
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Wet-expandable capsules made from partially modified cellulose†
Katarzyna
Mystek
 *a,
Hailong
Li
a,
Torbjörn
Pettersson
a,
Hugo
Françon
a,
Anna J.
Svagan
a,
Per A.
Larsson
a and
Lars
Wågberg
*ab
*a,
Hailong
Li
a,
Torbjörn
Pettersson
a,
Hugo
Françon
a,
Anna J.
Svagan
a,
Per A.
Larsson
a and
Lars
Wågberg
*ab
aDepartment of Fibre and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 56–58, 100 44 Stockholm, Sweden. E-mail: mystek@kth.se; wagberg@kth.se
bDepartment of Fibre and Polymer Technology, Wallenberg Wood Science Center, KTH Royal Institute of Technology, Teknikringen 56–58, 100 44 Stockholm, Sweden
First published on 22nd June 2020
Abstract
Preparation of lightweight and biocompatible hollow capsules holds great promise for various advanced engineering applications. Here, we use a heterogeneously modified structure of cellulose, which on the molecular level increases the flexibility of the capsule shell, to form hollow capsules. These capsules expand in the wet state when they are exposed to an external stimulus, in the present case a decreased external pressure. The capsules were prepared by a dropwise precipitation of a propane-saturated solution of cellulose partially modified to dialcohol cellulose, dissolved in a mixture of N,N-dimethylacetamide and lithium chloride, into a non-solvent. The mechanical properties of the capsules were determined by measuring the expansion of the capsules upon a controlled decrease in external pressure. In addition, indentation measurements using atomic force microscopy were used to independently quantify the moduli of the capsule walls. The results show that the wet, modified cellulose capsules are much softer and, upon the same pressure change, expand significantly more than those made from unmodified cellulose. The greatest expansion observed for the modified capsules was 1.9 times the original volume, which corresponds to a final density of the expanded capsules of about 14 kg m−3. These capsules therefore hold great potential to form green and lightweight foam-like materials.
Introduction
The development of new, innovative materials and technologies based on renewable and sustainable resources is now recognized as a major scientific and societal challenge. The world-wide availability of biopolymers, as well as an increased awareness of environmental consequences related to a high consumption of oil and oil-based materials over the last decades, has led to a greater emphasis on a new generation of green products that can meet the requirements of the modern society. One of the most readily available biopolymers is cellulose which, due to its chemical structure, possesses interesting properties, including excellent mechanical properties and good chemical stability under very harsh conditions. As already demonstrated, the use of cellulose-based materials in new or highly refined products has increased considerably in recent decades.1 By using cellulose in the form of fibers, nanofibrils, nanocrystals and even solutions, it is possible to create materials with unique and/or targeted mechanical and structural properties. Moreover, the evolution of techniques that allow a large array of chemical functionalizations and better material shaping facilitate the use of cellulose in, for example: textiles,2,3 packaging,4 biomedicine5,6 and electronic devices.7–10Dissolution of cellulose followed by its regeneration in a non-solvent represent a simple and interesting approach to design novel materials, such as films, membranes, filaments, hydrogels, aerogels, solid beads, and core–shell structured capsules.11–13 Traditionally, regenerated cellulose has been used in the form of transparent films, such as cellophane, or in the form of endless filaments made from the viscose process.14 The latter has, however, received increasing competition from new solvents such as N-methyl morpholine oxide in the Lyocell process.2 Chemical treatment of the native cellulose fibers, before or after their dissolution in a suitable solvent, allows efficient modulation of the properties of the final material. The product can be further functionalized to obtain advanced characteristics, such as increased hydrophobicity, antibacterial activity and flame retardancy.15,16
Materials made of regenerated cellulose generally display useful material properties, such as large specific surface area, good adsorption and absorption capacity, low density and high mechanical strength.17,18 Dissolution of cellulose in a mixture of N,N-dimethylacetamide (DMAc) and lithium chloride has led to the development of homogeneous materials with different shapes and to address hitherto unexplored applications. For example, solid cellulose beads, prepared using a solution solidification method, represent an interesting model system to investigate the degradation kinetics of amorphous cellulose.19 Since they have a nanometer-smooth surface, cellulose beads have also been used to study water–cellulose and cellulose–cellulose interactions.20–26 Similarly to the formation of beads, hollow capsules of cellulose can be formed if the cellulose solution is saturated with a suitable gas before its precipitation in the non-solvent.27,28 Carrick and coworkers27,28 showed that the porosity of the capsule walls, as well as their specific surface area and density could be easily controlled by adjusting the concentration of the cellulose solution, the type of dissolved gas and the nature of the non-solvent. The simplicity of capsule formation coupled with the ability to control their dimensions and the possibility for further functionalization make them suitable for different end-use applications such as the encapsulation of biologically active substances and dyes for drug delivery and coatings respectively.27 Furthermore, they are suitable for use as lightweight fillers in food packaging and board manufacturing.28
In this study, expandable capsules were prepared from chemically modified cellulose, showing higher ductility and allowing a greater capsule expansion when exposed to a decreased external pressure. The increased ability to expand of the capsule shells was attained by a partial conversion of the native cellulose in wood-derived fibers to dialcohol cellulose before dissolution in DMAc/LiCl. This type of chemical modification has been intensively studied, for example by Larsson et al.,29–32 and is based on the heterogeneous sodium periodate oxidation of native cellulose fibers followed by a sodium borohydride reduction. This partial conversion leads to changes in the flexibility of the modified glucan chains, the interaction between the fibril aggregates in the fiber wall and the structure of individual fibers. Hence, dialcohol cellulose acts as an internal plasticizer in the material. Consequently, the novelty of the present work is the formation of wet-expandable capsules by the regeneration of dissolved cellulose that has been partially converted to dialcohol cellulose. Upon a decrease in external pressure, these capsules expand without significant damage of the capsule wall. To the best of the author's knowledge, this is the first work that presents an innovative technique for quantifying the expansion of capsules of this type with high precision when the pressure around them is decreased. The low density of the capsules, their good mechanical properties, as well as their ability to expand and form 3D foam-like materials, makes them suitable for applications that require lightweight materials.
