
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnrichment of glycopeptides using environmentally friendly wood materials†
Yuye
Zhou‡
 a,
Xia
Sheng‡
b,
Jonas
Garemark
b,
Leila
Josefsson
a,
Licheng
Sun
cd,
Yuanyuan
Li
*b and
Åsa
Emmer
*a
a,
Xia
Sheng‡
b,
Jonas
Garemark
b,
Leila
Josefsson
a,
Licheng
Sun
cd,
Yuanyuan
Li
*b and
Åsa
Emmer
*a
aKTH Royal Institute of Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, Department of Chemistry, Division of Applied Physical Chemistry, Analytical Chemistry, Stockholm SE-100 44, Sweden. E-mail: aae@kth.se
bKTH Royal Institute of Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, Department of Fiber and Polymer Technology, Wallenberg Wood Science Center, Stockholm SE-100 44, Sweden. E-mail: yua@kth.se
cKTH Royal Institute of Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, Department of Chemistry, Division of Organic Chemistry, Stockholm SE-100 44, Sweden
dWestlake University, School of Science, Center of Artificial Photosynthesis for Solar Fuels, Hangzhou CN-310024, China
First published on 31st July 2020
Abstract
Glycoproteomics is one of the main routes to study protein post-translational modifications, and enrichment is one of the important steps. Many hydrophilic materials have been used for glycopeptide enrichment, however involving complex synthesis procedures, high consumption in cost, time, and chemicals. Here, balsa wood, a naturally available hydrophilic material with highly porous structure, was investigated for immunoglobulin G (IgG) glycopeptide enrichment in a micropipette set-up. Native balsa wood without any pretreatment provided a green alternative to a commercial HILIC product, exhibiting comparable, or even better, enrichment performance for IgG standard and human plasma IgG samples. After delignification, binding capacity and recovery could be improved due to increased hydrophilicity and porosity. The developed method utilizing wood materials introduced an environmentally friendly option for glycoproteomics, saving both costs and chemicals, while exhibiting high performance.
Introduction
Glycosylation is a common protein post translation modification, where the structure and functionality of proteins are altered by the presence of glycans. Many of the proteins secreted in the human body are glycoproteins, such as immunoglobulins.1,2 Variations in the protein glycosylation are indicative of the development of certain chronic or infectious diseases, such as Huntington's disease, Alzheimer's disease, progeroid syndromes, cancer, and various autoimmune/inflammatory diseases.3–9 Moreover, studies on the novel Coronavirus disease 2019 (CoVID-2019) show that the coronavirus spike glycoproteins are crucial for membrane infusion, receptor binding, promoting entry into cells, and are the main targets of the antibodies.10,11 Detection of glycoprotein concentration and/or protein glycan profile variations in an early stage of a disease could aid in selecting treatment, and facilitate recovery of the patients.One challenge encountered when using glycoproteins as biomarkers is the relatively minor changes in glycan profiles between a healthy and a disease state. Highly sensitive analytical approaches are therefore required.12 With the help of matrix-assisted laser desorption/ionization – time of flight mass spectrometry (MALDI-TOF-MS), the structural diversity of the glycopeptides can be determined with high analysis speed, high resolution and low sample consumption.13–15 However, the signal of glycopeptides could be suppressed in the presence of non-glycopeptides and/or salts in biological samples. Therefore, an efficient enrichment of glycopeptides that could be applied on complex biological samples should be included in the sample preparation procedure.
Hydrophilic interactions between glycopeptides and an affinity material is the key factor in most glycopeptide enrichment methods. In addition to the commercially available hydrophilic interaction liquid chromatography (HILIC) products, a lot of laboratory synthesized hydrophilic materials have been used for the enrichment of glycopeptides. This includes silica materials, magnetic nanocomposites, metal–organic frameworks, and porous polymeric monoliths etc. For most of the synthesized materials, cautious structure control has been carried out to create materials with high surface area, which is beneficial for glycopeptide adsorption. In addition, different functional groups, such as amino groups, carbonyl groups or metal nanoparticles, have further been coupled to the surface of the synthesized materials to improve the hydrophilicity.16–23 Although intensively studied, the use of the materials are limited by high cost, high chemical consumption, environmentally unfriendly starting materials, complex and time consuming procedures for both material preparation and glycopeptide enrichment, and low repeatability and reproducibility. Wood is one of the most abundant naturally available, cheap and green hydrophilic materials.24 The intrinsic hydrophilicity of wood is due to the high abundance of hydroxyl groups mainly from cellulose and hemicelluloses, which are two of the three main components of the cell wall. Cellulose is a polysaccharide consisting of 8000 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 glucose units, while hemicellulose has a degree of polymerization of around 200 and is constituted by carbohydrate polymers with heterogenous polysaccharide chains (Fig. 1).25 The hierarchical porous architecture of native wood brings an immense influence on molecular adsorption capacity and diffusion speed, due to the anisotropic pore structure and pore channels. The third main component of wood is lignin, which is relatively hydrophobic, and rich in aromatic subunits. The actual structure of lignin is still not completely determined, and monolignols are commonly used as representatives (Fig. 1). Literature shows that delignification could increase the hydrophilicity and porosity of wood.26
000 glucose units, while hemicellulose has a degree of polymerization of around 200 and is constituted by carbohydrate polymers with heterogenous polysaccharide chains (Fig. 1).25 The hierarchical porous architecture of native wood brings an immense influence on molecular adsorption capacity and diffusion speed, due to the anisotropic pore structure and pore channels. The third main component of wood is lignin, which is relatively hydrophobic, and rich in aromatic subunits. The actual structure of lignin is still not completely determined, and monolignols are commonly used as representatives (Fig. 1). Literature shows that delignification could increase the hydrophilicity and porosity of wood.26
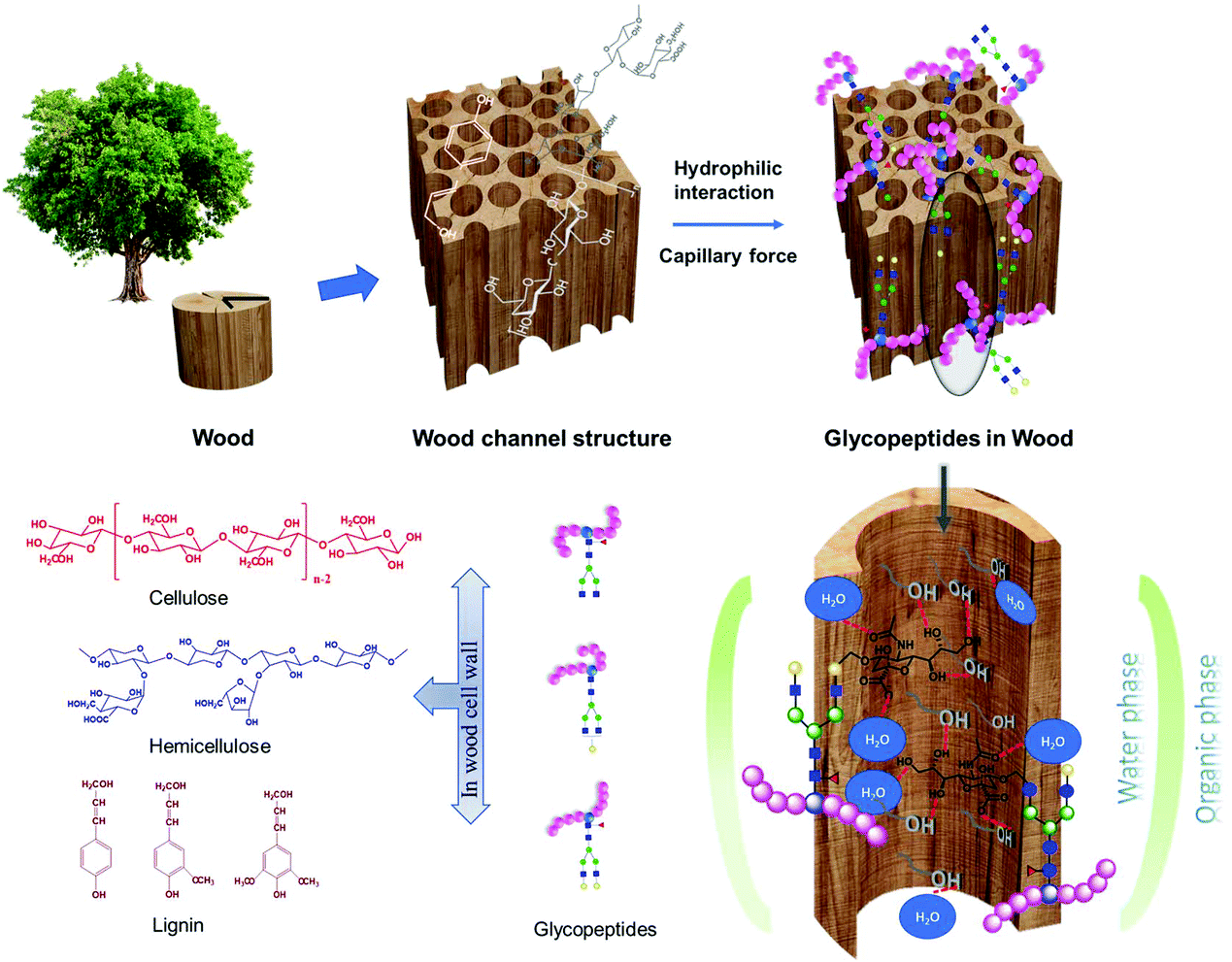 | ||
| Fig. 1 Schematic illustration of the channeled porous structure, main components of wood, and the hydrophilic interaction between glycopeptides and wood via the partitioning of glycopeptides between the organic-rich loading solution and the water-rich layer on the surface of porous wood material. | ||
In the present work, to the best of the authors’ knowledge, it is the first time wood materials have been utilized for immunoglobulin G (IgG) glycopeptide enrichment in a micropipette tip set-up, based on the hydrophilic interaction between the hydrophilic wood components and glycopeptides. A water-rich layer could be formed on the hydrophilic surface of the wood materials, aiding in the retention of glycopeptides, through the partitioning of hydrophilic glycopeptides between the bulk of organic-rich loading solution and the hydrophilic wood materials.27,28 The special ordered channel pore structure can also be beneficial for the enrichment by creating capillary forces facilitating glycopeptide adsorption (Fig. 1).28 The direct utilization of native wood without any pretreatment could prevent complex synthesis procedures, high costs, long preparation times, and the use of chemicals needed for many of the previously described lab synthesized materials. The tip set-up is also advantageous avoiding the long incubation times observed for those materials,16–23 significantly increasing the time-to-result efficiency. Both native balsa wood (N-wood), and delignified balsa wood (D-wood) were evaluated and compared. The results obtained using the wood materials were compared with a commercial HILIC product. The influence of hydrophilicity and porosity on the enrichment performance, as decided by selectivity, binding capacity, sensitivity and recovery were studied. Furthermore, the developed tip set-up using wood materials was applied on a sample of IgG extracted from human plasma in order to investigate its application for a real biological sample. The use of these environmentally friendly materials would not only save costs, time and chemicals involved in glycopeptide enrichment, but also extend the application of environmentally friendly materials, and green chemistry generally in glycoproteomics, and biological sample analysis.
