
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
p-Xylene from 2,5-dimethylfuran and acrylic acid using zeolite in a continuous flow system†
Jose Alirio
Mendoza Mesa
 ab,
Francesco
Brandi
a,
Irina
Shekova
a,
Markus
Antonietti
a and
Majd
Al-Naji
*a
ab,
Francesco
Brandi
a,
Irina
Shekova
a,
Markus
Antonietti
a and
Majd
Al-Naji
*a
aMax Planck Institute of Colloids and Interfaces, Department of Colloid Chemistry, 14476 Potsdam, Germany. E-mail: majd.al-naji@mpikg.mpg.de
bLaboratorio de Catálisis Heterogénea, Universidad Nacional de Colombia, 11001 Bogotá, Colombia
First published on 4th September 2020
Abstract
The continuous flow synthesis of p-xylene (pXL) via Diels–Alder cycloaddition of lignocellulosic biomass-derivable 2,5-dimethylfuran (DMF) and acrylic acid (AA) was performed over different type of zeolites, i.e. Beta, ZSM-5 and Y. Among the tested zeolites, Beta zeolite showed an optimum catalytic performance in pXL synthesis from DMF and AA. In this context, Beta zeolite with a Si/Al molar ratio of 150 which is abbreviated as Beta(150), resulted in complete DMF conversion with a pXL yield of 83% and by-product 2,5-dimethylbenzoic acid (DMBA) with a yield of 17%, at 473 K in 10.1 min residence time (τ), with excess AA (0.7 M). This high catalytic activity is attributed to the high specific surface area of 1180 m2 g−1 with a three-dimensional porous architecture with a pore diameter of 6.6 × 6.7 Å and an acid site density above 40 μmol g−1. The utilized Beta(150) showed a very stable performance up to 10 h time on stream with minor deactivation after 8 h of TOS, while the pXL yield remained above 70%. The original catalytic performance of Beta(150) in the conversion of DMF to pXL was restored by applying a regeneration step for the spent catalyst, which is simple in continuous flow reactors. Finally, this sustainable continuous flow process enables an efficient and selective pXL production from DMF and AA as a dienophile at lower reaction temperature (473 K) and shorter residence time (τ = 10.1 min) in comparison to a batch fashion.
Introduction
From the beginning of industrialization, our society has developed a heavy dependence on coal and fossil oils as a source of energy and chemicals.1–7 This has led to the well-known environmental side effects that we are facing today. In order to decrease the dependency on fossil resources, alternative methodologies to produce fine chemicals from renewable resources, e.g. lignocellulosic biomass, with low environmental impact are greatly needed.1–11 In this regard, a large number of publications have described the utilization of waste lignocellulosic biomass-derived compounds for the synthesis of fine chemicals, e.g. sugars, 5-hydroxymethylfurfural (HMF), 2,5-dimethylfuran (DMF) and levulinic acid (LA).12–19 Among these is DMF which is produced simply by the catalytic hydrogenation of HMF.1,15 DMF is not only a high-quality biofuel, but can be used as a precursor for the synthesis of a high value platform chemical, e.g. p-xylene (pXL).1,15,20–24In 2015, around 37 million metric tons of pXL were consumed mostly for the production of poly(ethylene terephthalate) (PET) and polyester fiber.23–26 There are reported studies on the synthesis of pXL from biomass-derived DMF, which include a Diels–Alder cycloaddition of ethylene to form the intermediate 1,4 dimethyl-7-oxabicyclo[2.2.1]hept-2-ene, followed by a dehydration step to pXL using different types of zeolites as catalysts.23–29 Usually, for this reaction batch systems are used, combined with high reaction temperatures (523 K–623 K), high ethylene pressures (5.0 MPa–7.0 MPa) and long reaction times (24 h–48 h).30 All these are less appealing in terms of green chemistry. Williams et al.30 have reported a 75% yield of pXL as a result of cycloaddition of ethylene to DMF using H–Y zeolite at 573 K. Moreover, diverse side products from a competitive reaction on H–Y zeolite were found under the reported reaction conditions, i.e. 2,5-hexanedione, pXL alkylation products and polycondensates of 2,5-hexanedione.30,31 H-Beta zeolite with a Si/Al ratio of 12.5 showed higher selectivity toward pXL compared to H-FAU and H-ZSM-5 at 523 K and 6.2 MPa of ethylene.30,32 This high performance of H-Beta is attributed to its resistance to deactivation and its ability to selectively catalyze the dehydration of Diels–Alder cycloaddition intermediates to pXL.30,32,33 Phosphorus modification of Beta zeolite led to an increase in the pXL yield from 75% to 97% at 523 K and 6.2 kPa of ethylene in the batch system.33 Similarly, Zr-, Sn- and Ti-Beta zeolites with Lewis acid sites were found to be highly active and selective for the conversion of DMF to pXL in the presence of ethylene (6.2 MPa) at 523 K.31,34 Of these, Zr-Beta exhibited the highest catalytic performance (DMF conversion = 99% and pXL selectivity = 90%).34 Also, heteropolyacids and sulfonic acids supported on SiO2 showed a high DMF conversion (94%) and pXL selectivity (85%), here due to enhanced Brønsted acid sites at 523 K, 2.0 MPa and 6 h of reaction time.35,36 From the above investigations, high acid site density combined with high surface area seems to be essential for catalysts used in the Diels–Alder cycloaddition of DMF/ethylene followed by dehydration of the intermediate product. Alternatively, several trials were made for the synthesis of aromatics by Diels–Alder cycloaddition using furans and ethanol (as in situ ethylene source) over zeolite as catalysts in a batch system.36–39 However, this approach requires high reaction temperatures to dehydrate ethanol to ethylene and leads to high amounts of by-products. An alternative route for the sustainable production of aromatics from furanics was proposed by replacing ethylene with different dienophiles such as acrylic acid (AA) or maleic anhydride (MA).26,40–42 In this green and safe route, the reaction can be performed in the liquid phase at lower reaction temperatures (423 K–473 K) and without external gas pressure. Furthermore, competing reactions that lead to the wide range of by-products are suppressed, and the selectivity towards the targeted aromatics is increased. However, the synthesis of pXL utilizing this route was rarely described, as it comes with an additional elimination step of the intermidates to improve the pXL yield. However, this additional step can be avoided by performing this in situ cascade reactions in continous flow system. As a move in this direction, the two step batch synthesis of pXL from DMF and AA catalyzed by scandium(III) triflate (Sc(OTf)3) in ionic liquids under mild reaction conditions was reported.43 It was found that 48% of pXL and 22% of 2,5-dimethylbenzoic acid (DMBA) are formed in the 1st step. The consecutive 2nd step includes the decarboxylation of the pXL and DMBA mixture which improves the yield of pXL to 57%.43 Similarly, the reaction between DMF and AA at room temperature was performed by using acidic ionic liquid ([Bmim]H2SO4) in a batch system.28 After the 1st step at room temperature, a pXL yield of 45 mol% was achieved. A decarboxylation step at 473 K was again added to improve the yield of pXL.28 From these multistep and homogenously-catalyzed pXL syntheses from DMF, we will move here to a more sustainable, heterogeneously catalyzed, continuous valorization of bio-based derivable DMF and AA to pXL and DMBA in the liquid phase (Scheme 1). The in situ triplet reaction includes Diels–Alder cycloaddition, dehydration and decarboxylation over different zeolites, i.e. Beta, ZSM-5 and Y, in a continuous flow-process under comparably mild reaction conditions at very high conversion and yields with a contact time of only 10.1 minutes (Scheme 1). We expect this integrated intrinsically benign and sustainable process to be of significant industrial potential for the production of pXL (Bp = 411.3 K) and DMBA (Bp = 559 K) from directly accessible products from biomass, as it might even reduce pXL costs due to the use of the flow system and simple separation of the products via a simple distillation step.
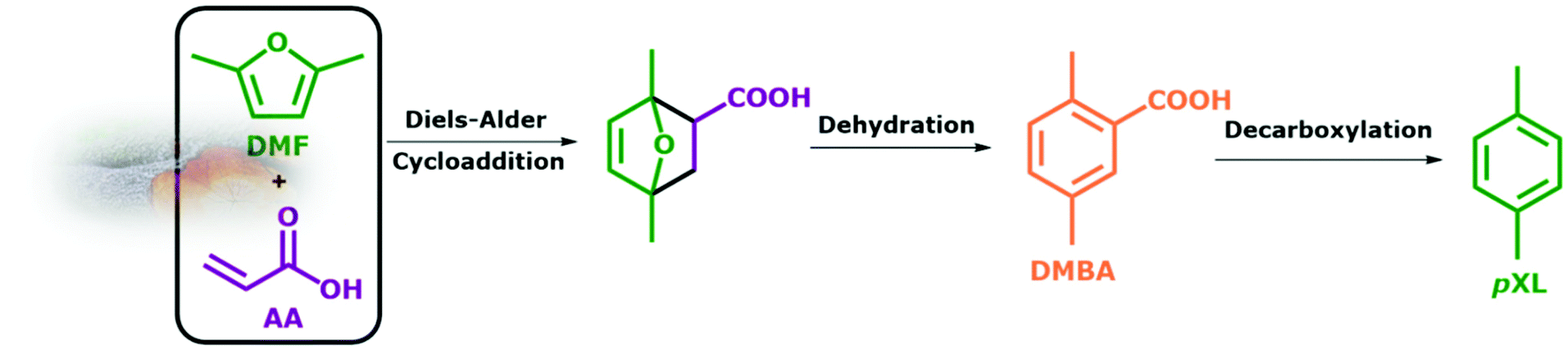 | ||
| Scheme 1 The proposed three-in-one continuous reaction of DMF and AA to pXL and DMBA using zeolite as a catalyst. | ||
Results and discussion
Catalyst characterization
The different utilized zeolites were abbreviated as follows: zeolite framework (ZSM-5, Beta or Y), followed by (Si/Al molar ratio), e.g., Beta(12.5). Prior to the catalytic experiments, all utilized H-form zeolites, i.e. ZSM-5(280), Beta(12.5, 90 and 150) and Y(80), were thoroughly characterized using N2-sorption, X-ray diffraction (XRD), elemental analysis via inductively coupled plasma optical emission spectroscopy (ICP-OES) and temperature-programmed desorption of ammonia (NH3-TPD). The experimental procedures of these characterization techniques are reported in the ESI.† The textural properties of the utilized zeolites are presented in Table 1. All catalysts were coded as follows: zeolite framework (Si/Al molar ratio).| Catalyst | V micro/cm3 g−1 | V total/cm3 g−1 | S micro/m2 g−1 | S total/m2 g−1 | Acid site density/μmol g−1 |
|---|---|---|---|---|---|
| ZSM-5(280) | 0.11 | 0.20 | 511 | 608 | 51 |
| Y(80) | 0.26 | 0.46 | 1133 | 1276 | 162 |
| Beta(150) | 0.26 | 0.38 | 1180 | 1278 | 48 |
| Beta(12.5) | 0.16 | 0.40 | 724 | 835 | 285 |
| Beta(90) | 0.20 | 0.39 | 1078 | 1160 | 120 |
| DeAl-Beta(150) | 0.20 | 0.32 | 867 | 962 | 28 |
| Na-Beta(150) | 0.19 | 0.29 | 829 | 908 | 29 |
| Hi-Beta(150) | 0.07 | 0.45 | 137 | 357 | 39 |
| Spent Beta(150) | 0.20 | 0.29 | 840 | 925 | 36 |
| Beta(150)_after 1st regeneration | 0.23 | 0.35 | 1006 | 1186 | 44 |
| Beta(150)_after 2nd regeneration | 0.20 | 0.33 | 989 | 1217 | 49 |
The zeolites, i.e. ZSM-5(280), Y(80) and Beta(12.5, 90 and 150), showed standard adsorption–desorption isotherms, as well as the typical XRD patterns and no remarkable or unusual characteristics were observed, cf. Fig. S2–S4 in the ESI.† The total specific surface area of all catalysts was found to be in a range between 350 and 1250 m2 g−1 corresponding mainly to the microporosity of the zeolites, showing the highest values for Y(80) and Beta(150), i.e. >1200 m2 g−1, owing to their porous architecture. The lowest values are found for ZMS-5(280) with 608 m2 g−1. In addition to the specific microporous surface area, Hi-Beta(150) is an exception which showed a high specific mesoporous surface area (592 m2 g−1) as a result of successful alkaline treatment of Beta(150) to generate an additional mesoporous structure.44–46
Reaction of DMF with AA to synthesize pXL on different zeolite frameworks
Based on the available studies on the synthesis of pXL from DMF and ethylene, zeolites with a high Si/Al ratio are favored in the dehydration reaction due to their high catalytic performance.47,48 In addition, it is well-known that improving zeolite hydrophobicity resulted in a thermodynamically favored water removal.48 Therefore, we analyzed the performance of three different H-form zeolite frameworks, namely ZSM-5(280), Beta(150) and Y(80), in the conversion of DMF to pXL using AA (Fig. 1). In the absence of zeolite, no significant DMF conversion as well as no pXL was detected (Fig. 1), i.e. the presence of zeolite as an acid catalyst is crucial for the dehydration of the intermediate to DMBA, followed by decarboxylation of DMBA to pXL. Among the tested catalysts, Beta(150) and Y(80) showed complete conversion of DMF at 473 K after 10.1 min residence time (τ), while ZSM-5(280) showed a poor performance (36% of DMF conversion), cf. Fig. 1. Additionally, Beta(150) and Y(80) showed AA conversion of ∼52%, whereas low AA conversion (25%) was found over ZSM-5(280), cf. Fig. S5 of the ESI.† Furthermore, pXL was found to be the main product, combined with the formation of DMBA as a by-product; Beta(150) exhibited the highest yield of pXL (83%) and DMBA (17%), and no further products could be identified. This pXL yield is the highest to be reported for DMF conversion with AA in comparison with 45% using [Bmim]HSO4 and 57% in two steps in the presence of Sc(OTf)3 and H3PO4.28,43 ZSM-5(280) and Y(80) gave pXL in yields of 31.5% and 66.9% and DMBA in yields of 1.2% and 32.3%, respectively. The high pXL yield (83%) using Beta(150) is attributed to the 12-membered-ring and three-dimensional porous structure with diameters (6.6 × 6.7 Å) slightly larger than the pXL cross section diameter (6.3 Å) and smaller than ortho (o)- and meta (m)-xylenes (∼6.8 Å), with no excess space (e.g. supercage) causing side reactions. Additionally, the Beta(150) structure selectively catalyzes the dehydration of the cycloadduct to pXL, i.e. small amounts of side reactions occur on it, as confirmed by TGA (Fig. 2), rendering it resistant to deactivation. This high catalytic performance of Beta zeolite with respect to other zeolites, i.e. ZSM-5(280) and Y(80), is in a good agreement with the previously reported studies on pXL synthesis from DMF.26,30,31,42 The low selectivity of Y(80) is attributed to the larger pore diameter (7.3 × 7.3 Å) which allows the formation of higher amounts of by-products with higher molecular weights (coking), as confirmed by TGA for spent Y(80) and ZSM-5(280), cf.Fig. 2. Finally, ZSM-5(280) represented the lowest performance due to its smaller pore size (5.1 × 5.5 Å) in comparison to Beta and Y zeolites. In addition, ZSM-5(280) and Y(80) suffer from a rapid deactivation after 5 hours of time on stream (TOS) due to the deposition of higher amounts of coke in comparison to Beta(150), as confirmed by TGA (Fig. 2). The formation mechanism of the deposited coke is proposed in Scheme S1.† It depends on the pore size of the zeolite framework,31,49 for instance when Y(280) was used, dimers and oligomers were observed on its surface, while ZSM-5(80) caused only the ring-opening of DMF, and alkylated aromatic and decarboxylated by-products, cf. ESI Scheme S1.† | ||
| Fig. 1 DMF conversion (XDMF) and pXL and DMBA yields (YpXL and YDMBA) after 2 h of time on stream in the absence of the catalyst and in the presence of three different zeolite frameworks, i.e. ZSM-5(280), Beta(150) and Y(80); reaction conditions: cDMF = 0.3 M, cAA = 0.7 M, T = 473 K, mcat = 2 g, Qeduct = 0.3 cm3 min−1, Vreactor = 3.04 cm3, psystem = 3 MPa and τ = 10.1 min. | ||
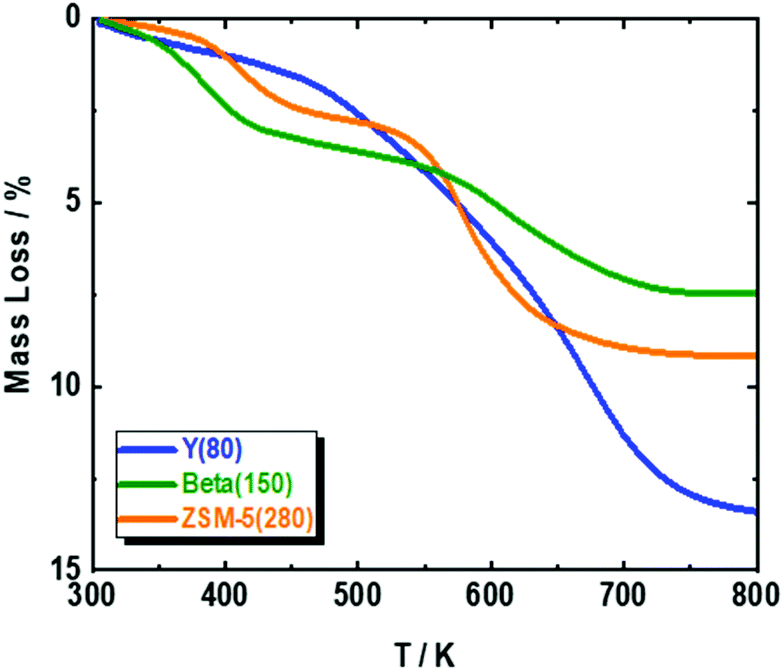 | ||
| Fig. 2 TGA of spent ZSM-5(280), Beta(150) and Y(80) in the conversion of DMF and AA to pXL; reaction conditions: cDMF = 0.3 M, cAA = 0.7 M, T = 473 K, mcat = 2 g, Qeduct = 0.3 cm3 min−1, Vreactor = 3.04 cm3, psystem = 3 MPa and τ = 10.1 min. | ||
Based on these results, Beta(150) was selected for further investigation on the influence of acid site density (using three different Si/Al molar ratios) and introduction of additional mesopores into the Beta zeolite framework. All tested Beta zeolites (12.5, 90 and 150) showed complete conversion of DMF (Fig. 3). Additionally, AA conversion was found to be between 50–60% for Beta zeolites with Si/AL molar ratios of 12.5, 90 and 150, respectively (Fig. S6 at ESI†). Beta(12.5) exhibited the lowest yield of pXL (40%) which could speculatively be attributed to its high density of acid sites that can stimulate other side reactions, e.g. cracking of n-heptane and ring-opening of DMF, leading to the formation of other side products. Moreover, it is well-known that the dehydration reaction is thermodynamically favored by increasing the hydrophobicity of the zeolite by using high Si/Al molar ratios.48 This improvement in the catalytic performance in comparison with Beta(12.5) can be clearly seen when using Beta(90) and Beta(150), which showed higher yield of pXL (77% and 80%), cf.Fig. 3.
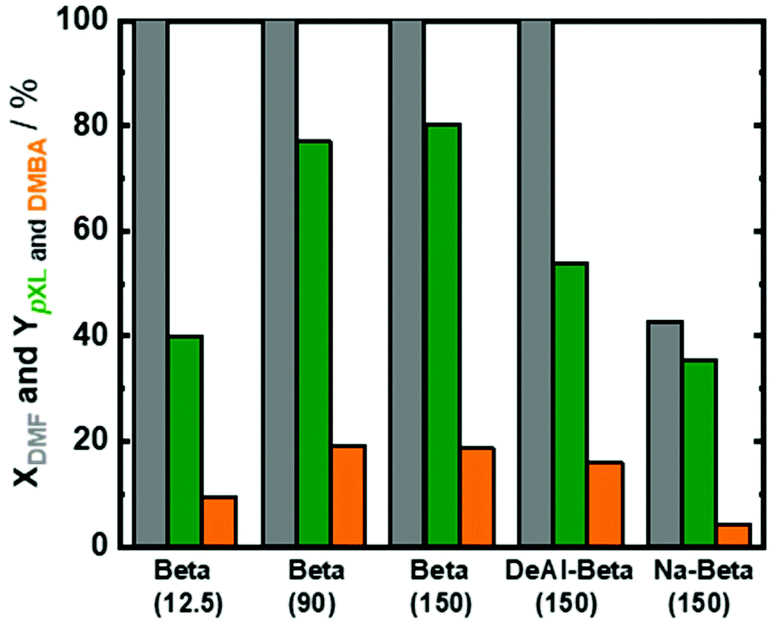 | ||
| Fig. 3 DMF conversion (XDMF) and pXL and DMBA yields (YpXL and YDMBA) after 2 h of time on stream over Beta zeolite with different Si/Al molar ratios (25, 90 and 150), as well as DeAl-Beta(150) and Na-Beta(150); reaction conditions: cDMF = 0.3 M, cAA = 0.7 M, T = 473 K, mcat = 2 g, Qeduct = 0.3 cm3 min−1, Vreactor = 3.04 cm3, psystem = 3 MPa, and τ = 10.1 min. | ||
In an attempt to improve the pXL yield, introduction of mesopores to the micropores in Beta(150) via alkaline treatment was performed, cf. the ESI† for the details on mesopore introduction. Hierarchical Beta(150) is coded as follows: Hi-Beta(150). The alkaline treatment was successful and Hi-Beta(150) showed, in addition to the presence of micropores, a high specific mesoporous surface area (294 m2 g−1) in comparison to Beta(150) which possesses a very low specific mesoporous surface area (Table 1 and Fig. S2 in the ESI†). Additionally, TEM images of Beta(150) and Hi-Beta(150) show a clear change in the morphology of Beta(150) after alkaline treatment, cf. ESI, Fig. S7.† The performance of Hi-Beta(150) in the conversion of DMF and AA to pXL was compared with Beta(150), viz. ESI Fig. S8.† Under the tested reaction conditions, DMF conversion completed, and no improvement in pXL and DMBA yields was observed by the introduction of mesopores to Beta(150) even after 5 h of time on stream (TOS), cf. ESI Fig. S8.† This suggests that under these reaction conditions the conversion of DMF with AA to pXL is free of diffusion limitations and additional mesopores do not have a positive impact on the pXL yield, which is in line with the previous study.30,32
In an attempt to improve the catalytic performance of Beta(150), it was subjected to two different treatments, i.e., the dealumination process and ion-exchange of Beta(150) with Na, respectively. Throughout the manuscript, the dealuminated Beta(150) and Na-ion exchanged Beta(150) are coded as follows: DeAl-Beta(150) and Na-Beta(150), respectively. The details of these treatments and experimental procedure are described in the ESI.† Furthermore, the textural properties of these synthesized DeAl-Beta(150) and Na-Beta(150) are reported in Table 1. Complete DMF conversion was achieved over DeAl-Beta(150), while the pXL yield reduced to 54% in comparison with non-treated Beta(150). In contrast, low AA conversion (<50%) was observed over the treated Beta(150), viz. Fig. S6 in the ESI.† This indicates that DeAl-Beta(150) facilitates the occurrence of side reactions that led to coke deposition on the catalyst. Whereas, Na-Beta(150) showed a significant decrease in DMF conversion from 100% to 42% with respect to the non-treated Beta(150), cf.Fig. 3. Also, the pXL yield dropped from 80% to 35% with Na-Beta(150), and the DMBA yield was reduced from 18% to 4% in the presence of Na-Beta(150), cf.Fig. 3. These results are attributed to the drop in the specific surface area and total density of acid sites (confirmed by N2-sorption and NH3-TPD) caused by the treatment of Beta(150) via Na-ion exchange (Table 1). Based on these results, Beta(150) was selected for further optimization of the reaction conditions. Moreover, the stability of Beta(150) under the optimum reaction conditions was evaluated. Finally, the performance of Beta(150) in the flow process and batch experiment was assessed.
Therefore, reaction temperature, residence time, DMF/AA molar ratio were adjusted using Beta(150) to maximize the formation of pXL from DMF. In addition to the reaction temperature mentioned above (473 K), two additional reaction temperatures were tested (423 K and 523 K). Over the whole analyzed temperature range, DMF was completely converted (Fig. 4), and the AA conversion was found to be ∼50% (Fig. S9 in the ESI†). As expected, at a low reaction temperature (423 K), a lower pXL yield (64%) was found, cf.Fig. 4. Unexpectedly, a drop in the pXL yield was also observed at a high reaction temperature (523 K). This point is correlated to the occurrence of side reactions for the reactants (DMF or AA), as well as reactions between products (pXL and DMBA) at such high temperatures (Scheme S1 in the ESI†). Noteworthily, no significant differences in the DMBA yield (15–19%) were observed over all tested reaction temperatures.
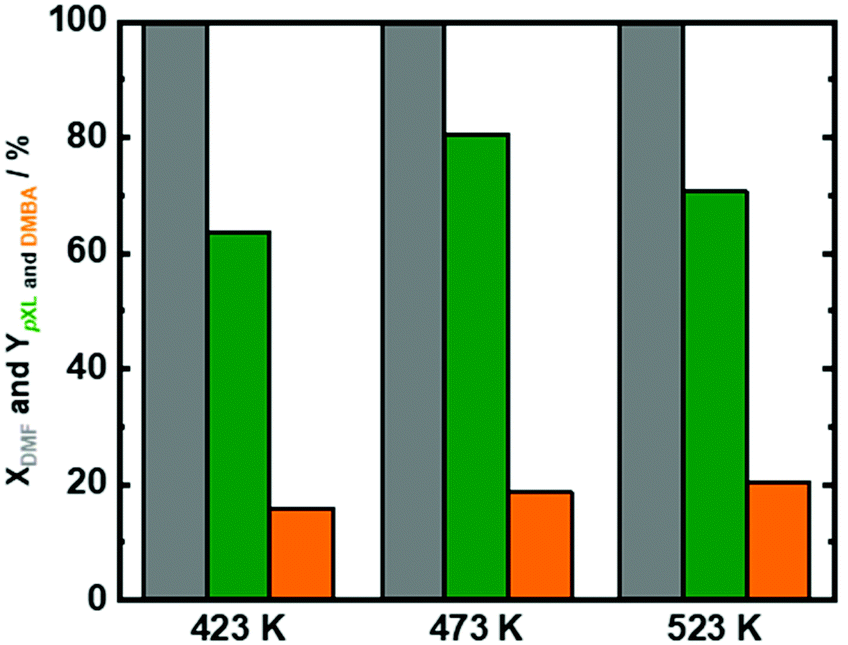 | ||
| Fig. 4 DMF conversion (XDMF) and pXL and DMBA yields (YpXL and YDMBA) after 2 h of time on stream as a function of reaction temperature (423 K, 473 K and 523 K); reaction conditions: cDMF = 0.3 M, cAA = 0.7 M, mcat = 2 g, Qeduct = 0.3 cm3 min−1, Vreactor = 3.04 cm3, psystem = 3 MPa and τ = 10.1 min. | ||
Based on the abovementioned results, a temperature of 473 K was selected, and the influence of residence time (τ) under these reaction conditions was investigated (Fig. 5). In addition to 10.1 min as the residence time, additional experiments with 3.04 min, 5.0 min and 30.4 min residence times were performed. At all utilized residence times, above 96% conversion of DMF combined with ∼50% AA conversion was recorded (Fig. 5 and Fig. S10 at ESI†). Furthermore, pXL and DMBA were the only products that were identified under these reaction conditions. Increasing the residence time from 3.04 min to 5.01 min and further to 10.1 min (Fig. 5), led to improved pXL yields from 59.3% to 70% and 80%, respectively. In contrast, the increase of residence time (t) from 3.04 min to 5.01 min to 10.1 min and 30.4 min is associated with the drop in the DMBA yield from 31.7% to 16% to 14% and 13%, respectively. The low pXL yield (59.3%) at a residence time of 3.01 min is due to shorter contact time between the substrate and Beta(150) which led to inefficient dehydration/decarboxylation of the cycloaddition product (Fig. 5). A further increase in residence time from 10.1 min to 30.4 min showed no significant change in the pXL yield (∼83%), as well as DMBA yields of ∼13%, cf.Fig. 5. These results further indicate that the formation of pXL is mainly through DMBA decarboxylation.
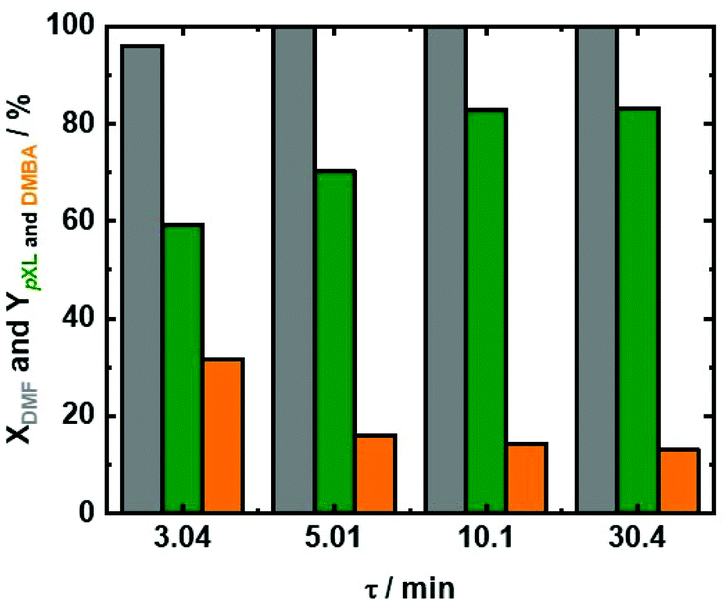 | ||
| Fig. 5 DMF conversion (XDMF) and pXL and DMBA yields (YpXL and YDMBA) after 2 hours of time on stream as a function of residence time (τ); reaction conditions: cDMF = 0.3 M, cAA = 0.7 M, T = 473 K, mcat = 2 g, Qeduct = 0.6 cm3 min−1, 0.3 cm3 min−1 and 0.1 cm3 min−1, Vreactor = 3.04 cm3, psystem = 3 MPa and τ = 5.0 min, 10.1 min and 30.4 min. | ||
Taking into consideration the minor differences in the catalyst performance in 10.1 min and 30.4 min and the 3 times higher space–time yield of pXL after 10.1 min, 10.1 min was selected to assess the influence of DMF/AA molar ratio variation. DMF conversion was completed at all utilized DMF/AA molar ratios (Fig. 6), i.e. 0.14, 0.27, 0.45, 1.0 and 1.1. Additionally, AA conversion was linearly increased from 23% to 80% with increasing DMF/AA molar ratio from 0.14 to 1 (Fig. 6). In contrast, the pXL yield was found to be slightly increased (from 77% to 80%) using DMF/AA molar ratios between 0.14, 0.27 and 0.45 (Fig. 6). Equimolar DMF and AA led to a decrease in the pXL yield from 80% to 61% (Fig. 6). A further reduction of the AA amount (DMF/AA molar ratio = 1.1), caused further decrease of pXL yield to 52% (Fig. 6). This behavior can be explained by the kinetics of bimolecular reactions (for the cycloaddition) which appears to be fast with an excess of AA. In addition, too high amounts of DMF lead to undesired side reactions, such as dimerization and oligomerisation of DMF, as well as DMF hydrolysis to 2,5-hexandione (proposed in Scheme S1†), which obviously lowers the yield of pXL. Finally, this observation suggests that the presence of an excess of AA is required to obtain a high pXL yield and selectivity (Fig. 6).
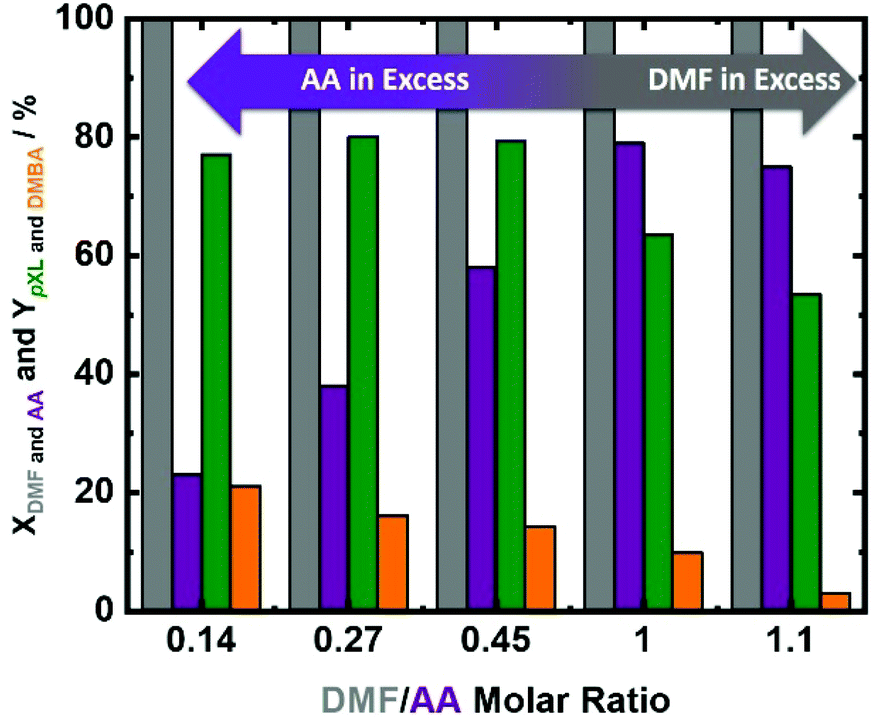 | ||
| Fig. 6 DMF and AA conversion (XDMF and AA) and pXL and DMBA yields (YpXL and YDMBA) after 2 h of time on stream as a function of DMF/AA molar ratio (0.14, 0.27, 0.45, 1.0 and 1.1); reaction conditions: cDMF = 0.09 M, 0.18 M, 0.30 M, 0.70 M and 0.76 M, cAA = 0.70 M, T = 473 K, mcat = 2 g, Qeduct = 0.3 cm3 min−1, Vreactor = 3.04 cm3, psystem = 3 MPa and τ = 10.1 min. | ||
Using Beta(150) under the optimum reaction conditions, i.e., cDMF = 0.3 M, cAA = 0.7 M, T = 473 K, mcat = 2 g, Qeduct = 0.3 cm3 min−1, Vreactor = 3.04 cm3, psystem = 3 MPa, and τ = 10.1 min, the turnover frequency (TOF) was calculated by considering the total acid site density of Beta(150). The calculated TOF was 48 h−1 with a pXL space–time yield (STYpXL) of 0.23 kgpXL h−1 kgcat−1. The calculated TOF (48 h−1) for Beta(150) in this reaction system (DMF and AA in a continuous flow system) was lower than the reported TOF values for Zr-Beta (121 h−1), Al-Beta (122 h−1), Sn-Beta (113 h−1) and Ti-Beta (65 h−1) for the conversion of DMF with ethylene in a batch system at 523 K.31 The low TOF value for Beta(150) in comparison to the reported TOF values for Zr-Beta, Al-Beta, Sn-Beta and Ti-Beta is due to a low operational temperature (473 K) of our system, as well as the different reaction mechanisms (the occurrence of an additional decarboxylation step of DMBA).
Catalyst stability
Under the optimized reaction conditions, the performance of fresh Beta150 under 10 hours of time on stream (TOS) was evaluated (Fig. 7). It can be seen that Beta(150) showed a stable performance with complete DMF conversion and pXL yield of ∼80%, after 10 hours of TOS (Fig. 7). In addition, AA conversion remained stable after 10 h of TOS (Fig. S11 at ESI†). However, a slight drop in the pXL yield after 8 hours of TOS was observed in parallel with a reduction in the specific surface area from 1278 m2 g−1 to 925 m2 g−1 and acid site density from 48 μmol g−1 to 36 μmol g−1 (Table 1 and Fig. S12 at ESI†). With the changes of the textural properties, we find the deposition of (∼20%) of carbonaceous species after 10 h of TOS, which is confirmed by TGA (Fig. S14 at ESI†). The implementation of regeneration cycles via thermal treatment of the used Beta(150) have been proposed as a strategy to extend the long-term use of the catalysts (the details of the regeneration procedure is described in the ESI†). Applying the regeneration steps resulted in successful recovery of the catalyst performance back to the level of the freshly used catalyst (Fig. 7). Characterization after each regeneration step revealed that the textural properties and crystalline structure were mostly restored (Table 1 and Fig. S12 in the ESI†). Of course, practical catalyst reactivation processes in flow reactors preferentially occur in the switched flow mode, e.g. under different solvents. We assume that the side products under optimized conditions are DMF oligomers only, which are insoluble in the used solvent (n-heptane).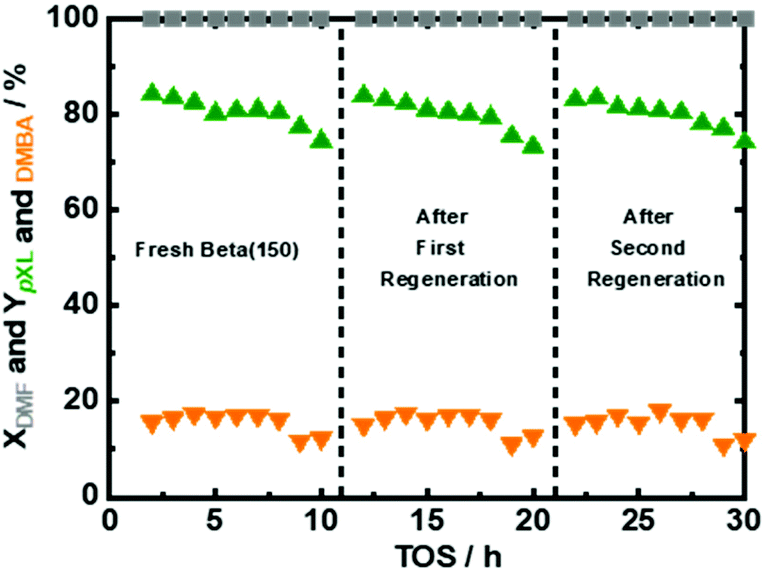 | ||
| Fig. 7 DMF conversion (XDMF) and pXL and DMBA yields (YpXL and YDMBA) as a function of time on stream (TOS) for fresh Beta(150) after two regeneration steps; reaction conditions: cDMF = 0.3 M, cAA = 0.7 M, mcat = 2 g, Qeduct = 0.3 cm3 min−1, Vreactor = 3.04 cm3, psystem = 3 MPa, and τ = 10.1 min. | ||
Catalytic performance in flow and batch reactors
It is clear that a contact time of 10.1 min for full conversion in the continuous flow process is very short when compared to previous reports on those using the batch system, and we attribute this to the perfect heat transfer and reaction profile in flow reactors. To underline this point, the catalytic performance was analyzed in two types of reactors, i.e. the continuous flow system vs. batch system. We exposed the same amount of Beta(150) to catalytic experiments for 8 h in both systems (Fig. 8). As reported above Beta(150) in a continuous flow system showed quantitative conversion of DMF with a pXL yield of 80% and a DMBA yield of 10%. Moreover, Beta(150) in a continuous flow system showed higher AA conversion (∼60%) than that in the batch system (40%), viz. Fig. S14 in the ESI.† The batch experiment using Beta(150) revealed a low DMF conversion (40%) and pXL and DMBA yield of 34% and 4%, respectively. The low DMF conversion and low pXL and DMBA yields in the batch system are attributed to rapid catalyst deactivation because the products are not flushed out, but remain on the catalyst. The flow system offers a short contact time (10.1 min) as well as a fresh supply of the reaction solution, which allows a longer operation time without deactivation. Furthermore, TGA characterization of the spent Beta(150) in the batch reactor showed 47 wt% coke or deposits while the experiment conducted in a continuous flow reactor showed a weight loss of 10 wt% (Fig. S14 in the ESI†). These experiments clearly support the view that the flow system is favored to run the present system economically and in a sustainable fashion.12,50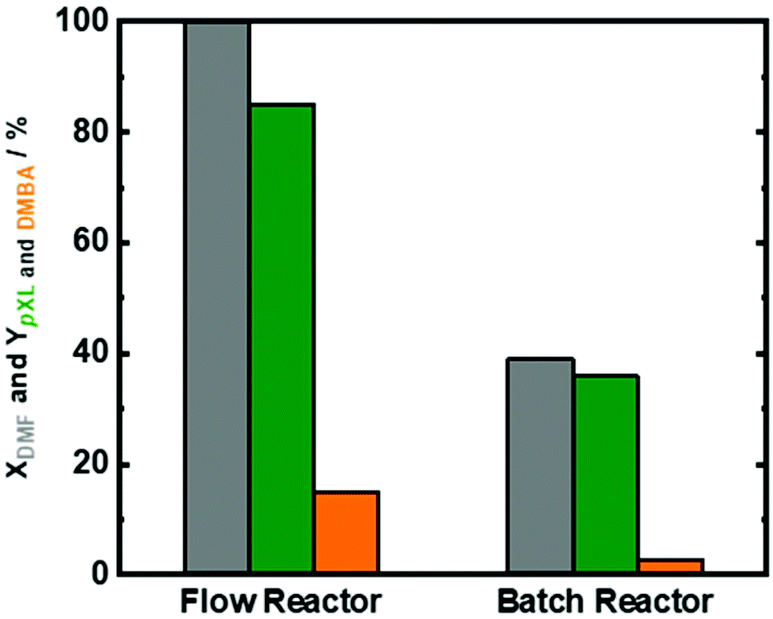 | ||
| Fig. 8 DMF conversion (XDMF) and pXL and DMBA yields (YpXL and DMBA) over Beta(150) after 8 hours in a continuous flow system and batch reactor; reaction conditions in the continuous flow system: cDMF = 0.3 M, cAA = 0.7 M, T = 473 K, mcat = 2 g, Qeduct = 0.3 cm3 min−1, Vreactor = 3.04 cm3, psystem = 3 MPa, and t = 10.1 min; reaction conditions in the batch reactor: cDMF = 0.3 M, cAA = 0.7 M, T = 473 K, mcat = 2 g, n = 400 rpm, p = 3 MPa of N2. | ||
Conclusions
In summary, a three-in-one reaction, Diels–Alder cycloaddition of 2,5-dimethylfuran to acrylic acid, dehydration to form the phenyl-ring, and final decarboxylation to p-xylene, was successfully performed in a continuous flow system over Beta zeolite, using n-heptane as a solvent. This reaction requires a catalyst with specific parameters to obtain the optimum performance. We found that the combination of high specific surface area (mainly microporous) with a 3D pore architecture of 6.6 × 6.7 Å size, and a medium to high acid site density in the range of 50 μmol g−1 in a zeolite promote the two follow up reactions, while alternative self-condensation reactions are suppressed. Furthermore, an optimal balance between the 2,5-dimethylfuran and acrylic acid molar ratios is likely important for efficient p-xylene production. As a result of this process, a highly valuable product mixture consisting of p-xylene and 2,5-dimethylbenzoic acid is obtained which can be simply separated based on their boiling points. This benign process provides an alternative and efficient pathway to produce p-xylene from renewable resources, e.g. lignocellulosic biomass, which could substitute its production pathway from fossil oils within the biorefinery scheme. To apply this process on a large scale, a detailed microkinetics study of this reaction is greatly required, which will provide more insight into the underlying mechanism. Finally, the evaluation of the final cost of the products through detailed techno-economic studies will reveal the industrial relevance of the proposed strategy.Experimental section
Different types of zeolites (Beta, ZSM-5 and Y) in H-form were supplied by Clariant and Zeolyst. The material details that have been used in this work are described in the ESI.† All utilized catalysts were thoroughly characterized via N2-sorption, XRD, NH3-TPD and TGA. The experimental details and procedures of the characterization techniques are also described in the ESI.†The reaction of 2,5-dimethylfuran (DMF) in the presence of acrylic acid (AA) was studied in a homemade continuous-flow fixed bed reactor similar to the one described in our previous studies (Fig. S1A in the ESI†).12,17 In a typical experiment, a solution of DMF (0.3 M) with acrylic acid (0.7 M) in n-heptane was fed using a HPLC pump through a stainless steel tubing. A heating unit consisting of a pre-heated tube, tubular reactor and thermocouple was placed in a homemade aluminum heating mantle (Fig. S1B at ESI†). A stainless steel tubular reactor from Swagelok with an inner diameter (ID) = 4.6 mm and length (l) = 25 cm was used to perform the catalytic experiments. Prior to sampling, internal liquid pressure was set at 3.0–3.5 MPa using a back-pressure regulator. For qualitative and quantitative analyses, a sample of ca. 2.0 cm3 was collected and analyzed via gas chromatography (GC-MS) and high performance liquid chromatography (HPLC). More details about the experimental setup, procedures and reactants and product analysis can be found in the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the Max Planck Society. Jose Alirio Mendoza Mesa thanks the “Gobernación del Tolima” and Colfuturo for a research scholarship granted through Convocatoria 727-2017 of Colciencias. Thanks are also extended to Jessica Brandt, Rona Pitschke and Antje Völkel of the Max Planck Institute of Colloids and Interfaces for elemental analysis, TEM and TGA measurements, respectively. The effort of the electrical and mechanical workshops at the Max Planck Institute of Colloids and Interfaces is greatly acknowledged. Our special thanks goes to Valerio Cersani for the design and idea of the table of contents. Open Access funding provided by the Max Planck Society.References
- D. Esposito and M. Antonietti, Chem. Soc. Rev., 2015, 44, 5821–5835 RSC.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS.
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef CAS.
- C. K. W. Arthur, J. Ragauskas, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef.
- D. A. S. Edward, L. Kunkes, R. M. West, J. C. Serrano-Ruiz, C. A. Gärtner and J. A. Dumesic, Science, 2008, 322, 417–421 CrossRef.
- Zeolites and Zeolite-like Materials, ed. W. S. T. Ennaert, J. Dijkmans, M. Dusselier and B. F. Sels, Elsevier, Amesterdam, 2016 Search PubMed.
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2011, 4, 83–99 RSC.
- P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 11980–11987 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- B. Kumru, J. Mendoza Mesa, M. Antonietti and M. Al-Naji, ACS Sustainable Chem. Eng., 2019, 7, 17574–17579 CrossRef CAS.
- F. Brandi, M. Bäumel, V. Molinari, I. Shekova, I. Lauermann, T. Heil, M. Antonietti and M. Al-Naji, Green Chem., 2020, 22, 2755–2766 RSC.
- D. Zhao, D. Rodriguez-Padron, K. S. Triantafyllidis, Y. Wang, R. Luque and C. Len, ACS Sustainable Chem. Eng., 2020, 8, 3091–3102 CrossRef CAS.
- S. Mhadmhan, A. Franco, A. Pineda, P. Reubroycharoen and R. Luque, ACS Sustainable Chem. Eng., 2019, 7, 14210–14216 CrossRef CAS.
- M. Braun and M. Antonietti, Green Chem., 2017, 19, 3813–3819 RSC.
- M. Al-Naji, M. Popova, Z. Chen, N. Wilde and R. Gläser, ACS Sustainable Chem. Eng., 2019, 8, 393–402 CrossRef.
- M. Al-Naji, B. Puertolas, B. Kumru, D. Cruz, M. Baumel, B. Schmidt, N. V. Tarakina and J. Perez-Ramirez, ChemSusChem, 2019, 12, 2628–2636 CrossRef CAS.
- M. Al-Naji, J. Van Aelst, Y. Liao, M. d'Hullian, Z. Tian, C. Wang, R. Gläser and B. F. Sels, Green Chem., 2020, 22, 1171–1181 RSC.
- M. Al-Naji, A. Yepez, A. M. Balu, A. A. Romero, Z. Chen, N. Wilde, H. Li, K. Shih, R. Gläser and R. Luqueb, J. Mol. Catal. A: Chem., 2016, 417, 145–152 CrossRef CAS.
- H. Xiao, B. Hou, P. Zeng, A. Jiang, X. Hou and J. Liu, Fuel, 2017, 192, 53–59 CrossRef CAS.
- M. Wei, S. Li, J. Liu, G. Guo, Z. Sun and H. Xiao, Fuel, 2017, 192, 208–217 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2014, 16, 950–963 RSC.
- A. Maneffa, P. Priecel and J. A. Lopez-Sanchez, ChemSusChem, 2016, 9, 2736–2748 CrossRef CAS.
- V. J. Margarit, E. M. Gallego, C. Paris, M. Boronat, M. Moliner and A. Corma, Green Chem., 2020, 22, 5123–5131 RSC.
- K. Smith, M. Pampel and P. Feng, IHS Chemical Chemical Economics Handbook–Xylenes, 2015 Search PubMed.
- A. E. Settle, L. Berstis, N. A. Rorrer, Y. Roman-Leshkóv, G. T. Beckham, R. M. Richards and D. R. Vardon, Green Chem., 2017, 19, 3468–3492 RSC.
- Z. Lin, M. Ierapetritou and V. Nikolakis, AIChE J., 2013, 59, 2079–2087 CrossRef CAS.
- L. Ni, J. Xin, K. Jiang, L. Chen, D. Yan, X. Lu and S. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 2541–2551 CrossRef CAS.
- Y. T. Cheng, Z. Wang, C. J. Gilbert, W. Fan and G. W. Huber, Angew. Chem., Int. Ed., 2012, 51, 11097–11100 CrossRef CAS.
- C. L. Williams, C.-C. Chang, P. Do, N. Nikbin, S. Caratzoulas, D. G. Vlachos, R. F. Lobo, W. Fan and P. J. Dauenhauer, ACS Catal., 2012, 2, 935–939 CrossRef CAS.
- C.-C. Chang, H. Je Cho, J. Yu, R. J. Gorte, J. Gulbinski, P. Dauenhauer and W. Fan, Green Chem., 2016, 18, 1368–1376 RSC.
- C.-C. Chang, S. K. Green, C. L. Williams, P. J. Dauenhauer and W. Fan, Green Chem., 2014, 16, 585–588 RSC.
- H. J. Cho, L. Ren, V. Vattipalli, Y.-H. Yeh, N. Gould, B. Xu, R. J. Gorte, R. Lobo, P. J. Dauenhauer, M. Tsapatsis and W. Fan, ChemCatChem, 2017, 9, 398–402 CrossRef CAS.
- J. Yu, S. Zhu, P. J. Dauenhauer, H. J. Cho, W. Fan and R. J. Gorte, Catal. Sci. Technol., 2016, 6, 5729–5736 RSC.
- X. Feng, C. Shen, C. Tian and T. Tan, Ind. Eng. Chem. Res., 2017, 56, 5852–5859 CrossRef CAS.
- Y. P. Wijaya, H. P. Winoto, Y.-K. Park, D. J. Suh, H. Lee, J.-M. Ha and J. Jae, Catal. Today, 2017, 293–294, 167–175 CrossRef CAS.
- I. F. Teixeira, B. T. Lo, P. Kostetskyy, M. Stamatakis, L. Ye, C. C. Tang, G. Mpourmpakis and S. C. Tsang, Angew. Chem., Int. Ed., 2016, 55, 13061–13066 CrossRef CAS.
- L. Ye, I. Teixeira, B. T. W. Lo, P. Zhao and S. C. Edman Tsang, Chem. Commun., 2017, 53, 9725–9728 RSC.
- I. F. Teixeira, B. T. W. Lo, P. Kostetskyy, L. Ye, C. C. Tang, G. Mpourmpakis and S. C. E. Tsang, ACS Catal., 2018, 8, 1843–1850 CrossRef CAS.
- S. Thiyagarajan, H. C. Genuino, M. Sliwa, J. C. van der Waal, E. de Jong, J. van Haveren, B. M. Weckhuysen, P. C. Bruijnincx and D. S. van Es, ChemSusChem, 2015, 8, 3052–3056 CrossRef CAS.
- S. Thiyagarajan, H. C. Genuino, J. C. van der Waal, E. de Jong, B. M. Weckhuysen, J. van Haveren, P. C. Bruijnincx and D. S. van Es, Angew. Chem., Int. Ed., 2016, 55, 1368–1371 CrossRef CAS.
- E. Mahmoud, J. Yu, R. J. Gorte and R. F. Lobo, ACS Catal., 2015, 5, 6946–6955 CrossRef CAS.
- L. Ni, J. Xin, H. Dong, X. Lu, X. Liu and S. Zhang, ChemSusChem, 2017, 10, 2394–2401 CrossRef CAS.
- P. Y. Dapsens, C. Mondelli and J. Perez-Ramirez, Chem. Soc. Rev., 2015, 44, 7025–7043 RSC.
- J. Van Aelst, D. Verboekend, A. Philippaerts, N. Nuttens, M. Kurttepeli, E. Gobechiya, M. Haouas, S. P. Sree, J. F. M. Denayer, J. A. Martens, C. E. A. Kirschhock, F. Taulelle, S. Bals, G. V. Baron, P. A. Jacobs and B. F. Sels, Adv. Funct. Mater., 2015, 25, 7130–7144 CrossRef CAS.
- D. Verboekend, N. Nuttens, R. Locus, J. Van Aelst, P. Verolme, J. C. Groen, J. Perez-Ramirez and B. F. Sels, Chem. Soc. Rev., 2016, 45, 3331–3352 RSC.
- R. Otomo, T. Yokoi and T. Tatsumi, Appl. Catal., A, 2015, 505, 28–35 CrossRef CAS.
- H. Kobayashi, H. Yokoyama, B. Feng and A. Fukuoka, Green Chem., 2015, 17, 2732–2735 RSC.
- Y. Mathieu, A. Corma, M. Echard and M. Bories, Appl. Catal., A, 2014, 469, 451–465 CrossRef CAS.
- R. Gerardy, D. P. Debecker, J. Estager, P. Luis and J. M. Monbaliu, Chem. Rev., 2020, 120(15), 7219–7347 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc01517b |
| This journal is © The Royal Society of Chemistry 2020 |