Experimental
Fibers
A dissolving grade pulp (Domsjö Dissolving Plus) was provided by Domsjö Fabriker AB, Sweden. The cellulose content was previously reported to be 93%, and the charge of the fibers was 29 μeq g−1 with an average degree of polymerization of the cellulose of 780.27 To remove any unwanted metal ions and to have carboxyl groups present in the fibers in their sodium form, the fibers were first washed under acidic conditions (pH 2) and then with 1 mM NaHCO3 under slightly alkaline conditions (pH 9), followed by excessive washing with deionized water.Chemicals
N,N-Dimethylacetamide (≥99.5%), lithium chloride (anhydrous ≥99%), sodium borohydride (≥96%), hydroxylamine hydrochloride (99%), sodium phosphate monobasic monohydrate (98%) were purchased from Sigma-Aldrich, Sweden. Sodium metaperiodate (99%), 2-propanol (99.9 vol%), ethanol (96 vol%), methanol (≥98.5 vol%) were purchased from VWR International AB, Sweden. Propane gas was purchased from AGA Gas AB, Sweden. The chemicals were used as delivered without further purification.Preparation of cellulose fibers partially converted to dialcohol cellulose
Modification of the fibers was performed using a previously published procedure.33 In short, an aqueous suspension of fibers, with a consistency of 20 g L−1, were reacted with 1.35 g of NaIO4 for each gram of fiber, in the presence of 2-propanol (6.3 vol%). The mixture was left in the dark for two hours at a temperature of 50 °C. The reaction was stopped by filtration and washing with water until the conductivity of the filtrate was lower than 5 μS cm−1. The fibers were then re-dispersed in water to a consistency of 8 g L−1, followed by reduction of the dialdehydes by adding 0.5 g NaBH4 per gram of fiber in the presence of 0.01 M NaH2PO4 to prevent a too high pH. After two hours, the reaction was stopped by filtration followed by washing with water until a filtrate conductivity of less than 5 μS cm−1 was achieved. The degree of cellulose oxidation was determined to be 22% by reacting the aldehyde groups formed with hydroxylamine hydrochloride to release a stoichiometric amount of protons that were then titrated with sodium hydroxide.34 Reactions were performed in triplicate.Preparation of cellulose solutions
Unmodified cellulose or dialcohol-modified cellulose fibers were dissolved in DMAc/LiCl after a solvent exchange to replace water with DMAc. First, 3 g of the water-swollen fibers were suspended in 96 vol% ethanol, with an exchange to fresh ethanol three times per day for two days. Then, the ethanol-swollen fibers were suspended in DMAc, filtered and re-suspended in DMAc four times over a period of two days. Before initiating the dissolution of the fibers, 195 mL of DMAc was pre-heated in an oil bath to 110 °C (for 30 min) and 14 g of LiCl, that had been dried in an oven at 105 °C, was then added to the DMAc and allowed to cool to 60 °C (a point where all the salt was dissolved). Finally, the DMAc-suspended cellulose fibers were added, to a final consistency of 1.5 wt%. To ensure a proper dissolution of the fibers, the dispersion was stirred overnight at room temperature.Preparation of capsules
Spherical capsules were formed using the solution solidification method established by Carrick et al.27 First, 20 mL of the dialcohol-modified cellulose solution was saturated with propane gas by flowing propane through the solution, which was placed in a sealed vessel, for one hour. Three different pressures of propane were used: 1.5, 2.5 and 4.0 bar and therefore the capsules formed in each will be denoted as DCC_1.5, DCC_2.5 and DCC_4.0 respectively. Then, the propane-saturated solution was added drop-wise to a non-solvent consisting of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 (by volume) water–methanol mixture, using stainless steel needles with an inner diameter of 1.2 mm; resulting in precipitation of gas-filled, millimeter-sized, spherical capsules. After the precipitation, approximately 70 capsules of each type were left in excess non-solvent for 24 hours to leach out the DMAc/LiCl, before placing them in fresh non-solvent to further remove residual DMAc/LiCl. This washing procedure, which was performed under gentle agitation on an orbital shaker, was repeated three times a day for two days, using 250 mL of fresh non-solvent in each exchange. A similar procedure was used to make native cellulose capsules (NCC_2.5); only for these capsules the propane pressure was set to 2.5 bar, the solution was diluted to 1 wt% with DMAc/LiCl, the non-solvent consisted of a 2:1 water–methanol mixture and the final washing was done in water.
:1.2 (by volume) water–methanol mixture, using stainless steel needles with an inner diameter of 1.2 mm; resulting in precipitation of gas-filled, millimeter-sized, spherical capsules. After the precipitation, approximately 70 capsules of each type were left in excess non-solvent for 24 hours to leach out the DMAc/LiCl, before placing them in fresh non-solvent to further remove residual DMAc/LiCl. This washing procedure, which was performed under gentle agitation on an orbital shaker, was repeated three times a day for two days, using 250 mL of fresh non-solvent in each exchange. A similar procedure was used to make native cellulose capsules (NCC_2.5); only for these capsules the propane pressure was set to 2.5 bar, the solution was diluted to 1 wt% with DMAc/LiCl, the non-solvent consisted of a 2:1 water–methanol mixture and the final washing was done in water.
Viscosity of solutions
The kinematic viscosity of native cellulose and dialcohol-modified cellulose solutions was determined using an iVisc capillary viscometer with a thermostat ET 15 S (LAUDA, Germany) for temperature control. The measurements were performed using an EGV 925 capillary at a temperature of 23 °C. Three measurements were performed for each cellulose solution.Determination of molecular mass of the unmodified and modified cellulose
The molar mass distributions (expressed as weight-average molar mass, Mw, and dispersity, Đ) were determined using size exclusion chromatography (SEC) after tricarbanilation of cellulose. Regenerated films of unmodified cellulose and dialcohol-modified cellulose were prepared by precipitating the cellulose solutions in a high-humidity environment, i.e. by allowing the solutions to absorb moisture. Two glass Petri dishes with 25 mL of cellulose and modified cellulose solution, respectively, were placed in a desiccator containing 450 mL of water–methanol (1:1.2) mixture. After one week of incubation at room temperature, the films were taken out and washed with the non-solvent mixture of water–methanol (1:1.2). This washing step, which was performed under gentle agitation on an orbital shaker, was done for one week changing the solution three times a day, using 250 mL of fresh non-solvent. After that, the cellulose in the films was converted to tricarbanilates, dissolved in tetrahydrofuran (approx 0.5 mg ml−1) and filtered (PTFE syringe filter 0.2 mm).35,36 The SEC system consisted of columns connected in series: a PLgel 10 μm Guard column (50 × 7.5 mm) followed by three PLgel 10 μm MIXED-B LS (300 × 7.5 mm) columns (Agilent, USA). The calibration of the system was conducted using polystyrene standards with known molecular weights ranging from 3000 to 7270000 g mol−1. For detection, a Waters 2414 refractive index (RI) detector (Waters, USA) and a Knauer Smartline 2500 UV detector (Knauer Wissenschaftliche Geräte GmbH, Germany) were used. The UV absorbance was determined at 280 nm. The molar mass distribution of the cellulose tricarbanilate was calculated using Cirrus GPC/SEC software v.3.1 (Varian Inc., USA). The integration limits used were the times for elution of the 3000 and 7270000 g mol−1 polystyrene standard, respectively. To get the molar weight of pure cellulose without the tricarbanilation, the values from the SEC measurement were divided by 3.2 (i.e. assuming full degree of substitution). These measurements were performed by RISE Bioeconomy, Stockholm, Sweden.