Results and discussion
Characterization of N-wood and D-wood
Wood materials possess highly ordered macro and nano pores building up a channel structure, where the network of pores affects diffusion and rates of molecule adsorption to the materials. Delignification was applied to native wood to increase the hydrophilicity as well as the porosity. Sodium chlorite (NaClO2) is the reagent applied in the delignification process. Although widely used, the chemical reactions are still not well understood in many aspects. It is generally accepted that ClO2 is generated from NaClO2, and oxidizes the phenolic groups in lignin molecules, generating phenoxy radicals. The phenoxy radicals further react with ClO2, resulting in either ring openings of the lignin molecule to muconic acids, or side chain cleavage to quinone. Recently, Tarvo and co-workers, simulated the reactions between ClO2 and lignin, and the possible reactions can be found in Fig. S1.†29In order to observe the porous wood structure in detail, scanning electron microscope (SEM) was used to characterize the wood before and after delignification, as shown in Fig. 2. By delignification, the lignin content was decreased from 24.9% to 2.9%.30 Nanoscale pores were generated (marked with blue arrows in Fig. 2f) in the cell wall corners due to delignification in lignin rich parts, while the honeycomb-like structure, and nanocellulose alignment were still well-preserved. Balsa is the wood with the lowest density in nature, and the flexibility was further increased by delignification. The flexibility of the material is an important advantage for the set-up, where a 200 μL pipette tip was used for sample preparation. A N2 absorption/desorption technique was used to characterize the two materials. No obvious micro- or meso-pores (Fig. S2†) were detected. Nonetheless, macropores could be observed with the SEM analysis. Brunauer–Emmett–Teller (BET) specific surface area was increased from 0.9 m2 g−1 (N-wood) to 4.6 m2 g−1 (D-wood), and pore volume was increased from 3.69 × 10−4 cm3 g−1 (N-wood) to 1.38 × 10−3 cm3 g−1 (D-wood). Two-dimensional (2D) small-angle X-ray scattering (SAXs) measurement results from previous work further supported that the nano pores increased in the structure, with a more pronounced pore size distribution around 4.8 nm for D-wood.30 In addition, porosity was increased after delignification, from 92% for N-wood to 94% for D-wood.
 | ||
| Fig. 2 Morphology of the two wood-based materials: SEM images of N-wood: (a) cross section, (b) channel surface section, and (c) corner wall; D-wood: (d) cross section, (e) channel surface section, and (f) corner wall. The insets are images of the top view of 1 cm2 wood materials. | ||
Both wood materials are highly hydrophilic, as water droplets were immediately absorbed by the wood materials, in less than 3 s for N-wood (Video S1,† and Fig. 3a, b, c), and in less than 1 s for D-wood (Video S2,† and Fig. 3d, e). The hydrophilicity was further increased after delignification showing decreased contact angle in Fig. 3a and d. Meanwhile, the water uptake value was increased from 2 g g−1 to 9 g g−1 after delignification (Fig. 3f). The increased liquid absorption speed and capacity is due to increased hydrophilicity and porosity after delignification.30
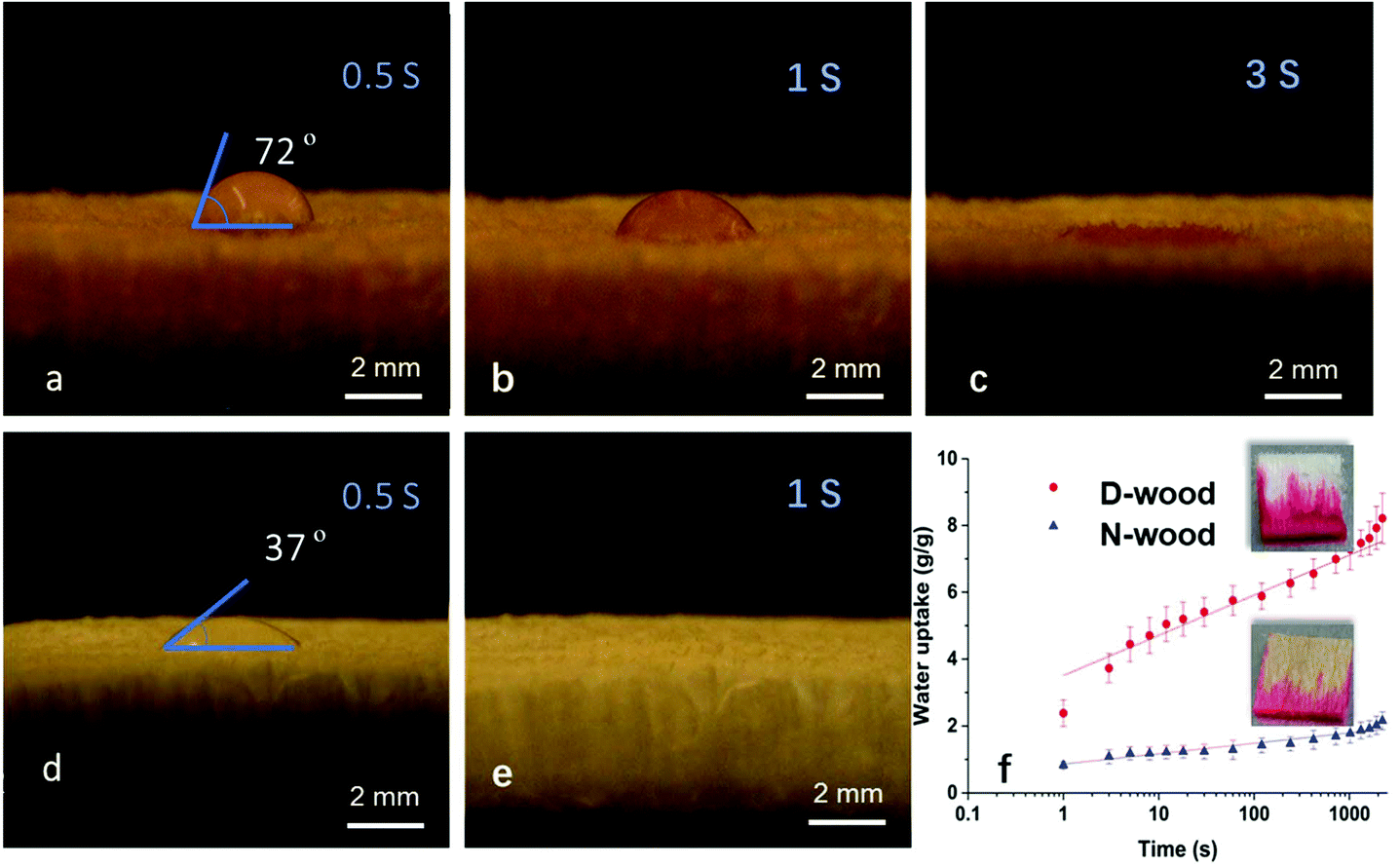 | ||
| Fig. 3 Images of water droplet absorption along the cross section with time, (a–c) N-wood, (d and e) D-wood; (f) water uptake data vs. time, and the Rhodamine 6G dye solution absorption images along the growth channel section in 2 s. | ||
Optimization of IgG glycopeptide enrichment conditions using N-wood tips
The commonly used loading solutions for glycopeptide enrichment are organic–aqueous solutions containing more than 80% of organic solvent, and acetonitrile (ACN) is most often applied.31 The proportion of organic solvent in the loading solution is an important factor since it could not only affect the retention of glycopeptides, but also the hydrophilic non-glycopeptides, on the hydrophilic materials.19 To minimize the co-retention of non-glycopeptides, ion-pairing reagents, such as trifluoroacetic acid (TFA) has been widely used as an additive in the loading solution. This could neutralize the highly charged non-glycopeptides, and increase the difference in hydrophilicity between glycopeptides and non-glycopeptides.32 Therefore, in the present work, loading solutions with different proportions of ACN (83%, 86% and 89%), and TFA (0.1%, 1% and 3%) in H2O were assessed using wood tips to optimize the selectivity towards glycopeptides (Table S1†).Preliminary tests were carried out with 10 μg IgG digests. It could be seen from Fig. S3† that only around 10 non-glycopeptides with much lower intensities than for the glycopeptides were detected when loading solutions contained 83% and 86% ACN. When 89% ACN was used, similar numbers of non-glycopeptides could be seen in the spectra but with much higher intensities, compared to that of 83% and 86% ACN. One of the detected non-glycopeptide (m/z 2136, highlighted in Fig. S3†) even displayed a similar intensity as the main enriched glycopeptides, regardless of the TFA proportion. The intensities of enriched glycopeptides were observed to increase when the ACN proportion was increased from 83% to 89%. This trend was the same for all the studied TFA proportions. The influence of the ACN proportion on glycopeptide intensities was further studied on 5 μg IgG digests, and a considerable increase in glycopeptide intensities could be obtained when ACN proportion was 86%, instead of 83% (Fig. S4†). Higher proportions of ACN in the loading solution could improve the retention of glycopeptides to N-wood, but the retention of some non-glycopeptides would also increase if a too high ACN proportion was used, resulting in interferences in MALDI-TOF-MS spectra. Therefore, an ACN proportion of 86% in the loading solution is recommended.
