
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLevoglucosan: a promising platform molecule?
Ivaldo
Itabaiana Junior
 ab,
Marcelo
Avelar do Nascimento
c,
Rodrigo Octavio Mendonça Alves
de Souza
c,
Anthony
Dufour
d and
Robert
Wojcieszak
*a
ab,
Marcelo
Avelar do Nascimento
c,
Rodrigo Octavio Mendonça Alves
de Souza
c,
Anthony
Dufour
d and
Robert
Wojcieszak
*a
aUniv. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: Robert.wojcieszak@univ-lille.fr
bDepartment of Biochemical Engineering, School of Chemistry, Federal University of Rio de Janeiro, 21941910, Rio de Janeiro, Brazil
cBOSS Group – Biocatalysis and Organic Synthesis Group, Chemistry Institute, Federal University of Rio de Janeiro, 21941909, Rio de Janeiro, Brazil
dLRGP, CNRS, Université de Lorraine, ENSIC, 1 rue Grandville, 54000 Nancy, France
First published on 29th June 2020
Abstract
Lignocellulosic biomass is the most abundant carbon source and it is a base of the whole biorefinery concept. Levoglucosan (1,6-anhydro-β-D-glucopyranose) (LG) is an anhydrous sugar formed as a major product during pyrolysis of cellulose. LG might be a promising chemical platform. It can be converted to different high added-value chemicals such as levoglucosenone, 5-hydroxymethylfurfural and styrene directly or through a glucose intermediate via chemical, catalytic and biochemical processes. In this critical review we focus not only on the pyrolytic methods for the synthesis of levoglucosan, but above all also on the recent scientific progress in the chemical and biochemical transformation of levoglucosan to highly valued compounds. The catalytic performances of the heterogeneous and enzymatic catalytic systems used for different reactions of LG conversion are reviewed. Specific properties of the active sites, roles of additives, and solvents and conditions are discussed in detail. We conclude on recommendations to further improve LG conversion to high added-value products.
 Ivaldo Itabaiana Junior | Professor Ivaldo Itabaiana Junior obtained his PhD in pharmaceutical science from the Federal University of Rio de Janeiro, Brazil where he is now a full professor. He has experience in biocatalysis, biotransformation and the use of biomass for the production of bio-products of industrial interest. He has extensive collaboration with CNRS Lille, France working on the development of a new concept of hybrid catalysis for the transformation of biomass residues, such as pyrolysis oils, lignin and microbial hydrolysis derivatives into products with higher added value. His research interests are biocatalysis, enzymatic biotechnology (lipases, oxidoreductases, laccases), enzyme immobilization and drug delivery. |
 Marcelo Avelar do Nascimento | MSc Marcelo Avelar is graduated in Applied Chemistry at the Federal Fluminense University (UFF, 2016) and obtained his master's degree in Chemistry in 2018 from the Program of Post-Graduation in Chemistry (PGQu) from the Federal University of Rio de Janeiro (UFRJ). He is currently a PhD student at PGQu and is supervised by professors Ivaldo Itabaiana Jr. and Rodrigo M. A. de Souza. Marcelo has experience in the biocatalysis of levoglucosan and the immobilization of enzymes for application in batch and continuous flow systems. |
 Rodrigo Octavio Mendonça Alves de Souza | Prof. Dr Rodrigo O. M. A. de Souza has a degree from the Pharmacy School of Federal University of Rio de Janeiro (UFRJ) and PhD from the Institute of Natural Products Research. He has been coordinating the Biocatalysis and Organic Synthesis group at the Chemistry Institute of UFRJ since 2010 with Prof. Leandro S. M. Miranda, and has been working on process development for API synthesis as well as introducing continuous-flow methodologies on biocatalytic processes. |
 Anthony Dufour | Dr Anthony Dufour is a research scientist at CNRS (France) working on the thermochemical conversion of biomass and wastes. He did his PhD on hydrogen production from biomass gasification for the National Gas Company (now ENGIE) in collaboration with several French research centers. He joined CNRS Nancy in 2008. His main research interests are biomass pyrolysis, reactivity of carbons, catalysis for tar reforming or bio-oil hydrodeoxygenation, development of pyrolysis, liquefaction and gasification reactors. He is currently associate editor of Energy & Fuels (ACS) and a member of the editorial boards of Journal of Analytical & Applied Pyrolysis. |
 Robert Wojcieszak | Dr Robert Wojcieszak is the senior researcher at the Centre National de la Recherche Scientifique (CNRS) in Lille, France. He obtained his Ph.D. in Molecular Chemistry from Henri Poincare University in 2006 and subsequently held a Postdoctoral Fellowship in the IMCN at the Catholic University of Louvain, Belgium. Then, he was a FAPESP fellow at the University of São Paulo, Brazil. His research interests cover materials science and catalytic materials design. In particular he is working on the development of new advanced catalysts for biomass transformation especially for the oxidation and hydrogenation of bio-based compounds and hybrid catalysis. |
1. Introduction
The need for natural resources has influenced the development of human society throughout history. Due to the increase of life expectancy, the world population has been growing alarmingly, bringing as immediate consequences the increase of industrial and agricultural activities as a way to supply the energy and food demands.1,2 As a direct consequence of this development, the generation of industrial waste has also been increasing, as well as the concern about the environmental impact caused, and the development of new practices to reduce residue generation and new approaches for valorization. In this trend, the integration between researchers and industry has been crucial to the search for short-term solutions. The unsustainability of fossil fuels requires a shift towards the use of renewable sources to meet future energy and chemical needs. Scientific advances in renewable technologies are promising, but no technology can completely replace fossil fuels and fine chemicals.3 Therefore, the use of each resource is necessary to gradually reduce the dependence on these fuels. Biomass is currently the largest source of renewable carbon-based energy. Lignocellulosic biomass, consisting of cellulose (40–50%), hemicellulose (15–25%) and lignin (15–30%) (Fig. 1), is the most abundant source of carbon-based molecules.4–6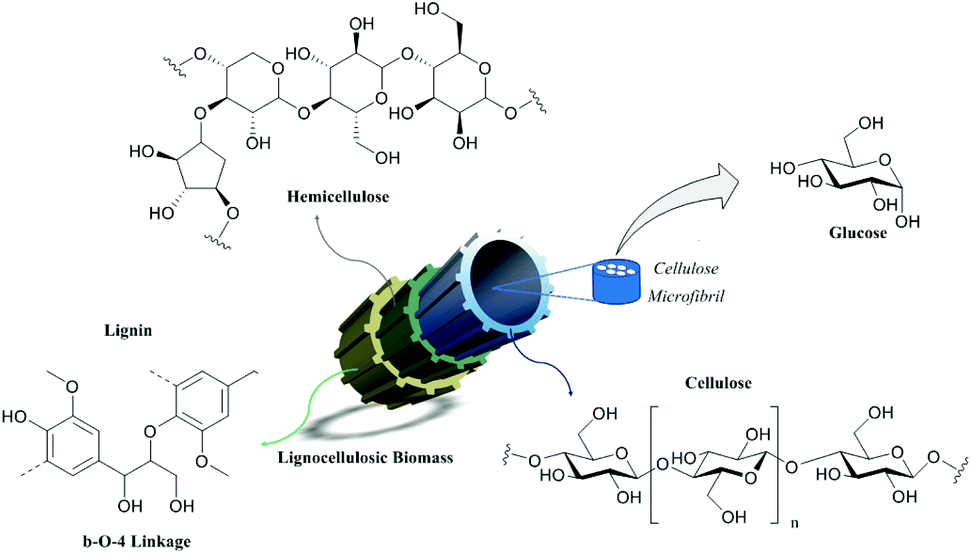 | ||
| Fig. 1 Schematic overview of the lignocellulosic biomass structure. | ||
This fact is often overlooked, as most of this biomass is used non-commercially for energy and heating, which is inefficient in terms of productivity, in addition to generating more pollution. Due to its chemical and structural complexity, biomass can be used in a better way through the synthesis of bio-based fuels or value-added chemicals.7,8
Lignocellulosic biomass can be fractionated by pulping processes like the Kraft or Organosolv processes in order to valorize more selectively each of its macromolecules.9,10 Lignin and hemicelluloses can be further valorized to produce green chemicals by various chemical or biochemical processes.10 Cellulose is a polydispersed linear polysaccharide consisting of β-1,4-glycosidic linked D-glucose units (so-called anhydroglucose units). It can be converted to various chemical platforms such as furans derivatives, glucose and itaconic acid.
The purpose of this review is to focus on the production of levoglucosan from cellulose and on its conversion to various added-value chemicals. Levoglucosan (LG) is a 1,6-anhydro derivative of beta-D-glucopyranose, which is mainly formed by cellulose or starch pyrolysis. Cellulose is preferred over starch because it does not compete with food supply.
The industrial interest in a LG production and other anhydro sugars has recently increased.11 This growing interest is due to a number of factors. LG is potentially useful in the chemical industry for the manufacture of plastics, surfactants, pharmaceuticals, propellants and resins, and as a saving substitute for materials such as sorbitol. Furthermore the process of the levoglucosan synthesis involves pyrolysis. Pyrolysis does not use any solvent or enzyme to depolymerize cellulose. It is easily up-scalable facilitating commercial applications.12 Furthermore, other by-products formed during cellulose pyrolysis can also have considerable commercial value (such as furan derivatives).13
Fig. 2 presents an overview of levoglucosan conversion through various downstream chemical platforms. The hydrolysis of LG generates the fermentable sugar glucose. Glucose can be taken up by microorganisms and follow fermentative pathways of biotransformation or be modified by chemical, enzymatic, homogeneous or heterogeneous catalysis.14 In this review we focus on the catalytic and biocatalytic transformation of LG. Various highly valued products can be obtained as represented in Fig. 2. The main objective of the present review is to go beyond a simple description of the process of LG valorization. We discuss the key factors that influence the catalytic performances of the heterogeneous and enzymatic catalytic systems used for different reactions. In order to efficiently improve the reaction yields for a given process, specific properties of the active sites, roles of additives, and solvents and conditions are discussed in each approach. We summarize the most important factors to be taken into account in the further industrial applications of these processes. The first part of this review presents the production of LG via pyrolytic processes. The second part is devoted to the conversion of LG using biochemical processes, for which promising results have been obtained over the last decade. The following part deals with the chemical transformation for which general trends linking the catalyst composition and structure are established.
 | ||
| Fig. 2 Examples of highly valued products obtained by chemical and biochemical transformation. | ||
2. Production of levoglucosan by cellulose fast pyrolysis
LG is a major product of cellulose pyrolysis. Cellulose pyrolysis has already been reviewed extensively.15–20 The purpose of this section is only to give a brief overview of LG production from cellulose pyrolysis with the main important parameters impacting LG production, its selectivity and side product formation. The production of LG by cellulose pyrolysis has been largely reported in the literature. However, the LG yields vary in a wide range from 5 to 80 per cent carbon (%C) depending on the experimental conditions, the type of the reactor and the nature of the lignocellulosic substrate, as reviewed by Maduskar et al.21 This wide range of LG yields can easily be explained by the fundamental mechanisms of cellulose. Pyrolysis of cellulose presents a complex interplay between heat transfer, phase changes, reactions and mass transfer.15,22,23 Upon heating, cellulose particles not only undergo depolymerization reactions, but also phase changes (formation of liquids and gas). Then various secondary reactions can occur in the liquid or gas-phase. Products (liquid or vapor) are then transferred to the gas surrounding cellulose particles.An initial step during cellulose fast pyrolysis is the production of an intermediate reactive liquid from depolymerized cellulose (see Fig. 3).24 This intermediate liquid undergoes complex competitive phenomena, namely: reactions in the liquid phase, evaporation (forming a vapor phase) and mass transfer through bubble formation and aerosol ejection.15,23,25–27
 | ||
| Fig. 3 Schematic representation of primary and secondary reactions in cellulose pyrolysis. Reprinted with permission from ref. 28. | ||
Firstly, heat is brought by the pyrolysis reactor to the surface of the cellulose particles. Heat is usually brought to the outer surface of cellulose particles by a hot sand (in a fluidized bed reactor), a hot surface (in micro analytical reactors) and/or by convection of a hot gas and radiation. The reactor must provide a high heat flux density (W m−2) to particle surfaces in order to promote the reaction of LG formation.24,29 The optimal temperature of the heat source to promote LG formation is around 450–500 °C (for a fluidized bed reactor).30,31 But the true temperature of pyrolysis reactions remains unknown during fast pyrolysis and is lower than the temperature of the heat source.18 The first step of depolymerization is endothermic (consuming heat), the evaporation of intermediate liquids is also endothermic but crosslinking reactions which form char (solid rich in carbon) are exothermic.25,32
Secondly, upon heating, cellulose undergoes various chemical reactions. Mechanisms of cellulose pyrolysis are proposed since the early twentieth century.19 The primary conversion of cellulose occurs principally from 300 °C to 450 °C. These mechanisms are favored or inhibited by the operating conditions (temperature of the heat source, heat flux density) or the presence of inorganics. Cellulose can be converted by three main pathways:
(1) Transglycosylation17,33,34 which leads to the formation of anhydro-sugars (levoglucosan, cellobiosan, etc.);
(2) Dehydration35–37 which induces the formation of unsaturated bonds;
(3) Open-ring/fragmentation38–40 reactions leading to the formation of light oxygenated compounds.
The compounds formed from cellulose (furans, methylglyoxal) are produced by the combination of these different mechanisms. For instance, the formation of furans needs open-ring reactions and dehydration in order to form furan rings from the glucopyranose structure. LG is formed by transglycosylation reactions. Important novel findings have been revealed on the fundamental reactions by theoretical chemistry (ab initio calculation, density functional theory-DFT model). The modeled reactants are small surrogates of cellulose like glucose, cellobiose or cellotriose due to the high computing cost of this type of DFT modeling.41 It becomes now recognized that the main reactions of cellulose pyrolysis occur through a concerted mechanism.42Fig. 4 presents an important recent result from DFT modeling to explain notably LG formation during cellulose fast pyrolysis.