Determination of capsule dimensions
An optical microscope was used to measure the dimensions of the capsules in their wet and dry states. The dimensions obtained were then used, together with their weight, to calculate the density of the capsules and the density of capsule wall. The dimensions were assessed for at least 10 capsules and were measured by analysing the images in Image J. The weight of the wet capsules was measured on an analytical balance and the average values reported are based on weighing at least 10 capsules. Due to the low mass of the dry capsules, the same conventional analytical balance was not deemed accurate enough. Therefore, the dry mass was determined using a Mettler Toledo TGA/DSC 1 STARe system, the weight was recorded for 10 min at a temperature of 25–30 °C. Six capsules of each type were measured. Morphology and thickness of the wet cellulosic shell were studied using a Carl Zeiss Merlin Field-Emission Scanning Electron Microscope, equipped with a Quorum Technologies PP3000 T cryo-preparation system. The capsules were first frozen in liquid nitrogen for five minutes and cryo-fractured with a steel scalpel. The fractured capsules were then flash-frozen in liquid nitrogen and placed, without any intermediate thawing, in the main chamber of the SEM. Imaging was performed for at least two capsules of each type. The morphology and thickness of the dry cellulosic shell were studied with a Hitachi S-4800 field-emission scanning electron microscope operating at high vacuum. Prior to imaging, to eliminate charging effects of the samples, all samples were coated with Pt–Pd in a Cressington 208HR High Resolution Sputter Coater for 25 seconds (∼2 nm layer thickness). To characterize the thickness and macrostructure of the capsule wall, the capsules were frozen in liquid nitrogen and then crushed. The mean wall thickness was assessed by measuring 10 capsules, at three positions in each capsule.Characterization of the expansion of wet capsules by a reduced external pressure
The capsules expansion was studied by exposing the wet cellulose capsules to a decreased external pressure, using a custom-made experimental setup facilitating quantitative and reproducible experiments (Fig. 1). The equipment is composed of a large pre-chamber connected to a vacuum pump at one side and to a small sample chamber at the other side (in this case a glass Pasteur pipette that had been sealed by melting the narrow end with a flame). A pre-determined reduced pressure could be adjusted in the pre-chamber using a leak valve and quantified with a pressure sensor. This pressure was then exposed to the small chamber with the sample to be tested. To control the rate of the pressure decrease in the sample chamber, another leak valve, placed between the pre- and sample chambers, was used. A decrease in pressure in the sample chamber can be reached quickly since its volume is significantly (about 1000 times) smaller than the volume of the pre-chamber. A pressure difference (ΔP) used to describe the expansion experiments is calculated as a difference between the atmospheric pressure and the pressure set in the pre-chamber. The ideal pressure difference between the inside and outside of the capsule is naturally slightly less since the pre-chamber is not infinitely larger than the sample chamber. At the largest possible pressure difference in the system, i.e. 1 mbar in the pre-chamber and 1000 mbar in the sample chamber, the deviation was 22 mbar (a ΔP of 999 mbar would in fact be 977 mbar). The dimensions of the spheres were continuously monitored by using a high-speed camera (IDT N4M-S3), or a Dino-Lite Premier AM4113TL camera. A fresh capsule was used for each datum in the expansion-pressure difference plot points.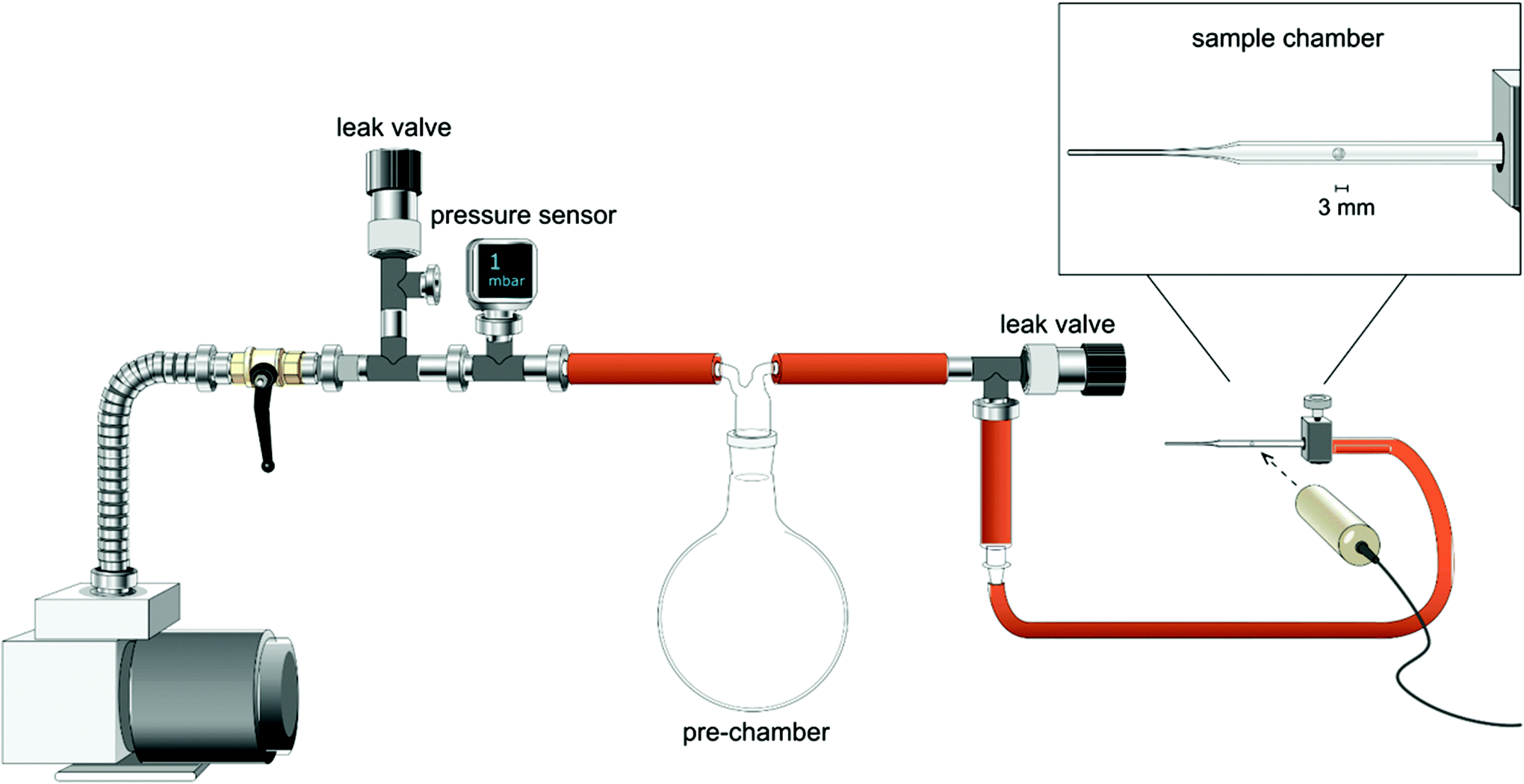 | ||
| Fig. 1 Schematic illustration of the custom-made experimental setup used to induce a reduced external pressure and simultaneously observe the resulting capsule expansion. By using a much larger volume of the pre-chamber than for the sample chamber, it is possible to change the pressure without losing pressure control, by opening/closing the valves between the pre-chamber and the sample chamber the rate of pressure change can also be controlled. | ||
Determination of the elastic modulus of the capsule wall by indentation
The elastic modulus of the capsule walls was determined by indentation experiments conducted with the aid of a MultiMode IIIa atomic force microscope (AFM) (Veeco Instrument, Santa Barbara, CA) with a PicoForce scanner. A spherical gold microparticle with a radius of 16.5 μm was used as the indentation probe.37,38 The particle was glued onto a tip-less cantilever that was calibrated for its normal spring constant using a method based on thermal fluctuation and hydrodynamic damping.39,40 To minimize capillary forces between the capsule and probe, both the probe and the cantilever were hydrophobized with mercaptododecane thiol (1 mM in ethanol for 12 h). The indentation measurements were then performed, at 23 °C and 50% RH, during the drying of the capsule. The measurements were limited to a maximum elastic modulus of about 50 MPa by the selection of spring constant of the cantilever (NSC12 tipless, MikroMasch, nominal length 110 μm). Force curves were recorded as the probe approached and retracted from the capsule at a speed of 6 μm s−1. The elastic modulus was calculated for the force range from 100 to 400 nN of the approach using a linearized Derjaguin–Muller–Toporov model, taking into account the adhesive contribution to the applied load during the indentation.23,25Results
Characterization of native and partially modified cellulose and their solutions
Native cellulose fibers were partially modified to dialcohol cellulose by performing a sodium periodate oxidation followed by a borohydride reduction. To quantify the degree of cellulose modification, the amount of aldehyde groups introduced after periodate oxidation was determined through a stoichiometric reaction with hydroxylamine hydrochloride. After 2 h of oxidation, the aldehyde content was determined to be 2.61 ± 0.03 mmol g−1 fibers, which equates to a degree of oxidation of 22% of all the cellulose in the fibers. Native cellulose and dialcohol-modified cellulose fibers were then dissolved in DMAc/LiCl to prepare solutions with the same concentration of 1.5 wt%. Kinematic viscosities of the solutions were determined and reported in Table 1. The results indicate that the chemical modification of the cellulose fibers causes a significant decrease in the kinematic viscosity of the solutions from 2265 mm2 s−1 to 25 mm2 s−1. The respective molecular mass of both types of cellulose was determined by SEC (with a refractive index detector) and the results show that the molecular mass changes from 259000 to 81000 mol g−1 for the native and modified cellulose samples, respectively. Moreover, as shown in Table 1, with the partial conversion of cellulose to dialcohol cellulose, a decrease in dispersity is observed. Chromatograms achieved for both of the samples are summarized in Fig. S1.†
| Properties | Native cellulose | Dialcohol-modified cellulose |
|---|---|---|
| Kinematic viscosity (mm2 s−1) | 2265 ± 1 | 25 ± 1 |
| M w (mol g−1) | 259000 |
81000 |
| M n (mol g−1) | 39000 |
23000 |
| Đ | 6.7 | 3.5 |
Formation and physical properties of capsules from modified cellulose
The dissolution of the modified cellulose fibers in DMAc/LiCl resulted in a transparent and homogeneous polymer solution, which could be used to form hollow capsules containing dialcohol-modified cellulose in the shell. The formation of hollow capsules was enabled by saturating the cellulosic solution with propane prior to precipitation. As the modified cellulose is regenerated in the non-solvent, where the solubility of propane is much lower, gas nucleation could be observed taking place inside the precipitating cellulose solution. As previously reported, a decreased polarity of the non-solvent results in lower encapsulation of propane in capsules from regenerated native cellulose.28 Consequently, the composition of the non-solvent was optimized to form perfectly spherical structures while maximizing the efficiency of gas encapsulation. From our investigations, it was found that a non-solvent consisting of a 1:1.2 water–methanol mixture resulted in spherical capsules with a minimal loss of propane gas.
