef CAS.
- Y. Wang, Y. M. Kim and R. Langer, J. Biomed. Mater. Res., Part A, 2003, 66, 192–197 CrossRef PubMed.
- W. Cai and L. Liu, Mater. Lett., 2008, 62, 2171–2173 CrossRef.
- M. R. Whittaker, C. N. Urbani and M. J. Monteiro, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6346–6357 CrossRef CAS.
- Q. Zhang, N. Vanparijs, B. Louage, B. G. De Geest and R. Hoogenboom, Polym. Chem., 2014, 5, 1140–1144 RSC.
- M. Fujita, Y. Iizuka and A. Miyake, J. Therm. Anal. Calorim., 2017, 128, 1227–1233 CrossRef CAS.
- C. LâĂŹHostis, E. Fredon, M.-F. Thévenon, F.-J. Santiago-Medina and P. Gérardin, Holzforschung, 2020, 74, 351–361 Search PubMed.
- F.-Y. Lin, M. Yan and E. W. Cochran, Macromolecules, 2019, 52, 7005–7015 CrossRef CAS.
- R. Scherf, L. S. Müller, D. Grosch, E. G. Hübner and W. Oppermann, Polymer, 2015, 58, 36–42 CrossRef CAS.
- A. Matsumoto, A. Okamoto, S. Okuno and H. Aota, Angew. Makromol. Chem., 1996, 240, 275–284 CrossRef CAS.
- P. J. Flory, J. Am. Chem. Soc., 1952, 74, 2718–2723 CrossRef CAS.
- A.-M. Caminade, D. Yan and D. K. Smith, Chem. Soc. Rev., 2015, 44, 3870–3873 RSC.
- M. Yan, F.-Y. Lin and E. W. Cochran, Polymer, 2017, 125, 117–125 CrossRef CAS.
- E. Malmström and A. Hult, J. Macromol. Sci., Part C: Polym. Rev., 1997, 37, 555–579 CrossRef.
- M. Yan, Y. Huang, M. Lu, F.-Y. Lin, N. B. HernÃąndez and E. W. Cochran, Biomacromolecules, 2016, 17, 2701–2709 CrossRef CAS PubMed.
- A. Matsumoto, Y. Kitaguchi and O. Sonoda, Macromolecules, 1999, 32, 8336–8339 CrossRef CAS.
- H. Gao, K. Min and K. Matyjaszewski, Macromolecules, 2009, 42, 8039–8043 CrossRef CAS.
- S. Jin, K. Li, Q. Gao, W. Zhang, H. Chen and J. Li, Carbohydr. Polym., 2020, 116141, DOI:10.1016/j.carbpol.2020.116141.
- M. Bochenek, N. Oleszko-Torbus, W. Wałach, D. Lipowska-Kur, A. Dworak and A. Utrata-Wesołek, Polym. Rev., 2020, 1–51 Search PubMed.
- R. A. Venditti and J. K. Gillham, J. Appl. Polym. Sci., 1997, 64, 3–14 CrossRef CAS.
- V. Bershtein, L. Egorova, P. Yakushev, P. Sysel, R. Hobzova, J. Kotek, P. Pissis, S. Kripotou and P. Maroulas, Polymer, 2006, 47, 6765–6772 CrossRef CAS.
- M. Van Gurp and J. Palmen, Rheol. Bull., 1998, 67, 5–8 Search PubMed.
- J. J. Paredes, R. Jara, S. M. Shaler and A. Van Heiningen, For. Prod. J., 2008, 58, 56–62 CAS.
- H. Chen and N. Yan, Ind. Crops Prod., 2018, 113, 1–9 CrossRef CAS.
- G.-Z. Xu, Y.-G. Eom, B.-H. Lee and H.-J. Kim, J. Korean Wood Sci. Technol., 2010, 38, 414–420 CrossRef.
- A. Grigoriou, Wood Sci. Technol., 2000, 34, 355–365 CrossRef CAS.
- A. Gooch, N. S. Murphy, N. H. Thomson and A. J. Wilson, Macromolecules, 2013, 46, 9634–9641 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/D0GC01831G |
| This journal is © The Royal Society of Chemistry 2020 |