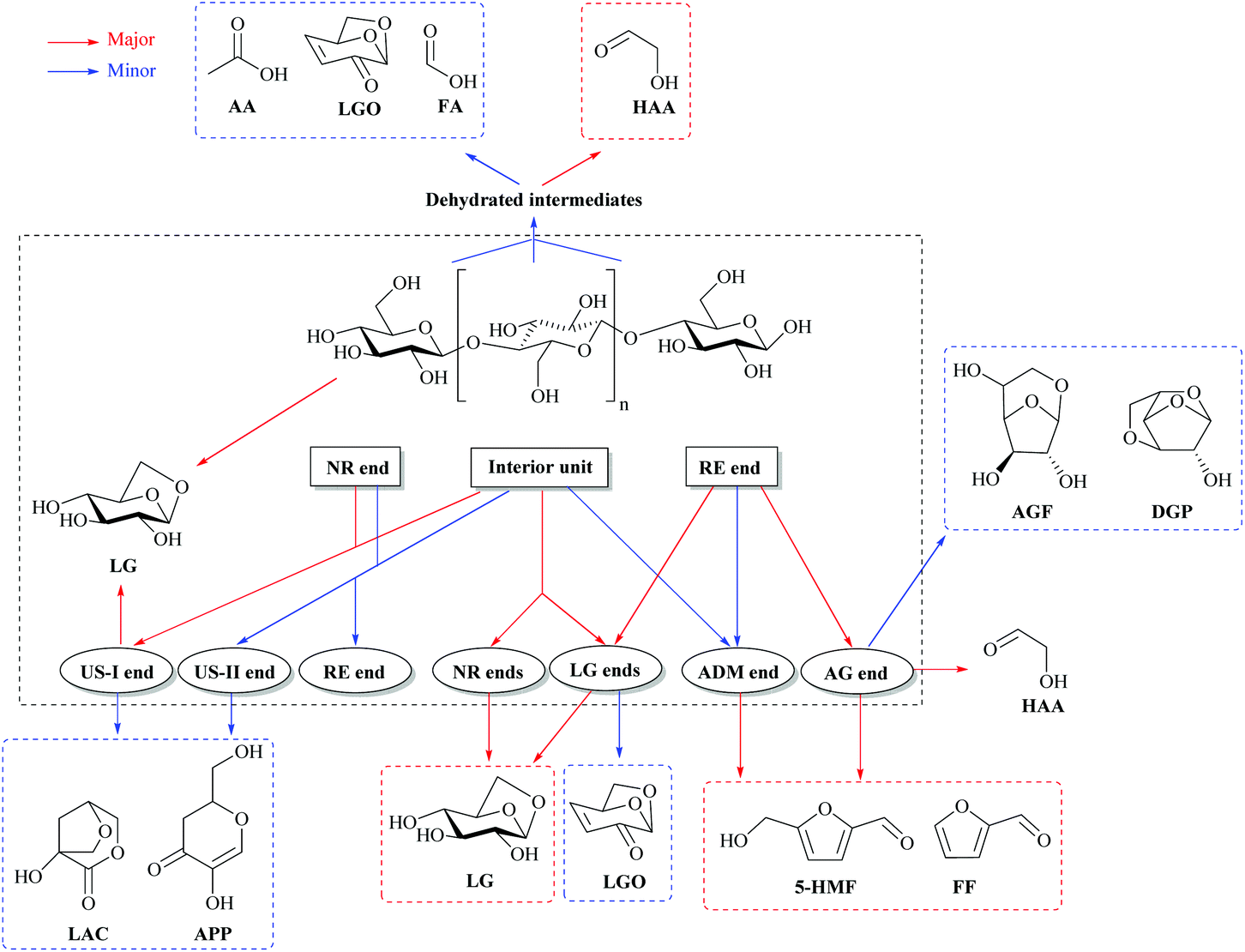 | ||
| Fig. 4 Major fast pyrolysis pathways for glucose-based carbohydrates and cellulose pyrolysis. Adapted with permission from ref. 41. | ||
Cellulose presents three main types of units: the indigenous interior units, reducing end (RE end) and non-reducing end (NR end) (Fig. 4).41,43 Each cellulose chain has one RE end, one NR end and hundreds or thousands of interior units depending on their degree of polymerization (DP). The amount of the interior units is proportional to DP. When DP increases, reactions occurring at the interior unit and NR end are favored vs. the RE end, resulting in distinct pyrolytic product distribution.41 The LG end and NR end tend to generate LG in a chain de-propagation pattern, which results in the formation of LG as the predominant product. As a result, the larger the DP is, the more LG will be formed.41
A catalytic mechanism was proposed by Dauenhauer et al.44 wherein vicinal hydroxyl groups from neighboring cellulose sheets promote transglycosidic C–O bond activation and therefore LG formation. Catalytic promotion of LG with reactive hydroxyl groups may be promoted in mass-transport limited experiments where the intermediate liquids generated by cellulose fast pyrolysis are important.21
The degree of polymerization and also the crystallinity of cellulose and its allomorph type impact LG formation. Wang et al.45 have shown that at temperatures between 350 and 450 °C, higher yields of mono-anhydrosugars (levoglucosan and levoglucosenone) were obtained with the samples with higher crystallinity during fast pyrolysis (by Py-GC/MS). On the other hand, Shanks et al.46 have demonstrated that the crystallinity index, DP, and feedstock type had a negligible influence on the resulting product distribution. Whether the cellulose sequence was arranged by crystallinity, DP, or feedstock source, no trend could be found in the yields of a single product. The results suggested that H-bonding and van der Waals forces do not play a significant role in the primary reactions of cellulose thermal deconstruction under fast pyrolysis conditions. Therefore, there is not yet a consensus on the effect of the DP and crystallinity of cellulose on LG yields. But the important effect of minerals on LG formation is a consensus.
Indeed, it is well known that inorganics (or nutrients, minerals, especially potassium, sodium, magnesium, etc., present naturally in biomass) have an important impact on cellulose pyrolysis product distribution due to their inherent catalytic effects.38,47–49
The fundamental effects of minerals (such as potassium, sodium, etc.) have been studied in detail both experimentally38,47–51 and by DFT modeling.52–54 Minerals catalyze the reactions that led to the formation of low molecular weight species from cellulose as compared to those leading to anhydro-sugars like LG. A mechanism explaining the effect of potassium on volatile product formation has been proposed (Fig. 5).55
 | ||
| Fig. 5 Simplified mechanism of cellulose pyrolysis highlighting the main degradation pathways and key volatile markers. Adapted with permission from ref. 55. | ||
Minerals inhibit LG formation by transglycosylation reactions and promote the formation of cyclopentenones by rearrangement (aldol reaction) and dehydration of carbohydrates.49,55 For this reason, it has been proposed to demineralize lignocellulosic biomass in order to increase the LG yield.38,56 Woody biomass presents a relatively small content in inorganics but this low content is sufficient to inhibit LG production. Several review articles on removing minerals from biomass and the corresponding effect on the pyrolysis of cellulose have been published.57,58 Only a brief example is provided here. It has been shown that leaching pinewood with an acid-rich bio-oil fraction prior to pyrolysis of the biomass effectively removed the alkali ions initially present in biomass.56 No significant consumption of acetic acid during the washing step was observed. Rinsing of the biomass after acid washing was required for maximal levoglucosan production, to remove the washing liquid containing the dissolved minerals. By this method, Oudenhoven et al. have produced 17 wt% LG (based on anhydrous wood mass) after demineralization of pine wood by the acid-rich fraction of the as-produced bio-oil.
The residence time of the reactive intermediate liquid in hot particles also considerably impacts the product formation and notably the LG yield. Indeed, LG is a highly reactive compound. Therefore its fast evacuation from hot particles is of tremendous importance in order to reduce its secondary reactions, notably to char. This important mechanism (char formation from liquid LG) is not captured by the simplified scheme as proposed in Fig. 3.28 It has been shown that char formation from cellulose highly depends on intermediate liquid (and LG) removal from the particle.23,25,26,28 The effect of sample size has been studied in detail, showing a decrease of LG yield and an increase of char yield when the particle thickness increases. The increase of biomass particle size reduces apparent heating rates, but also impedes the mass transfer of products from the inside of particles to their outer surface (and surrounding gas). Higher pressure in bigger particles may promote the ejection of the intermediate in the form of aerosols.26 The particle thickness plays a role in LG evacuation, but also the velocity of carrier gas surrounding cellulose should be high in order to promote the evacuation of volatiles.32
Once LG is evacuated from the particles, it can undergo further secondary reactions in the vapor-phase (or in liquid aerosols). For this reason, it is of high importance to design a fast pyrolysis reactor with a short residence time of gas in order to maximize LG mass yields.59 LG gas-phase conversion has been studied in detail.60,61 Methanol, acetaldehyde, glyoxal and furans are some secondary products of LG conversion.60 It is admitted that secondary reactions are low for a gas-phase residence time of about 1 s at 500 °C.
To summarize, best on the fundamental knowledge of cellulose pyrolysis, LG yield can be optimized by:
(1) the use of small particles (to promote both intra-particle heat and mass transfers);
(2) demineralization of woody biomass by acid leaching;
(3) fast heating rate (as in a fluidized bed reactor);
(4) a moderate reactor temperature of about 500 °C;
(5) a low residence time of vapours of about 1 s at 500 °C.
Despite the numerous studies on the fundamentals of LG formation, the works on the production of LG in a significant yield in bigger and continuous reactors are still scarce.22,38,48 Up to 38 wt% of LG (based on the mass of anhydrous cellulose) has been produced by Piskorz et al. in a continuous pilot fluidized bed at 0.5 s gas residence time and at 500 °C sand temperature on Avicel cellulose (with 5 mass % sulfuric acid at 90 °C for 5.5 hours).38 42 wt% (based on cellulose) has been reported by P. R. Patwardhan in a fluidized bed at 500 °C (1.3 s gas residence time).48
3. Purification of levoglucosan
One of the main challenges in the production of LG by cellulose pyrolysis is the further separation and purification of the obtained mixture in the pyrolysis oil. Generally LG is condensed with other cellulose pyrolysis products (aldehydes, furans, acids) into a viscous bio-oil. If real lignocellulosic biomass is used, other compounds from lignin and hemicelluloses are also present in the bio-oils. Pyrolysis bio-oils contain hundreds of organic compounds.62Due to its complex and reactive structure, LG is difficult to separate and purify. In addition, classically used methods such as liquid chromatography or solvent extraction are quite expensive and difficult to scale up for a further industrial application.11
The first purification stage is a staged condensation of vapours at the outlet of the pyrolysis reactor in order to selectively condense the compounds present in the vapour phase based on their boiling points. Therefore, the condensation system allows recovering the heavy oil rich in LG (for further conversion) separated from the light oil (aldehydes, alcohols, carboxylic acids).63,64 The light fractions can be valorised for other purposes, such as the pre-treatment of biomass for its demineralization in order to increase the LG yield after fast pyrolysis.56
The levoglucosan production was concentrated, by staged condensation, in a single fraction up to a concentration of 37 wt% in the first condenser oil by Oudenhoven et al.56 Various fractions with distinct chemical and physical properties were obtained by staged condensation by Pollard et al.63 The sugar fractions were recovered with the phenolic compounds as so called “heavy ends”. The condensed bio-oil with a rich LG fraction needs further purification for downstream chemical or biological conversion. In order to enhance the fermentation of the LG fraction by fungi and yeasts, LG rich bio-oils (produced from wood pyrolysis) have been purified in three different ways: (1) an aqueous extract (lignin removed); (2) activated charcoal treated (lignin and aromatics removed); and (3) acid hydrolysate (lignin and aromatics removed with the levoglucosan hydrolyzed to glucose). It was clear that yeast and fungi were capable of utilizing levoglucosan present in wood pyrolysate liquor after this detoxification of bio-oils.65
The differences in water solubility can be exploited to recover a sugar-rich aqueous phase and a phenolic-rich raffinate (formed from lignin pyrolysis). Rover has demonstrated that the sugars (mainly LG) were extracted effectively at over 93 wt% with two waterwashes.64
A vacuum pyrolysis process was operated between 1958 and 1970 at the Krasnodar hydrolysis plant (USSR) for the production of purified levoglucosan (75 kg h−1). Levoglucosan was crystallized using acetone. Detailed information on the exact process is however lacking.66 To the best of our knowledge, Brown et al. have presented recently the first study on the production of purified LG crystals by a combination of liquid/liquid extraction, adsorption and crystallization. This methodology is schematically represented in Fig. 6.11
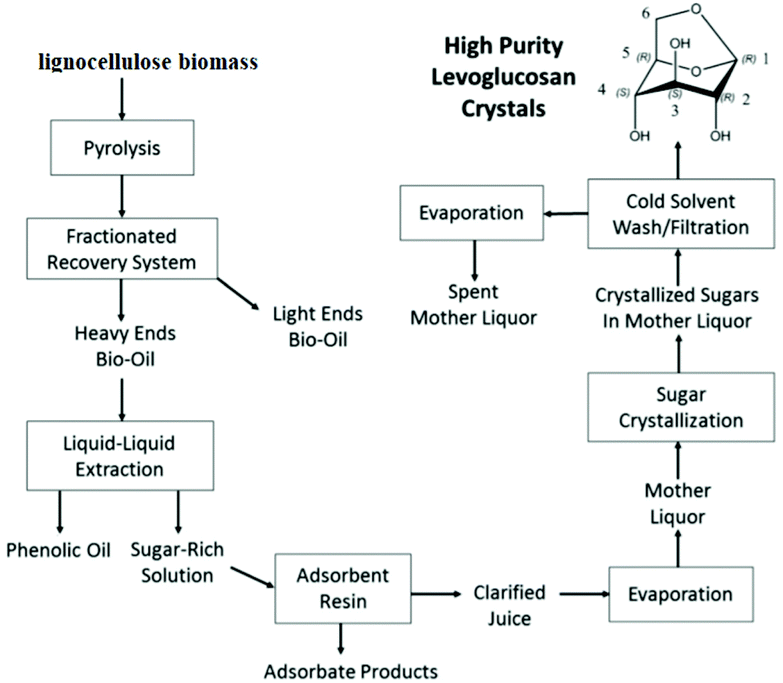 | ||
| Fig. 6 Methodology for the production and purification of LG from lignocellulosic biomass. Reprinted with permission from ref. 11. | ||
The combination between bio-oil fractionation, liquid–liquid extraction and filtration using acid resins yielded 81.2 wt% of total sugars. The LG part in this fraction was 44.7 wt%. The phenolic fraction was successfully removed using filtration with Sepabeads SP207 resins. After the solvent evaporation the crystallization permitted to obtain 24.7% of LG recovery from the mother liquor with a high purity level of 102.5% (±3.109% at the 99% confidence level). The authors performed also the techno-economic analysis. They indicated that high purity crystals of LG could be produced at a cost of $1333 per MT, which is much lower than the current market price for comparable LG purity crystals.11
The reduction of inhibitory compounds and enhancement of LG or the efficient separation between them have been the major challenges in bio-oil valorization. As stressed before, almost all kinds of pretreatment can lead to inhibitory compounds.67,68 The microbial growth is affected by the presence of inhibitors in reaction media, which also impact the fermentation steps. Phenolic compounds are considered as higher inhibitors due to their ability to interact with biological membranes.69 Furfural and HMF have also been described as potential inhibitors as synergic effects with acetic, formic and phenolic acids.70 Many articles have reported several detoxification methods to overcome these issues.71–73 These strategies can be physicochemical detoxification of hydrolysate, solvent extraction, microbial engineering and plant improvements for reduced lignin content.