Three separate solutions of dissolved dialcohol cellulose were saturated with propane at three different pressures (1.5, 2.5 and 4.0 bar) and precipitated by drop-wise addition into the optimized non-solvent (Fig. 2). During the precipitation, the applied gas pressure was shown to significantly influence the process of gas phase nucleation in the capsule (Fig. 2c). When, for example, a pressure of 1.5 bar was used, the gas bubble was formed one to two minutes after the solution precipitation. Whereas at pressures of 2.5 and 4.0 bar, the gas nucleation was much faster and the capsules were observed floating on the surface of the non-solvent a few seconds after contact with the non-solvent.
 | ||
| Fig. 2 Schematic image of the formation of capsules: (a) drop-wise precipitation of a propane saturated cellulose solution, (b) and (c) gas phase nucleation in the capsules, (d) hollow capsules floating on the surface of the non-solvent. | ||
Fig. 3 shows a photograph of the prepared dialcohol-modified cellulose capsules (each cuvette contains about 50 capsules). The figure clearly shows that more gas was encapsulated upon saturation at a higher pressure, i.e. when more gas is dissolved in the cellulose solution. The largest difference was observed when the pressure was increased from 1.5 to 2.5 bar. Capsules formed after saturation at a pressure of 4 bar (DCC_4.0) had similar dimensions to those saturated at 2.5 bar (DCC_2.5).
 | ||
| Fig. 3 Dialcohol-modified cellulose capsules prepared using propane pressures of (a) 1.5 bar, (b) 2.5 bar and (c) 4.0 bar. Each cuvette contains about 50 capsules. The scale bar corresponds to a length of 10 mm. | ||
Table 2 shows the dry and wet structural properties of the capsules made from modified cellulose. Wet capsules had external diameters ranging between 2.8 and 3.6 mm when prepared at different propane pressures, whereas the corresponding values after drying were 2.2 and 2.9 mm. In order to characterize the properties of the capsules, a precise determination of the thickness of the capsule wall is essential. For this purpose, cryo-fracturing and cryo-SEM were used, which gave sharp cuts, facilitating reliable thickness data for the wet capsule wall. The results show that there is a significant variation in wall thickness within a capsule (Fig. 4). However, there was, in line with the difference in size seen in Fig. 3, also a significant difference in the average wall thickness between DCC_1.5 and DCC_2.5, while the wall thickness of DCC_4.0 was similar to that of DCC_2.5. The calculated densities of the capsules in the dry state, as well as their standard deviations are based on an average weight of six capsules and at least 10 different measurements of the radius. As shown in Table 2, the density of the capsules decreased with increasing propane pressure and the lowest density (15 kg m−3) was obtained for DCC_4.0. The densities of the dry capsule walls were calculated to be in the range of 1038 to 1894 kg m−3. These large variations are due to the fact that the density calculations are very sensitive to the wall thickness determination, and since there is a considerable variation in the average capsule wall thickness (for each type of capsule), there will also be a large variation in the estimated density.
 | ||
| Fig. 4 Cryo-SEM images of the wet capsule wall of dialcohol-modified cellulose capsules prepared using three different saturation pressures: (a) and (b) 1.5 bar, (c) and (d) 2.5 bar, (e) and (f) 4.0 bar; the left and right columns display the thickest and thinnest parts of the capsule wall, respectively. Note that the scale of (a) and (b) is different from the other images. The protrusions observed on the surface of capsules are ice crystals formed during the specimen preparation. | ||
| Abbreviation | Weight (mg) | Diameter (mm) | Wall thickness (μm) | Capsule density (kg m−3) | g H2O per g cellulose |
|---|---|---|---|---|---|
| DCC_1.5wet | 6.15 ± 0.52 | 2.83 ± 0.10 | 200–310 | — | 26 ± 2 |
| DCC_1.5dry | 0.23 ± 0.01 | 2.25 ± 0.05 | 13 ± 4 | 39 ± 3 | — |
| DCC_2.5wet | 8.24 ± 0.92 | 3.52 ± 0.14 | 35–100 | — | 30 ± 3 |
| DCC_2.5dry | 0.27 ± 0.01 | 2.71 ± 0.11 | 6 ± 2 | 26 ± 3 | — |
| DCC_4.0wet | 5.40 ± 0.42 | 3.57 ± 0.13 | 35–100 | — | 26 ± 2 |
| DCC_4.0dry | 0.20 ± 0.02 | 2.88 ± 0.13 | 8 ± 2 | 15 ± 2 | — |
The structure and surface morphology of the dry capsule walls were imaged with SEM, and Fig. 5 shows images of a DCC_2.5. These images indicate a condensed and uniform wall structure within a capsule. Furthermore, the results show that the surface exhibits a homogeneous morphology (Fig. 5b). Similar observations were made for the capsules prepared from modified cellulose at saturation pressures of 1.5 and 4.0 bar.
 | ||
| Fig. 5 SEM images of the a dry DCC_2.5, where (a) shows the capsule, (b) surface morphology of the capsule shell, (c) the wall cross-section after freeze-fracturing, and (d) the wall cross-section at a higher magnification. | ||
Expansion of capsules under reduced external pressure
The expansion experiments were conducted in a custom-made equipment with high-precision pressure control (Fig. 1); using the 2.5 bar-propane-saturated unmodified cellulose and dialcohol-modified cellulose capsules. Since wet capsules from unmodified and modified cellulose have similar sizes and wall thicknesses (Table 2 and Fig. S2†), a fair comparison can be made between the two.For the first set of experiments, the capsules were expanded having the leak valve between the pre-chamber (where the pressure is set) and the sample chamber fully open, i.e. the rate of the pressure change in the sample chamber is “fast” (Fig. 6a). It was observed that a 50 mbar decrease in the sample chamber caused an increase in the capsule volumes of 10% for NCC_2.5 and about 30% for DCC_2.5. During the subsequent measurements, which were performed at incrementally higher pressure differences (ΔPs), no further expansion of the capsules was observed. Images taken during the experiments reveal that the capsules are slightly deformed in their initial state due to capillary forces created by the water meniscus between the wet capsule and the wall of the sample chamber (Fig. S3a†). When setting the rate of pressure change to “fast”, the vapor pressure (and hence the relative humidity) in the sample chamber is rapidly reduced (within less than five seconds) since the partial pressure in the pre-chamber is much lower than in the vapor saturated sample chamber. This drop in relative humidity decrease the size and radius of the water meniscus (Fig. S3b†), which consequently lowers the capillary forces acting on the capsules. With this decrease of the capillary force, the capsules go from a slightly deformed state to their non-deformed state. This transition from a slightly more ellipsoidal shape to a spherical shape is therefore the presumed reason for the initial expansion. This initially deformed state makes the evaluation of the expansion more complicated, but, at the same time, this phenomenon shows that the dialcohol-modified cellulose capsules are softer than the native cellulose capsules, being more deformed upon a similar capillary force. The actual pressure-induced expansion of the non-deformed capsules seems to occur at a pressure difference of 350 mbar and 500 mbar for DCC_2.5 and NCC_2.5, respectively. The data at higher pressure differences show that the dialcohol-modified cellulose capsules are softer and undergo greater expansion than those made of unmodified cellulose. DCC_2.5 underwent a volumetric expansion of 1.6 times the initial volume before failure, while NCC_2.5 expanded 1.2 times (these expansion values were determined after subtraction of the initial volume expansion discussed above). The capsules burst when the pressure in the sample chamber is decreased by more than 650 mbar and 900 mbar for the modified and unmodified capsules, respectively.
