
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cell-free biocatalytic syntheses of L-pipecolic acid: a dual strategy approach and process intensification in flow†
David
Roura Padrosa‡
 ab,
Ana I.
Benítez-Mateos‡
ab,
Liam
Calvey
b and
Francesca
Paradisi
*ab
ab,
Ana I.
Benítez-Mateos‡
ab,
Liam
Calvey
b and
Francesca
Paradisi
*ab
aDepartment of Chemistry and Biochemistry University of Bern, Freiestrasse 3, Bern, Switzerland. E-mail: francesca.paradisi@dcb.unibe.ch
bSchool of Chemistry, University of Nottingham, University Park, NG7 2RD, Nottingham, UK
First published on 21st July 2020
Abstract
As an alternative to the traditional chemical synthesis or in vivo production of L-pipecolic acid, we have developed two ex vivo strategies using purified and immobilised enzymes for the production of this key building block. Firstly, a transaminase capable of lysine ε-deamination was coupled with a novel pyrroline-5-carboxylate reductase, yielding 60% conversion at the 50 mM scale with free enzymes and in situ recycling of the cofactor. A second, simpler, redox neutral system was then constructed by combining the pyrroline-5-carboxylate reductase with a lysine-6-dehydrogenase. This bienzymatic system, with catalytic amount of free cofactor yielded >99% of pipecolic acid in batch and, following co-immobilisation of both enzymes, it was applied as a packed-bed reactor in continuous flow achieving again a molar conversion of >99% with 30 min residence time, and a space–time yield up to 2.5 g L−1 h−1. The sustainability of the system was further improved by a catch-and-release strategy to purify the product, and recovery and recycling of the cofactor.
Introduction
Pipecolic acid (PA) is a natural non-proteinogenic α-amino acid found in a plethora of biologically active molecules.1,2 It is incorporated in natural products with anticancer or antibiotic activity (Swainsonine, Virginiamycin S1, and Rapamycin)3–6 and it serves as a precursor to some simple manmade bioactive molecules, such as amide anaesthetic drugs (Mepivacaine and Ropivacaine),7 (Fig. 1A). In addition, pipecolic acid can be used as chiral organocatalyst in stereoselective Mannich reactions,8,9 as a building block in peptidic catalysts,10 as well as diketopiperzine scaffolds.11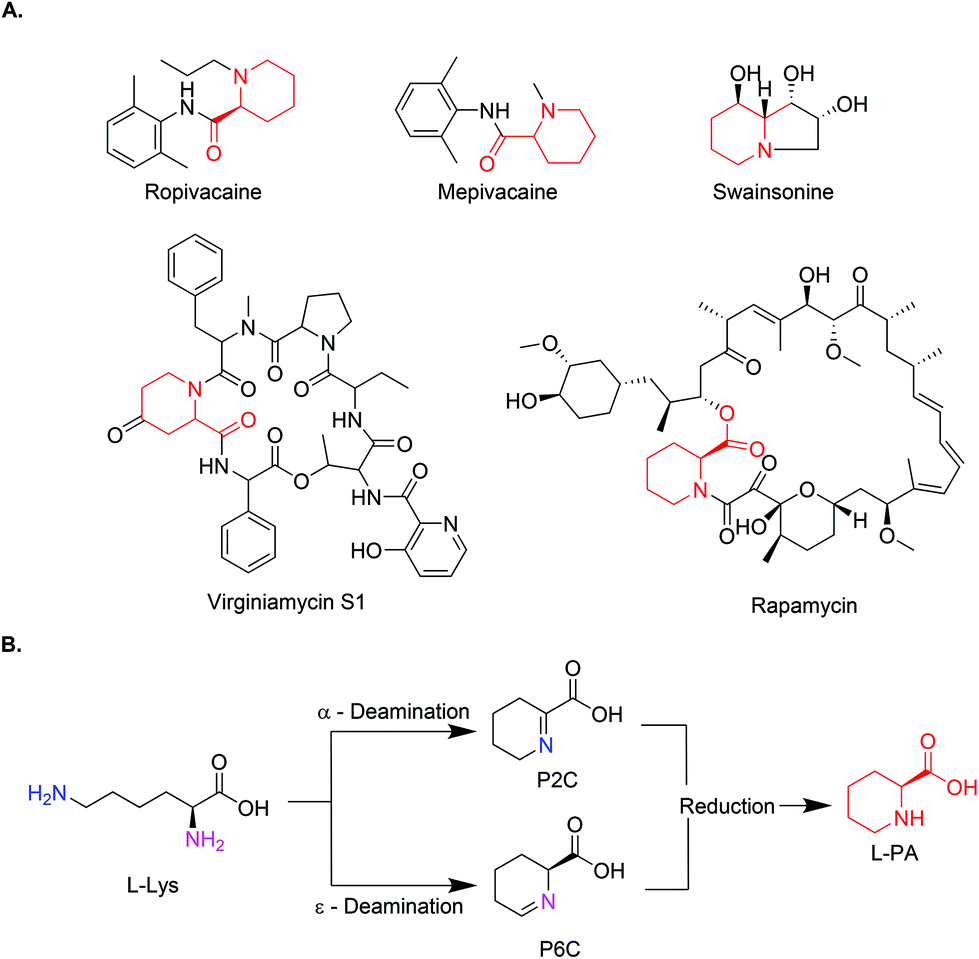 | ||
| Fig. 1 (A) Chemical structures of L-PA containing drugs (L-PA identified in red). (B) General reaction scheme of the biosynthetic pathway for the production of L-PA. | ||
Despite its simplicity, L-PA as an optically pure molecule, is orders of magnitude more expensive than the homologous five-member ring L-proline, naturally impacting on the cost of the final drugs. Its preparation by available chemical methods normally involves harsh conditions and hazardous reagents despite the most recent efforts to move towards greener strategies.12–14 Moreover, classical synthetic approaches require multi step synthesis, with protection and deprotection steps, which affect the overall yield. Ginesta et al., developed a 5 step synthesis of pipecolic acid in very good enantiomeric excess but with only ∼40% overall yield.15 More recently, two single-step strategies have been reported. In the first one, the direct synthesis of derivatives of pipecolic acid was achieved by stereoselective aldol reaction, but again low yields were obtained (30%).16 Alternatively, photocatalysis using a TiO2 catalyst allowed the production of racemic and L-PA with moderate enantiomeric excess but still 40% final yield.17
As an alternative, new approaches mimicking the natural pathways have gained momentum in the last years. PA pathways have been studied for years now in plants and animals, due to their relationship with lysine metabolism defects. The conversion from L-lysine to PA normally occurs in a two-step process, where L-lysine is deaminated either in the α or ε-position by a lysine dehydrogenase or lysine oxidase, leaving the corresponding aldehyde which spontaneously cyclises in water. The resulting product (Δ1-piperidine-2-carboxylic acid (P2C) or Δ1-piperidine-6-carboxylic acid (P6C)) is then reduced by a specific reductase to yield PA (Fig. 1B). Some cell-free examples using these combinations of enzymes have been reported.18–20 While in the ε-deamination the stereochemistry of the intermediate (at the α-carbon) is preserved throughout, in the α-deamination path, the chirality is lost and reintroduced in the reduction step by stereospecific imine reductases.
In addition to the two-step reaction catalysed by two different enzymes, a lysine cyclodeaminase, has been recently shown to perform both the deamination and the subsequent reduction of the cyclic intermediate, but in this case the reaction is very slow, achieving 90% conversion on a 5 mM scale in 60 hours.20–24In vivo, the combination of a lysine-6-dehydrogenase (Lys6DH) with a pyrroline-5-carboxylate reductase (P5C) has been successfully implemented in Corynebacterium glutamicum, with a productivity of 15 g L−1 over 70 h fermentations.25 In another example of whole cell production, Cheng et al. achieved a higher yield (46.7 g L−1) over 36 h fermentations by creating and artificial pathway, co-expressing 4 recombinant proteins in E. coli.26 However, many biocatalytic methods are still far from meeting industrial requirements, and chemical methods offering higher yields are often preferred.27
Interestingly, from an enzymatic perspective, even though the first reaction in the synthesis of PA from L-lysine consists of a deamination, no transaminase based approach has been described to date, albeit lysine-ε-aminotransferases have been identified in bacteria, fungi and animals, and even some ω-transaminases have shown activity with lysine.28–30 Recently, lysine has been suggested as the amino donor in whole cells expressing an S-selective transaminase.31
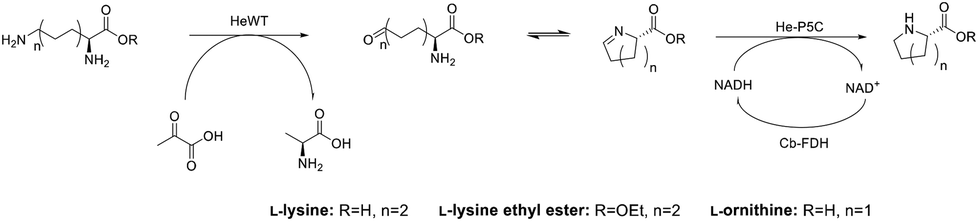
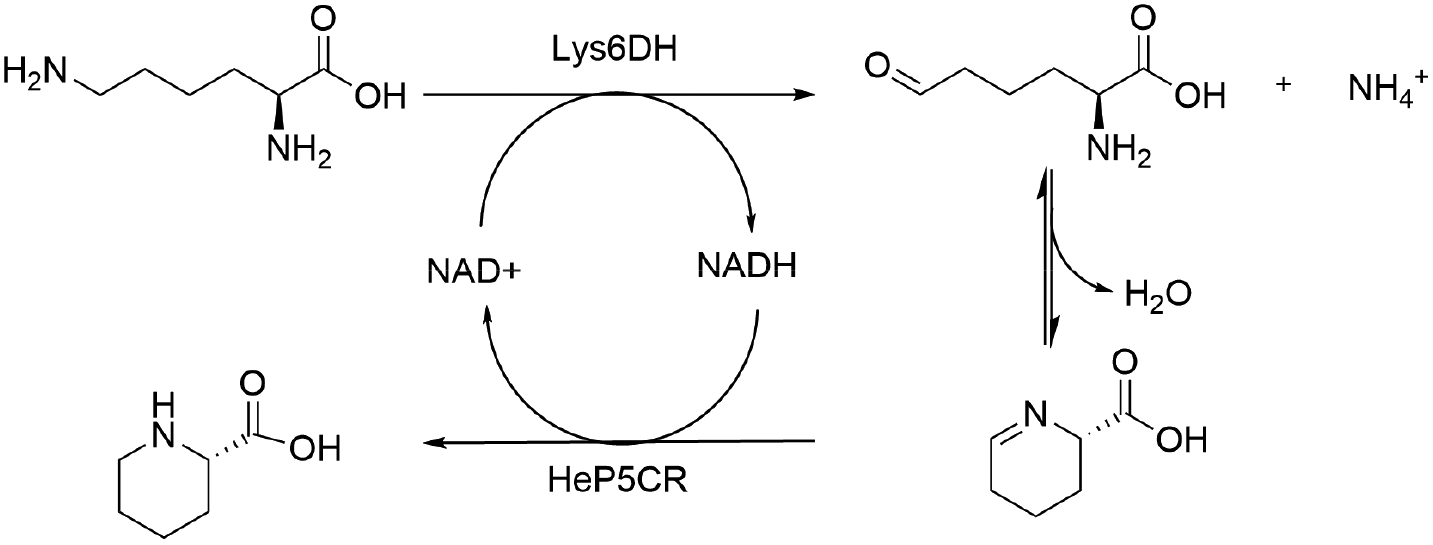
Here, we describe the successful application in batch of a transaminase-based synthesis of L-PA and derivates, using HeWT,32 in combination with a newly cloned pyrroline-5-carboxylate reductase (He-P5C), both from the halotolerant organism Halomonas elongata. As an alternative, to simplify the cascade and move towards a redox neutral approach we developed an additional cascade using lysine-6-dehydrogenase (Gs-Lys6DH) and He-P5C. The resulting system could be used in batch with both free and co-immobilised enzymes, as well as with a concurrent immobilisation of the cofactor to avoid the need of external addition of NAD(H). Finally, the latter system was implemented as a packed bed reactor in a continuous flow system with catalytic amount of free cofactor. Here, the purification of L-PA and the reuse of the cofactor was accomplished in continuous by using a scavenger column.
Results and discussion
Multi-enzymatic cascade combining HeWT and He-P5C