In order to obtain better selectivity, the number of washing cycles after sample loading was increased from initially 3 repetitions to 5 or 8 repetitions. 86% ACN with 0.1%, 1% and 3% TFA were used as loading solutions for further optimization of the TFA proportion, applied on 7.5 μg IgG digests. Almost all non-glycopeptides could be eliminated with 5 washing repetitions when using 86% ACN with 0.1% and 1% TFA, but approximately 10 non-glycopeptides appeared in the spectra when using 3% TFA (Fig. S5†). Besides being widely used as an ion-pairing reagent in glycopeptide enrichment, TFA is also used in polysaccharide hydrolysis, because of its high acidity. High contents of TFA in the loading solution could hence cause decrystallization of cellulose and hydrolysis of polysaccharides. At the same time, partial esterification of cellulose happens, leading to decreased wood hydrophilicity.33,34 Here, this could explain the decreased selectivity observed when TFA proportion was increased to 3%. There was no obvious improvement in selectivity when the number of washing cycles was increased from 5 to 8 repetitions. The total volume of 1 mL of loading solution was enough to wash out the non-glycopeptides having low interaction with N-wood, while the non-glycopeptides having strong interaction with N-wood could not be washed out with repeated washing but remained in the material. For better selectivity, and considering green chemistry principles,35,36 5 washing repetitions is preferred, and 3% TFA is not recommended. Since similar glycopeptide intensities were observed when loading solutions were 86% ACN/0.1% TFA and 86% ACN/1% (Fig. S3 and S5†), both loading solutions could be utilized for glycopeptide enrichment using N-wood tips.
Optimization of IgG glycopeptide enrichment conditions using D-wood tips
Studies on loading solutions were also carried out on D-wood tips for optimization. Similar to using N-wood tips, higher intensities of glycopeptides could be obtained when using 86% ACN instead of 83% ACN. 3% TFA was not recommended due to decreased selectivity, compared to 1% TFA (Fig. S6†). When the loading solution was 86% ACN/3% TFA, more than 20 non-glycopeptides were detected in the spectra, and two of these had higher intensities than most of the detected glycopeptides. In contrast, both the number and intensities of detected non-glycopeptides were decreased to less than half when 86% ACN/1% TFA was used. Thus, loading solutions of 86% ACN/0.1% TFA and 86% ACN/1% were further investigated for optimization. When applied on 7.5 μg IgG digests, there were no obvious differences in selectivity or glycopeptide intensities between these two loading solutions, or between different numbers of washing repetitions (Fig. S7†). However, in the case of 5 μg IgG digests, much higher intensities of glycopeptides could be obtained when the loading solution was 86% ACN/1% TFA, and tips were washed using 5 repetitions (Fig. S7†). Since coelution of non-glycopeptides could be observed, an extra H2O wetting step was added before preconditioning, to investigate if higher selectivity could be achieved. Almost all non-glycopeptides could be separated from glycopeptides after adding this step into the enrichment procedure using the D-wood tips, and this modified procedure could also be used on N-wood tips (Fig. S8†). Finally, elution solutions of H2O and 1% TFA were investigated and compared, where more non-glycopeptides and higher intensities of non-glycopeptides were observed in the spectra using 1% TFA as elution solution (Fig. S9†). Therefore, H2O is recommended for elution.In conclusion, 86% ACN/1% TFA/13% H2O was selected as loading solution for both N-wood and D-wood tips. Preconditioning was carried out by wetting the tips with H2O, and then preconditioning with loading solution. After sample loading, the tips were washed 5 times with loading solution, and finally eluted with H2O.
Selectivity of N-wood tips, D-wood tips and HILIC tips
10 μg IgG digests were applied on N-wood tips, D-wood tips and commercial HILIC tips, respectively, to investigate how many glycopeptides could be enriched from digests by using each material. MALDI-TOF-MS spectra of enriched glycopeptides using the different tips are shown in Fig. 4. The glycopeptides detected after enrichment with each type of material are listed in Table S2.† There were totally 36 (with 16 glycan compositions), 38 (with 16 glycan compositions), and 35 (with 15 glycan compositions) glycopeptides identified using N-wood tips, D-wood tips, and HILIC tips, respectively. When using N-wood tips, 16 IgG1 glycopeptides (peptide sequences:![[E with combining low line]](https://www.rsc.org/images/entities/char_0045_0332.gif)
![[Q with combining low line]](https://www.rsc.org/images/entities/char_0051_0332.gif)
![[Y with combining low line]](https://www.rsc.org/images/entities/char_0059_0332.gif)
![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif)
![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif) and TKPR), and 20 IgG2 glycopeptides (peptide sequences:
and TKPR), and 20 IgG2 glycopeptides (peptide sequences: ![[F with combining low line]](https://www.rsc.org/images/entities/char_0046_0332.gif) and TKPR) could be enriched and detected. There were 18 IgG1 and 20 IgG2 enriched glycopeptides detected using D-wood tips, while 18 IgG1 and 17 IgG2 glycopeptides using HILIC tips. Thus, the number of enriched glycopeptides was slightly higher using D-wood tips, compared to N-wood tips and HILIC tips.
and TKPR) could be enriched and detected. There were 18 IgG1 and 20 IgG2 enriched glycopeptides detected using D-wood tips, while 18 IgG1 and 17 IgG2 glycopeptides using HILIC tips. Thus, the number of enriched glycopeptides was slightly higher using D-wood tips, compared to N-wood tips and HILIC tips.
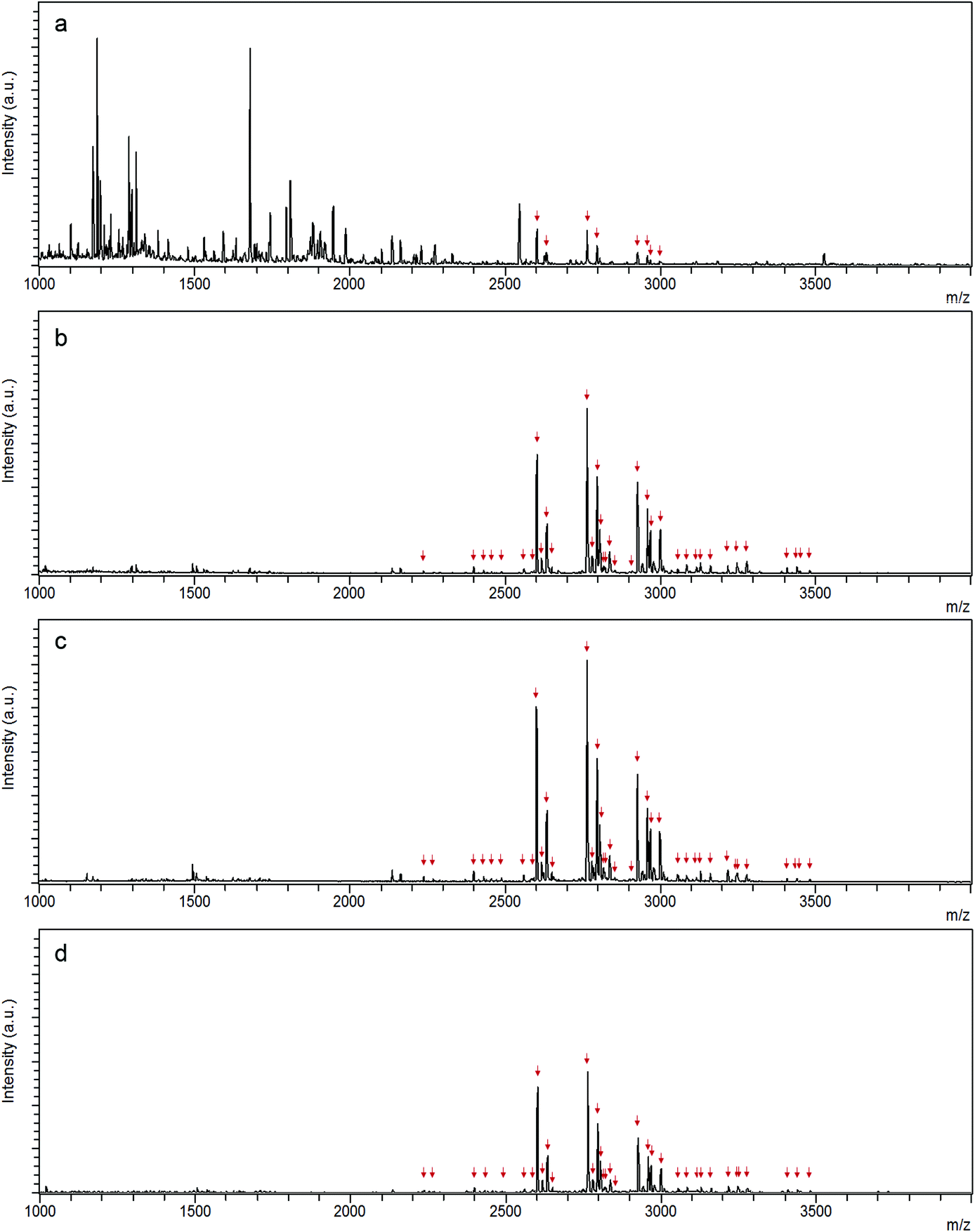 | ||
| Fig. 4 MALDI-TOF-MS spectra of (a) IgG digest, and enriched IgG glycopeptides using (b) N-wood tip, (c) D-wood tip and (d) HILIC tip. 10 μg IgG digest was loaded on each tip, and 0.5 μg sample was applied on each spot. Glycopeptides are marked with red arrows. | ||
The S/N ratios of enriched glycopeptides using different tips were also evaluated, and the average S/N values of the 10 most abundant glycopeptides are shown in Fig. S10.† Glycopeptides enriched using D-wood tips had the highest S/N values. The affinity of glycopeptides to the wood material was improved considerably after delignification, especially for IgG2 glycopeptides, such as m/z 2602, 2674, 2805, 2926 and 2967. N-wood also exhibited higher S/N values for enriched glycopeptides, except glycopeptide m/z 2634 having slightly lower S/N value, compared to the commercial HILIC product.
In order to further investigate the selectivity of glycopeptide enrichment using different tips, sample mixtures of IgG digest and bovine serum albumin (BSA) digest with different weight mixing ratios were loaded onto the three kinds of tips for comparison.18,37 Sample mixtures with the following IgG digest/BSA digest (IgG/BSA) weight mixing ratios were enriched: 1/10, 1/50, 1/100 and 1/150, corresponding to molar ratios of 1/23, 1/115, 1/230 and 1/345. After mixing the IgG digest with the BSA digest, a lot of non-glycopeptides could be detected in the elution fractions together with glycopeptides, no matter what kind of tip was used for enrichment (Fig. S11 and S12†). As shown in Fig. 5 and Table S3,† both the number of detected glycopeptides and the S/N values decreased with increasing proportion of BSA digest in the mixture. When IgG/BSA was 1/50, 10 glycopeptides were detected when using N-wood tips, and 9 and 7 with D-wood tips and HILIC tips, respectively (Fig. S11†). The numbers were decreased to 9, 9, and 7 for N-wood, D-wood and HILIC tips when IgG/BSA was 1/100 (w/w) (Fig. S12†). Only 8, 5 and 4 glycopeptides showed average S/N > 3 when loaded with IgG/BSA 1/150 (w/w), using N-wood, D-wood and HILIC tips, respectively (Fig. S12†). N-wood and D-wood tips exhibited better selectivity compared to HILIC, with both higher numbers and larger average S/N values of enriched glycopeptides. Due to the decrease in selectivity when large amounts of BSA digest were added, it is crucial to remove abundant unrelated proteins when analyzing real biological samples.
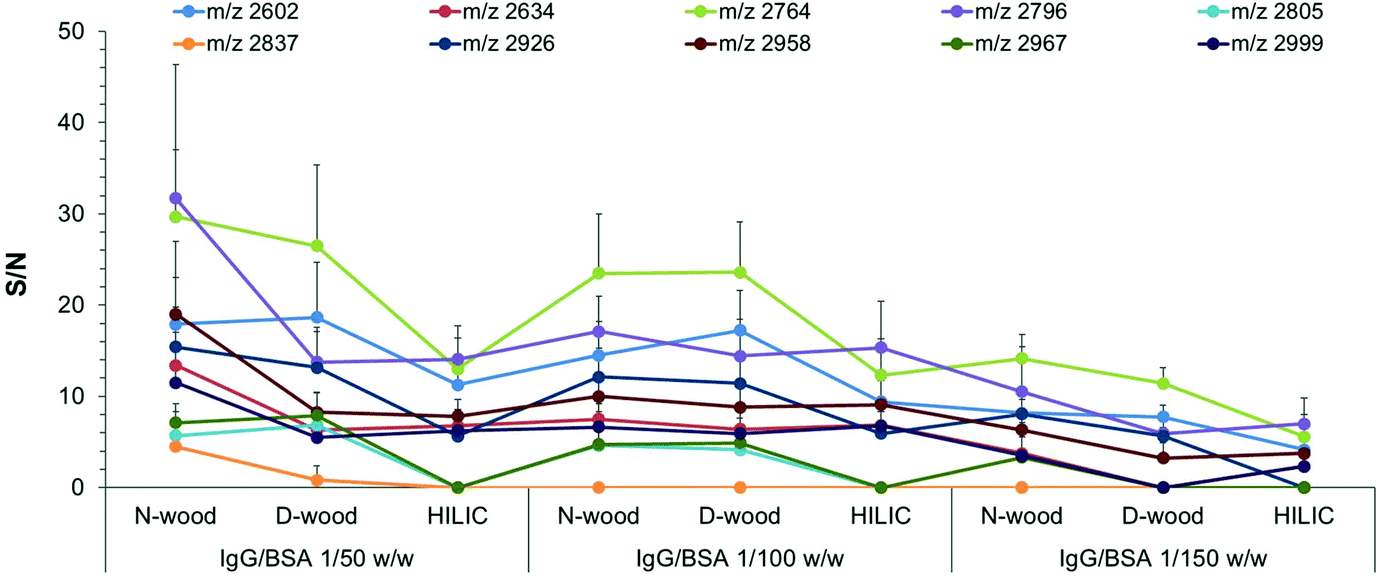 | ||
| Fig. 5 Average S/N values of enriched glycopeptides using different tips when loading the same amount of IgG digest (0.5 μg, 165 fmol on each spot) with different amounts of BSA digest (25 μg, 50 μg, 75 μg), corresponding to IgG/BSA weight ratios of 1/50, 1/100 and 1/150. 12 spots from 3 replicates were analyzed with MALDI-TOF-MS. | ||
Binding capacity of N-wood and D-wood tips
The binding capacities of N-wood and D-wood tips regarding IgG digests were assessed by loading sequentially increasing amounts of IgG digests to the tips,19 4 to 10 μg for N-wood tips, and 4 to 12 μg for D-wood tips. S/N values of 10 abundant glycopeptides present in the elution fractions were evaluated (Fig. 6 and Tables S4, S5†). Most of the selected IgG2 glycopeptides (m/z 2602, 2764, 2805, 2926, 2967) gave maximum average S/N values when 8 μg IgG digests were loaded on N-wood tips. However, the S/N values for IgG1 glycopeptides (m/z 2634, 2796, 2837, 2958, 2999) were slightly increased when the loading amount was increased to 10 μg using N-wood tips. The results were somewhat different when D-wood tips were evaluated. The S/N values of IgG1 glycopeptides reached a maximum at 11 μg, while S/N values of IgG2 glycopeptides were slightly increased when 12 μg IgG digests were loaded. Therefore, the binding capacities were assumed to be 8 μg mg−1 using N-wood tips, and 11 μg mg−1 using D-wood tips. It could also be seen in Fig. 6, that higher amounts of glycopeptides could be enriched, eluted and detected using D-wood tips when loading the same amount of IgG digest, compared to N-wood tips.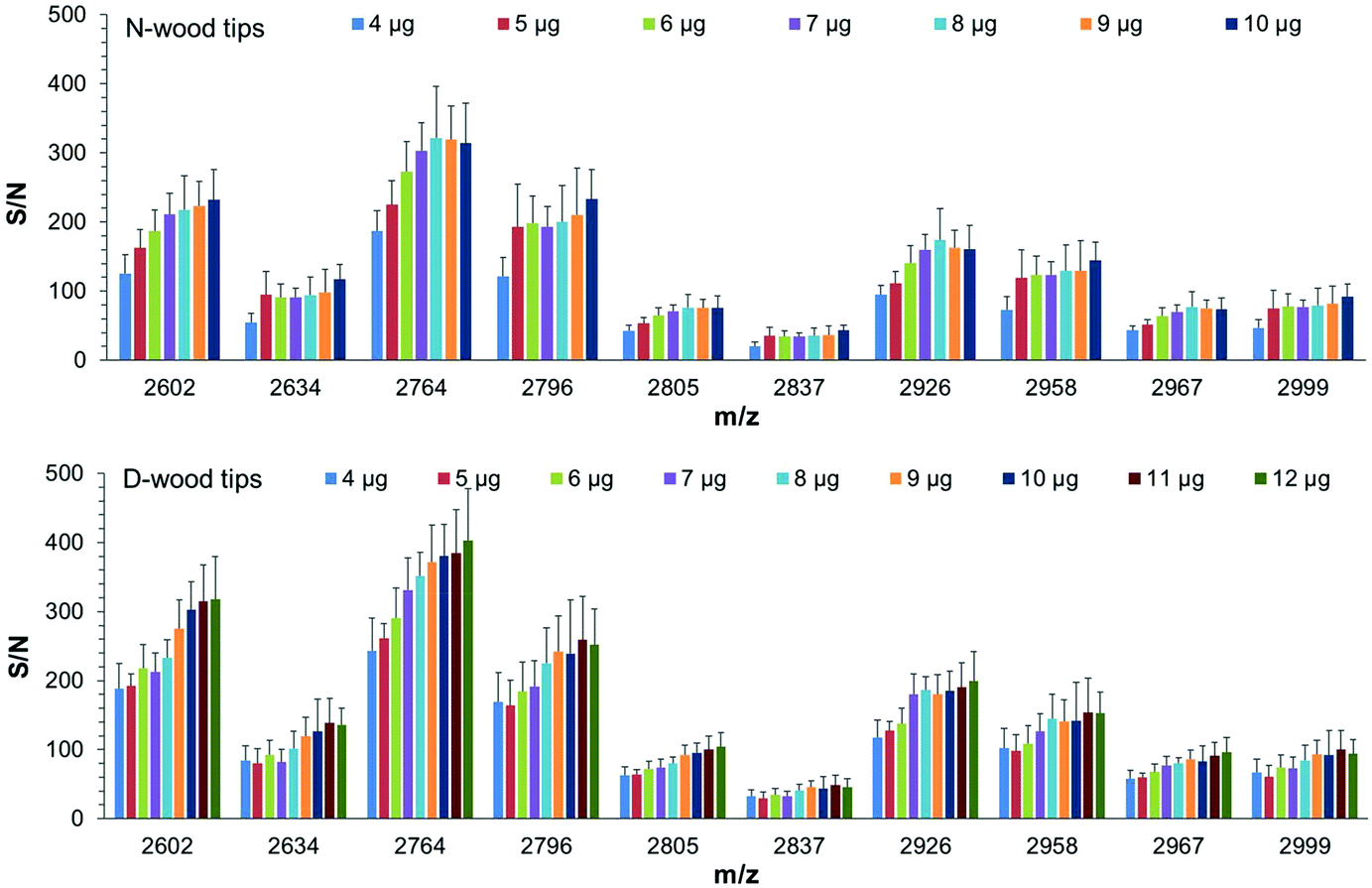 | ||
| Fig. 6 Average S/N values of enriched glycopeptides using different tips when loading different amounts of IgG digests. Top: N-wood tips, 4 to 10 μg IgG digests, Bottom: D-wood tips, 4 to 12 μg IgG digests. 12 spots from 3 replicates were analyzed with MALDI-TOF-MS. | ||
Sensitivity obtained using N-wood, D-wood and HILIC tips
High sensitivity is important in analysis of biological samples due to the low abundance of some proteins in biological matrices. In the present work, N-wood, D-wood and HILIC tips were loaded with different amounts of IgG digests, 300 ng, 150 ng and 75 ng (corresponding to final amounts of 100 fmol, 50 fmol and 25 fmol on each spot on the MALDI target plate). The number of detected glycopeptides and the S/N data were investigated for different amounts of enriched glycopeptides applied on the MALDI target plate, and summarized in Fig. 7 (Fig. S13, S14, Table S6†). At 100 fmol, 12 glycopeptides could be detected, with N-wood tips having the highest S/N values, followed by D-wood tips, and then HILIC tips. At 50 fmol, 10, 10 and 9 glycopeptides could be detected for N-wood, D-wood and HILIC tips respectively. Similar S/N values could be obtained for most of the enriched glycopeptides when comparing N-wood and D-wood tips, while the values were lower when HILIC tips were used. At 25 fmol, 7 of 9, 7 of 9 and 3 of 6 detected glycopeptides showed average S/N values >3 for N-wood, D-wood and HILIC tips, respectively. Furthermore, 30 ng IgG digests (10 fmol applied on the MALDI target plate) were also evaluated on N-wood and D-wood tips. However, only 2 glycopeptides showed average S/N > 3 using N-wood tips, and 4 using D-wood tips. Overall, N-wood and D-wood tips exhibited better sensitivity than commercial HILIC tips.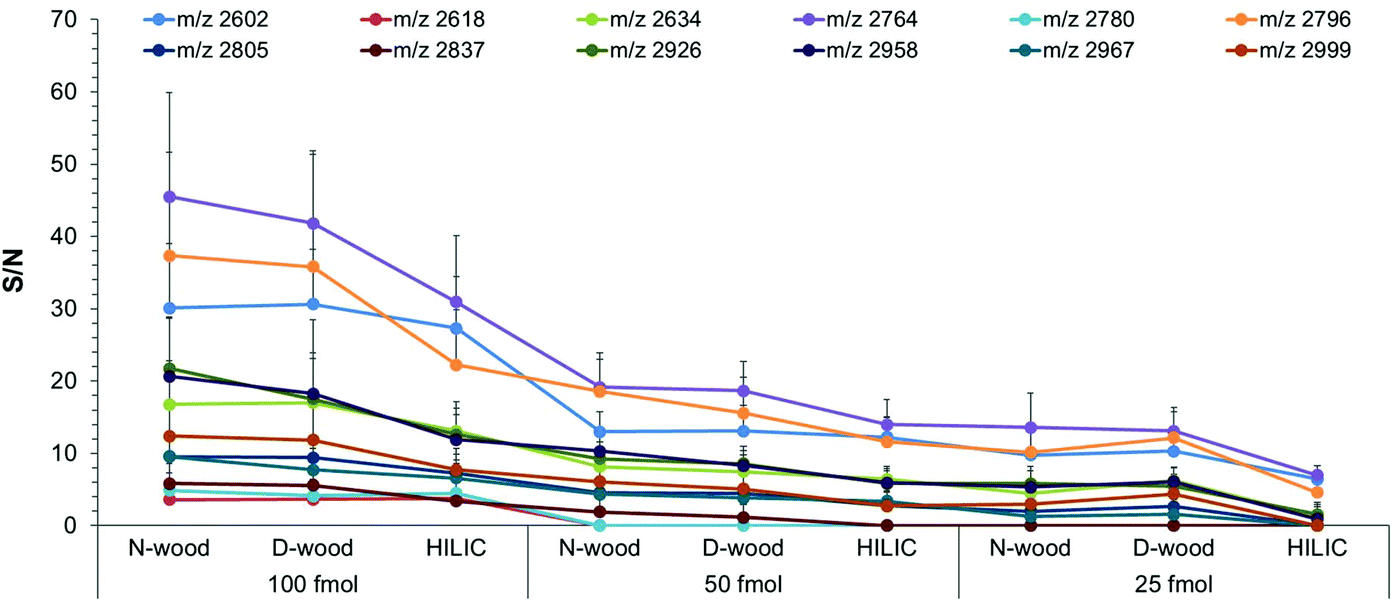 | ||
| Fig. 7 Average S/N of enriched and detected glycopeptides using different tips loaded with different amounts of IgG digests, 100 fmol, 50 fmol and 25 fmol on spot, respectively. 12 spots from 3 replicates were analyzed using MALDI-TOF-MS. | ||
Recovery obtained using N-wood and D-wood tips
In order to facilitate the application of N-wood and D-wood tips for glycopeptide enrichment from biological samples, glycopeptide enrichment recovery was also studied according to a previously discussed method using MALDI-TOF-MS for relative quantification (Table S7 and Fig. S15†).21,38 Briefly, the recoveries were estimated for three main enriched IgG1 glycopeptides using a synthesized label-free IgG1 glycopeptide as internal standard (S1.† Experimental section). The recoveries were 56% (m/z 2634), 72% (m/z 2796) and 83% (m/z 2958) using N-wood tips, and 80% (m/z 2634), 88% (m/z 2796) and 91% (m/z 2958) using D-wood tips (Tables S8 and S9†). It was observed that the recovery of glycopeptides varied for different glycosylation structures, and the recovery was increased with the increasing mass of the glycopeptides, regardless of which kind of tips that were used. A possible explanation is that glycopeptides with higher mass contain larger glycan chains, which makes the glycopeptide more hydrophilic, and thus gains a higher affinity towards wood materials compared to glycopeptides with smaller glycans. Recovery data also demonstrated that higher recoveries could be obtained when D-wood was used for glycopeptide enrichment, compared to N-wood. This agreed with the results obtained regarding binding capacity, where increased S/N values were observed for glycopeptides when using D-wood tips instead of N-wood tips, applying the same amount of IgG digest.Comparing N-wood with D-wood tips
Compared with N-wood tips, better affinity for glycopeptides was demonstrated utilizing D-wood tips, showing higher S/N values for enriched glycopeptides, higher binding capacity and higher recoveries. This could be explained by the increased hydrophilicity and porosity acquired by delignification, resulting in improved binding of glycopeptides. To further study the chemical interaction between the wood materials and the glycopeptides, attenuated total reflection-Fourier transformed infrared spectroscopy (ATR-FTIR) was employed to characterize the materials before and after loading IgG digest samples. Fig. 8a shows a FTIR spectrum of N-wood with the typical peaks as reported in literature.39 Compared to N-wood, the characteristic lignin vibration C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak at 1593 cm−1 (marked with yellow oval in Fig. 8) was reduced as expected for D-wood.40 Meanwhile, the glycopeptides adsorbed in both N-wood and D-wood tips could be determined with the typical C–N stretching and N–H bending at 1558 and 1540 cm−1 (Fig. 8, marked with black arrows). In Fig. 8d, the multiple peaks in the region of 1600–1700 cm−1 represent amide CO stretching associated with different peptide structures.41 This agrees with the MALDI-TOF-MS spectra of enriched glycopeptides using D-wood tips, with some of the non-glycopeptides showing up together with the glycopeptides. When adding BSA digest to the IgG digest, much higher intensities of non-glycopeptides, and slightly lower intensities of enriched glycopeptides were obtained using D-wood tips, compared to using N-wood tips. The selectivity of the wood material was slightly decreased after delignification, probably due to more nanopores generated (Fig. 2f), and increased liquid absorption capacity after delignification (Fig. 3f). The capillary force enhances the liquid diffusion, which could facilitate the adsorption of more non-glycopeptides when the amount of glycopeptides is much lower than the binding capacity, and thus cause interference in the detection of glycopeptides.
O peak at 1593 cm−1 (marked with yellow oval in Fig. 8) was reduced as expected for D-wood.40 Meanwhile, the glycopeptides adsorbed in both N-wood and D-wood tips could be determined with the typical C–N stretching and N–H bending at 1558 and 1540 cm−1 (Fig. 8, marked with black arrows). In Fig. 8d, the multiple peaks in the region of 1600–1700 cm−1 represent amide CO stretching associated with different peptide structures.41 This agrees with the MALDI-TOF-MS spectra of enriched glycopeptides using D-wood tips, with some of the non-glycopeptides showing up together with the glycopeptides. When adding BSA digest to the IgG digest, much higher intensities of non-glycopeptides, and slightly lower intensities of enriched glycopeptides were obtained using D-wood tips, compared to using N-wood tips. The selectivity of the wood material was slightly decreased after delignification, probably due to more nanopores generated (Fig. 2f), and increased liquid absorption capacity after delignification (Fig. 3f). The capillary force enhances the liquid diffusion, which could facilitate the adsorption of more non-glycopeptides when the amount of glycopeptides is much lower than the binding capacity, and thus cause interference in the detection of glycopeptides.
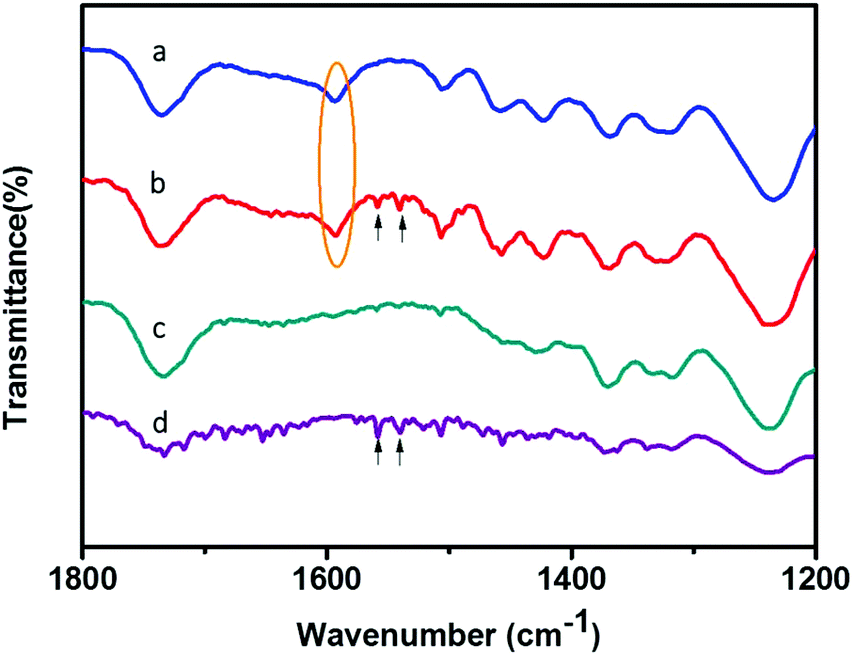 | ||
| Fig. 8 FTIR spectra of (a) N-wood, (b) N-wood after loading 10 μg IgG digest, (c) D-wood, (d) D-wood after loading 10 μg IgG digest. | ||
IgG Glycopeptide enrichment from human plasma
To illustrate the usefulness of the wood tips for real biological samples, IgG extracted from human plasma was applied on N-wood, D-wood and HILIC tips. After enrichment, 25, 25 and 24 glycopeptides could be detected using N-wood, D-wood and HILIC tips respectively (Fig. S16, S17 and Table S10†). Compared to IgG standard samples, glycopeptides with only one peptide sequence for each IgG subclass were enriched and detected, due to the fact that denaturation, reduction and alkylation steps were included before digestion, giving higher efficiency of the digestion, and less missed cleavages. There were 10 IgG 1 (peptide sequence: EEQYNSTYR) glycopeptides detected from human plasma IgG samples after enrichment for all three materials. 15 IgG 2 (peptide sequence: EEQFNSTFR) glycopeptides could be detected using N-wood and D-wood tips, and 14 using HILIC tip. The total number of glycosylation compositions of detected human plasma IgG glycopeptides was 15 for N-wood and D-wood tips, and 14 for HILIC tips, similar to the total numbers of glycan compositions observed in IgG standard samples.Conclusions
A micropipette set-up using wood materials was to the authors’ knowledge successfully utilized for the first time for IgG glycopeptide enrichment, with both IgG standard samples, and IgG samples extracted from human plasma. Tips packed with native wood without any pretreatment exhibited comparable performance, or better, compared with commercial HILIC tips. It could prevent complex synthesis procedures, decrease time and chemicals consumed, as well as costs involved for lab synthesized materials, saving chemicals, energy, and providing high time-to-result efficiency. The binding capacity and recoveries could be improved by delignification due to the increased hydrophilicity and porosity of the material. Glycopeptide enrichment based on wood materials could not only provide an alternative to commercial HILIC products with high performance and low costs, but also shows environmental sustainability, and high potential for scaling up. We believe that wood materials could be further utilized in applications regarding the analysis of glycoprotein biomarkers. It could also help broaden the way for environmentally friendly methods in the study of biological samples.Experimental section
Chemicals
Immunoglobulin G from human serum (IgG, 56834-25MG), albumin from bovine serum (BSA, A3059), trypsin from bovine pancreas (T1426), acetonitrile (ACN), trifluoroacetic acid (TFA), DL-dithiothreitol (DTT), iodoacetamide (IAA), sodium chlorite (NaClO2), Eppendorf LoBind® microcentrifuge tubes (Lobind tube), and all salts used in the present work were purchased from Sigma-Aldrich (Stockholm, Sweden). H2O used in the present work was purified to have a resistivity of 18.2 MΩ cm at 25 °C with a Millipore Synergy® 185 system (Bedford, MA, USA). 2,5-Dihydroxybenzoic acid (DHB) and α-cyano-4-hydroxycinnamic acid (HCCA) were obtained from Bruker Daltonics (Bremen, Germany). Anonymized and pooled human plasma from volunteers was stored in −80 °C before use. DEAE Affi-Gel® Blue Gel (Affi-Gel) was from Bio-RAD (Hercules, USA).Wood delignification
Balsa wood (Ochroma pyramidale, 160 kg m−3, Wentzels Co. Ltd, Sweden) was dried at 105 ± 3 °C for 24 h prior to chemical extraction. Wood samples were longitudinally cut to a size of 20 mm × 20 mm × 3 mm (L × W × T). After drying, the samples were extracted using NaClO2 (1 wt%) with acetate buffer solution (pH 4.6) at 80 °C for 6 h according to literature.30 The extracted samples were then washed carefully with H2O, followed by freeze-drying before further application.Wood material characterization
The SEM images of wood samples were obtained using a Field-Emission Scanning Electron Microscope (Hitachi S-4800, Japan). The acceleration voltage was 1 kV. All samples were coated with platinum–palladium before characterization with SEM. Brunauer–Emmett–Teller (BET) N2 absorption (Micromeritics ASAP 2020) was carried out to obtain data on pore size distribution. Wood samples were first degassed at 70 °C for 5 h, and then analyzed at −196 °C by N2 physisorption. Water absorption videos and figures were recorded using a Dino-Lite digital microscope (AM4000/AD4000 series, 1.3 Megapixel, magnification 10×), 5 μL water droplets were applied. The absorption capability of water was calculated by the mass of water uptake divided by the original mass of the wood sample in g, measured three times, according to a previously reported method.42 The density of the wood material was the function of the weight between the dried samples (105 °C overnight, 30 min under vacuum), and samples before drying.43 The total porosity (f) as a function of the solid density of holocellulose (1500 kg m−3), and the solid density of balsa wood can be described by the equation:44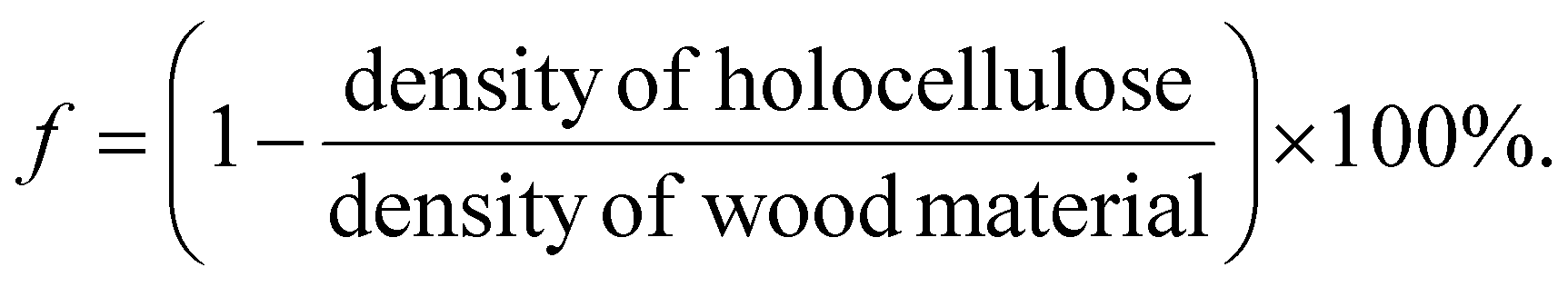
ATR-FTIR spectra were obtained using Thermo Nicolet 380 spectrometer (Nicolet, Wisconsin, USA).
Human plasma IgG extraction
50 μL Affi-Gel was preconditioned with 200 μL of Tris-HCl buffer (20 mM, pH 8.0). Human plasma (5 μL) was diluted with Tris-HCl buffer (45 μL) and loaded onto preconditioned Affi-Gel. Plasma and Affi-Gel were mixed for 30 min at 1000 rpm in Eppendorf ThermoMixer® C (Hamburg, Germany). After centrifugation, the supernatant containing human plasma IgG was collected for further use. Human plasma IgG sample was denatured at 95 °C for 10 min, reduced by DTT (final concentration 10 mM) at 60 °C for 30 min, and alkylated by IAA (final concentration 25 mM) in dark at 37 °C for 30 min in a thermomixer. Denatured, reduced and alkylated human plasma IgG samples were then desalted using 100 μL bed sized Pierce® C18 tips (Thermo Fisher Scientific, Rockford, USA). Collected fractions were concentrated to 10 μL before trypsin digestion.Trypsin digestion
Trypsin was mixed with IgG samples and ammonium bicarbonate solution (pH = 8.0, final concentration 10 mM) for digestion at 37 °C for 17 h. For IgG standard samples, the digestion mixture had a final IgG concentration of 1 mg mL−1, weight ratio of 30/1 (IgG/trypsin). For human plasma IgG samples, 2 μg trypsin were added into 10 μL obtained aliquots, the final volume was 20 μL. For all samples, digestion was quenched by incubating at 75 °C for 5 min.Glycopeptide enrichment using wood and HILIC tips
1 mg N-wood or D-wood was pushed into a 200 μL micropipette tip (Eppendorf® epT.I.P.S. standard 2–200 μL, EP0030000870, Sigma-Aldrich, Stockholm, Sweden) (Fig. S18†), and stored before use (in sensitivity tests, 0.5 mg material was used). HILIC tips were purchased from Thermo Scientific (HyperSep™ Tip 60109-214, Rockwood, United States). Before loading sample solution, the tips were preconditioned with 100 μL H2O 5 times, and 100 μL loading solution 5 times (Table S1†). Sample solutions were prepared by adding ACN, TFA and H2O into IgG digests to have a total volume of 100 μL with the same ACN/TFA proportion as in the loading solution. Samples were loaded by pipette aspiration for 20 times. After that, the tips were washed several times using the loading solution, and then samples were eluted with 100 μL H2O. Finally, the elution fractions were dried using Eppendorf concentrator 5301 (Hamburg, Germany), and reconstituted with H2O to 5 μL for MALDI-TOF-MS (Fig. S19†).IgG glycopeptide profiling using MALDI-TOF-MS
0.25 μL of the enriched samples were spotted onto a MALDI plate (MTP AnchorChip 384 BC, Bruker Daltonics, Bremen, Germany). 0.25 μL of 20 mg ml−1 DHB matrix (in ACN/0.1% TFA 30/70 v/v, TA30) was added onto the top of the sample spots after they had dried. For each sample, 12 spots from 3 replicates were prepared and analyzed. UltrafleXtreme MALDI TOF/TOF mass spectrometer was used in positive reflectron mode for glycopeptide profiling. The laser (Smartbeam™-II laser, 355 nm, UV) intensity was set at 80%, and each mass spectrum was summed from 5000 shots with a frequency of 1000. In order to have homogeneous spot sampling, random walk (partial sample) with 100 laser shots per raster spot was applied. Calibrations were performed using peptide calibration standard (Bruker Daltonics, Bremen, Germany) before sample analysis.Human plasma IgG glycopeptide identification using MALDI-TOF-MS/MS
The glycan compositions of enriched IgG glycopeptides from standard samples were obtained by comparing with previous work.19,21 In the case of human plasma IgG glycopeptides, MALDI-TOF-MS/MS spectra of main glycopeptides were analyzed in combination with the glycan annotation tool incorporated in the FlexAnalysis software (Version 3.4, Bruker, Bremen, Germany). In MS/MS mode, HCCA (saturated in TA30) was used as matrix, 80% of total laser intensity was set for parent ion and 90% for fragmentation. In total, 10000 shots including 2000 shots for parent ion, and 8000 shots for daughter ions were accumulated. IgG N-linked glycopeptides have a common biantennary core structure consisting of heptasaccharides. Within this core structure, two N-acetylglucosamine (GlcNAc) residues are linked to each other, one of them is linked to asparagine (N) and the other is linked to a mannose, followed by two mannose branches, and a GlcNAc residue to each mannose. Individual glycans can have one or several monosaccharides attached to this core structure, such as fucose, galactose and sialic acid.45 This common structure could generate a characteristic peak pattern in MS/MS spectra, with series of fragmentations including [M + H]+ − 17, [M + H]+, [M + H]+ + 83 and [M + H]+ + 120 Da.46 The peptide mass within the glycopeptide could be calculated by searching for this characteristic pattern in the MS/MS spectrum. Glycan composition can be annotated using the tool in FlexAnalysis to calculate the saccharides in MS/MS spectra. For some IgG glycopeptides, the glycan compositions could be obtained both by using the annotation tool (Fig. S16†), and comparison with enriched glycopeptides from IgG standard samples.