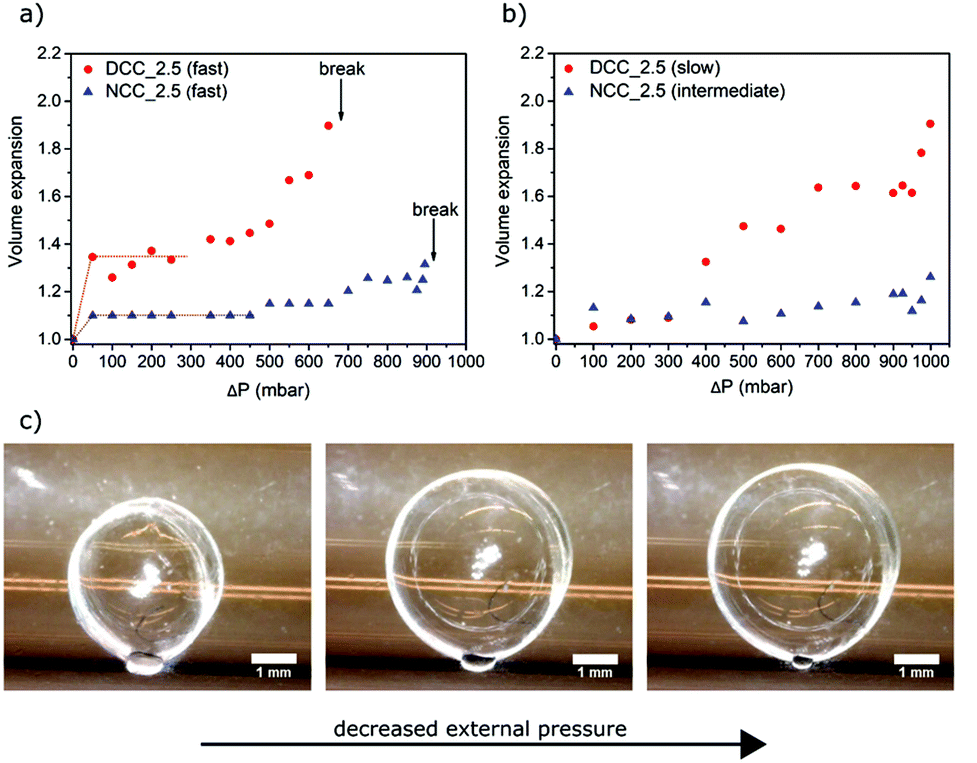 | ||
| Fig. 6 Wet expansion of the 2.5 bar propane saturated modified cellulose and the unmodified cellulose capsules (DCC_2.5 and NCC_2.5, respectively) at various pressure differences where the rate of pressure change in the main chamber is (a) “fast” for both samples, (b) “slow” for DCC_2.5 and “intermediate” for NCC_2.5. Each point represents one capsule. (c) Series of images of the expansion of a DCC_2.5 (“slow” rate) at approximate ΔPs of 0, 700 and 1000 mbar. | ||
The capsules were also tested at slower rate of pressure decrease by adjusting the setting of the leak valve placed between the pre-chamber and the sample chamber. These experiments allowed the comparison of the expansion of the capsules across pressure differences from 1 mbar to 999 mbar. It was also possible to study the influence of the expansion rate of the wet capsules on the pressure at which they failed mechanically. Fig. 6b shows that, by optimizing the rate of pressure change, it is possible to achieve even greater expansion of the modified capsules (expansion of 1.9 times the original volume) without rupturing the capsule wall. This indicates that the prepared capsules display a viscoelastic behavior within the range of strain-rates studied, as their strain-at-break clearly depends on the strain rate. With the leak valve in the “slow” position, the volume of DCC_2.5 increases gradually with ΔP. There is no observable initial expansion, suggesting that when a slower pressure decrease is used, the relative humidity in the sample chamber does not change as much upon opening the valve as when the faster rate in pressure change is used. Consequently, when the valve is opened, there is less or even no change in the size of the water meniscus formed between the wet capsule and the sample chamber (Fig. S4†). Moreover, no expansion at all is observed if the rate of pressure change is extremely slow (data not shown), this is a result of the capsule wall not being an ideal gas barrier in its wet state. The rate of the pressure change in the main chamber to generate the expansion data shown in Fig. 6b was hence optimized for both of the samples, explaining why unmodified cellulose and modified cellulose capsules were tested at “intermediate” and “slow” rates respectively. A more detailed assessment of the rates of the pressure decrease can be found in Fig. S5.†Fig. 6c shows a series of photos taken during the expansion of a wet DCC_2.5. This expansion was achieved by a “slow” decrease of the pressure surrounding the capsule to a ΔP of about 1000 mbar.
In order to determine the change in thickness of the capsule wall due to the expansion, the fully expanded DCC_2.5 were left to dry at the decreased pressure. They were then imploded by rapidly increasing the pressure in the sample chamber, dry cellulose is an excellent gas barrier,41 and the thickness of the wall could be determined by SEM. These measurements indicate that the drying in the expanded state resulted in a dry capsule wall thickness of 1.7 ± 0.1 μm compared to 6 ± 2 μm when dried in the non-expanded state.
The images also show that the rupture of the cellulosic shell formed rather rough failure surfaces, which can be observed as cavities in Fig. 7.
 | ||
| Fig. 7 SEM image of a dry wall cross-section of the expanded DCC_2.5. | ||
Elastic modulus of the wet capsule walls
The elastic modulus of the capsule wall was measured as a function of drying time (i.e. decreasing moisture content) by indentation experiments using colloidal-probe AFM (Fig. 8). The results indicate that DCC_1.5 and DCC_2.5 show no significant difference in elasticity of the shell during the first 15 minutes of their drying, suggesting that they have the same molecular structure and solid contents. In both cases, the elastic modulus was constant and lower than 0.4 MPa. After more than 15 minutes of drying, the values of the elastic modulus were seen to gradually increase; DCC_1.5 reached a modulus of 40 MPa in 24 minutes, whereas DCC_2.5 reached the same modulus in only 19 minutes. In contrast, the modulus of NCC_2.5 increased from the first minute of drying (Fig. 8b) and a value of 50 MPa, the limit of the method, was reached after six minutes. | ||
| Fig. 8 (a) Illustration of the AFM indentation experiments performed. Elastic modulus of the cellulosic shell of DCC_1.5, DCC_2.5 and NCC_2.5 as function of drying time for (b) short times and (c) long times. | ||
Discussion
Partial modification of cellulose to dialcohol cellulose and its effect on the properties of the DMAc/LiCl solution
Partial sodium metaperiodate oxidation of cellulose followed by reduction with sodium borohydride allows for the modulation of the hygro- and thermo-mechanical properties of the polymer, leading to a higher strainability and facilitation of a wide array of processing routes for the modified material.30,32,42 This type of cellulose modification has received an increased interest over the last years due its intriguing material properties, which also has spurred a significant progress in the development of methods of the periodate and borohydride regeneration, possibly facilitating an economical and environment-friendly scale-up of the process.43–45 Films and sheets made from partially modified cellulose fibers and nanofibrils exhibit increased flexibility. In this case it is proposed that an amorphous shell of dialcohol cellulose is formed around a crystalline cellulose core.29 Additionally, the extensibility of the dialcohol materials can be further enhanced by increases in temperature and moisture. As an example, it was shown that by increasing the relative humidity from 50% to 70–80%, i.e. increasing the moisture content of the modified sheet to 12–16%, or increasing the temperature from 23 °C to about 115 °C, the extensibility of dialcohol cellulose (with a degree of modification of about 40%) can be increased from 40% to 70–80%.32It is well established that dissolution of cellulose fibers and a further regeneration of the non-ordered solution will significantly influence the properties of the final material.46,47 However, to the best of our knowledge, no studies related to dissolution and regeneration of cellulose partly converted to dialcohol cellulose have been reported. It was therefore necessary to perform a detailed analysis of how the modification affected the properties of solutions of the modified cellulose. Table 1 presents significant differences in the kinematic viscosity when native and partially modified dialcohol cellulose fibers are dissolved in DMAc/LiCl; showing that they have viscosities of 2265 mm2 s−1 and 25 mm2 s−1, respectively. This difference can be attributed to a decreased molecular mass and/or an enhanced mobility of the polymeric chains after the reaction. Since the viscosity after modification is lowered by a factor of almost 100, a contribution from both of the factors is most probable. In order to evaluate this, the molecular mass of the unmodified and modified cellulose was measured. The results shown in Table 1 demonstrate that, upon modification, the molecular mass decreased by a factor of three, which is equal to an average of two scissions per polymer chain. This suggests that the main contributing factor to the lower viscosity is not a reduced molecular weight but an increased molecular mobility. A previous study of the viscosity-molecular weight relationship for cellulose indicates that a decrease in Mw by a factor of three cannot result in as significant a reduction in viscosity as was observed here (a 100 fold decrease).48 In addition, the molecular mass distribution showed lower polydispersity (Fig. S1†).
Hiraoki et al.49 observed a similar phenomenon after TEMPO-mediated oxidation of cellulose fibers, where the alkali treatment resulted in the formation of a more homogeneous population of cellulose chains. In the aforementioned study the reported a decrease of Mw and Mn values from 382000 g mol−1 and 74200 g mol−1 to 148000 g mol−1 and 60500 g mol−1, respectively.
Formation of hollow capsules of modified cellulose
Capsules were made from a propane-saturated solution of cellulose partially modified to dialcohol cellulose dissolved in DMAc/LiCl. As mentioned earlier, it was shown by Carrick et al.27 that the choice of gas dictates the size of the capsule and the morphology of the capsule wall. Propane is highly soluble in DMAc/LiCl while being poorly soluble in water and water–methanol mixtures. This enhances the amount of gas that can be nucleated and contained inside the cellulose droplet during the regeneration process and thereby facilitates the formation of a dense capsule wall. In the present investigation, a 22% conversion of cellulose to dialcohol cellulose was obtained. It is shown that the developed protocol of the capsules formation has a reasonable flexibility, and by tuning the non-solvent composition can be adapted to the cellulose solution with a lower molecular weight and viscosity. The 22% conversion rate in fibers is known to provide dry materials with both high strainability and good overall mechanical properties.30 A higher level of modification would presumably increase the deformability of the capsules used in this study,42 but would also be likely to reduce the viscosity of the dialcohol cellulose solution even further. This would complicate the formation of the hollow capsules since the non-solvent used for cellulose regeneration would need to have a lower surface tension than the current mixture. However, a lower surface tension of the non-solvent is associated with a higher solubility of propane gas and therefore a lower tendency for gas encapsulation.28 Furthermore, since pure dialcohol cellulose is soluble in water, the formation of propane-filled capsules made from highly converted cellulose would be challenging in water-containing non-solvents.50Using a blend of native and dialcohol cellulose (since a heterogeneous fiber modification is performed), our results indicate that the flexible dialcohol cellulose chains are trapped in-between the native cellulose molecules during the precipitation of the capsule shell. By this method, the modified cellulose is able to provide deformability to the formed shell, while the native cellulose prevents the dialcohol cellulose molecules from dissolution during the fast regeneration process. The degree of cellulose modification in the present investigation and the selected precipitating solvent mixture was found to be ideal for precipitating the modified cellulose capsules (improper combinations do not result in spherical capsules). A full theoretical description of the balance between degree of cellulose modification and composition of the precipitating solvent for maximum yield during the precipitation is currently under investigation. The volume of gas encapsulated in the capsules was altered by changing the saturation pressure of propane; three different pressures were used: 1.5, 2.5 and 4.0 bar. The properties of the capsules presented in Table 2 show that there is a significant difference in the dimensions and wall thicknesses of the 1.5 bar- and 2.5 bar-saturated capsules. In contrast, the properties of the 2.5 bar- and 4.0 bar-saturated capsules are quite similar. However, the lower density of DCC_4.0 indicates that there is an increase in the volume of gas ultimately confined inside these capsules, which could not be observed when measuring their size using optical microscopy. As also shown in Fig. 4, the wall thickness varies within a wet capsule for all of the samples. It was an expected phenomenon, since the buoyancy of the formed capsules makes it difficult to fully control the gas nucleation in the capsules. This variation in thickness can naturally have implications on the mechanical properties of the capsule. Thinner regions are not only more sensitive to defects, but will also have to carry a greater stress, and the risk of capsule failure would increase.
The utilization of a water–methanol mixture as the non-solvent facilitates formation of capsules with an efficient encapsulation of propane. Furthermore, it has been shown that water–methanol mixtures are environmentally favorable compared to pure alcohol or highly elaborate solvents (e.g. tetrahydrofuran, cyclohexanone). The higher the water content, the lower the environmental impact of the mixture. However, at the same time, methanol has a lower boiling point and a lower heat of evaporation than water, and consequently less energy is consumed for its evaporation.51
Expansion and mechanical properties of the capsules
The ability to regenerate cellulose partly converted to dialcohol cellulose and to successfully form capsules from it, suggests that the dialcohol cellulose was effectively incorporated into the capsule walls. The ability to do so is attributed to molecular entanglement and the poor solubility of dialcohol cellulose in the water–methanol mixture. Retention of flexible dialcohol cellulose would in turn mean that the capsule wall is softened and more strainable in the wet state, which is a prerequisite for the formation of expandable capsules. Furthermore, to ultimately make capsules that are permanently expanded after removing the expanding force, the material should also have a low yield strain in relation to the total deformation. AFM indentation measurements were used to probe the softness of the capsules. The experiments were conducted for the wet dialcohol-modified cellulose (DCC_1.5 and DCC_2.5) and unmodified cellulose (NCC_2.5) capsules. For fully wet DCC_1.5 and DCC_2.5, an elastic modulus of 0.01 MPa was measured, while for wet NCC_2.5 a corresponding value of 0.23 MPa was obtained (Fig. 8). The modified capsule wall is more than one order of magnitude softer than the unmodified and little force is needed to achieve plastic deformation. The softening of the modified capsule wall can be attributed to a lower modulus of the wet cellulosic wall after partial modification, but also to a higher water-holding capacity of dialcohol-modified cellulose. According to the results from Table 2, DCC_1.5 and DCC_2.5 exhibited a water content of 26 and 30 g g−1 cellulose, respectively, whereas a water content of 14 g g−1 cellulose was calculated for NCC_2.5. This indicates that a lower dry content is contributing to the lower observed modulus. For drying times greater than three minutes (if a “slow” capsule expansion rate is used) the difference in the elastic modulus of the native and dialcohol-modified cellulose walls becomes more pronounced. This is attributed to the increasing stiffness of the unmodified cellulosic wall during drying, i.e. removal of plasticizing water. A smaller and thicker-walled capsule, such as DCC_1.5, is expected to dry slower than a larger, thin-walled capsule of the same mass. Since the non-solvent (water–methanol mixture) acts as a plasticizer, the wall exhibits a lower elastic modulus for a relatively longer time. It is also observed that the elastic modulus of the wall of DCC_1.5, which is lower than DCC_2.5, starts to rapidly increase earlier in the drying process, indicating that the rate of the shell drying affects the elastic modulus of the capsules. The elastic modulus calculated for the native cellulose wall (0.23 MPa) is higher than that determined by Carrick et al. for wet capsules made from CO2-saturated native cellulose solutions (0.09 MPa); or by Karlsson et al. for wet native cellulose beads (0.04 MPa).23,27 This difference can be due to use of different characterization techniques and most likely due to changes in the preparation conditions (e.g. type of gas and non-solvent).With a low elastic modulus of 0.01 MPa, the dialcohol-modified capsules would easily expand beyond the yield stress of the material, for dry dialcohol cellulose materials observed to be less than 2%,31 when submitted to a significant pressure difference across the capsule wall. Fig. 6a shows that modified cellulose capsules exhibit significantly higher strainability than those made of native cellulose. Under the same pressure difference (ΔP of 650 mbar), DCC_2.5 expand to 1.6 times their original volume, while NCC_2.5 only undergo a 1.1 fold expansion. At pressure differences larger than 650 mbar, a mechanical failure of the dialcohol-modified capsules is observed. Since less expansion and no failure was observed in the native cellulose capsules, this shows that the partial conversion of native cellulose to dialcohol cellulose resulted in a material that is more easily deformed. Furthermore, the wet native cellulose capsules ruptured when a pressure difference of 900 mbar was reached, with a maximum volume expansion of no more than 1.2 times the original volume. The deformations observed are likely to occur in the plastic deformation regime (further supported by wrinkling of the capsules upon cycling of the external pressure; Fig. S6†). The expansion data shown in Fig. 6a can therefore be used to estimate the strain-hardening modulus of the wet cellulosic shells, considering the expansions when changing ΔP from 350 to 650 mbar and from 500 to 900 mbar for DCC_2.5 and NCC_2.5, respectively. For this purpose, the wet capsules were modelled as thin-walled spheres, which undergo linear expansion as the surrounding pressure is decreased. Using the constitutive equations of isotropic linear deformation, one can derive a relationship between the radial expansion of the capsule and the pressure difference across the capsule shell.52 This can be used to describe a linear deformation of the wet capsule wall (see ESI†). By doing this, the NCC_2.5 samples displayed a (strain-hardening) modulus of 9.7 MPa whereas the shell of DCC_2.5 was found to be softer, having a modulus of 3.2 MPa.
The viscoelastic properties of the capsules will also lead to different expansion responses of the capsules at different rates of pressure-change. This can be observed in Fig. 6a and b, as the volume expansion curves exhibit different trends depending on the rate of the pressure change in the sample chamber, i.e. “fast” or “slow” (described in Fig. S5†). With a “slow” rate, a volume expansion of 1.9 times was achieved for DCC_2.5 at a pressure difference of almost 1000 mbar, without observing any mechanical failure of the capsule. When the “fast” rate of the pressure change was used for the same type of capsule; the expansion took place so rapidly (within seconds) that the cellulosic shell responded in a less viscous and more brittle manner to the stimulus, causing a mechanical failure of the capsules. By using optimized expansion conditions, modified cellulose capsules can expand to nearly double their original volume. Since the expansion represents a plastic deformation of the capsule wall, capsules do no shrink back to their initial size after drying, which would result in the density of dry modified capsules being reduced from 26 kg m−3 to 14 kg m−3. The expansion of DCC_4.0 was not tested, however, due to their dimensions being similar to DCC_2.5 but their densities being lower (15 kg m−3) in the unexpanded state (Table 2), it is reasonable to assume that the final density of the expanded dialcohol-modified cellulose capsules can be decreased to 8 kg m−3. This is comparable to the lightest cellulosic structures reported, such as highly porous aerogels made from cellulose nanofibers.53 Furthermore, if the capsules are packed together, an ultra-lightweight (density of about 5 kg m−3) foam-like material could be formed.54
Besides the low density, dry modified capsules exhibit high stability under ambient conditions, which means that they probably can be stored for years without any structural change. Additionally, due to their good mechanical properties and their low gas permeability, as well as high transparency, the capsules can potentially be used as sustainable lightweight fillers in packaging applications or be used in shock-absorbent materials. The low gas diffusivity of the dry capsule wall needs, however, to be considered when removing the decreased external pressure, as a rapid pressure change can cause a collapse of the capsule due to a significant pressure difference across the capsule wall.
Conclusions
Millimeter-sized hollow cellulosic capsules with unique and well-defined properties were prepared by the dropwise regeneration of a solution of cellulose partially converted to dialcohol cellulose. This solution contained propane gas dissolved at different pressures and made use of a specially designed non-solvent to control the capsule size and wall properties. The degree of cellulose conversion to dialcohol cellulose was 22%, which resulted in capsule walls displaying higher water-holding capacity, as well as increased softness and flexibility in comparison to capsules made with unmodified cellulose. The amount of gas encapsulated was controlled by altering the saturation pressure of propane, where the most significant difference in the dimensions and wall thicknesses of the capsules was observed when the saturation pressure was increased from 1.5 to 2.5 bar. AFM indentation measurements showed that the wet shells of 2.5 bar-saturated dialcohol-modified capsules have elastic moduli of 0.01 MPa, while unmodified cellulose capsules had the moduli of 0.23 MPa in the wet state. Expansion experiments were conducted by exposing the wet capsules to a decreased external pressure using an equipment designed and built in-house. These experiments highlighted the importance of the rate of the pressure change on the expansion process, as capsules displayed a larger expansion at a slower rate (with a maximum volume expansion of 1.9 times for capsules containing dialcohol-modified cellulose). Using the linear data from volume expansion measurements, strain-hardening moduli were calculated to be 3.2 MPa and 9.7 MPa for the modified and unmodified cellulose capsules, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Formas Swedish Research Council for Environment, Agriculture and Spacial Planning (942-2016-12) and Nouryon AB are gratefully acknowledged. Dr Cheng Choo Lee, Umeå Core Facility for Electron Microscopy (UCEM) and National Microscopy Infrastructure, NMI (VR-RFI 2016-00968) are acknowledged for providing the Cryo-SEM images. The authors would like to thank Dr Johan Erlandsson for essential discussions on the expansion experiments, as well as Dr Andrew Marais for his help with the photographs. Lars Wågberg also acknowledges the Knut and Alice Wallenberg Research Foundation for financial support through the Wallenberg Wood Science Centre (WWSC). The authors would like to thank “Miscible Text” for proof reading the language of the manuscript.Notes and references
- X. Qiu, S. Hu, X. Qiu and S. Hu, Materials, 2013, 6, 738–781 CrossRef CAS PubMed.
- C. Woodings, Regenerated cellulose fibres, Woodhead Publishing Ltd, 2001 Search PubMed.
- S. B. Stanković, D. Popović and G. B. Poparić, Polym. Test., 2008, 27, 41–48 CrossRef.
- G. Rodionova, M. Lenes, Ø. Eriksen and Ø. Gregersen, Cellulose, 2011, 18, 127–134 CrossRef CAS.
- G. Helenius, H. Bäckdahl, A. Bodin, U. Nannmark, P. Gatenholm and B. Risberg, J. Biomed. Mater. Res., Part A, 2006, 76A, 431–438 CrossRef CAS PubMed.
- R. Kolakovic, L. Peltonen, A. Laukkanen, J. Hirvonen and T. Laaksonen, Eur. J. Pharm. Biopharm., 2012, 82, 308–315 CrossRef CAS PubMed.
- G. Nyström, A. Marais, E. Karabulut, L. Wågberg, Y. Cui and M. M. Hamedi, Nat. Commun., 2015, 6, 7259 CrossRef PubMed.
- M. Hamedi, E. Karabulut, A. Marais, A. Herland, G. Nyström and L. Wågberg, Angew. Chem., Int. Ed., 2013, 52, 12038–12042 CrossRef CAS PubMed.
- J. Erlandsson, V. López Durán, H. Granberg, M. Sandberg, P. A. Larsson and L. Wågberg, Appl. Mater. Today, 2016, 5, 246–254 CrossRef.
- H. Françon, Z. Wang, A. Marais, K. Mystek, A. Piper, H. Granberg, A. Malti, P. Gatenholm, P. A. Larsson and L. Wågberg, Adv. Funct. Mater., 2020, 30, 1909383 CrossRef.
- J. Zhang, J. Wu, J. Yu, X. Zhang, J. He and J. Zhang, Mater. Chem. Front., 2017, 1, 1273–1290 RSC.
- S. Wang, A. Lu and L. Zhang, Prog. Polym. Sci., 2016, 53, 169–206 CrossRef CAS.
- C. Carrick, P. A. Larsson, H. Brismar, C. Aidun and L. Wågberg, RSC Adv., 2014, 4, 19061–19067 RSC.
- H. A. Krässig, Cellulose-Structure, Accessibility and Reactivity, Gordon and Breach Science Publishers, 1996 Search PubMed.
- C. Wan, Y. Lu, Y. Jiao, C. Jin, Q. Sun and J. Li, Carbohydr. Polym., 2015, 118, 115–118 CrossRef CAS PubMed.
- S. W. Chook, C. H. Chia, S. Zakaria, M. K. Ayob, N. M. Huang, H. M. Neoh, M. He, L. Zhang and R. Jamal, Cellulose, 2014, 21, 4261–4270 CrossRef CAS.
- R. Wu, P. Huang and B. He, BioResources, 2016, 11(2), 4146–4158 CAS.
- C. Wan, Y. Jiao, S. Wei, L. Zhang, Y. Wu and J. Li, Chem. Eng. J., 2019, 359, 459–475 CrossRef CAS.
- B. Pettersson and K.-E. L. Eriksson, Anal. Biochem., 2000, 285, 220–224 CrossRef CAS PubMed.
- C. Carrick, S. A. Pendergraph and L. Wågberg, ACS Appl. Mater. Interfaces, 2014, 6, 20928–20935 CrossRef CAS PubMed.
- C. Carrick, L. Wågberg and P. A. Larsson, ACS Macro Lett., 2014, 3, 1117–1120 CrossRef CAS.
- R. M. P. Karlsson, P. T. Larsson, P. Hansson and L. Wågberg, Biomacromolecules, 2019, 20, 1603–1612 CrossRef CAS PubMed.
- R. M. P. Karlsson, P. T. Larsson, S. Yu, S. A. Pendergraph, T. Pettersson, J. Hellwig and L. Wågberg, J. Colloid Interface Sci., 2018, 519, 119–129 CrossRef CAS PubMed.
- A. Träger, G. Klein, C. Carrick, T. Pettersson, M. Johansson, L. Wågberg, S. A. Pendergraph and A. Carlmark, Cellulose, 2019, 26, 1467–1477 CrossRef.
- J. Hellwig, R. M. P. Karlsson, L. Wågberg and T. Pettersson, Anal. Methods, 2017, 9, 4019–4022 RSC.
- K. Mystek, M. S. Reid, P. A. Larsson and L. Wågberg, Ind. Eng. Chem. Res., 2020, 59, 2968–2976 CrossRef CAS.
- C. Carrick, M. Ruda, B. Pettersson, P. T. Larsson and L. Wågberg, RSC Adv., 2013, 3, 2462–2469 RSC.
- C. Carrick, S. B. Lindström, P. T. Larsson and L. Wågberg, Langmuir, 2014, 30, 7635–7644 CrossRef CAS PubMed.
- P. A. Larsson, L. A. Berglund and L. Wågberg, Biomacromolecules, 2014, 15, 2218–2223 CrossRef CAS PubMed.
- P. A. Larsson, L. A. Berglund and L. Wågberg, Cellulose, 2014, 21, 323–333 CrossRef CAS.
- P. A. Larsson and L. Wågberg, Green Chem., 2016, 18, 3324–3333 RSC.
- E. Linvill, P. A. Larsson and S. Östlund, Mater. Des., 2017, 128, 231–240 CrossRef CAS.
- V. López Durán, P. A. Larsson and L. Wågberg, Carbohydr. Polym., 2018, 182, 1–7 CrossRef PubMed.
- P. A. Larsson, M. Gimåker and L. Wågberg, Cellulose, 2008, 15, 837–847 CrossRef CAS.
- M. Kihlman, F. Aldaeus, F. Chedid and U. Germgård, Holzforschung, 2012, 66, 601–606 CAS.
- U. Drechsler, S. Radosta and W. Vorwerg, Macromol. Chem. Phys., 2000, 201, 2023–2030 CrossRef CAS.
- H. Li, K. Mystek, L. Wagberg and T. Pettersson, Soft Matter 10.1039/D0SM00866D.
- W. A. Ducker, T. J. Senden and R. M. Pashley, Nature, 1991, 353, 239–241 CrossRef CAS.
- J. E. Sader, J. W. M. Chon and P. Mulvaney, Rev. Sci. Instrum., 1999, 70, 3967–3969 CrossRef CAS.
- T. Pettersson, N. Nordgren, M. W. Rutland and A. Feiler, Rev. Sci. Instrum., 2007, 78, 093702 CrossRef PubMed.
- Q. Yang, H. Fukuzumi, T. Saito, A. Isogai and L. Zhang, Biomacromolecules, 2011, 12, 2766–2771 CrossRef CAS PubMed.
- W. Kasai, T. Morooka and M. Ek, Cellulose, 2014, 21, 769–776 CrossRef CAS.
- S. Koprivica, M. Siller, T. Hosoya, W. Roggenstein, T. Rosenau and A. Potthast, ChemSusChem, 2016, 9, 825–833 CrossRef CAS PubMed.
- H. Liimatainen, J. Sirviö, H. Pajari, O. Hormi and J. Niinimäki, J. Wood Chem. Technol., 2013, 33, 258–266 CrossRef CAS.
- T. Kemmitt and G. J. Gainsford, Int. J. Hydrogen Energy, 2009, 34, 5726–5731 CrossRef CAS.
- B. Medronho and B. Lindman, Adv. Colloid Interface Sci., 2015, 222, 502–508 CrossRef CAS PubMed.
- A. Abbott and A. Bismarck, Cellulose, 2010, 17, 779–791 CrossRef CAS.
- C. L. McCormick, P. A. Callais and B. H. Hutchinson, Macromolecules, 1985, 18, 2394–2401 CrossRef CAS.
- R. Hiraoki, Y. Ono, T. Saito and A. Isogai, Biomacromolecules, 2015, 16, 675–681 CrossRef CAS PubMed.
- T. J. Painter, Carbohydr. Res., 1988, 179, 259–268 CrossRef CAS.
- C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–993 RSC.
- P. Kelly, Solid mechanics part I. I: An Introduction to Solid Mechanics, 2014 Search PubMed.
- P. Gupta, B. Singh, A. K. Agrawal and P. K. Maji, Mater. Des., 2018, 158, 224–236 CrossRef CAS.
- H. M. Jaeger and S. R. Nagel, Science, 1992, 255, 1523–1531 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEC chromatograms for regenerated native cellulose and dialcohol cellulose solutions determined by using RI and UV detection, Cryo-SEM images of the wet shell of NCC_2.5, images of dialcohol-modified cellulose capsules tested using “fast” and “slow” rates of pressure change, curves describing the decrease in ambient pressure in the sample chamber over time at different positions of the leak valve, images showing the behavior of the modified capsule when the external pressure is cycled, and calculations of the moduli of the capsules’ shell. See DOI: 10.1039/d0gc01523g |
| This journal is © The Royal Society of Chemistry 2020 |