While the transaminase from Halomonas elongata has been extensively studied and displayed a broad substrate scope,32 its ability to deaminate lysine at the ε-position was unknown. Initial studies on HeWT mediated deamination of lysine were performed using pyruvate, glyoxylate and benzaldehyde as amino acceptors. The reactions were followed by monitoring both the depletion of the substrate and the formation of the corresponding amine. On a 10 mM scale, HeWT was capable of deaminating both enantiomers of lysine with moderate yields (∼50%), suggesting that the ε-amino group rather than the α-position, is deaminated. In fact, the deamination of the α-amino group would require HeWT to accommodate double-bulky primary amines in the active site, which for this enzyme has never been observed.To further shift the equilibrium of the first reaction and to complete the cascade, a new pyrroline-5-carboxylate reductase was also identified in the genome of Halomonas elongata. The protein, named He-P5C, was successfully cloned and expressed (ESI section 1, Fig. S1†), and had activity with P6C. While previously reported bacterial P5C reductases showed a higher specificity for NADPH,33,34 He-P5C exhibited dual cofactor affinity, with a KM of 0.013 ± 0.004 for NADH and 0.17 ± 0.03 for NADPH (Fig. S2†), suggesting that, in fact, the preferred electron donor is NADH. For our purpose, the dual affinity for both cofactors offers flexibility on the choice of cofactor regeneration system, which is required to make the overall reaction sustainable.
The cascade reaction was then assayed on a 10 mM scale using only HeWT and He-P5C with, initially, equimolar amount of cofactor and no recycling system. The presence of the second enzyme, He-P5C, improved significantly the yield of lysine deamination (while yielding L-PA), indicating an effect on the overall reaction equilibrium (Fig. S3†). From the different tested conditions, the best results were obtained with benzaldehyde and pyruvate as amino acceptors, with ∼60% and ∼40% L-lysine deamination yields respectively (Fig. S4A†). Only with pyruvate, equimolar amount of L-PA is formed, as He-P5C was found to be inactivated by benzaldehyde, benzylamine and DMSO (Fig. S5†). When using D-lysine, the aldehyde intermediate was formed in a similar yield to L-lysine but no production of D-pipecolic acid was observed, confirming the high stereoselectivity of He-P5C previously observed for similar reductases.35–37 These conversions were maintained when scaling up L-lysine to 50 mM (Fig. S4B†). However, the depletion of D-lysine was significantly lower at higher scale, possibly indicating an inhibition effect of the substrate.
To construct a more cost-efficient cascade, a glucose dehydrogenase from Bacillus megaterium with dual cofactor specificity, but preference for NADP+ (Bm-GDH)38,39 and a NAD-dependant formate dehydrogenase from Candida boidinii (Cb-FDH)40 were trialled in combination with the two enzymes, 50 mM L-lysine and just 0.1 eq. of the suitable cofactor. While with BmGDH the efficiency of the second step yielded ∼50% of the final product, with Cb-FDH total conversion of the intermediate was observed (Table S1†). Further improvement was obtained in the first step of the reaction (lysine deamination) by increasing the pH of the reaction from 8 to 10 (Fig. S6†). At higher pH, significant production of L-PA was observed only with Cb-FDH as recycling system, while Bm-GDH was rapidly inactivated.38
With fully optimised conditions, the cascade was expanded to two additional substrates (Table 1). The system produced almost equimolar amount of the two six member rings, with respect to the cyclic imine intermediate. In the case of L-ornithine, the substrate was fully deaminated but only 27.5 mM (55%) of the corresponding cyclic amine (L-proline) was formed, probably due to the reduced activity of He-P5C over long periods (Fig. S7†). Despite the efficiency of the second step, the equilibrium of the deamination was only marginally improved (Fig. S8†).
|
|
||
|---|---|---|
| Substrate | Imine intermediate (%) | Cyclic amine (%) |
| L-Lysine | 60 ± 2 | 57 ± 2 |
| L-Lysine ethyl ester | 55 ± 3 | 49 ± 3 |
| L-Ornithine | 99 ± 1 | 55 ± 2 |
Redox neutral cascade: coupling Gs-Lys6DH and He-P5C
To overcome the limitation of the transaminase-based cascade, a NAD-dependent lysine dehydrogenase from Geobacillus stearothermophilus (Gs-Lys6DH) was selected instead to perform the first catalytic step due to its high reported activity and excellent stability.41 As the He-P5C has a higher preference for NADH, the complementarity of the two enzymes is optimal, with Gs-Lys6DH requiring the oxidised form for the deamination step and He-P5C the reduced one for P6C reduction, establishing a closed cofactor regeneration system.The cofactor recycling capability of the cascade was tested at increasing concentration of L-lysine keeping NAD+ at 1 mM (Table 2). Complete conversion was observed both at 10 and 50 mM substrate concentration in 24 h, while on a 100 mM scale 62% of L-lysine was converted during the first 24 h with no further increase.
|
|
||
|---|---|---|
| Substrate concentration (mM) | Conversion to L-PA | |
| 4 h | 24 h | |
| 10 mM | >99 ± 3 | >99 ± 2 |
| 50 mM | 26 ± 2 | >99 ± 5 |
| 100 mM | 12 ± 4 | 62 ± 3 |
To increase the reusability of the biocatalysts, immobilisation of both Gs-Lys6DH and He-P5C was achieved on various supports. Although the immobilisation yield of Gs-Lys6DH reached >90% with all the supports tested, the best results in terms of recovered activity (immobilised specific activity [U mg−1]/free specific activity [U mg−1]) × (100) were obtained with the most hydrophilic matrix (Table S2†). Specifically, more than 90% of recovered activity was achieved when using agarose microbeads as support. In contrast, the activity of He-P5C dropped dramatically upon immobilisation. To better understand the factors governing such loss of activity, a model of the enzyme structure was studied (Fig. S9†). The distortion of the He-P5C dimeric structure induced by covalent bonding could explain its low recovered activity, since the catalytic site is located at the interface of the two monomers. It has been previously reported that subunit dissociation of immobilised enzymes can be prevented by pre- or post-immobilisation polymer coating, such as with poliethyleneimine (PEI).42,43 In our case, the addition of PEI during the immobilisation of He-P5C increased the recovered activity up to 21% (Fig. S10 and S11†).
Self-sufficient biocatalytic module with co-immobilised NAD+
Previous work reported a strategy for the reversible co-immobilisation of cofactors through amine groups on the support to retain the cofactor within their microenvironment.44–46 This approach would be ideally suited to this system allowing for the reuse and recycle of the cofactors directly on the resin avoiding the need for external supply. In our case however, despite several attempts (ESI section 5†), only marginal success was obtained.Likely, the requirement for high ionic strength in the buffer to maintain He-P5C activity causes the lixiviation of the of the cofactor over time. On the other hand, when low ionic strength is used, while the cofactor is retained, a rapid loss of catalytic efficiency of the system is observed (Fig. S12 and S13†).
Flow biocatalysis: process intensification
While cofactor co-immobilisation on the resin could not be exploited, the immobilised bi-enzymatic system with catalytic amounts of cofactor showed excellent performance in batch and the enzyme mediated synthesis of L-PA on a 10 mM scale was moved to a continuous flow PBR (packed-bed reactor) (Table 3). The immobilised biocatalyst was packed into a 2 mL-column which was fed with L-lysine. The optimization of the flow conditions was achieved yielding complete conversion at 37 °C using just 0.1 equivalent of cofactor and 30 minutes residence time (Table S4†). While in batch conditions the turnover number (TN) of each free enzyme was 0.04 μmol min−1 mg−1, under optimised flow conditions it increased to 0.33 μmol min−1 mg−1 which is almost a 10-fold increase. Moreover, the stability of the biocatalyst was sustained for more than 24 hours continuous operation at different flow conditions retaining more than 95% of its initial activity.|
|
||
|---|---|---|
| PBR | Scavenger column | |
| L-PA production | L-PA purification | NAD+ recovery |
| >99% | 97.3% | 87.4% |
| 12.8 mg | 12.5 mg | 4.3 mg |
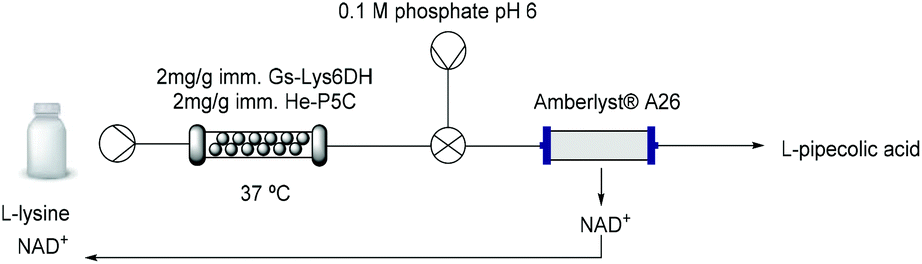
Although this system is efficient with a catalytic amount of NAD+, the implementation of cofactor-dependent enzymes is still limited on a larger scale in flow, as the costly cofactors are lost downstream.47,48 In order to maximize the efficiency of the system, we applied an in-line catch-and-release strategy enabling the separation of the L-PA product from the cofactor (Table 3 and Fig. S14†). Using a single scavenger column packed with Amberlyst® A26, the L-PA produced continuously over 180 min was purified with a 97% yield, while the cofactor was efficiently trapped by the A26 resin. The NAD+ could be easily recovered by flushing the column with a slightly acidic buffered solution (pH 6) achieving a recovery yield of more than 87% (Fig. S15†). Afterwards, the cofactor solution was recirculated into the system and re-used for a new flow reaction obtaining similar conversions. Finally, the A26 column was regenerated with 1 M NaOH for 10 min and reused in a new catch-and-release reaction, enhancing the overall sustainability of the system.
Conclusion
Greener strategies for the synthesis of important pharmaceutical building blocks, such as L-pipecolic acid, are essential to progress towards a sustainable society. In this sense, the use of enzymes and specially of immobilised biocatalysts is gaining momentum. In this work, immobilised cell-free biocatalysts have been used for the first time to produce L-pipecolic acid using cheap L-lysine as substrate. The final product is 2000-fold more expensive with respect to the current cost of the starting material.49 Therefore, the implementation of cost-efficient approaches towards the synthesis of such a high value chemical, which could be produced on-demand with high degree of purity, on a simple and portable system such as a 2 mL reactor, appears extremely timely. Both a transaminase (HeWT) and a lysine-6-dehydrogenase (Gs-Lys6DH) have been proved to efficiently perform the specific ε-deamination of the substrate. For the reduction of the resulting cyclic imine, a novel P5C from Halomonas elongata has been identified as an excellent biocatalyst due to its catalytic activity and dual cofactor specificity (accepting both NADH and NADPH). Remarkably, the calculated KM for NADH is the lowest reported to date for this type of enzymes. The promiscuous cofactor affinity offers not only flexibility on the choice of recycling system, but also impacts on its stability and cost-efficiency. NAD(H) is nearly 20 times less expensive than NADP(H) and has been shown to be more stable. Moreover, when He-P5C was immobilised together with Gs-Lys6DH, a redox-neutral cascade was achieved and successfully applied as packed-bed reactor in flow. The calculated space–time yield for this system is 2.5 g L−1 h−1 which is the highest reported so far. In batch, co-immobilisation of the cofactor on the same microbead was also achieved. However, the low stability of the enzymes in lower ionic strength buffers which are required to retain the cofactor on the solid phase, hampered its application. As an alternative, a catch-and-release strategy was introduced to simultaneously separate and reuse the cofactor and efficiently purify the target product.Experimental section
Materials
6BCL Agarose was acquired from Agarose Beads Technologies. Polyethyleneimine branched 270![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da (PEI270), L-lysine hydrochloride, D-lysine hydrochloride, L-lysine ethyl ester dihydrochloride, L-ornithine, L-pipecolinic acid, pyridoxal 5′-phosphate monohydrate were purchased from Sigma Aldrich. Polyethyleneimine 50% aq. solution branched 60000 Da (PEI60) and Amberlyst® A26 were acquired from Thermo Fisher Scientific. All the methacrylate-based supports (EC-EP/S, EP403/S, EC-HFA/S, HFA403/S, EP113/S resins) were donated by Resindion. Nicotinamide adenine dinucleotide (NAD+) was purchased from Apollo Scientific. All other reagents were of analytical grade unless otherwise specified.
000 Da (PEI270), L-lysine hydrochloride, D-lysine hydrochloride, L-lysine ethyl ester dihydrochloride, L-ornithine, L-pipecolinic acid, pyridoxal 5′-phosphate monohydrate were purchased from Sigma Aldrich. Polyethyleneimine 50% aq. solution branched 60000 Da (PEI60) and Amberlyst® A26 were acquired from Thermo Fisher Scientific. All the methacrylate-based supports (EC-EP/S, EP403/S, EC-HFA/S, HFA403/S, EP113/S resins) were donated by Resindion. Nicotinamide adenine dinucleotide (NAD+) was purchased from Apollo Scientific. All other reagents were of analytical grade unless otherwise specified.
Protein expression and purification
The plasmid harbouring He-P5C was transformed into E. coli BL21(DE3) Star from Thermo Fisher Scientific. 1 L flasks containing 300 mL of Terrific Broth media were inoculated with 1 mL of an overnight culture in LB and left growing at 37 °C and 180 rpm until OD600 reached 0.7–0.9. At that point, the expression was induced with 1 mM IPTG and the cultures left to grow for 16 h at 30 °C and 180 rpm. For Gs-Lys6DH, a single colony of E. coli BL21 (DE3) Star cells previously transformed with the corresponding plasmid, were inoculated in autoinduction media ZYP-5052. The cells were left to grow at 37 °C and 180 rpm for 20 h. Cells were collected by centrifugation at 4500 rpm and resuspended in phosphate buffer (50 mM), NaCl (0.3 M), imidazole (30 mM) pH 8. Cells were disrupted by sonication with pulses of 5 seconds ON and 5 seconds off at 40% amplitude for 8 min. The insoluble fraction was separated by centrifugation at 14500 rpm for 45 min. The supernatant, after filtration with 0.45 μm filters, was loaded into a Ni-NTA column using an AKTA-pure FPLC. The protein was eluted with phosphate buffer (50 mM), NaCl (0.3 M) and imidazole (300 mM) pH 8. He-P5C was dialysed twice against 50 mM phosphate buffer pH 8 while Gs-Lys6DH was dialysed also two times against phosphate buffer (10 mM) pH 8 containing DTT (0.1 mM) and glycerol (100 g L−1) as described before.41
Activity assay
For Gs-Lys6DH, HeWT and Cb-FDH, the activity assays were performed as indicated previously.32,41,50 He-P5C activity assay was performed with L-thiazolidine-4-carboxylic acid (10 mM), NAD+ (1 mM) in phosphate buffer (50 mM) pH 8 at 25 °C. One unit of enzymatic activity was defined as the reduction of 1 nmol of cofactor per minute and mg of enzyme. For the immobilised enzyme, the activity was measured using the same conditions as the free enzyme. Typically, from 20 to 50 mg of biocatalyst were added to a 5 mL reaction pot. At regular interval of times during 10 min, a sample was taken to measure the absorbance at 340 nm. In the case of the immobilised enzyme, the specific activity of the biocatalyst (U g−1) was defined as the reduction or consumption of 1 nmol of the cofactor per minute and g of biocatalyst.Enzyme biotransformation
Batch reactions with pure soluble enzymes were performed at 37 °C with a suitable amount of enzyme in 0.5 mL of reaction mix. Buffers, substrate and enzyme concentrations were varied as described in this manuscript. The reaction was monitored by HPLC. To allow for the detection of both substrates and products, FMOC derivatization was used. In short, to 100 μL of sample with a maximum concentration of substrate (10 mM), 200 μL of borate buffer (0.1 M) pH 9 were added followed by the addition of 400 μL of FMOC (15 mM) in acetonitrile. The reaction was left for 5 min to proceed and then 100 μL of the mix were added to 450 μL of 0.1% HCl and 450 μL of acetonitrile for injection. The samples were analysed by HPLC (Dionex UltiMate 3000, Waters X-Bridge C18 (3.5 μm, 2.1 × 100 mm), 0.8 mL min−1, measuring at 210 nm, 250 nm and 265 nm) using a gradient method from 40:60 to 95:5 (H2O:MeCN 0.1%TFA) over 4 minutes with a flow rate of 0.8 mL min−1. The retention times of the different compounds were: lysine-FMOC 4.11 min, lysine ethyl esther-FMOC 4.02 min, ornithine-FMOC 4.00, proline-FMOC 1.98 min, pipecolic acid-FMOC 3.15 min, alanine-FMOC 1.58 min. Molar conversions were calculated through a standard curve of the product or the substrate.
Enzyme immobilisation
The immobilisation of the enzymes, either in methacrylic or agarose resins, was performed as described previously.51 In short, 1 g of resin was treated with 2 mL of modification buffer (sodium borate (0.1 M) and iminodiacetic acid (2 M) in phosphate buffer (50 mM) pH 8.5) under gentle shaking for 2 h at room temperature. The resin was then filtered, washed three times with distilled water and mixed with 5 mL of the metal solution (1 M sodium chloride and 5 mg mL−1 of cobalt chloride in 50 mM phosphate buffer pH 6) for another 2 h. Following the same procedure, the sample was washed 3 times with distilled water and then 2 mL of the protein solution were added. The sample was kept under agitation during 4 h. When He-P5C immobilisation was carried out with the addition of PEI, 4 mL of enzyme solution containing PEI (2 g) were used. The support was then filtered and washed thoroughly with desorption buffer (EDTA (50 mM) and NaCl (0.5 M) in phosphate buffer (20 mM) pH 7.4) and washed with distilled water. Finally, 4 mL of blocking buffer (glycine (3 M) in phosphate buffer (50 mM) pH 8.5) were added and the suspension was left under agitation for 20 hours (in case of further PEI-coating of immobilised biocatalysts, blocking step was avoided). The beads were washed, collected and conserved in an appropriate volume of storage buffer at 4 °C.Co-immobilisation of NAD+
The previously immobilised biocatalyst was coated with PEI as previously described.44 Briefly, 10 mL of a solution containing PEI (100 mg) in phosphate buffer (50 mM) at pH 8 were added to the resin (1 g). The suspension was incubated overnight. Then, the suspension was thoroughly washed with buffer. Ionic adsorption of cofactors was achieved by incubating the PEI-coated resin with 10 mL of NAD+ (10 mM) in phosphate buffer (10 mM) at pH 8. The suspension was kept under agitation for 1 hour at room temperature. Afterwards, the resin was filtered and washed with the same buffer. The immobilisation yield of the cofactor was calculated by measuring the absorbance of the supernatant at 260 nm in a 96-well plate reader.Flow-reactions in a packed-bead reactor
The continuous flow reactions were performed by using a R2 + /R4 flow reactor from Vapourtec equipped with an Omnifit glass column (6.6 mm i.d × 100 mm lenght) and filled with the biocatalyst (2–2.5 g). A first washing step with buffer was performed at flow rate of 0.2 mL min−1 to equilibrate the column. Then, a substrate solution containing L-lysine (10 mM) and NAD+ (at the specified concentration) in potassium phosphate buffer (50 mM) at pH 8.0 was pumped towards the column PBR with the biocatalyst (PBR volume: 1.6–2 mL) at 0.1 mL min−1 for two column volumes. Afterwards, the flow rate was varied in order to obtain the desired residence time of the reaction. The resulting flow product was analysed by HPLC following the protocol describe above. In case of co-immobilised cofactor, the substrate solution was prepared in phosphate buffer (10 mM) at pH 8.0.In-line purification of L-PA and NAD+
Under optimised reaction conditions (substrate solution: NAD+ (1 mM), L-lysine (10 mM) in phosphate buffer (50 mM) at pH 8; T°: 37 °C; R.T.: 30 min; molar conversion: >99%), a scavenger column (1–1.7 mL) containing Amberlyst® A26 was connected to the PBR to perform the in-line purification of L-PA, retaining the cofactor into the column (Fig. S12 and S13†). Then, a washing step with phosphate buffer (50 mM) pH 8.0 was performed for 3 column volumes at R.T. 30 min. Finally, the cofactor was desorbed from the scavenger column and recovered by flushing a phosphate buffer solution (100 mM) at pH 6 for 10 column volumes at R.T. 10 min. The NAD+ recovered from both the washing and the desorption steps was collected and reused in a new flow reaction by mixing with a more concentrated solution of L-lysine. L-PA was quantified by HPLC as described above. NAD+ was detected by measuring the absorbance at 260 nm in the EPOCH microplate reader (Agilent).Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This project was supported by the Welcome Prime Award (F.P.) from the University of Nottingham. The authors wish to thank Resindion S.R.L. for donating the EC-EP/S, EP403/S, EC-HFA/S, HFA403/S, EP113/S resins.Notes and references
- H. Návarová, F. Bernsdorff, A.-C. Döring and J. Zeier, Plant Cell, 2012, 24, 5123–5141 CrossRef PubMed.
- Y. –F. Chang, J. Neurochem., 1978, 30, 347–354 CrossRef CAS PubMed.
- V. Medicine and A. Northwest, J. Microbiol. Biotechnol., 2017, 27, 1897–1906 CrossRef PubMed.
- J. W. Reed, M. B. Purvis, D. G. I. Kingston, A. Biot and F. Gosselé, J. Org. Chem., 1989, 4, 1161–1165 CrossRef.
- S. R. Park, Y. J. Yoo, Y. Ban and Y. J. Yoon, J. Antibiot., 2010, 434–441 CrossRef CAS PubMed.
- T. Schwecke, J. F. Aparicio, I. Molnár, A. König, L. E. Khaw, S. F. Haydock, M. Oliynyk, P. Caffrey, J. Cortés and J. B. Lester, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7839–7843 CrossRef CAS PubMed.
- B. Adger, U. Dyer, G. Hutton, M. Woods, C. S. Park and M. Road, Tetrahedron Lett., 1996, 37, 6399–6402 CrossRef CAS.
- P. H. Y. Cheong, H. Zhang, R. Thayumanavan, F. Tanaka, K. N. Houk and C. F. Barbas, Org. Lett., 2006, 8, 811–814 CrossRef CAS PubMed.
- M. Agirre, A. Arrieta, I. Arrastia and F. P. Cossío, Chem. – Asian J., 2019, 14, 44–66 CrossRef CAS PubMed.
- T. Schnitzer and H. Wennemers, J. Am. Chem. Soc., 2017, 139, 15356–15362 CrossRef CAS PubMed.
- Nonappa, K. Ahonen, M. Lahtinen and E. Kolehmainen, Green Chem., 2011, 13, 1203–1209 RSC.
- L. A. Watanabe, S. Haranaka, B. Jose, M. Yoshida, T. Kato, M. Moriguchi, K. Soda and N. Nishino, Tetrahedron: Asymmetry, 2005, 16, 903–908 CrossRef CAS.
- A. Cant and A. Sutherland, Synthesis, 2012, 44, 1935–1950 CrossRef CAS.
- I. T. Horváth and P. T. Anastas, Chem. Rev., 2007, 107, 2167–2168 CrossRef PubMed.
- X. Ginesta, M. A. Perica and A. Riera, Tetrahedron Lett., 2002, 43, 779–782 CrossRef CAS.
- S. Bas, R. Kusy, M. Pasternak-suder and C. Nicolas, Org. Biomol. Chem., 2018, 16, 1118–1125 RSC.
- Y. Zhai and S. S. C. Chuang, Org. Process Res. Dev., 2018, 1636–1643 CrossRef CAS.
- T. Fujii, M. Mukaihara, H. Agematu and H. Tsunekawa, Biosci. Biotechnol. Biochem., 2002, 66, 622–627 CrossRef CAS PubMed.
- M. Yasuda, M. Ueda, H. Muramatsu, H. Mihara and N. Esaki, Tetrahedron: Asymmetry, 2006, 17, 1775–1779 CrossRef CAS.
- S. M. Byun, S. W. Jeong, D. H. Cho and Y. H. Kim, Biotechnol. Bioprocess Eng., 2015, 20, 73–78 CrossRef CAS.
- H. Ying, J. Wang, Z. Wang, J. Feng, K. Chen, Y. Li and P. Ouyang, J. Mol. Catal. B: Enzym., 2015, 117, 75–80 CrossRef CAS.
- Y. Tani, R. Miyake, R. Yukami, Y. Dekishima, H. China, S. Saito, H. Kawabata and H. Mihara, Appl. Microbiol. Biotechnol., 2015, 99, 5045–5054 CrossRef CAS PubMed.
- M.-L. Wu, J.-H. Chen, C.-T. Ho and T.-C. Huang, J. Agric. Food Chem., 2007, 55, 1767–1772 CrossRef CAS PubMed.
- J. V. K. Jensen and V. F. Wendisch, Microb. Cell Fact., 2013, 12, 1–10 CrossRef PubMed.
- F. Pérez-García, J. Max Risse, K. Friehs and V. F. Wendisch, Biotechnol. J., 2017, 12, 1–12 Search PubMed.
- J. Cheng, Y. Huang, L. Mi, W. Chen, D. Wang and Q. Wang, J. Ind. Microbiol. Biotechnol., 2018, 45, 405–415 CrossRef CAS PubMed.
- V. Vranova, L. Lojkova, K. Rejsek and P. Formanek, Chirality, 2013, 25, 823–831 CrossRef CAS PubMed.
- H. H. Lo, S. K. Hsu, W. De Lin, N. L. Chan and W. H. Hsu, Biotechnol. Prog., 2005, 21, 411–415 CrossRef PubMed.
- B. A. Kern, D. Hendlin and E. Inamine, Antimicrob. Agents Chemother., 1980, 17, 679–685 CrossRef CAS PubMed.
- J. C. Fothergill and J. R. Guest, J. Gen. Microbiol., 1977, 99, 139–155 CrossRef CAS PubMed.
- S. Grigoriou, P. Kugler, E. Kulcinskaja, F. Walter, J. R. King, P. Hill, V. F. Wendisch and E. O'Reilly, Green Chem., 2020, 22, 4128–4132 RSC.
- L. Cerioli, M. Planchestainer, J. Cassidy, D. Tessaro and F. Paradisi, J. Mol. Catal. B: Enzym., 2015, 120, 141–150 CrossRef CAS.
- D. Petrollino and G. Forlani, Amino Acids, 2012, 43, 493–497 CrossRef CAS PubMed.
- E. M. Christensen, S. M. Patel, D. A. Korasick, A. C. Campbell, K. L. Krause, D. F. Becker and J. J. Tanner, J. Biol. Chem., 2017, 292, 7233–7243 CrossRef CAS PubMed.
- Z. Meng, Z. Lou, Z. Liu, M. Li, X. Zhao, M. Bartlam and Z. Rao, J. Mol. Biol., 2006, 359, 1364–1377 CrossRef CAS PubMed.
- B. Nocek, C. Chang, H. Li, L. Lezondra, D. Holzle, F. Collart and A. Joachimiak, J. Mol. Biol., 2005, 354, 91–106 CrossRef CAS PubMed.
- Y. Yang, S. Xu, M. Zhang, R. Jin, L. Zhang, J. Bao and H. Wang, Protein Expression Purif., 2006, 45, 241–248 CrossRef CAS PubMed.
- K. Yamamoto, G. Kurisu, M. Kusunoki, S. Tabata, I. Urabe and S. Osaki, J. Biochem., 2001, 129, 303–312 CrossRef CAS PubMed.
- H. E. Pauly and G. Pfleiderer, Physiol. Chem., 1975, 356, 1613–1624 CrossRef CAS PubMed.
- Q. Guo, L. Gakhar, K. Wickersham, K. Francis, A. Vardi-Kilshtain, D. T. Major, C. M. Cheatum and A. Kohen, Biochemistry, 2016, 55, 2760–2771 CrossRef CAS PubMed.
- M. Heydari, T. Ohshima, N. Nunoura-kominato and H. Sakuraba, Appl. Environ. Microbiol., 2004, 70, 937–942 CrossRef CAS PubMed.
- J. M. Bolivar, J. Rocha-Martín, C. Mateo and J. M. Guisan, Process Biochem., 2012, 47, 679–686 CrossRef CAS.
- L. Trobo-Maseda, A. H. Orrego, S. Moreno-Pérez, G. Fernández-Lorente, J. M. Guisan and J. Rocha-Martin, Appl. Microbiol. Biotechnol., 2018, 102, 773–787 CrossRef CAS PubMed.
- S. Velasco-Lozano, A. I. Benítez-Mateos and F. López-Gallego, Angew. Chem., Int. Ed., 2017, 56, 771–775 CrossRef CAS PubMed.
- A. I. Benítez-Mateos, E. San Sebastian, N. Ríos-Lombardía, F. Morís, J. González-Sabín and F. López-Gallego, Chem. Eur. J., 2017, 23, 16843 CrossRef PubMed.
- A. I. Benítez-Mateos, M. L. Contente, S. Velasco-Lozano, F. Paradisi and F. López-Gallego, ACS Sustainable Chem. Eng., 2018, 6, 13151–13159 CrossRef.
- W. Liu and P. Wang, Biotechnol. Adv., 2007, 25, 369–384 CrossRef CAS PubMed.
- F. Hollmann, I. W. C. E. Arends and K. Buehler, ChemCatChem, 2010, 2, 762–782 CrossRef CAS.
- Sigma-Aldrich catalogue: L-pipecolic acid (P2519) 524 £ per g. L-lysine monohydrochloride (L5626) 23£ per 100 g.
- H. Schütte, J. Flossdorf, H. Sahm and M. –R. Kula, Eur. J. Biochem., 1976, 62, 151–160 CrossRef PubMed.
- C. Mateo, V. Grazú, B. C. C. Pessela, T. Montes, J. M. Palomo, R. Torres, F. López-Gallego, R. Fernández-Lafuente and J. M. Guisán, Biochem. Soc. Trans., 2008, 35, 1593–1601 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc01817a |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2020 |