Some strategies in adsorption methods have been studied aiming to remove inhibitors from bio-oil or its hydrolysate.65,74,75 Yu and Zhang76 after a screening of several adsorbents for the improvement of ethanol yield by bio-oil hydrolysate found that Ca(OH)2 for neutralization and diatomite for adsorption are the best systems, generating a concentration of 0.4 g ethanol per g glucose from Saccharomyces Cerevisiae 2.399. Zhuang et al.77 applied activated carbon to retain inhibitors after pyrolysis of cotton cellulose. The LG was applied as a substrate for direct fermentation for citric acid production by mutant Aspergillus niger CBX-209. The strain produced higher citric acid concentrations, comparable to that obtained from commercial LG (70 g L−1) in five days. Wang et al.78 also applied activated carbon (4 °C overnight) for the hydrolysate detoxification of bio-oil of loblolly pine, obtaining expressively decreases of furfural and acetic acid.
The overliming is also a current method applied to remove efficiently phenolic derivatives, furans and aldehydes from bio-oil. Lian et al.79 investigated the production of styrene by an engineered styrene-producing E. coli strain from the sugars detoxified by overliming (100 g L−1 pyrolytic sugar was added in 18.5 g L−1 solution of Ca(OH)2) and laccase from Trametes versicolor treatment. The modified strain was able to produce around 240 mg L−1 styrene from pure LG, similar to 251 ± 3 mg L−1 produced from glucose. Chi et al.80 studied the best conditions to improve the fermentability of pyrolytic sugars by engineered E. coli KO11+lgk. Among several systems the overliming with 18.5 g L−1 of Ca(OH)2 at 60 °C for 4 h was the best conditions. A NaOH overliming treatment has been also proposed and provided the most successful results with no removal of sugars during the treatment. Ethanologenic E. coli were able to utilize pyrolytic sugars at 2 wt% after NaOH overliming. However, this weight percent is too low to produce commercially viable ethanol and requires additional research.64 For this reason, further purification must be looked for.
Another simple method that has gained significant notoriety in the detoxification of bio-oil is solvent extraction whose main feature is to separate pyrolytic sugars and inhibitors by polarity difference through liquid–liquid extraction. Several authors have described the separation of inhibitors by extraction with linoleic acid, hexane, ethyl acetate, n-butanol and water. Among them, ethyl acetate is the most suitable to extract inhibitors of low polarity from bio-oil. Chang et al.81 found that on increasing the ratio between ethyl acetate and bio-oil most inhibitors could be extracted at an 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 ratio, an ethanol productivity of 0.93 g L−1 h−1 in batch fermentation was found for E. coli ACCC 11177. The efficiency of ethyl acetate was also pointed out by Lian et al.75 who found ethyl acetate to be the best solvent to extract phenolic compounds in the organic phase, leaving LG and acetic acid in the aqueous phase. The group of Luque et al.82 suggested that hydrolysis of bio-oil, followed by neutralization and extraction with ethyl acetate was the best system to generate glucose and remove the major amounts of inhibitors. Islam et al.83 obtained 8.62 g L−1 of ethanol from detoxified hydrolysate of waste cotton bio-oil. For the process the hydrolysate was adsorbed on activated carbon, neutralized with Ba(OH)2 and extracted with ethyl acetate. Besides ethyl acetate, n-butanol has been showing expressive results on the extraction of phenolic compounds, as evidenced by the group of Hassan et al.,84 who found that a proportion of 1.8:1 between n-butanol and the aqueous phase of bio-oil was able to remove furfural, HMF and acetic and formic acid. Chan and Duff85 when investigating the most suitable solvent system for extraction of bio-oil hydrolysate found that a mixture of tri-n-octylamine in 1-octane (25% v/v) removed more than 90% of acetic acid and retained all amount of glucose, expressing high selectivity. The combination of solvents was also explored by Wang et al.86 where the mixture of hexane and linoleic acid could reduce furfural by 61% and acetic acid by 61%.
:0.5 ratio, an ethanol productivity of 0.93 g L−1 h−1 in batch fermentation was found for E. coli ACCC 11177. The efficiency of ethyl acetate was also pointed out by Lian et al.75 who found ethyl acetate to be the best solvent to extract phenolic compounds in the organic phase, leaving LG and acetic acid in the aqueous phase. The group of Luque et al.82 suggested that hydrolysis of bio-oil, followed by neutralization and extraction with ethyl acetate was the best system to generate glucose and remove the major amounts of inhibitors. Islam et al.83 obtained 8.62 g L−1 of ethanol from detoxified hydrolysate of waste cotton bio-oil. For the process the hydrolysate was adsorbed on activated carbon, neutralized with Ba(OH)2 and extracted with ethyl acetate. Besides ethyl acetate, n-butanol has been showing expressive results on the extraction of phenolic compounds, as evidenced by the group of Hassan et al.,84 who found that a proportion of 1.8:1 between n-butanol and the aqueous phase of bio-oil was able to remove furfural, HMF and acetic and formic acid. Chan and Duff85 when investigating the most suitable solvent system for extraction of bio-oil hydrolysate found that a mixture of tri-n-octylamine in 1-octane (25% v/v) removed more than 90% of acetic acid and retained all amount of glucose, expressing high selectivity. The combination of solvents was also explored by Wang et al.86 where the mixture of hexane and linoleic acid could reduce furfural by 61% and acetic acid by 61%.
Due to its polarity, safety and economic viability, water has also been widely used to remove inhibitors in crude or hydrolysed bio-oil. Aqueous systems have been applied with relative ease in the precipitation of compounds of reduced polarity, such as some phenolic derivatives.87,88
Several proportions between water and bio-oil have been reported, as well as the application of different temperature ranges. Bennett et al.89 found that a proportion between water–bio-oil 62:100 at 34 °C and 22 min of extraction was able to concentrate LG up to 87 g L−1 Li et al.90 also optimized the water extraction conditions to concentrate LG of bio-oil from Loblolly Pinewood. High yields of LG (>12 wt%) were obtained with a 1.3:1 water–bio-oil ratio for 20 min at 25 °C. In the experiments of the group of Chan and Duff,85 water was crucial to achieve a LG yield of 4.98 wt% from bio-oil. Lian et al.74 applied different temperatures for aqueous extraction of LG from bio-oil, where more than 93 wt% of sugars in bio-oil were extracted by coupling hot water extraction at 60 °C for 1 h and further extraction with cold-water at 4 °C overnight. Sukhbaatar et al.91 attested that a system of bio-oil with a 1:1 ratio was not selective for isolation of LG from the aqueous phase, but was an efficient combination to separate water-soluble constituents from other inhibitors. Wang et al.92 proposed a combination of strategies for LG purification where after bio-oil adsorption with activated carbon, water extraction and overliming with Ca(OH)2 and Soxhlet extraction, LG was recovered from the straw bio-oil with a yield of 78% and >95% of purity.
As we showed above, the catalytic valorization of LG is highly dependent on the by-products present in the pyrolysis oil. To the best of our knowledge, there is still an important lack of study on the effect of by-products and LG purity on the deactivation and selectivity of catalysts.
4. Biotechnological transformations of levoglucosan
The LG present in the bio-oil can follow the fermentation pathway for the direct production of ethanol, and lipids, or be hydrolyzed to glucose as a carbon source for several microorganisms for the production of high-added value compounds such as organic acids, solvents and biopolymers. In all cases, several detoxification methods have been applied.93 Several papers have reported the most diverse types of pre-treatment so that lignocellulosic biomass can provide the highest possible yields of LG. In fact, the kind of pre-treatment applied has an influence on the final characteristics of the biomass to be pyrolyzed, impacting the process both economically, in the downstream stage, as well as in the efficiency of the pyrolytic process applied in obtaining the final LG, since the appropriate pre-treatment method can increase the yield of the desired product in addition to reducing the amount of the final inhibitory by-products in the bio-oil.94 With the increase in the generation of residual lignocellulosic biomass coupled with the advances in the development of cost-effective pre-treatment and pyrolysis processes nowadays, increased yields in LG have been achieved.89,95 This approach, albeit in a subtle way, has aroused the interest of researchers in the development of processes for the full use of this anhydrous sugar.96 The LG present in the bio-oil can be reused directly, through fermentative processes using microorganisms able to directly assimilate LG as a source of organic carbon. For this, the presence of the enzyme LG kinase (LGK), present in some filamentous fungi and yeasts and LG Dehydrogenase (LGDH) present in some bacteria strains in a wild form is essential, or should be acquired, through protein engineering tools.97–100 Another suitable approach is the hydrolysis of LG to glucose, through chemical or enzymatic catalysis, where the product can be fully assimilated by different strains and culminates in the production of several compounds of biotechnological interest. For these purposes it is essential the entire process of obtaining LG is efficient in removing microbial metabolism inhibitors. Finally, LG can also be separated and applied in biotechnological routes for the production of several compounds with high added value, either directly or through conversion to glucose and other sugars by the application of chemical, and enzymatic catalysts, or by hybrid catalysis.4.1. Direct fermentation of LG from bio-oil
Although the direct application of bio-oil as a microbial substrate is both an economically and environmentally favorable approach, there are many challenges to overcome, both on laboratory or industrial scales, since several metabolic inhibitors may be present. Therefore, the coupling between biomass pre-treatment and the subsequent detoxification of the bio-oil may influence the assimilation of sugars by different microbial strains, in addition to the biotransformation of phenolic compounds. Besides, the correct understanding of inhibitors’ effect on growth and fermentation is necessary for the choice of the more adequate detoxification system to be applied in each matrix.Among all classes of inhibitors, furan derivatives and carboxylic acids are typical hinderers of microbial growth, especially fungi and yeasts, where furfural, HMF and acetic acid when in critical concentrations are able to change the pH of the medium and the solubility of fermentable sugars, generating cellular stress as well as decreasing the growth rate and suppression of the activity of key-enzymes, culminating in reducing the productivity of the metabolites of interest.101 In addition, several studies have reported that some phenolic compounds are more toxic to microbial cells, where mainly phenolic derivatives of low molecular weight are able to penetrate through the microbial membrane, causing osmotic stress and disturbing the balance of the lipid bilayer, generating cell death.102 There are also several potentially inhibitory compounds whose mechanisms of action have not yet been elucidated, despite knowing that they can act in a synergistic or antagonistic way with those previously mentioned. Thus, several bio-oil detoxification techniques must be applied in order to improve efficiency in the fermentation stage.
4.2. Metabolic engineering for levoglucosan assimilation
Although most microbial metabolism inhibitors can be removed from the medium by various detoxification methods, or even be avoided according to the type of pretreatment applied to the biomass before the pyrolysis process, these approaches are not selective for all inhibitors. In addition, the coupling of pre-treatment and detoxification techniques can increase not only the number of steps, but also the final costs, in addition to leading to a non-ecologically favorable approach. Thus, understanding the interaction between inhibitors and microbial metabolism has been a current field of study. The construction of strains resistant to high concentrations of inhibitors can also promote improvements in the metabolic systems of fungi, yeasts and bacteria for better direct uptake of LG and introduce LG into the glycolytic pathway for the production of the most diverse products of interest.The microbial metabolism of LG was first described in 1991, with the identification and isolation of the enzyme LG kinase (LGK) from the yeast Sporobolomyces Salmonicolor103,104 in a screening of several species of yeasts and fungi that had previously been shown to be functional for LG assimilation. From then on, several strains capable of assimilating LG directly have been isolated in recent decades including yeasts (genera Candida, Rhodotorula, Saccharomyces, Cryptococcus and Schwanniomyces), fungi (genera Geotrichum, Asperigillus, Neurospora, Penicillium, Phanerochaete, Alternaria, Eupenicillium) and bacteria (genera Arthrobacter, Escherichia and others).65,97,103–107
The metabolic pathways of LG differ between prokaryotes and eukaryotes. On bacterial systems, LG can be converted into glucose in three different steps (Fig. 7): NAD+-dependent dehydrogenation by the enzyme levoglucosan LG dehydrogenase (LGDH) leading to the production of 3-keto-levoglucosan LG (1,6-anhydro-β-D-ribo-hexopyranos-3-ulose, 3-keto LG), intramolecular hydrolysis producing 3-keto D-glucose, and NADH-dependent reduction, leading the production of final D-glucose.108,109
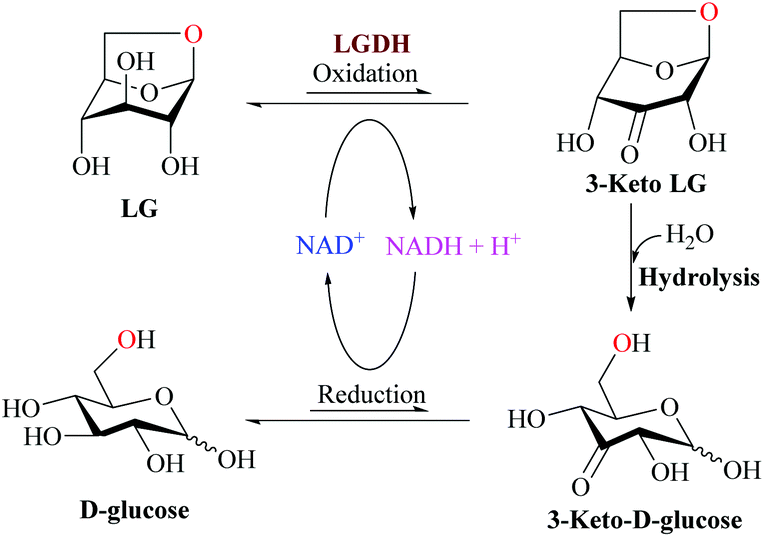 | ||
| Fig. 7 Proposal of LG metabolism by bacterial systems. Adapted from ref. 108. | ||
Although the glucose biosynthesis is already well described, the whole bacterial LGDH pathway is still unknown as the target genes for the metabolic pathway have not been completely identified. Recently Sugiura et al.108 identified and cloned the lgdh gene from the bacterium Pseudarthrobacter phenanthrenivorans and characterized the recombinant LGDH. The enzyme showed weak activities (4%) toward L-sorbose and 1,5-anhydro-D-glucitol. The reverse (reductive) reaction using 3-keto-levoglucosan and NADH exhibited significantly lower Km and higher kcat values than those of the forward reaction. The crystal structures (Fig. 8) of LGDH revealed that LGDH has a typical fold of Gfo/Idh/MocA family proteins, similar to those of scyllo-inositol dehydrogenase, aldose–aldose oxidoreductase, 1,5-anhydro-D-fructose reductase, and glucose–fructose oxidoreductase.