Conflicts of interest
There are no conflicts to be declared.Acknowledgements
The authors gratefully acknowledge the scholarship from China Scholarship Council (No. 201600160033), and a research grant from Royal Swedish Academy of Sciences (ES2019-0015). Also, thanks for the financial support from the Swedish Research Council (2017-00935, 2017-05349), the Swedish Energy Agency, the Knut & Alice Wallenberg Foundation, the European Research Council Advanced Grant (No. 742733), Wood NanoTech, the National Basic Research Program of China (973 Program, 2014CB239402).References
- A. Gonzalez-Quintela, R. Alende, F. Gude, J. Campos, J. Rey, L. M. Meijide, C. Fernandez-Merino and C. Vidal, Clin. Exp. Immunol., 2008, 151, 42–50 CrossRef CAS PubMed.
- M. Papakonstantinou, G. Dryllis, M. Efstathopoulou, D. Vlachopanou, M. Kechriotis and S. Valsami, J. Biomed., 2019, 4, 35–43 CrossRef.
- S. T. Gizaw, T. Koda, M. Amano, K. Kamimura, T. Ohashi, H. Hinou and S. Nishimura, Biochim. Biophys. Acta, 2015, 1850, 1704–1718 CrossRef CAS.
- Y. Miura and T. Endo, Biochim. Biophys. Acta, 2016, 1860, 1608–1614 CrossRef CAS PubMed.
- X. Li, X. Wang, Z. Tan, S. Chen and F. Guan, Front. Oncol., 2016, 6, 33 Search PubMed.
- M. Dalziel, M. Crispin, C. N. Scanlan, N. Zitzmann and R. A. Dwek, Science, 2014, 343, 1235681 CrossRef.
- I. Gudelj, G. Lauc and M. Pezer, Cell. Immunol., 2018, 333, 65–79 CrossRef CAS PubMed.
- H. Liu, N. Zhang, D. Wan, M. Cui, Z. Liu and S. Liu, Clin. Proteomics, 2014, 11, 11–14 CrossRef PubMed.
- Y. Kizuka, S. Kitazume and N. Taniguchi, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 2447–2454 CrossRef CAS PubMed.
- A. C. Walls, Y.-J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Cell, 2020, 180, 1–12 CrossRef.
- S. Kumar, V. K. Maurya, A. K. Prasad, M. L. B. Bhatt and S. K. Saxena, Virusdisease, 2020, 31, 13–21 CrossRef PubMed.
- Y. Li, Y. Tian, T. Rezai, A. Prakash, M. F. Lopez, D. W. Chan and H. Zhang, Anal. Chem., 2011, 83, 240–245 CrossRef PubMed.
- W. Morelle, V. Faid, F. d. r. Chirat and J.-C. Michalski, in Glycomics-Methods and protocols, ed. N. G. Karlsson and N. H. Packer, Humana Press, Totowa, NJ, 2009, vol. 534, ch. 1, pp. 3–21 Search PubMed.
- K. Mills, A. W. Johnson, O. Diettrich, P. T. Clayton and B. G. Winchester, Tetrahedron: Asymmetry, 1999, 11, 75–93 CrossRef.
- Y. J. Kim, A. Freas and C. Fenselau, Anal. Chem., 2001, 73, 1544–1548 CrossRef PubMed.
- Y. Xu, Z. Wu, L. Zhang, H. Lu, P. Yang, P. A. Webley and D. Zhao, Anal. Chem., 2009, 81, 503–508 CrossRef PubMed.
- W. Huan, J. Zhang, H. Qin, F. Huan, B. Wang, M. Wua and J. Li, Nanoscale, 2019, 11, 10952–10960 RSC.
- C. Xia, F. Jiao, F. Gao, H. Wang, Y. Lv, Y. Shen, Y. Zhang and X. Qian, Anal. Chem., 2018, 90, 6651–6659 CrossRef PubMed.
- L. Zhang, S. Ma, Y. Chen, Y. Wang, J. Ou, H. Uyama and M. Ye, Anal. Chem., 2019, 91, 2985–2993 CrossRef PubMed.
- M. S. Sajid, F. Jabeen, D. Hussain, Q.-t.-A. A. Gardner, M. N. Ashiq and M. Najam-ul-Haq, J. Sep. Sci., 2020, 43, 1348–1355 CrossRef.
- Y. Zhou, Y. Xu, C. Zhang, A. Emmer and H. Zheng, Anal. Chem., 2020, 92, 2151–2158 CrossRef.
- J. Bai, Z. Liu, H. Wang, X. You, J. Ou, Y. Shen and M. Ye, J. Chromatogr. A, 2017, 1498, 37–45 CrossRef PubMed.
- F. Jiao, F. Gao, H. Wang, Y. Deng, Y. Zhang, X. Qian and Y. Zhang, Anal. Chim. Acta, 2017, 970, 47–56 CrossRef.
- L. A. Berglund and I. Burgert, Adv. Mater., 2018, 30, e1704285 CrossRef PubMed.
- M. Zhu, J. Song, T. Li, A. Gong, Y. Wang, J. Dai, Y. Yao, W. Luo, D. Henderson and L. Hu, Adv. Mater., 2016, 28, 5181–5187 CrossRef CAS PubMed.
- C. Piao, J. E. Winandy and T. F. Shupe, Wood Fiber Sci., 2010, 42, 490–510 CAS.
- Y. Xue, J. Xie, P. Fang, J. Yao, G. Yan, H. Shen and P. Yang, Analyst, 2018, 143, 1870–1880 RSC.
- C. C. Chen, W. C. Su, B. Y. Huang, Y. J. Chen, H. C. Tai and R. P. Obena, Analyst, 2014, 139, 688–704 RSC.
- V. Tarvo, T. Lehtimaa, S. Kuitunen, V. Alopaeus, T. Vuorinen and J. Aittamaa, J. Wood Chem. Technol., 2010, 30, 230–268 CrossRef CAS.
- Y. Li, Q. Fu, S. Yu, M. Yan and L. Berglund, Biomacromolecules, 2016, 17, 1358–1364 CrossRef CAS PubMed.
- K. Alagesan, S. K. Khilji and D. Kolarich, Anal. Bioanal. Chem., 2017, 409, 529–538 CrossRef CAS.
- S. Mysling, G. Palmisano, P. Højrup and M. Thaysen-Andersen, Anal. Chem., 2010, 82, 5598–5609 CrossRef CAS PubMed.
- D. Fengel and G. Wegener, in Hydrolysis of Cellulose: Mechanisms of Enzymatic and Acid Catalysis, American Chemical Society, 1979, vol. 181, ch. 7, pp. 145–158 Search PubMed.
- Monika, IOSR–JAC, 2018, 11, 71–74 CAS.
- S. L. Y. Tang, R. L. Smithb and M. Poliakoff, Green Chem., 2005, 7, 761–762 RSC.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- B. Liu, B. Wang, Y. Yan, K. Tang and C. F. Ding, Rapid Commun. Mass Spectrom., 2020, 34, e8607 CAS.
- R. Roy, E. Ang, E. Komatsu, R. Domalaon, A. Bosseboeuf, J. Harb, S. Hermouet, O. Krokhin, F. Schweizer and H. Perreault, J. Am. Soc. Mass Spectrom., 2018, 29, 1086–1098 CrossRef CAS.
- V. Emmanuel, B. Odile and R. Celine, Spectrochim. Acta, Part A, 2015, 136 Pt C, 1255–1259 CrossRef PubMed.
- R. Agrawal, A. Satlewal, M. Kapoor, S. Mondal and B. Basu, Bioresour. Technol., 2017, 224, 411–418 CrossRef CAS.
- L. Chen, J. Feng, D. Yang, F. Tian, X. Ye, Q. Qian, S. Wei and Y. Zhou, Chem. Sci., 2019, 10, 8171–8178 RSC.
- Q. Fu, F. Ansari, Q. Zhou and L. A. Berglund, ACS Nano, 2018, 12, 2222–2230 CrossRef CAS PubMed.
- J. Garemark, X. Yang, X. Sheng, O. Cheung, L. Sun, L. A. Berglund and Y. Li, ACS Nano, 2020, 14, 7111–7120 CrossRef CAS PubMed.
- L. J. Gibson and M. F. T. Ashby, The Structure of Cellular Solids. Cellular Solids: Structure and Properties, 2nd edn, Cambridge University Press, Cambridge, 1997 Search PubMed.
- G. Alter, T. H. M. Ottenhoff and S. A. Joosten, Semin. Immunol., 2018, 39, 102–110 CrossRef CAS PubMed.
- K. Sparbier, A. Asperger, A. Resemann, I. Kessler, S. Koch, T. Wenzel, G. N. Stein, L. Vorwerg, D. Suckau and M. Kostrzewa, J. Biomol. Tech., 2007, 18, 252–258 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc01467b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |