
Efficient and selective catalytic hydrogenation of furanic aldehydes using well defined Ru and Ir pincer complexes†

Abstract
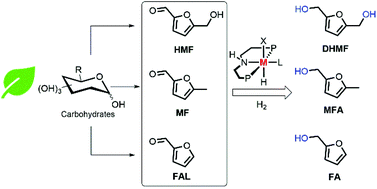
We report the homogeneous catalytic hydrogenation of biomass derived furanic aldehydes to furfuryl alcohols using low loadings of PNP metal complexes under mild conditions. Our strategy represents an efficient and selective approach to the direct hydrogenation of furan derivatives to promising platform chemicals.
- This article is part of the themed collection: 2020 Green Chemistry Hot Articles
 Please wait while we load your content...
Please wait while we load your content...