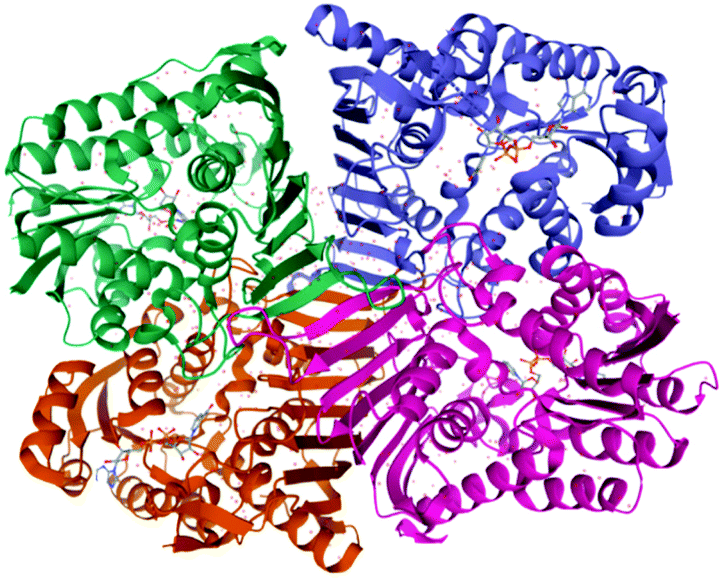 | ||
| Fig. 8 3D structure of LGDH from Pseudarthrobacter phenanthrenivorans evidencing a biological tetramer structure. Data obtained from PDB.110,111 | ||
The LGDH active site extensively recognized the LG molecule with six hydrogen bonds, and the C3 atom of LG was closely located to the C4 atom of NADH nicotinamide, evidencing a C3-specific oxidation for the first time.108
Despite the recent advances in the LGDH pathway, the eukaryotic system for LG metabolism is well established. Biochemical studies have indicated that the metabolism of LG in fungi and yeasts is initiated by the direct phosphorylation to glucose 6-phosphate by LGK followed by the glycolytic pathway (Fig. 9).97,112
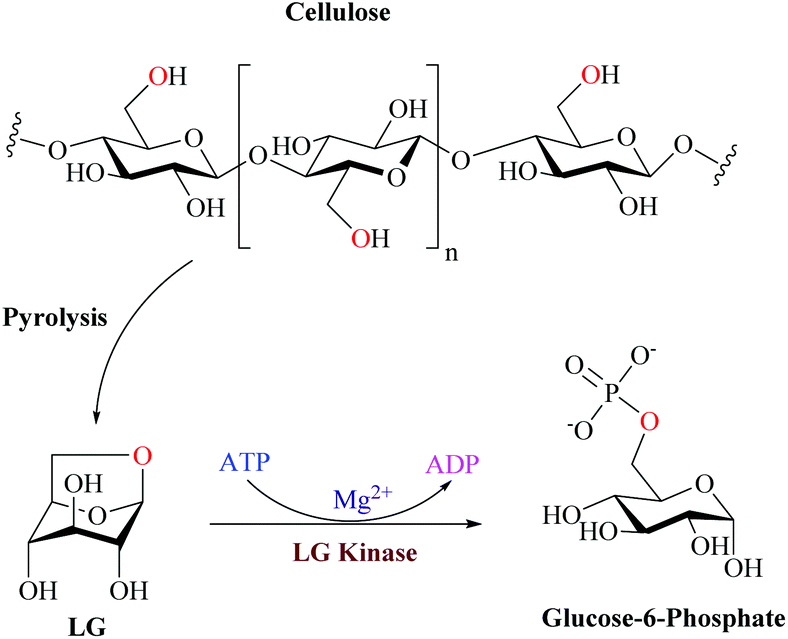 | ||
| Fig. 9 Eukaryotic enzymatic cleavage and phosphorylation through the action of LGK to produce G6P. Adapted from ref. 112. | ||
Previous studies of LGK substrate selectivity demonstrated activity for only LG and no activity for galactosan or maltosan and only very weak activity for mannosan (1% relative activity) that also contain the same 1,6-anhydro intramolecular linkage as LG, demonstrating that the nature of the pyranose frame is also important for substrate recognition by LGK.104 Some biotechnological products have already been obtained by wild type strains which evidenced the presence of LGK coupled to intrinsic metabolic ability: organic acids such as itaconic, malic and citric were obtained directly from LG by Aspergillus terreus, Aspergillus oryzae and Aspergillus niger CBX-209 respectively.77,107,113,114 Lian et al.74 achieved a similar level of lipid accumulation to glucose fermentation by LG as a substrate for Rhodotorula glutinis ATCC204091 and Rhodosporidium Toruloides ATCC10788.
Despite the previous promising results, wild types strains are not more efficient than industrial biocatalysts in LG uptake. The concentrations of LG assimilated by these species are highly inferior to those of glucose as a substrate, demonstrating that there is competition between metabolic pathways. Furthermore, mainly in bacterial systems, LG transporters and the mechanisms of inhibition of the LGDH metabolic pathway are still unknown. Due to this, traditional industrial strains have been targeted for metabolic engineering approaches.115 In the research of Lian et al.,79 engineered E. coli produced 240 mg L−1 styrene from pure LG (Fig. 10), which was similar to that from glucose (251 mg L−1).
 | ||
| Fig. 10 Production of styrene from LG by engineered E. coli. Adapted from ref. 79. | ||
Codon-optimized Lipomyces Starkeyi LG kinase was expressed in E. coli KO11 for effective pure LG utilization to convert detoxified pyrolytic sugars to ethanol.80,116 However, the experimental yield was much lower than the theoretical yield. Kim et al.117 expressed lgk gene from Lipomyces Starkeyi in Corynebacterium glutamicum for succinic acid production from LG, giving a higher yield (0.25 g succinate per g LG) from LG as compared to that from glucose. Although in the last few years the mechanisms of LGK have been discovered and several molecular techniques have facilitated protein engineering, the limited LG assimilation and the persistent low catalytic activity of LGK on engineered strains are still challenging. Furthermore, a rational multifaceted strategy that combines structural, biochemical and protein engineering efforts is needed to improve LG consumption in organisms. The group of Klesmith et al.118 described the method of FluxScan to enable the mapping of sequence determinants to the pathway of LG assimilation in E. coli strains. As results were mapped 15 beneficial mutations supported a 15-fold improvement in growth rate and a greater than 24-fold improvement in enzyme activity relative to the starting pathway.
Numerous studies have been reported that the combination of hydrolysis of lignocellulosic bio-oil and subsequent fermentation to ethanol is feasible. This is possible because through this application, glucose is released not only from LG, but from other oligomers resulting from pyrolysis, in addition to the release of other fermentable sugars. Chang et al.81 found that the fermentation of bio-oil by E. coli ACCC 11177 gave an ethanol yield of 0.41 g g−1 and a productivity of 0.93 g L−1 h−1. Yu and Zhang76,95 reported that the diluted acid hydrolysate of bio-oil (containing 31.6 g L−1 glucose) was fermented by S. Cerevisiae, giving an ethanol concentration of 14.2 g L−1 with a higher yield than the pure glucose control. Bennett et al.89 found that under the optimal hydrolysis conditions (125 °C, 0.5 M H2SO4 and 44 min), the hydrolysis of LG (extracted from a softwood bio-oil via phase separation) gave a maximum glucose yield of 216%. The yield of 216% based on the original levoglucosan proved that other precursors of glucose were also present in the aqueous phase during hydrolysis. Without any detoxification process, S. Cerevisiae T2 successfully fermented the hydrolysate to ethanol with both high yield (0.46 g ethanol per g glucose) and high productivity (0.55 g L−1 h−1). The yield was comparable to the theoretical yield from pure glucose (0.51 g ethanol per g glucose). In the research of Sukhbaatar et al.91 the hydrolysis of bio-oil (76.6 g L−1 LG) was performed under the optimal hydrolysis conditions (125 °C, 0.5 M H2SO4, 44 min).89 The hydrolysate rich in glucose (114.19 g L−1) was fermented by Saccharomyces patorianus, obtaining a considerably high ethanol yield (98% of the theoretical yield). In the work of Lian et al.,75 after acid hydrolysis and detoxification, the aqueous phase was converted to ethanol with a yield of 0.473 g ethanol per g glucose by S. cerevisiae, and to lipids with a yield of 0.167 g lipids per g glucose by Cryptococcus curvatus and Rhodotorula glutinis.119 The maximum ethanol concentration (32 g L−1) and the yield of ethanol (0.473 g ethanol per g glucose) were very close to those from pure glucose (35 g L−1 and 0.50 g ethanol per g glucose), indicating that the hydrolysate of bio-oil was an attractive substrate for fermentation.
The integration between the pre-treatment of biomass, pyrolysis, and detoxification techniques, followed by fermentation with wild or genetically modified strains has been used by researchers to obtain biotechnological products. In fact, ethanol is still the most obtained bio-product. However, lipids and organic acids have also been reported (Table 1). In a recent publication,120 the main products obtained through this combined approach were compiled.
| Feedstock | Pretreatment | Detoxification method | Strain | Scale of the process | Bioproduct | Yield | Ref. |
|---|---|---|---|---|---|---|---|
| Pinewood particles | H3PO4 solution, 100 °C, 1 h | Extraction with n-butanol, hydrolysis with H2SO4, neutralization with NaOH and CaCO3, filtration reverse osmosis | Saccharomyces pastorianus | Flasks | Ethanol | 0.5 g of ethanol per g of glucose | 90 |
| Poplar wood | Acid wash | Extraction with ethyl acetate and biodiesel, hydrolysis with H2SO4, neutralization with Ba(OH)2 and detoxification with activated carbon | Saccharomyces cerevisae ATCC 00062 | Flasks | Ethanol | 0.473 g of ethanol per g of glucose | 74 |
| Bio-oil | — | LG hydrolysis with H2SO4 and overliming with Ca (OH)2 | Saccharomyces cerevisae T2 | Flasks | Ethanol | 0.19 g ethanol per g glucose at 50% volume hydrolyzate and 0.45 (g ethanol per g glucose) at 40% volume hydrolyzate | 84 |
| Loblolly pine | Acid pretreatment | Water extraction, hydrolysis with H2SO4 and activated carbon | Saccharomyce pastorianus ATCC 2345 | Flasks | Ethanol | 0.4 g of ethanol per g of glucose; 79% yield | 120 |
| Waste cotton | — | H2SO4 (0.2 M) hydrolysis, Ba(OH)2 neutralization, ethyl acetate extraction, activated carbon | Saccharomyce cerevisiae 2.399 | Stirred fermenter | Ethanol | 14.78 g L−1 of ethanol | 121 |
| Waste cotton | H2SO4 hydrolysis, Ba(OH)2 neutralization, ethyl acetate extraction | Escherichia coli ACCC 11177 | Fermenter | Ethanol | 0.41 g ethanol per g glucose | 80 | |
| Red oak wood | Water extraction, overliming with NaOH | Escherichia coli | Flasks | Ethanol | 0.9 g L−1 ethanol | 122 | |
| Pure LG | — | — | Engineered Escherichia coli KO11 | Flasks | 0.35 g ethanol produced per g LG | 115 | |
| Red oak wood | — | Fractionating bio-oil, water extraction, Ca(OH)2 treatment | Engineered Escherichia coli KO11 | Flasks | 0.24 g ethanol per g LG | 79 | |
| Cotton cellulose | — | — | Aspergillus níger CBX 209 | Flasks | Citric acid | 87.5% of citric acid citric acid | 76 |
| LG | — | — | Engineered Escherichiacoli | Flasks | Styrene | 0.24 g L−1 styrene; 0.021 g g LG | 118 |
| Pure LG | — | — | Rhodotorula glutinis | Flasks | Lipid | R. glutinis: 2.7 g lipid per L medium (20 g L−1) | 73 |
| Poplar wood | LGA extracted with ethyl acetate, hydrolysis with H2SO4, neutralization with Ba(OH)2, detoxification with activated carbon | Cryptococcus curvatusor R. glutinis | Flask | Lipid | C. curvatus: 0.167 g lipids per g sugar; R. glutinis 0.089 g lipid per g glucose | 74 |
4.3. Applications of pure LG
Currently, several studies have turned their attention to the application of pure LG in different approaches for the production of high added value compounds. In fact, the use of LG in the absence of pyrolysis inhibitors and by-products may provide new biotechnological possibilities. However, the molecule also imposes barriers. LG is not highly hydrophilic, its dissolution in organic solvents usually applied in the organic synthesis can be limited.121 In addition, it is already known that LG has instability in some organic solvents, leading to the formation of side products, decreasing the efficiency of some catalytic systems. Hu et al.122 analyzed the effect and the influence of several solvents on the stability of LG during acid catalysis at 70 °C using Amberlyst 70, and demonstrated that the nature of the solvent strongly impacts the product distribution and polymer formation in acid-catalyzed conversions. Alcohols could inhibit the growth of soluble polymers, different from water. Chloroform is not a suitable solvent, although acetone reacted with LG, resulting in polymerization. DMSO catalyzed the conversion of a complex series of derivatives of LG (Fig. 11) including HMF, the corresponding sulfur and 2,5-furandicarboxaldehyde, that according to Climent et al.123 is a platform molecule for value-added chemicals. | ||
| Fig. 11 Possible pathways of LG in DMSO. Definition of the abbreviation: DAGP: 1,4:3,6-dianhydro-α-D-glucopyranose; HMF: 5-(hydroxymethyl) furfural; MMFC: 5-[(methylthio) methyl]-2-furancarboxaldehyde. Adapted from ref. 122. | ||
Although the aqueous medium is favorable for LG and enzyme systems, some transformations are also limited due to the low solubility of hydrophobic substrates. Thus, medium polarity solvent systems, ionic liquids, application of immobilized enzymes and catalysts resistant to drastic conditions are the strategies currently used to counteract these barriers in LG biocatalysis.
Sugar esters are notably known as good biosurfactants and biolubricants, with several industrial applications. The esterification reaction of several sugars has been performed both in chemical and enzymatic approaches.124 By biocatalysis, esterification and transesterification reactions constitute another major class of enzymatic biomass processing. In general, lipases (triacylglycerol hydrolases, EC 3.1.1.3) are the most suitable class of enzymes. Among some advantages, lipases show activity in organic solvents, do not need co-factors, are enzymes abundant in nature and catalyze a reaction with high region-, chemo- and enantioselectivity.125 In addition, because they recognize a high range of substrates, they can be modeled through immobilization or metabolic engineering tools to adapt to reaction conditions. The large amount of hydroxyl groups makes sugars very polar, making them difficult to dissolve with long carbon chain acyl donors.
The first work involving the esterification of LG was published in 1973 where among several sugars, LG was esterified with different acid chlorides or acid anhydrides in pyridine. As a result we evidenced that in the reaction medium LG reaches different conformations, leading to the formation of intramolecular hydrogen bonds, resulting in different hydroxyl reactivities, generating different products according to the acid chloride used.126 The same rules were found in the work of Ward and Shafizadeh.127 However, different from the previous work, linear chain acyl donors (octanoyl and hexadecanoyl chloride) were used as reagents. For this reason, there was less steric impediment between the reagents, generating mixtures of mono and diesters as well as regioisomers.
Due to these characteristics, just a few papers have been reported on the enzymatic acylation of LG. However, in LG the positions 1 and 6 present anacetalic function and the remaining secondary hydroxyl groups are in the axial position. Therefore, LG is much less reactive than glucose or other sugars both to chemical or enzymatic attack. This peculiar feature can be extended to the regioselective acylation of a single secondary OH function that represents a very challenging task since the presence of three hydroxyls in its structure allows the formation of monoesters and diesters at positions C2, C3 and C4.128 The pyranose ring of LG can adopt two conformations: inverted chair and boat type.129 In the chair conformation, the intramolecular interactions, due to hydrogen bonding occurring between the hydroxyl of C3 in the axial position and the oxygen in C6, also in axial, contribute to a greater stabilization of the molecule, increasing its population, but decreasing its reactivity as a nucleophile.99,130 The same interaction, hydrogen donor–acceptor, can occur with the hydroxyls in C2 and C4, according to Fig. 12A1–A3 (Fig. 12).
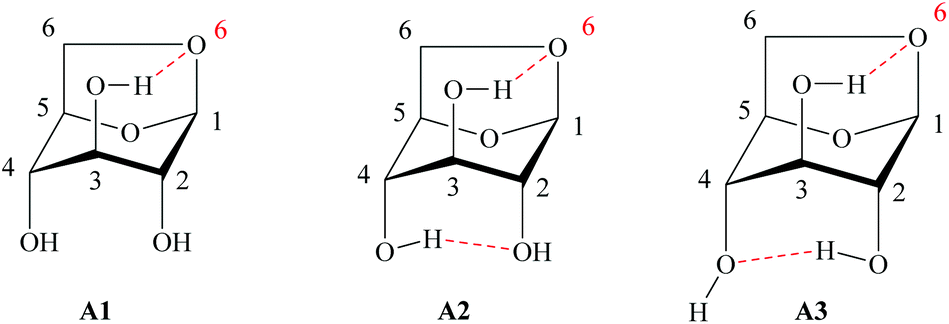 | ||
| Fig. 12 Possible conformational structures for LG in the axial position.99,130 | ||
Galletti et al.129 studied the lipase-catalysed acylation of LG by esterification and transesterification reactions with a wide range of acyl donors and both free and immobilized lipases. As green solvents CH3CN and ionic liquids were investigated and reaction times of 5 days were also applied. The commercial lipases Novozym 435 (lipase B from Candida Antarctica immobilized on acrylic resin) and PS (lipase from Burkholderia cepacia) were more suitable biocatalysts (Fig. 13).
 | ||
| Fig. 13 Lipase-catalysed acetylation of LG in CH3CN and ionic liquids with vinyl acetate and acetic acid. Adapted from ref. 100 and 129. | ||
On LG acetylation in CH3CN (Fig. 13), the product 4-O-acetyl LG (blue structure) was the major compound obtained with all tested immobilized lipases. By transesterification, higher selectivities were found when the ionic liquid 1-methoxyethyl-3-methylimidazolium in dicyanamide ([MOEMIm] [dca]) was added to the system. It is also important to note that the water formed by the esterification reaction was adsorbed through molecular sieves. Because acetate is a small soluble molecule in solvents of medium polarity, there was great diffusion through the LG molecule, which may explain the mixture of compounds obtained. However, in larger carbon chains, the best results were only obtained by transesterification, indicating hysterical and solubility limitation. However, no assay to detect the presence of diesters has been performed. Recently our research group investigated the LG acylation by transesterification with ethyl esters of long chain (laurate, oleate and palmitate) catalyzed by Novozyme® 435, PS IM (immobilized lipase from Burkholderia cepacia) and the homemade biocatalyst CaLB_epoxy (lipase CaLB immobilized on epoxy resin by hydrophobic adsorption).131 As a result, all biocatalysts generated mostly monoesters, where N435 was more selective to produce lauric esters (99% at 50 °C) and PSIM to produce oleic esters (97% at 55 °C) while CaLB_epoxy was more selective to produce oleic esters of LG (83% at 55 °C) (Fig. 14). In this work the final products were carefully identified by NMR: 1H, 13C, HMBC, HSQC, 1H,1H-COSY (“Correlation Spectroscopy”) and NOEdiff (NOE “difference spectroscopy”) spectra, FTIR spectrum ν (cm−1, KBr), HRMS (High-Resolution Mass Spectrometry) and GC–MS. These experiments clearly showed that there were marked differences between reactivities on C2 and C4 hydroxyl groups and also the type of binding and the support chosen for enzyme immobilization can influence the selectivity of the enzyme so that the hydroxyl groups can be selectively transesterified, since CaLB_epoxy and N435 have the same enzyme (CALB), but immobilized on different supports, generating different interactions between the enzyme and support and consequently different spatial conformations, revealing different preferences in the reaction under study.131
 | ||
| Fig. 14 General procedure for lipase-catalyzed transesterification reaction of LG (LG). Conditions: 2 ethyl esters of a: lauric, b: palmitic, c: stearic, and d: oleic acids, LG monoester aliphatic chains, and 4 ethanol.131 | ||
5. Levoglucosan catalytic transformation
The hydrolysis of LG generates the fermentable sugar glucose, and therefore the lignocellulosic material has great potential as a renewable feedstock. Moreover, due to its peculiar structure, LG can also follow other important reactions for the obtaining of several high valued products. In the following topics we have discussed the catalytic and biocatalytic transformations of LG.In recent years, economic and environmental concerns have encouraged the use of heterogeneous catalysts to carry out various organic transformations, as they facilitate catalytic separation of products, are less vulnerable to contamination and allow easy regeneration.2,132 Thus, in general, these catalysts make processes clean, safe, highly efficient and low cost. LG is a platform molecule that permits to obtain various high value-added value products as discussed below.
During the pyrolysis of cellulose, two anhydrous sugars could be obtained: LG and LGO. The yield of LG is generally much higher than that of LGO as discussed above. LGO has been a highly desired molecule as it is the intermediate of numerous important compounds (Fig. 15).
 | ||
| Fig. 15 Possible products obtained from LGO. | ||
Thus, direct catalytic transformation of LG to LGO (Fig. 16) instead of performing selective pyrolysis directly to LGO (low yield) is of high importance.
 | ||
| Fig. 16 Schematic reaction pathways of LG dehydration using heterogeneous catalysts. | ||
Recently Oyola-Rivera et al. showed that propylsulfonic acid functionalized silica materials significantly increase the LGO yield.133 They showed that Brønsted acid catalysts permit to obtain up to 59% of LGO yield. The synthesis of LGO via dehydration reactions was already studied in the past, but almost exclusively using homogeneous acids. Sulfuric acid was found to be very efficient in the conversion of cellulose to LGO in aprotic solvents.7,133,134 The authors compared the catalytic performances of various homogeneous acids: formic, hydrochloric, sulfuric and phosphoric acids. The best results in terms of LGO yield were obtained using sulfuric acid as a catalyst. As expected the catalytic results depended strongly on the reaction medium. Polar aprotic solvents were studied and THF was found to be the most efficient. This was attributed to the lower degradation rate in this solvent.133 Sulfuric and 1-propanesulfonic acids as well as propylsulfonic acid functionalized silica were also studied by the group of Cardona-Martínez.133 A LGO selectivity of 68% was obtained using the former catalyst. They stated that the water production during the dehydration reaction strongly affects the catalytic activity of the acid sulfonic groups due to the acid proton stabilization. However, they showed that this negative effect could be minimized by using a solid acid catalyst with hydroxyl groups on the surface. This permits to obtain higher LG conversion and LGO selectivity.133 The authors used relatively harsh conditions (210 °C and 69 bar He) that should limit the industrial implementation of this process. The new catalysts able to work at atmospheric pressure and lower temperature are needed.
LG is an example of an anhydrosugar, and thus it could be easily converted to glucose by acid-catalyzed hydrolysis. The hydrolysis of LG to glucose follows a first order reaction, with the activation energy of 114 kJ mol−1. The most used mineral acid for the hydrolysis of LG is sulfuric acid.135 It was found that the conversion and LG yield strongly depend on the temperature and sulfuric acid concentration (Fig. 17).
 | ||
| Fig. 17 Effect of the sulphuric acid concentration of kinetics of LGO synthesis. Hydrolysis at 120 mM of H2SO4 (■), 250 mM of H2SO4 (▲) and 500 mM of H2SO4 (●). All experiments performed at 110 C. Reprinted with permission from ref. 135. | ||
The conversion of LG to GLC in water using solid and homogeneous acid catalysts such as Amberlyst 16 and sulfuric acid is also possible. Abdilla-Santes et al.136,137 studied the effects of reaction temperature, initial LG concentration and catalyst loading. The highest glucose yield of 98% was obtained for the Amberlyst 16 catalyst at 115 °C.136 The authors studied also the kinetic parameters of this reaction using a first order approach and taking into account the diffusion limitations inside of the Amberlyst pores. The activation energy was calculated to be 132.3 ± 10.1 kJ mol−1.
Acid catalysts were also used by other groups to study the conversion of LG to various products. Murzin's group138 studied the H-MCM-36 mesoporous and H-MCM-22 microporous materials. The yield of the oxygenated products and their distribution were strongly influenced by the acidity of the zeolite materials. Oxygenated compounds were the main liquid products (aldehydes and furfural) and their formation was higher over MCM-36 than over MCM-22.138 Only acetone formation was higher over MCM-22. The severe deactivation of the catalyst was the most important issue. However, as stated by the authors, the possible regeneration of the catalysts permits to restore the good activity.138
Wang et al.139 studied the synergetic effect of a Lewis acid and base on LG conversion to lactic acid. The authors prepared a series of acid–base catalysts based on Sn-β catalysts exchanged with various alkali cations (Na, K, Mg, Ca). They showed that the exchange with alkaline earth cations is effective in promoting the retro-aldol condensation. Lewis acids can stabilize the oxygen atom of the deprotonated alkoxide. As demonstrated by the authors, the lactic acid yield can reach up to 66% on the Sn-β-Ca catalyst. The activity of the modified catalyst follows the order: Sn-β < Sn-β-Na < Sn-β-K < Sn-β-Mg, Sn-β-Ca. They demonstrated also that this synergetic effect shows universality toward enhanced retrol-aldol condensation.139
Lactic acid was also prepared by a simple universal method using a La(OTf)3 water resistant Lewis acid.140 They demonstrated that under moderate conditions (250 °C, 1 h) levoglucosan was transformed to lactic acid with the total yield was 75%. This yield was comparable to that obtained with other sugars such as glucose and xylose (74 and 64% respectively). The real advantage of this method is that no hydrolysis step is needed before the levoglucosan conversion to lactic acid.
Sorbitol is one of the 12 major building blocks according to the US Department of Energy.141 The global demand for sorbitol is growing at about 2–3% per year and reached over 2.5 million metric tons in 2018.142 It is widely used as an additive in many industrial products, particularly in the food, cosmetics and paper industries, and in the synthesis of various fine chemicals. Over the past decade, sorbitol has also been proposed as a feedstock for the production of biofuels and hydrogen by water-phase reforming. This polyol is generally produced by the catalytic hydrogenation of glucose.143 However, it could be also obtained with high yield from LG using heterogeneous catalysts. During this reaction two reactions are important: hydrolysis of LG to glucose and the hydrogenation of glucose to sorbitol. Hydrogenolysis and the formation of smaller diols such as glycerol and propylene glycol from sorbitol should be avoided, as well as subsequent acid-catalysed conversions of glucose to HMF and levulinic acid. Even if the acid hydrolysis of the LG is already well documented in the literature, a highly corrosive medium and post-reaction neutralization steps are required to remove the acid. Thus it is important to develop a heterogeneous catalyst for the one-step conversion of LG to sorbitol without using additional homogeneous catalysts.144
Only a few studies on the LG hydrogenation have been reported.144,145 Bindwal et al.146 reported that LG could be hydrogenated to ethylene glycol (4.7 wt%), 1,2-propanediol (28.6 wt%) and 1,4-butanediol (1.8 wt%) in water (140 °C, 20 bar H2 pressure) using a Ru based catalyst. However, in their study sorbitol was not detected, which strongly indicated that hydrogenolysis is the main reaction pathway.144 Indeed, in the proposed reaction pathways the first step is the hydrolysis of LG to glucose on acid sites, followed by selective hydrogenation of glucose. The subsequent hydrogenolysis of sorbitol to lower diols could be also observed. The authors used supported bifunctional catalysts possessing both acidic sites (hydrolysis reaction) and noble metal particles (hydrogenation step) and observed excellent performance of the Ru/CMK-3 catalyst. This efficacy was due to the low rate of secondary hydrogenolysis reactions forming low-molecular weight polyols.
Heterogeneous bifunctional catalysts have also been applied for the direct conversion of disaccharides to sugar alcohols. In this case, the disaccharide first hydrolysed to a monosaccharide and then hydrogenated on metal active sites to the corresponding sugar alcohol. A strong analogy with the proposed pathway for sorbitol synthesis from LG could be proposed. The CMK-3 support applied for the hydrogenation of LG is characterized by a large surface area and high acidity due to the highly oxygen-functionalized surface.144 The proposed mechanism for the selective hydrogenation of LG to C6 polyols is given in Fig. 18. The main route involves firstly the hydrolysis of LG to glucose (catalysed by acid sites), followed by the hydrogenation of glucose to sorbitol (catalysed by Ru). Unfortunately the formation of undesired products via a C–C cleavage reaction has also been observed indicating the catalytic hydrogenolysis reaction of intermediates and/or products.144
 | ||
| Fig. 18 Reaction pathways of the levoglucosan hydrolysis and successive hydrogenation. Adapted from ref. 144. | ||
The reactivity scheme implies that the formed sorbitol is converted to glycerol and 1,2-propanediol. It is also worth noting that neither 5-hydroxymethylfurfural (HMF) nor levulinic acid were detected in the reaction mixture. This indicates that the rate of the glucose hydrogenation to sorbitol is much faster than the dehydration reaction catalyzed on acid sites.144
Only rare works have been reported on the production of gluconic acid from LG. Santhanaraj et al.147 first reported a method of producing gluconic acid from a very pure solution of LG. They adapted a two-step process in which LG is firstly converted to glucose by hydrolysis catalyzed by acid sites of the Amberlyst-15 resins. The formed glucose was then separated in another reactor and partially oxidized to gluconic acid over a Pd/C catalyst in high pH medium. Hydrolysis of levoglucosan to glucose was reported earlier using sulfuric acid as a Brønsted acid catalyst.148 Unfortunately, it was difficult to achieve good performances by simply integrating the two above-mentioned catalysts into a single reactor. The main difficulty lies in the control of selectivity, as the reaction pool is interspersed with various reagents – products (intermediates) that are simultaneously influenced by two distinct catalytic functions.148 Recently the selective synthesis of gluconic acid from LG was reported on a bifunctional gold catalyst supported by Cs2.5H0.5PW12O40 without a pH control. With concerted exploitation of the active gold and acidic sites gluconic acid was obtained with a high selectivity of 93.1% with almost complete conversion.148 The gluconic acid yield obtained on this catalyst was much higher compared to homologues catalysts supported on phosphated TiO2, sulfated ZrO2 and HZSM-5. LG conversion increased with the reaction time, reaching 93.6% after 180 min of the reaction. Selectivity to gluconic acid followed the same trend. However, a longer reaction time resulted in the degradation of gluconic acid into by products such as glycolic and glyoxylic acids.148 The effect of O2 pressure was also studied. In the absence of oxygen the conversion of LG reached 64.7% while the selectivity of only 18.5% to gluconic acid was observed. As expected glucose was the main product (selectivity of 76.7%) observed in this case. The maximum gluconic acid selectivity (93.1%) was observed for the reaction conducted under 0.5 MPa of oxygen. A further increase in the oxygen pressure resulted in a decrease in gluconic acid selectivity.148 The effect of the temperature of the reaction on gluconic selectivity is shown in Fig. 19.
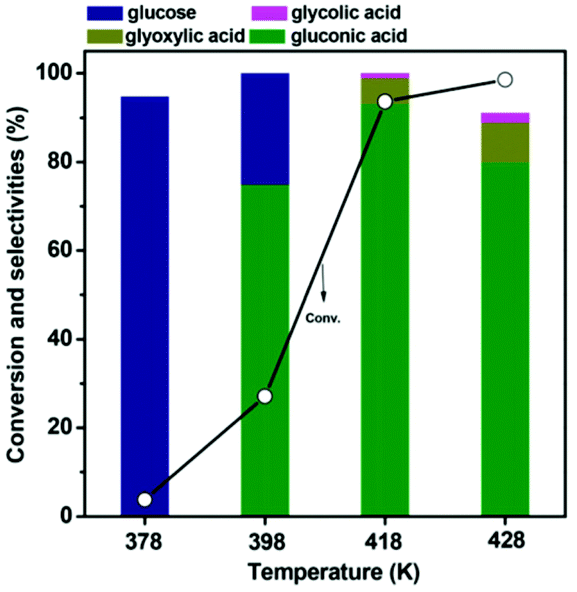 | ||
| Fig. 19 Effect of the reaction temperature on the catalytic performance of the Au/Cs2.5H0.5PW12O40 catalyst. Reaction conditions: 77.1 mM LG, 0.15 g catalyst, 20 ml H2O, 0.5 MPa O2, 3 h. Reprinted with permission from ref. 148. | ||
At 100 °C, LG conversion was only 3.8% with glucose as the major product formed. This clearly indicates that hydrolysis of LG to glucose takes place. At 120 °C, LG conversion reached 27.1%. Both glucose and gluconic acid formations were observed (gluconic acid selectivity of 74.9%). At high temperature LG conversion of 93.6% was obtained with the selectivity to gluconic acid of 93.1%. Other by-products, such as HMF and levulinic acid, originating from the dehydration and hydration reactions were not observed. However, a higher reaction temperature leads to a strong decrease in the gluconic acid selectivity and an increase of undesirable products.148
HMF is one of the most important platform molecules and it is formed by the dehydration of certain sugars. It consists of a furan ring, containing both aldehyde and alcohol functional groups.141,149,150 HMF can be converted to various important products such as 2,5dimethylfuran (DMF, used as a biofuel) and 2,5-furandicarboxylic acid (FDCA, highly important in green polymer synthesis), a possible substitute for terephthalic acid (Fig. 20).149
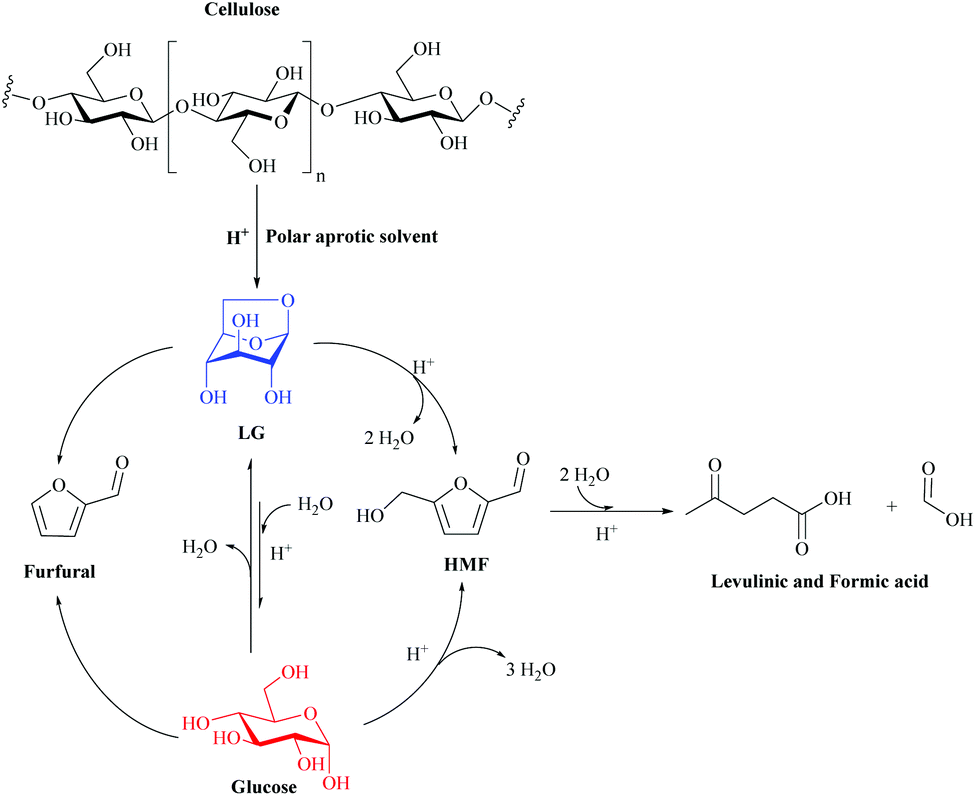 | ||
| Fig. 20 Proposed reaction scheme for the production of HMF from cellulose in polar aprotic solvents under acidic conditions. Adapted from ref. 152. | ||
LG then undergoes a double dehydration step to produce HMF. The water produced in this reaction can react with LG to produce glucose. HMF can also be rehydrated with water over an acid catalyst to produce Levulinic acid and formic acid. Once glucose is formed, it can also undergo dehydration to produce HMF, as well as degradation to produce humid species. Furfural has also been detected in carbon yields below 7%. Furfural has been reported to be a by-product of the decomposition of LG or glucose. Separate decomposition studies with HMF in THF and sulfuric acid have shown that HMF is relatively stable in the non-aqueous environment with conversions below 8% at 190 °C after 180 min. Other studies have also reported that THF prevents further degradation of furfural and HMF.151
Separate experiments with LG in THF under acidic conditions confirmed that HMF and levulinic acid could be produced directly from LG. Hu et al. also reported that LG undergoes dehydration to form HMF in the presence of Amberlyst 70 in organic solvents, but it is important to note that their reaction systems were not completely free of water due to the ionic resin that was not dried prior to the reaction (about 57% water content).122 The yield of HMF was higher when glucose was used as the raw material compared to LG, but not more than 3%. Polar aprotic solvents, including γ-valerolactone (GVL), acetone and THF, showed significantly higher yields of HMF from cellulose compared to ethyl acetate, water and ethanol, as shown in Fig. 20. The HMF yield increased in the following order: ethanol < water < ethyl acetate < GVL < acetone = THF.152
The system proposed in Fig. 20 has several distinct advantages over other existing processes for the production of HMF from cellulose, including a 20-fold reduction in acid use, a 20-fold increase in reaction rate (compared to aqueous systems), the ability to use cheaper raw materials (lignocellulosic biomass), operation at lower reaction temperatures, and improved stability of the HMF product in the solvent. In addition, reagents and products can be separated from the solvent using conventional petrochemical separation technology. This type of system also does not require the use of Lewis acids to promote the isomerization of glucose to fructose as a preliminary step in the production of HMF. Future advances in this technology promise to further improve the performance of HMF by developing a more detailed mechanistic understanding of how solvents affect acid-catalyzed chemistry, combined with the study of LG chemistry. This study opens new avenues to develop highly efficient and commercially feasible processes for converting cellulosic biomass into platform chemicals using polar aprotic solvents.152
LG can also be transformed via esterification using homogeneous acids as a catalyst.127,153,154 Ward et al.127 studied the esterification of LG with a fatty acid using HCl. As reported by the authors, the high reactivity of the hydroxyl groups in position 1 is not sufficient to obtain high yields of mono-esters by simply increasing the chain length of the acid. Steric hindrance is not enough to prevent polyesterification of the LG molecule.127 One step continuous esterification of LG with ethanol coupled with direct hydrogenation was studied on a bifunctional zeolite catalyst.155 The main products were ethyl levulinate and other various ethyl esters with carbon numbers greater than three. The authors concluded that a strong acid functionality is mandatory to efficiently hydrolyze the LG and subsequently performed esterification, dehydration, and ring opening reactions. In addition the metal functionality is necessary for hydrogenation reaction. The authors stated that the best results were observed for Ru/H-ZSM5 at 180 °C with elevated yields to ethyl levulinate and ethyl valerate esters. The acid zeolite H-ZSM5, with the SiO2/Al2O3 ratio of 50, efficiently transformed LG and acetic acid to ethyl levulinate (EL) and ethyl acetate (EA) with 4% and 85% yields, respectively.155
Regio/site-selective acylation of levoglucosan was achieved using the [Fe(acac)3] (acac = acetylacetonate) catalyst.156 The authors studied the acetylation of various glycosides containing a cis-vicinal diol. The authors showed that the Fe(III) active site formed cyclic dioxolane-type intermediates with vicinal diols of the substrates. The efficient and selective acylation of one hydroxyl group was subsequently achieved by adding acylation reagents in the presence of diiso-propylethylamine (DIPEA). Most of the tested substrates were selectively acylated at the equatorial hydroxyl groups in high yields (>80%). 84% was reached in the case of levoglucosan.156
Levoglucosan was also used as the substrate for the preparation of porous polymers with three dimensional nanochannels.157 Cationic ring-opening polymerization of levoglucosan was performed in the nanochannels of [Cu3(benzene-1,3,5-tricarboxylate)]n. After the removal of the host frameworks polysaccharide particles as replicas of the original molds were obtained. After 144 hours of polymerization, high surface area polymers were obtained (150 m2 g−1). The application of the produced polymer on drug delivery protocols demonstrated that the simple polymerization method was efficient to control the polymer particle size and porosity.157
The physical and biological properties of the sugars could be modified using fluorination. This was the object of the work reported by Quiquempoix et al.158 They described a 6-step synthesis of 2,3,4-trideoxy-2,3,4-trifluoroglucose starting from levoglucosan with a final yield of 24%. The crucial step involved a selective difluorination of 2,4-di-O-tosyl levoglucosan and the deoxyfluorination of 2,4-dideoxy-2,4-difluorolevoallosan. In this step, a rare F⋯H(O)⋯F bifurcated hydrogen bond was clearly observed. But as reported by the authors, even if this particular conformation was also present in the crystalline state, the clearly dominated intermolecular O–H⋯O bond was observed in the crystal packing of this compound.158
6. Perspectives
Although several efforts have been made for the valorization of lignocellulosic biomass in combined approaches, there are still few studies in which enzymatic catalysis goes hand in hand with chemical catalysis for the better use of biomass. In enzymatic catalysis, ethanol has still been the main product of interest when protein-engineering maneuvers are applied to improve strains for the use of LG or its glucose hydrolysate, since the number of papers published for this purpose is still very expressive compared to other products. However, in recent years, LG derivatives have been obtained through microbial biotransformation and biocatalysis, such as LG esters and organic acids.In chemical catalysis, several strategies have been combined for the production of furfural derivatives, LGO and organic acids, where temperature, nature of the catalyst and stability have been crucial to obtain the desired selectivity to molecules of interest.
According to the works discussed in this review, it is foreseeable that some methodological gaps will be filled, such as the coupling of biochemical and chemical steps in the biomass hydrolysis or the application of hybrid catalysts in obtaining products of high added value as a way to better use the residual biomass. Protein engineering has advanced in the development of techniques for improving the metabolic engineering of microbial strains, for the construction of engineered strains capable of both capturing larger amounts of LG as well as performing unusual metabolic pathways to obtain several other derived compounds. Therefore, there is the prospect of greater integration between the chemical, biochemical and enzymatic approaches.
7. Conclusions
There are many advantages of pyrolytic cellulose transformation to levoglucosan compared to the classical hydrolysis to glucose. It is well documented that enzymatic hydrolysis of cellulose allows high selectivity to glucose to be obtained. However, the main drawback of this process concerns the relatively high operational cost because of the enzyme cost and high complexity in their recycling. The pyrolysis process does not use solvents or enzymes to depolymerize cellulose. This reduces the wastes and risk of water pollution. It could also be easily scaled up facilitating its commercial application. It is an inexpensive technology and a wide variety of feedstocks can be used. Moreover, other products formed during cellulose pyrolysis can also have considerable commercial value (such as furan derivatives).The main advantage of producing levoglucosan from cellulose is the direct access to other valuable products such as HMF-platform, levoglucosenone, cyrene and HBO. Fig. 21 presents the comparison between the main products that could be obtained from levoglucosan and glucose. As an example, the absence of the isomerization step to access the furan derivative platform molecules such as HMF and 2,5-furandicarboxylic acid (FDCA) is of high importance. FDCA is a highly promising building block for the resin and polymer industries and it is advocated as a green replacement for (fossil-based) terephthalate. The main issue in the FDCA synthesis is the glucose to fructose isomerization step. HMF is usually obtained from fructose, which must be very pure. However, the dehydration and then the oxidation processes for obtaining FDCA are not always very selective and more stable by-products are formed. In the case of levoglucosan the HMF synthesis could be easily done in a one-step reaction of LG with dimethyl sulfoxide (DMSO).
 | ||
| Fig. 21 Main reaction pathways for levoglucosan and glucose transformations. | ||
In addition, as discussed above in Section 4, in the LG molecule the positions 1 and 6 present an acetalic function and the remaining secondary hydroxyl groups are in the axial position. Therefore, LG is much less reactive than glucose or other sugars both to chemical and enzymatic catalysts. This peculiar feature can be beneficial in the regioselective acylation of a single secondary OH function that represents a very challenging task since the presence of three hydroxyls in its structure allows the formation of monoesters and di-esters at the positions C2, C3 and C4. This gives access to the specific building blocks for the synthesis of chiral intermediates and polymers. Due to the specific rigid structure the pyranose ring is locked in the 1C4 conformation, which implies several consequences such as easy control of the regiochemical and stereochemical properties of axial hydroxyls, the thermal stability is considerably increased and the reduced number of OH groups, as compared to glucose, enhances its solubility in organic solvents.159
The increase in the life expectancy of the world population and the finitude of fossil fuels have driven the valorization of biomass as a means of obtaining energy and products with high added value, leading to trans disciplinary approaches where the combination of pre-treatment, pyrolysis, separation, (bio) catalysis and protein engineering has been an important tool for the obtaining of LG, which can be directly transformed into various value-added products, through heterogeneous catalysis, biocatalysis or biotransformation, or be hydrolyzed to glucose and further applied as a starting material of a range of other products. In each of these approaches, there are several obstacles to overcome, such as the combination of steps to reduce microbial growth inhibitors, the low catalytic efficiency of the LGK and LGDH enzymes in the recombinant strains, and even the median selectivity of the applied chemical catalysts from obtaining some compounds derived from LG, such as LGO and furfural. Although several efforts have been made, the improvement of methodologies and the construction of new catalysts are still important for the better valorization of biomass and the increased yield in LG.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Mr Gustavo Saraiva Silveira and Mr Yassine Benrezkallah for the help with the bibliography research. This study was supported by the French government through the Programme Investissement d'Avenir (I-SITE ULNE/ANR-16-IDEX-0004 ULNE) managed by the Agence Nationale de la Recherche. LIA CNRS France-Brazil “Energy & Environment” and Métropole Européen de Lille (MEL) for “CatBioInnov” project are also acknowledged.References
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- R. Wojcieszak, F. Santarelli, S. Paul, F. Dumeignil, F. Cavani and R. V. Gonçalves, Sustainable Chem. Processes, 2015, 3, 9 CrossRef.
- A. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Nature, 2016, 531, 215–219 CrossRef CAS PubMed.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- L. Jiang, Z. Fang, Z. Zhao, A. Zheng, X. Wang and H. Li, Renewable Sustainable Energy Rev., 2019, 105, 215–229 CrossRef CAS.
- S. G. Wettstein, D. M. Alonso, E. I. Gürbüz and J. A. Dumesic, Curr. Opin. Chem. Eng., 2012, 1, 218–224 CrossRef CAS.
- F. Cao, T. J. Schwartz, D. J. McClelland, S. H. Krishna, J. A. Dumesic and G. W. Huber, Energy Environ. Sci., 2015, 8, 1808–1815 RSC.
- J. M. O. Scurlock and D. O. Hall, Biomass, 1990, 21, 75–81 CrossRef.
- J. J. Bozell, S. K. Black, M. Myers, D. Cahill, W. P. Miller and S. Park, Biomass Bioenergy, 2011, 35, 4197–4208 CrossRef CAS.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- M. R. Rover, A. Aui, M. M. Wright, R. G. Smith and R. C. Brown, Green Chem., 2019, 21, 5980–5989 RSC.
- R. H. Venderbosch and W. Prins, Biofuels, Bioprod. Biorefin., 2010, 4, 178–208 CrossRef CAS.
- C. M. Lakshmanan and H. E. Hoelscher, Ind. Eng. Chem. Prod. Res. Dev., 1970, 9, 57–59 CrossRef CAS.
- M. A. Bergauff, T. J. Ward, C. W. Noonan, C. T. Migliaccio, C. D. Simpson, A. R. Evanoski and C. P. Palmer, J. Exposure Sci. Environ. Epidemiol., 2010, 20, 385–392 CrossRef CAS PubMed.
- M. B. Pecha, J. I. M. Arbelaez, M. Garcia-Perez, F. Chejne and P. N. Ciesielski, Green Chem., 2019, 21, 2868–2898 RSC.
- M. J. Antal Jr. and G. Varhegyi, Ind. Eng. Chem. Res., 1995, 34, 703–717 CrossRef.
- V. Mamleev, S. Bourbigot, M. Le Bras and J. Yvon, J. Anal. Appl. Pyrolysis, 2009, 84, 1–17 CrossRef CAS.
- J. Lédé, Fuel, 2002, 81, 1269–1279 CrossRef.
- J. Lédé, J. Anal. Appl. Pyrolysis, 2012, 94, 17–32 CrossRef.
- S. Wang, G. Dai, H. Yang and Z. Luo, Prog. Energy Combust. Sci., 2017, 62, 33–86 CrossRef.
- S. Maduskar, V. Maliekkal, M. Neurock and P. J. Dauenhauer, ACS Sustainable Chem. Eng., 2018, 6, 7017–7025 CrossRef CAS.
- R. J. M. Westerhof, S. R. G. Oudenhoven, P. S. Marathe, M. Engelen, M. Garcia-Perez, Z. Wang and S. R. A. Kersten, React. Chem. Eng., 2016, 1, 555–566 RSC.
- A. Dufour, B. Ouartassi, R. Bounaceur and A. Zoulalian, Chem. Eng. Res. Des., 2011, 89, 2136–2146 CrossRef CAS.
- O. Boutin, M. Ferrer and J. Lédé, J. Anal. Appl. Pyrolysis, 1998, 47, 13–31 CrossRef CAS.
- E. M. Suuberg, I. Milosavljevic and V. Oja, Symp. (Int.) Combust., [Proc.], 1996, 26, 1515–1521 CrossRef.
- J. Montoya, B. Pecha, F. C. Janna and M. Garcia-Perez, J. Anal. Appl. Pyrolysis, 2017, 124, 204–218 CrossRef CAS.
- A. R. Teixeira, K. G. Mooney, J. S. Kruger, C. L. Williams, W. J. Suszynski, L. D. Schmidt, D. P. Schmidt and P. J. Dauenhauer, Energy Environ. Sci., 2011, 4, 4306 RSC.
- S. M. Mettler, G. D. G. Vlachos and P. J. Dauenhauer, Energy Environ. Sci., 2012, 5, 7797–7809 RSC.
- C. I. De Jenga, M. J. Antal and M. Jones, J. Appl. Polym. Sci., 1982, 27, 4313–4322 CrossRef CAS.
- M. R. Rover, P. A. Johnston, L. E. Whitmer, R. G. Smith and R. C. Brown, J. Anal. Appl. Pyrolysis, 2014, 105, 262–268 CrossRef CAS.
- D. Radlein, J. Piskorz and D. S. Scott, J. Anal. Appl. Pyrolysis, 1991, 19, 41–63 CrossRef CAS.
- W. S. L. Mok and M. J. Antal, Thermochim. Acta, 1983, 68, 165–186 CrossRef CAS.
- R. J. Evans and T. A. Milne, Energy Fuels, 1987, 1, 123–137 CrossRef CAS.
- J. Piskorz, D. Radlein and D. S. Scott, J. Anal. Appl. Pyrolysis, 1986, 9, 121–137 CrossRef CAS.
- W. Chaiwat, I. Hasegawa, T. Tani, K. Sunagawa and K. Mae, Energy Fuels, 2009, 23, 5765–5772 CrossRef CAS.
- F. J. Kilzer and A. Broido, Pyrodynamics, 1965, 2, 151–163 CAS.
- J. Scheirs, G. Camino and W. Tumiatti, Eur. Polym. J., 2001, 37, 933–942 CrossRef CAS.
- J. Piskorz, D. S. A. G. Radlein, D. S. Scott and S. Czernik, J. Anal. Appl. Pyrolysis, 1989, 16, 127–142 CrossRef CAS.
- G. N. Richards, J. Anal. Appl. Pyrolysis, 1987, 10, 251–255 CrossRef CAS.
- D. K. Shen and S. Gu, Bioresour. Technol., 2009, 100, 6496–6504 CrossRef CAS PubMed.
- Q. Lu, B. Hu, Z.-X. Zhang, Y.-T. Wu, M.-S. Cui, D.-J. Liu, C.-Q. Dong and Y.-P. Yang, Combust. Flame, 2018, 198, 267–277 CrossRef CAS.
- V. Seshadri and P. R. Westmoreland, J. Phys. Chem. A, 2012, 116, 11997–12013 CrossRef CAS PubMed.
- S. Matsuoka, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2014, 106, 138–146 CrossRef CAS.
- V. Maliekkal, S. Maduskar, D. J. Saxon, M. Nasiri, T. M. Reineke, M. Neurock and P. Dauenhauer, ACS Catal., 2018, 9, 1943–1955 CrossRef.
- Z. Wang, A. G. McDonald, R. J. M. Westerhof, S. R. A. Kersten, C. M. Cuba-Torres, S. Ha, B. Pecha and M. Garcia-Perez, J. Anal. Appl. Pyrolysis, 2013, 100, 56–66 CrossRef CAS.
- J. Zhang, M. W. Nolte and B. H. Shanks, ACS Sustainable Chem. Eng., 2014, 2, 2820–2830 CrossRef CAS.
- I. Y. Eom, K. H. Kim, J. Y. Kim, S. M. Lee, H. M. Yeo, I. G. Choi and J. W. Choi, Bioresour. Technol., 2011, 102, 3437–3444 CrossRef CAS PubMed.
- P. R. Patwardhan, D. L. Dalluge, B. H. Shanks and R. C. Brown, Bioresour. Technol., 2011, 102, 5265–5269 CrossRef CAS PubMed.
- Y. Le Brech, T. Ghislain, S. Leclerc, M. Bouroukba, L. Delmotte, N. Brosse, C. Snape, P. Chaimbault and A. Dufour, ChemSusChem, 2016, 9, 863–872 CrossRef CAS PubMed.
- D. Liu, Y. Yu, Y. Long and H. Wu, Proc. Combust. Inst., 2015, 35, 2381–2388 CrossRef CAS.
- S. A. Rollag, J. K. Lindstrom and R. C. Brown, Chem. Eng. J., 2020, 385, 123889 CrossRef.
- K. H. Kim, K. Jeong, S.-S. Kim and R. C. Brown, Green Chem., 2019, 21, 1099–1107 RSC.
- J. Huang, C. Liu, S. Wei, X. Huang and H. Li, J. Mol. Struct.: THEOCHEM, 2010, 958, 64–70 CrossRef CAS.
- X. Zhou, M. W. Nolte, H. B. Mayes, B. H. Shanks and L. J. Broadbelt, Ind. Eng. Chem. Res., 2014, 53, 13274–13289 CrossRef CAS.
- Y. Le Brech, L. Jia, S. Cissé, G. Mauviel, N. Brosse and A. Dufour, J. Anal. Appl. Pyrolysis, 2016, 117, 334–346 CrossRef CAS.
- S. R. G. Oudenhoven, R. J. M. Westerhof, N. Aldenkamp, D. W. F. Brilman and S. R. A. Kersten, J. Anal. Appl. Pyrolysis, 2013, 103, 112–118 CrossRef CAS.
- D. Radlein, Study of levoglucosan production – A review, in Fast pyrolysis of Biomass: A handbook, ed. A. V. Bridgwater, 2002, vol. 2, pp. 205–241 Search PubMed.
- P. M. Molton and T. F. Demmitt, Reaction mechanisms in cellulose pyrolysis: A literature review, Pacific Nortwest laboratories, Richland, Washington 99352, 1977 Search PubMed.
- J. Piskorz, P. Majerski, D. Radlein, A. Vladars-Usas and D. S. Scott, J. Anal. Appl. Pyrolysis, 2000, 56, 145–166 CrossRef CAS.
- E.-J. Shin, M. R. Nimlos and R. J. Evans, Fuel, 2001, 80, 1697–1709 CrossRef CAS.
- K. Norinaga, T. Shoji, S. Kudo and J.-I. Hayashi, Fuel, 2013, 103, 141–150 CrossRef CAS.
- J. Hertzog, V. Carre, Y. Le Brech, C. L. Mackay, A. Dufour, O. Masek and F. Aubriet, Anal. Chim. Acta, 2017, 969, 26–34 CrossRef CAS PubMed.
- A. S. Pollard, M. R. Rover and R. C. Brown, J. Anal. Appl. Pyrolysis, 2012, 93, 129–138 CrossRef CAS.
- M. R. Rover, P. A. Johnston, T. Jin, R. G. Smith, R. C. Brown and L. Jarboe, ChemSusChem, 2014, 7, 1662–1668 CrossRef CAS PubMed.
- E. M. Prosen, D. Radlein, J. Piskorz, D. S. Scott and R. L. Legge, Biotechnol. Bioeng., 1993, 42, 538–541 CrossRef CAS PubMed.
- G. Dobele, in Fast pyrolysis of biomass: A Handbook, ed. A. V. Bridgwater, CPL Press, Newburry UK, 2002, vol. 2, pp. 147–204 Search PubMed.
- J. T. Cunha, P. O. Soares, A. Romaní, J. M. Thevelein and L. Domingues, Biotechnol. Biofuels, 2019, 12, 20 CrossRef PubMed.
- K. Zhang, Z. Pei and D. Wang, Bioresour. Technol., 2016, 199, 21–33 CrossRef CAS PubMed.
- Y. H. Jung and K. H. Kim, Bioresour. Technol., 2017, 12, 9348–9356 CAS.
- E. C. van der Pol, E. Vaessen, R. A. Weusthuis and G. Eggink, Bioresour. Technol., 2016, 209, 297–304 CrossRef CAS PubMed.
- L. J. Jönsson, B. Alriksson and N.-O. Nilvebrant, Biotechnol. Biofuels, 2013, 6, 16 CrossRef PubMed.
- L. J. Jönsson and C. Martín, Bioresour. Technol., 2016, 199, 103–112 CrossRef PubMed.
- J. K. Ko, Y. Um, Y.-C. Park, J.-H. Seo and K. H. Kim, Appl. Microbiol. Biotechnol., 2015, 99, 4201–4212 CrossRef CAS PubMed.
- J. N. Lian, M. Garcia-Perez and S. L. Chen, Bioresour. Technol., 2013, 133, 183–189 CrossRef CAS PubMed.
- J. N. Lian, S. L. Chen, S. Zhou, Z. H. Wang, J. O'Fallon, C. Z. Li and M. Garcia-Perez, Bioresour. Technol., 2010, 101, 9688–9699 CrossRef CAS PubMed.
- Z. S. Yu and H. X. Zhang, Biomass Bioenergy, 2003, 24, 257–262 CrossRef CAS.
- X. L. Zhuang, H. X. Zhang and J. J. Tang, Biomass Bioenergy, 2001, 21, 53–60 CrossRef CAS.
- H. Wang, D. Livingston, R. Srinivasan, Q. Li, P. Steele and F. Yu, Appl. Microbiol. Biotechnol., 2012, 168, 1568–1583 CAS.
- J. N. Lian, R. McKenna, M. R. Rover, D. R. Nielsen, Z. Y. Wen and L. R. Jarboe, J. Ind. Microbiol. Biotechnol., 2016, 43, 595–604 CrossRef CAS PubMed.
- Z. Chi, M. Rover, E. Jun, M. Deaton, P. Johnston, R. C. Brown, Z. Wen and L. R. Jarboe, Bioresour. Technol., 2013, 150, 220–227 CrossRef CAS PubMed.
- D. Chang, Z. Yu, Z. U. Islam and H. Zhang, Appl. Microbiol. Biotechnol., 2015, 99, 4093–4105 CrossRef CAS PubMed.
- L. Luque, S. Oudenhoven, R. Westerhof, G. van Rossum, F. Berruti, S. Kersten and L. Rehmann, Biotechnol. Biofuels, 2016, 9, 242 CrossRef PubMed.
- Z. U. Islam, S. P. Klykov, Z. Yu, E. B. Hassan and H. Zhang, Appl. Biochem. Microbiol., 2018, 54, 58–70 CrossRef CAS.
- E. B. Hassan, H. Abou-Yousef and P. Steele, Fuel Process. Technol., 2013, 110, 65–72 CrossRef CAS.
- J. K. Chan and S. J. Duff, Bioresour. Technol., 2010, 101, 3755–3759 CrossRef CAS PubMed.
- H. Wang, D. Livingston, R. Srinivasan, Q. Li, P. Steele and F. Yu, Appl. Microbiol. Biotechnol., 2012, 168, 1568–1583 CAS.
- B. Scholze and D. Meier, J. Anal. Appl. Pyrolysis, 2001, 60, 41–54 CrossRef CAS.
- Q. H. Song, J. Q. Nie, M. G. Ren and Q. X. Guo, Energy Fuels, 2009, 23, 3307–3312 CrossRef CAS.
- N. M. Bennett, S. S. Helle and S. J. Duff, Bioresour. Technol., 2009, 100, 6059–6063 CrossRef CAS PubMed.
- Q. Li, P. H. Steele, B. K. Mitchell, L. L. Ingram and F. Yu, BioResources, 2003, 8, 1686–1680 Search PubMed.
- B. Sukhbaatar, Q. Li, C. X. Wan, F. Yu, E. B. Hassan and P. Steele, Bioresour. Technol., 2014, 161, 379–384 CrossRef CAS PubMed.
- J. Wang, Q. Wei, J. Zheng and M. Zhu, J. Anal. Appl. Pyrolysis, 2016, 122, 294–303 CrossRef CAS.
- L.-Q. Jiang, Z. Fang, Z.-L. Zhao, A.-Q. Zheng, X.-B. Wang and H.-B. Li, Renewable Sustainable Energy Rev., 2019, 105, 215–229 CrossRef CAS.
- R. Beauchet, F. Monteil-Rivera and J. M. Lavoie, Bioresour. Technol., 2012, 121, 328–334 CrossRef CAS PubMed.
- A. V. Bridgwater, S. Czernik and J. Diebold, Fast Pyrolysis of Biomass: A Handbook, Scientific Publishing Services Ltd., Newbury, UK, 1999 Search PubMed.
- M. Jahirul, M. Rasul, A. Chowdhury and N. Ashwath, Energies, 2012, 5, 4952 CrossRef CAS.
- H. Xie, X. Zhuang, Z. Bai, H. Qi and H. Zhang, World J. Microbiol. Biotechnol., 2006, 22, 887–892 CrossRef CAS.
- Z. Yu, J. Ning, L. Zhang and H. Zhang, Chin. Agric. Sci. Bull. (Chinese), 2011, 27, 342–349 Search PubMed.
- J. Dai, Y. Yu, X. Wan and H. Zhang, Food Ferment. Ind., 2010, 36, 6–9 CAS.
- J. P. Bacik, G. E. Whitworth, K. A. Stubbs, A. K. Yadav, D. R. Martin, B. A. Bailey-Elkin, D. J. Vocadlo and B. L. Mark, J. Biol. Chem., 2011, 286, 12283–12291 CrossRef CAS PubMed.
- D. Chiaramonti, M. Prussi, S. Ferrero, L. Oriani, P. Ottonello, P. Torre and F. Cherchi, Biomass Bioenergy, 2012, 46, 25–35 CrossRef CAS.
- J.-H. Choi, M.-S. Kang, C.-G. Lee, N.-H. L. Wang and S. Mun, J. Chromatogr., A, 2017, 1491, 75–86 CrossRef CAS PubMed.
- Y. Kitamura, Y. Abe and T. Yasui, Agric. Biol. Chem., 1991, 55, 515–521 CAS.
- Y. Kitamura and T. Yasui, Agric. Biol. Chem., 1991, 55, 523–529 CAS.
- T. Yasui, Y. Kitamura, K. Nakahara and Y. Abe, Agric. Biol. Chem., 1991, 55, 1927–1929 CAS.
- S. Dorsam, J. Kirchhoff, M. Bigalke, N. Dahmen, C. Syldatk and K. Ochsenreither, Front. Microbiol., 2016, 7, 2059 Search PubMed.
- S. Dorsam, J. Fesseler, O. Gorte, T. Hahn, S. Zibek, C. Syldatk and K. Ochsenreither, Biotechnol. Biofuels, 2017, 10, 242 CrossRef PubMed.
- M. Sugiura, M. Nakahara, C. Yamada, T. Arakawa, M. Kitaoka and S. Fushinobu, J. Biol. Chem., 2018, 293, 17375–17386 CrossRef CAS PubMed.
- K. Nakahara, Y. Kitamura, Y. Yamagishi, H. Shoun and T. Yasui, Biosci., Biotechnol., Biochem., 1994, 58, 2193–2196 CrossRef CAS PubMed.
- M. Sugiura, M. Nakahara, C. Yamada, T. Arakawa, M. Kitaoka and S. Fushinobu, J. Biol. Chem., 2018, 293, 17375–17386 CrossRef CAS PubMed.
- M. Sugiura, M. Nakahara, C. Yamada, T. Arakawa, M. Kitaoka and S. Fushinobu, RSCB Protein Data Bank, 2018, https://www.rcsb.org/structure/6A3F.
- J.-P. Bacik, J. R. Klesmith, T. A. Whitehead, L. R. Jarboe, C. J. Unkefer, B. L. Mark and R. Michalczyk, J. Biol. Chem., 2015, 280, 26638–26648 CrossRef PubMed.
- X. L. Zhuang, H. X. Zhang, J. Z. Yang and H. Y. Qi, Bioresour. Technol., 2001, 79, 63–66 CrossRef CAS PubMed.
- M. Nakagawa, Y. Sakai and T. Yasui, J. Ferment. Technol., 1984, 62, 201–203 CAS.
- H. Alper and G. Stephanopoulos, Nat. Rev. Microbiol., 2009, 7, 715–723 CrossRef CAS PubMed.
- D. S. Layton, A. Ajjarapu, D. W. Choi and L. R. Jarboe, Bioresour. Technol., 2011, 102, 8318–8322 CrossRef CAS PubMed.
- E. M. Kim, Y. Um, M. Bott and H. M. Woo, FEMS Microbiol. Lett., 2015, 362, 19 Search PubMed.
- J. R. Klesmith, J. P. Bacik, R. M. Michalczyk and T. A. Whitehead, ACS Synth. Biol., 2015, 4, 1235–1243 CrossRef CAS PubMed.
- X. Xiong, J. Lian, X. Yu, M. Garcia-Perez and S. Chen, J. Ind. Microbiol. Biotechnol., 2016, 43, 1551–1560 CrossRef CAS PubMed.
- A. P. Ingle, A. K. Chandel and S. S. Da Silva, Lignocellulosic Biorefining Technologies, Wiley-Blackwell, 1st edn, 2020 Search PubMed.
- M. Mochida and K. Kawamura, J. Geophys. Res.: Atmos., 2004, 109, D21202 CrossRef.
- X. Hu, L. Wu, Y. Wang, D. Mourant, C. Lievens, R. Gunawan and C.-Z. Li, Green Chem., 2012, 14, 3087 RSC.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 13, 520–540 RSC.
- F. K. Sutili, H. S. Ruela, S. G. F. Leite, L. S. d. M. Miranda, I. C. R. Leal and R. O. M. A. de Souza, J. Mol. Catal. B: Enzym., 2013, 85–86, 37–42 CrossRef CAS.
- M. A. Do Nascimento, L. E. Gotardo, R. A. C. Leão, A. M. De Castro, R. O. M. A. De Souza and I. Itabaiana, ACS Omega, 2019, 4, 860–869 CrossRef CAS.
- J. Martin Macleod and L. R. Schroeder, Carbohydr. Res., 1973, 30, 337–347 CrossRef.
- D. D. Ward and F. Shafizadeh, Carbohydr. Res., 1982, 108, 71–79 CrossRef CAS.
- B. K. Hunter, L. D. Hall and J. K. M. Sanders, J. Chem. Soc., Perkin Trans. 1, 1983, 1, 657 RSC.
- P. Galletti, F. Moretti, C. Samorì and E. Tagliavini, Green Chem., 2007, 9, 987 RSC.
- I. Uriarte, P. Écija, R. Lozada-Garcia, P. Çarçabal and E. J. Cocinero, ChemPhysChem, 2018, 19, 766–773 CrossRef CAS PubMed.
- M. Avelar do Nascimento, L. Ester Gotardo, E. Miguez Bastos, F. C. L. Almeida, R. A. C. Leão, R. O. M. A. de Souza, R. Wojcieszak and I. Itabaiana, Sustainability, 2019, 11, 6044 CrossRef.
- R. Wojcieszak, I. Cuccovia, M. Silva and L. M. Rossi, J. Mol. Catal. A: Chem., 2016, 422, 35–42 CrossRef CAS.
- O. Oyola-Rivera, J. He, G. W. Huber, J. A. Dumesic and N. Cardona-Martínez, Green Chem., 2019, 21, 4988–4999 RSC.
- J. He, M. Liu, K. Huang, T. W. Walker, C. T. Maravelias, J. A. Dumesic and G. W. Huber, Green Chem., 2017, 19, 3642–3653 RSC.
- S. Helle, N. M. Bennett, K. Lau, J. H. Matsui and S. J. B. Duff, Carbohydr. Res., 2007, 342, 2365–2370 CrossRef CAS PubMed.
- R. M. Abdilla-Santes, C. B. Rasrendra, J. G. M. Winkelman and H. J. Heeres, Chem. Eng. Res. Des., 2019, 152, 193–200 CrossRef CAS.
- R. M. Abdilla, C. B. Rasrendra and H. J. Heeres, Ind. Eng. Chem. Res., 2018, 57, 3204–3214 CrossRef CAS PubMed.
- M. Käldström, N. Kumar, T. Heikkilä and D. Y. Murzin, Appl. Catal., A, 2011, 397, 13–21 CrossRef.
- W. Hu, Z. Chi, Y. Wan, S. Wang, J. Lin, S. Wan and Y. Wang, Catal. Sci. Technol., 2020, 10, 2986–2993 RSC.
- D. Liu, K. H. Kim, J. Sun, B. A. Simmons and S. Singh, ChemSusChem, 2018, 11, 598–604 CrossRef CAS PubMed.
- C. P. Ferraz, M. Zieliński, M. Pietrowski, S. Heyte, F. Dumeignil, L. M. Rossi and R. Wojcieszak, ACS Sustainable Chem. Eng., 2018, 6, 16332–16340 CrossRef CAS.
- G. IMARC, Sorbitol Market: Global Industry Trends, Share, Size, Growth, Opportunity & Forecast 2019–2024, 2019, 2020 Search PubMed.
- L. Silvester, F. Ramos Neves, J. Thuriot-Roukos, S. Heyte, M. Araque, S. Paul and R. Wojcieszak, Catal. Today, 2019, 338, 72–80 CrossRef CAS.
- W. Yin, Z. Tang, R. H. Venderbosch, Z. Zhang, C. Cannilla, G. Bonura, F. Frusteri and H. J. Heeres, ACS Catal., 2016, 6, 4411–4422 CrossRef CAS.
- A. Sanna, T. P. Vispute and G. W. Huber, Appl. Catal., B, 2015, 165, 446–456 CrossRef CAS.
- A. B. Bindwal and P. D. Vaidya, Ind. Eng. Chem. Res., 2013, 52, 17781–17789 CrossRef CAS.
- D. Santhanaraj, M. R. Rover, D. E. Resasco, R. C. Brown and S. Crossley, ChemSusChem, 2014, 7, 3132–3137 CrossRef CAS PubMed.
- Y. Wan, L. Zhang, Y. Chen, J. Lin, W. Hu, S. Wang, J. Lin, S. Wan and Y. Wang, Green Chem., 2019, 21, 6318–6325 RSC.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS PubMed.
- R. Wojcieszak, C. Ferraz, J. Sha, S. Houda, L. Rossi and S. Paul, Catalysts, 2017, 7, 23 CrossRef.
- C. M. Cai, T. Zhang, R. Kumar and C. E. Wyman, Green Chem., 2013, 15, 3140 RSC.
- R. Weingarten, A. Rodriguez-Beuerman, F. Cao, J. S. Luterbacher, D. Martin Alonso, J. A. Dumesic and G. W. Huber, ChemCatChem, 2014, 6, 2229–2234 CrossRef CAS.
- R. W. Jeanloz, A. M. C. Rapin and S. I. Hakomori, J. Org. Chem., 1961, 26, 3939–3946 CrossRef CAS.
- D. Shapiro, Y. Rabinsohn, A. J. Acher and A. Diver-Haber, J. Org. Chem., 1970, 35, 1464–1467 CrossRef CAS PubMed.
- R. Hilten, J. Weber and J. R. Kastner, Energy Fuels, 2016, 30, 9480–9489 CrossRef CAS.
- J. Lv, J.-T. Ge, T. Luo and H. Dong, Green Chem., 2018, 20, 1987–1991 RSC.
- Y. Kobayashi, K. Honjo, S. Kitagawa and T. Uemura, ACS Appl. Mater. Interfaces, 2017, 9, 11373–11379 CrossRef CAS PubMed.
- L. Quiquempoix, Z. Wang, J. Graton, P. G. Latchem, M. Light, J. Y. Le Questel and B. Linclau, J. Org. Chem., 2019, 84, 5899–5906 CrossRef CAS PubMed.
- D. Radlein, in Bio-oil Production and Utilisation, ed. A. V. Bridgwater and E. N. Hogan, CPL Press, Newburry, UK, 1999, vol. 1, pp. 113–140 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |