DOI:
10.1039/D0GC01427C
(Paper)
Green Chem., 2020,
22, 5447-5462
Ionic cross-linked polyvinyl alcohol tunes vitrification and cold-crystallization of sugar alcohol for long-term thermal energy storage†
Received
24th April 2020
, Accepted 13th July 2020
First published on 13th July 2020
Abstract
A new sustainable material for storing heat and releasing it on demand has been demonstrated for long-term latent heat storage (LLHS). The material consists of a high-latent-heat sugar alcohol phase change material (PCM) dispersed within ionic cross-linked matrices of polyvinyl alcohol (PVA). This material's unique property is the inhibition of undesired crystallization of the PCM during cooling due to the strong intermolecular interactions of the polymeric matrices, which leads to vitrification instead of crystallization. The release of latent heat can be controlled due to the PCM's stability below its cold-crystallization, which is triggered by reheating, as demonstrated by differential scanning calorimetry (DSC), optical microscopy (OM) and in situ X-ray diffraction (XRD). The addition of an ionic citrate cross-linker further tunes the vitrification and cold-crystallization properties of the PCM. Homogeneity and the presence of hydrogen bonding of the cold-crystalizing PCM (CC-PCM) were studied by scanning electron microscopy-energy dispersive X-ray spectroscopy (SEM-EDX), Fourier transform infrared spectroscopy (FTIR) and XRD. Thermal stability was confirmed by thermogravimetric analysis (TGA) and 100 consecutive DSC heating–cooling cycles. The CC-PCM demonstrated high latent heat of fusion, up to 266 J g−1, depending on the composition. As a super-adsorbent, PVA was able to swell and hold the liquid PCM resulting in form-stability and leakage-preventive properties above the melting temperature. Taken together, these results confirm that PVA matrices are promising for the thermal and structural stabilization of sugar alcohol PCMs, overcoming unexpected heat release and phase separation, and withstanding repeated melting-cooling cycles for LLHS.
1. Introduction
Renewable energy resources, including unlimited, clean solar energy, have attracted growing interest to reduce the dependency on finite fossil fuel supplies and the associated environmental concerns such as greenhouse gas emissions and climate change. However, the difference between supply and demand in time and location can limit the application of renewable energy resources. Energy storage technologies can improve the efficiency of energy utilization and contribute to renewable energy implementation and subsequently environmental protection.1–4 Different storage technologies operate based on mechanical, electrical, chemical and thermal energies.1 Among these methods, thermal energy storage (TES) where heat is stored and released by changing the internal energy of thermal materials is emerging with growing momentum for solar and district heating, domestic hot water production and heat exchange within industries.5–8
TES includes sensible, sorption, and latent heat storage with underlying thermal materials being thermophysical and thermochemical.5,8 Materials in the thermochemical category utilize adsorptive reactions to reversibly store and release adsorption–desorption heats and currently suffer from cycling instability and technology immaturity.9 The thermophysical category is based on sensible and latent heats and typically requires thermal insulation to prevent heat losses to the environment.10 Although sensible heat storage is commonly used, the low energy density and required large volume of storage materials, e.g. water and soil, have challenged its application.7,9 Water heat storage for example usually needs an additional expansion tank for compensating the pressure changes.
Latent heat storage (LHS) based on phase change materials (PCMs) presents an attractive way of renewable TES. PCMs undergo phase transition by absorbing, storing and releasing large latent heat (enthalpy) of fusion at nearly constant phase transition temperatures. They offer effective storage of higher energy densities than sensible storage across a wider temperature range with smaller volumes, making them suitable for many applications, e.g. solar and low-energy buildings, space heating, domestic hot water, and thermal buffers.3,4,8 In addition, the latent heat charge at a constant temperature can benefit specific applications requiring constant working temperatures.6
Unlike short term LHS, long-term LHS (LLHS) operates based on supercooling PCMs, such as salt hydrates and sugar alcohols,7,8,10,11 further removing the need for thermal insulation to prevent heat losses to the environment.12 The main principle of LLHS with supercooling PCMs is to charge the material with heat through enthalpy of melting, to supercool it to a lower temperature for storage, and to release the stored heat on demand by trigging the crystallization. This includes the seasonal storage of solar heat from summertime until the cold wintertime or sectional storage to use the heat in clusters of storage units at different times.10 Sugar alcohols (polyols), in particular, provide melting temperatures mainly between 90 °C and 200 °C and high volumetric energy densities.5,6,13,14 However, undesired heat release due to thermodynamic metastability during storage time, leakage due to fluidity, and lack of a reliable triggering mechanism for crystallization can disturb the real world application of supercooled PCMs.8
Additives can be utilized to overcome these issues, including polymer-incorporated PCMs, which can address leakage and provide form stability in the melt state.15–23 Shape-stabilization hinders the shrinkage or expansion of the material during cycling. Glass transition can be used as a preventive method for metastability13,24 since a so-called “good glass-former”25 material normally retains the liquid state over a wide temperature range.26 For such systems, cold-crystallization, an exothermic phenomenon due to the crystallization occurring during heating below the melting temperature of the material,24 can be used as a triggering mechanism to recover the stored heat with desired controllability on demand. In this approach, first, the PCM absorbs the heat during melting and stores it as latent heat. Upon cooling, the liquid PCM undergoes a steady supercooling and finally vitrifies as a kinetically stable glass. The amorphous PCM is unable to relax into a crystal form until it is reheated above its glass transition region (Tg). On reheating, still below the melting temperature, a good glass former typically first turns from the glassy state into a supercooled liquid before transforming into a crystalline form26 (e.g. cold-crystallization) when the stored heat is released. We have recently demonstrated a successful scaling up of this novel methodology as a new class of reliable long-term latent heat storage, enabling the recovery of heat after at least three months of storage by erythritol dispersed in covalently crosslinked polyacrylate.27
To this end and for the first time, we enable the stable supercooling, vitrification (glass transition), cold-crystallization, and shape-stabilization of sugar alcohols through the incorporation of polyvinyl alcohol (PVA) polymer for LLHS. Most importantly, we further demonstrate that the developed glass-transition and cold-crystallization properties of our novel cold-crystalizing PCM (CC-PCM) can be tuned through the addition and concentration of sodium citrate salt acting as an ionic cross-linker. Particularly, the materials include supercooling erythritol (ERY) as the thermally active PCM and PVA as the supporting additive for glass formation. As an inexpensive polymer, PVA provides the desired features including hydrophilicity, biocompatibility, biodegradability and excellent chemical resistance resulting in its broad industrial applications.15,16 PVA has been previously employed for the physical gelation of salt-hydrate PCMs,15,16 but never for enabling the glass transition and cold-crystallization of sugar alcohol PCMs for LLHS. The design, preparation, and function of this novel material fulfil the principles of sustainability and green chemistry on the importance of the inherent characteristics such as renewability, biodegradability and nontoxicity of the precursors and the product, simplicity of the preparation process, and environmentally benign sustainable application i.e. long-term thermal energy storage from renewable resources e.g. solar energy.28
Existing PCMs for TES include paraffins,1,3,17,19 fatty acids1,3,29,30 and salt hydrates.1,3,8,10,15,16 However, these PCMs suffer from several drawbacks including flammability, fossil-fuel based origin, large volume change, instability and phase segregation issues,1,3 while, the CC-PCM developed herein is based on green sugar alcohol PCM with the advantages of bio-based origin, non-flammability, nontoxicity, non-corrosiveness, and affordable market price, making them suitable for sustainable applications.5,6,13,14,27 Most importantly, commonly used PCMs are mainly intended for short-term LHS, from a day up to a week, as they are unable to preserve the stored heat for longer times.10 On the other hand, the CC-PCM is intended for longer time storage of latent heat (from weeks to seasonal) far below the melting temperature through supercooling and glass transition tendency and the recovery of the stored heat by clod-crystallization.8,27 In this way we can overcome the mismatch between supply and demand for the utilization of intermittent renewable energy, particularly in colder climate with extreme seasonal variation of solar energy. Ideally, we can store the abundant solar heat from summertime for cold wintertime, thus mitigating the climate change by reducing the dependency on fossil fuel-based burning, for example, for domestic heating purposes.
In this study, we synthesized the CC-PCM via a simple and facile aqueous fabrication route avoiding the usage of toxic solvent and covalent hazardous crosslinkers or monomers. Instead, we applied safe additives, citric acid salt and polyvinyl alcohol, which are environmentally friendly biocompatible nontoxic precursors. We further studied various affecting factors such as the ratio of PCM/polymer and addition of ionic citrate cross-linker on the compositional, morphological, and crystalline properties of the CC-PCM via standard laboratory methods including Fourier transform infrared spectroscopy (FTIR), scanning electron microscopy-energy dispersive X-ray spectroscopy (SEM-EDX), and in situ temperature X-ray diffraction (XRD). Differential scanning calorimetry (DSC) was used to extensively analyze the long-term latent heat storage performance including the glass transition, cold-crystallization, and melting temperatures and the corresponding latent heat and specific heat capacities. The thermal stability and repeatability were ensured via thermogravimetric analysis (TGA) and highly repeatable DSC cycling.
2. Materials and methods
2.1. Material preparation
The CC-PCM was prepared though an efficient simple route using environmentally benign and biodegradable precursors. Polyvinyl alcohol (PVA, Mw 145![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, fully hydrolyzed, for synthesis) was supplied by Merck (Germany). Sodium citrate dihydrate (SC) (≥99%, food grade) was purchased from Sigma Aldrich (Austria). Erythritol (ERY, technical-food grade) was supplied by Suomen Luontaistukku Oy (Finland). Molecular structures of the applied materials are illustrated in Fig. 1. To prepare the CC-PCM, a known amount of PVA was dissolved in distilled water (WPVA/WH2O ∼ 0.04, W shows the mass) at 70 °C until a homogeneous hydrogel was obtained. Aqueous solutions of ERY (WERY/WH2O ∼ 1.5) at 70 °C were added to the PVA hydrogel (corresponding to the compositions provided in Table 1) under continuous mixing. For ionic-crosslinking of the system, SC was added to the ERY-PVA hydrogel while mixing for another two hours. The samples were dehydrated at 105 °C and subsequently melted at 120 °C prior to further thermal, morphological and compositional analyses. Table 1 is a compilation of the compositions of CC-PCM including the amount in weight percent (wt%) of ERY, PVA, and SC.
000, fully hydrolyzed, for synthesis) was supplied by Merck (Germany). Sodium citrate dihydrate (SC) (≥99%, food grade) was purchased from Sigma Aldrich (Austria). Erythritol (ERY, technical-food grade) was supplied by Suomen Luontaistukku Oy (Finland). Molecular structures of the applied materials are illustrated in Fig. 1. To prepare the CC-PCM, a known amount of PVA was dissolved in distilled water (WPVA/WH2O ∼ 0.04, W shows the mass) at 70 °C until a homogeneous hydrogel was obtained. Aqueous solutions of ERY (WERY/WH2O ∼ 1.5) at 70 °C were added to the PVA hydrogel (corresponding to the compositions provided in Table 1) under continuous mixing. For ionic-crosslinking of the system, SC was added to the ERY-PVA hydrogel while mixing for another two hours. The samples were dehydrated at 105 °C and subsequently melted at 120 °C prior to further thermal, morphological and compositional analyses. Table 1 is a compilation of the compositions of CC-PCM including the amount in weight percent (wt%) of ERY, PVA, and SC.
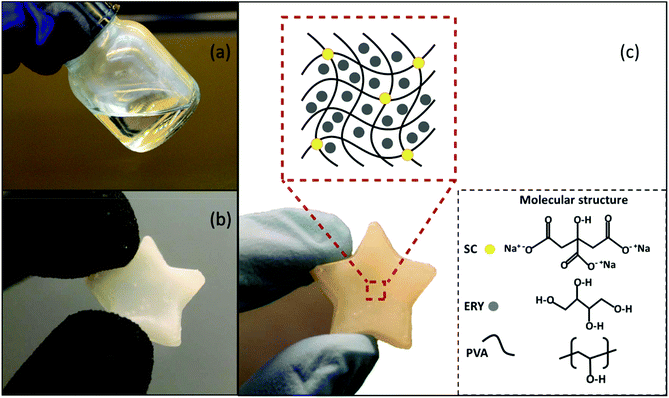 |
| | Fig. 1 Melting behavior of (a) pristine ERY and (b) CC-PCM organogel under two hours heat exposure at around 130 °C (composition A1); an observatory demonstration on shape-stabilization of the CC-PCM versus fluidity of the PCM. (c) A1 at room temperature with a schematic illustration of the PCM involvement with the polymeric network resulting in structural stabilization of CC-PCM. | |
Table 1 Compositions of the prepared CC-PCM samples including the weight fraction (wt%) of ERY, PVA, and SC
| Composition |
ERY |
PVA |
SC |
| A |
80.0 |
20.0 |
— |
| A1 |
80.0 |
17.2 |
2.8 |
| A2 |
80.0 |
15.0 |
5.0 |
| B |
75.0 |
25.0 |
— |
| B1 |
75.0 |
22.0 |
3.0 |
| B2 |
75.0 |
18.75 |
6.25 |
| C |
70.0 |
30.0 |
— |
| C1 |
70.0 |
27.0 |
3.0 |
| C2 |
70.0 |
22.5 |
7.5 |
2.2. Compositional analyses
Fourier transform infrared spectroscopy (FTIR) was employed to probe the functional groups and the intermolecular interactions between the PCM and the PVA chains. FTIR was conducted on an attenuated total reflection (ATR) PerkinElmer spectrometer (Spectrum Two Polymer QA/QC Analysis System) equipped with a diamond ATR crystal. The spectra were recorded within the range of 500–4000 cm−1 under 16 repetitious scans with 32 cm−1 resolution. The morphological structure of CC-PCM was observed by scanning electron microscopy (SEM) using an analytical field emission scanning electron microscope, JEOL JSM-7500FA, operated under vacuum and at 1.5 kV acceleration voltage. Energy-dispersive X-ray spectroscopy (EDX) was also performed at 10.0 kV acceleration voltage and 10 μA probe current for detecting the elemental composition of the sample. To prepare the sample, a thin film of CC-PCM was fixed on a metal stub with carbon tape and was then coated with 4 nm of gold (Au) palladium (Pd) alloy using a sputter coater (LECIA EM ACE600).
2.3. Crystallinity analysis
High-resolution powder X-ray diffraction (XRD) was applied to reveal the crystalline and amorphous structures of the CC-PCM and the precursors. XRD measurements were conducted at room temperature on a Rigaku SmartLab X-Ray diffractometer in parallel beam equipped with a monochromator, rotating anode X-ray source (9 kW, Cu Kα1) and HyPix-3000 2D detector. Prevention of crystallinity during cooling and crystallite growth (cold-crystallization) on reheating were analyzed by temperature controlled XRD. A modified Kapton-covered Linkam heating–cooling stage (HFSX-350-GI) was used for in situ temperature XRD in a nitrogen atmosphere within the −120 °C to 150 °C temperature range without a monochromator (CuKα) to analyze the glassy and crystalline states upon increasing the temperature and time. A thin layer of CC-PCM (A1) was placed on a silicon wafer and melted at 140 °C prior to the measurement. The program was set to first raise the temperature to 150 °C and hold for 10 min to ensure complete melting, and to collect the XRD pattern of the melt. The sample was then cooled to −120 °C under 20 K min−1 to collect the pattern of the glassy state. The XRD profile of the sample was recorded at different temperatures from this temperature on heating with 10 K min−1. A set of fast 2-minute scans were also collected within 0–20 °C with 1 K min−1 heating rate.
2.4. Optical microscopy
A polarized optical microscope (ZEISS SteREO Discovery.V12) equipped with a heating–cooling stage was used to observe and visualize the crystallization of pure PCM versus the cold-crystallization of CC-PCM. A thin film of the materials was placed between a glass slide and cover glass. The sample was melted at 140 °C on a heater prior to optical microscopy (OM). To demonstrate the crystallization of bulk PCM on cooling, the program was set to first raise the temperature from room temperature to 120 °C and hold at this temperature for 10 min to ensure complete melting. The samples were then cooled to 20 °C with 20 K min−1 and held for 1 min. To show the stable supercooling and vitrification of CC-PCM by cooling, after raising the temperature to 120 °C and holding for 10 min, the sample was cooled to −20 °C under 20 K min−1 and held for 2 min. The sample was lastly heated from −20 °C to 120 °C under 20 K min−1 for cold-crystallization.
2.5 Thermal energy measurements
Differential scanning calorimetry (DSC) is the most commonly used standard method to measure the latent heat storage and specific heat capacities of PCMs.3,31 The DSC measurements were performed on a Netzsch DSC204F1 Phoenix DSC instrument. We employed DSC to analyze the glass transition and phase transition temperatures, to determine the corresponding latent heat (enthalpy) of melting and cold-crystallization, and to measure the specific heat capacities for ERY and the developed CC-PCM. The glass-transition (Tg), cold-crystallization (Tcc) and melting (Tm) temperatures were determined by line fitting as the end of the glass transition region, the onset of cold-crystallization (exothermic peak) and the onset of melting (endothermic peak) on the heating DSC curves, respectively. The latent heat of cold-crystallization (ΔHcc) and melting (ΔHm) was measured as the corresponding area under endothermic melting and exothermic cold-crystallization DSC peaks. The ESI† includes further explanation on DSC measurements and an example DSC curve with the corresponding calculations shown in Fig. S1.† A dynamic temperature range within −60–150 °C was employed for the DSC measurement program. Each program included four consecutive heating–cooling cycles under mainly 5.0 K min−1 scan rate (q), constant for heating and cooling. A set of measurements were conducted to study the effect of the temperature scan rate on glass transition, where q was changed within 0.5–20.0 K min−1. To prepare the sample for DSC measurement, a 13–30 mg of PCM amount was placed in an aluminum crucible and sealed with a standard aluminum lid. The measurements were performed on at least three replicates. To study the repeatability and cycling properties, DSC melting-cooling cycles were repeated 100 times within the −40–150 °C temperature range under 5.0 K min−1 heating–cooling rate. Specific heat capacity (Cp) measurements were conducted within −60–150 °C under 5.0 K min−1 using the Sapphire correction Cp method. Furthermore, thermogravimetric analysis (TGA) was conducted on TA instruments (TGA Q500) from room temperature to 600 °C under 10 K min−1 ramp rate in a nitrogen gas atmosphere.
3. Results and discussion
3.1. Shape stability
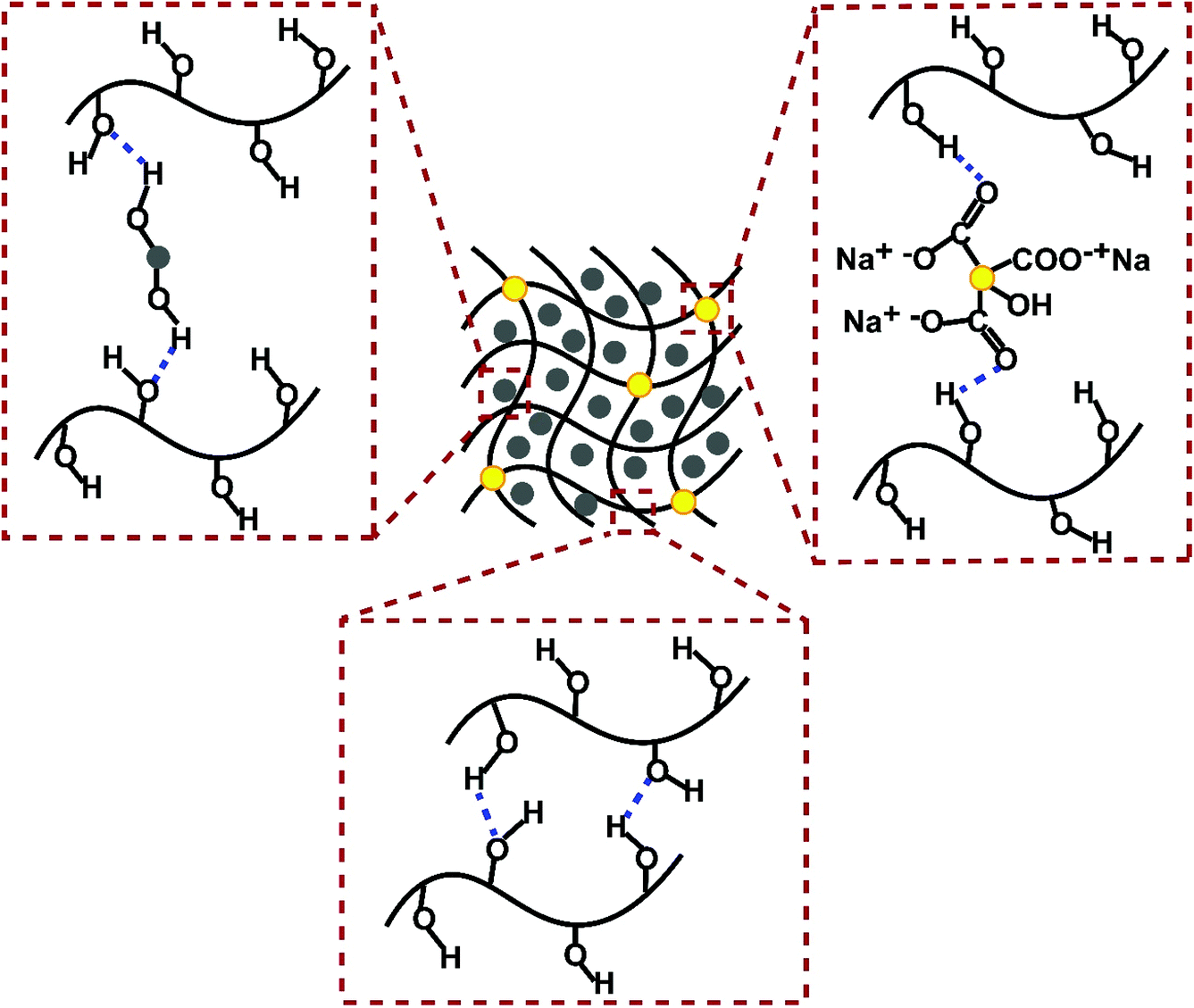
The melting behavior of pristine ERY and CC-PCM is quite distinct (Fig. 1). It is observed that ERY melts and becomes liquid, posing the risk of leakage and difficulties of handling the PCM in the melt state (Fig. 1a). In contrast, CC-PCM showed stabilized fluidity, inhibiting the leakage of the molten ERY in the PVA matrices due to numerous intermolecular interactions resulting in a solid-to-gel transformation rather than a typical solid-to-liquid transition (Fig. 1b). As a super-adsorbent, the PVA network can swell in melted polyol, acting as the solvent, and hold a large amount of polyol while preserving the physical structure.32 Therefore, the CC-PCM can be classified as an organogel,16,33 a class of soft materials with tunable mechanical properties. Structural stabilization originates from physical attractions (adsorptive forces) between polyol molecules and the PVA chains (e.g. numerous hydrogen bonds demonstrated by FTIR (Fig. 2) and illustrated in Scheme 1). At a more advanced hydrolysis state, PVA strongly tends to form hydrogen association and gels. As the PVA is not chemically cross-linked, it is considered as a physical gelation. Secondary interactions including van der Waals, hydrogen bonding, and ion-dipole contribute to physical gelation. Easy dissociation and the formation of new secondary bonds can restructure the physical network of the gel. Reversibility of these interactions can therefore result in tunable physical properties, for example, resiliency to mechanical damage which can improve the life cycle of the PCM.15,34
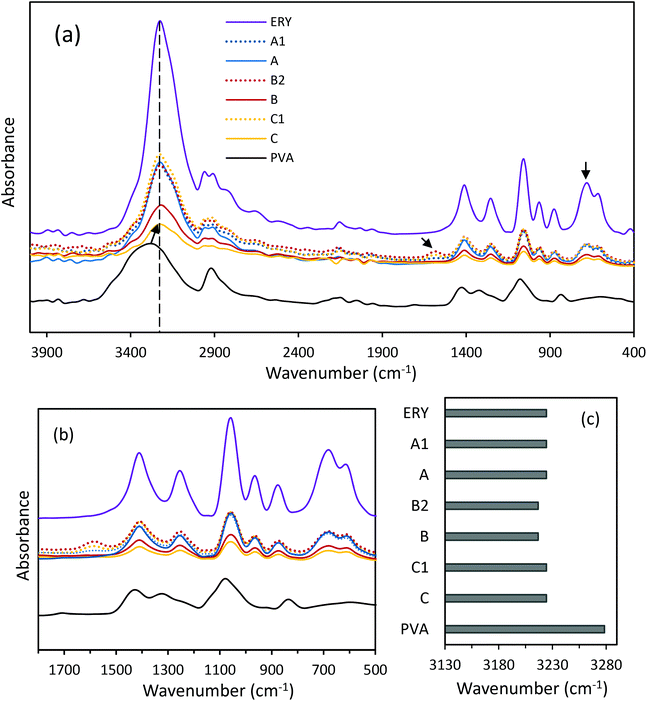 |
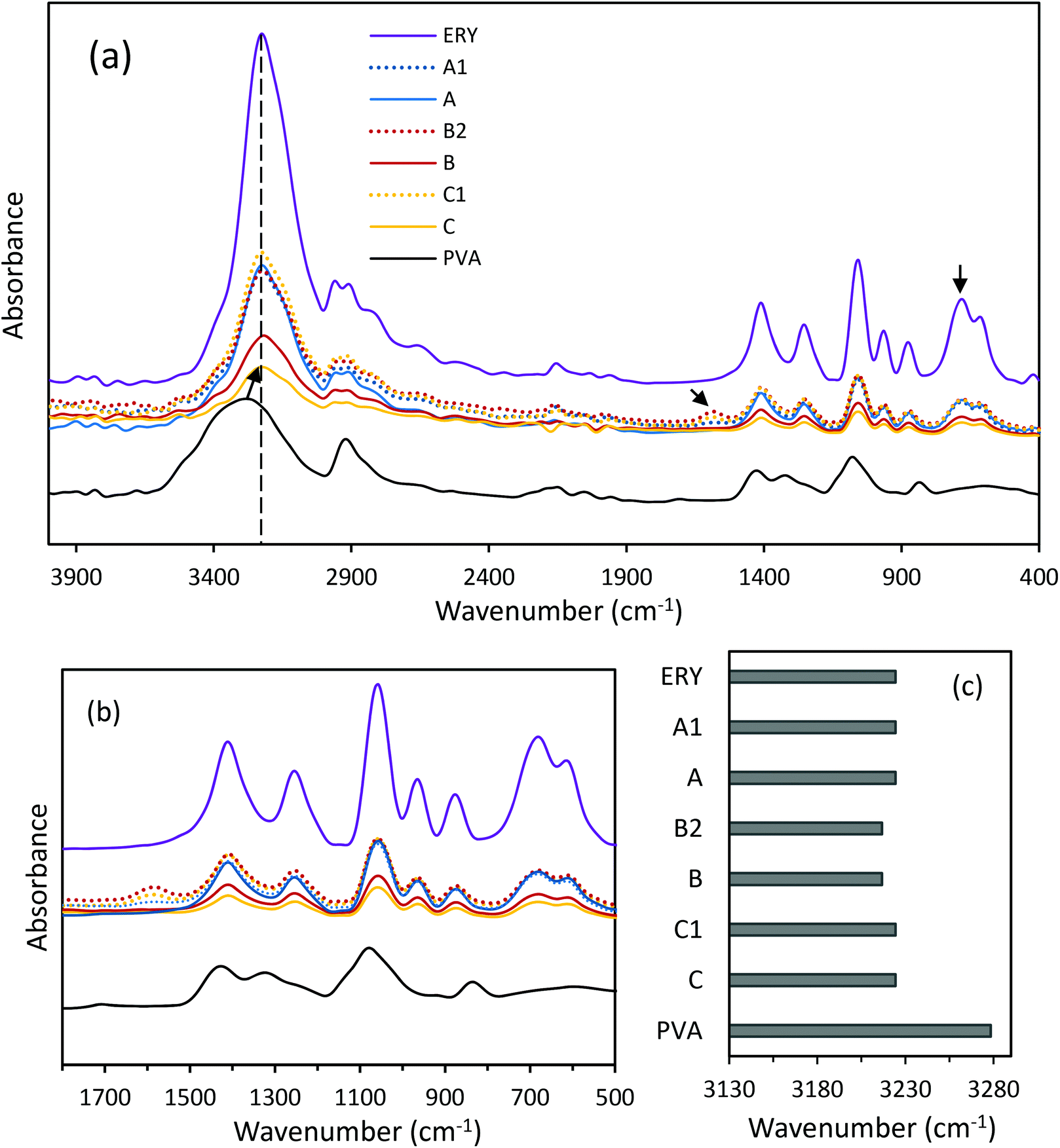
| | Fig. 2 (a) ATR-FTIR spectra of ERY, PVA and CC-PCM (different compositions), (b) enlarged ATR-FTIR spectra at low wavenumbers, and (c) wavenumber of the maximum of the –OH vibrational band. See Table 1 for the compositions. | |
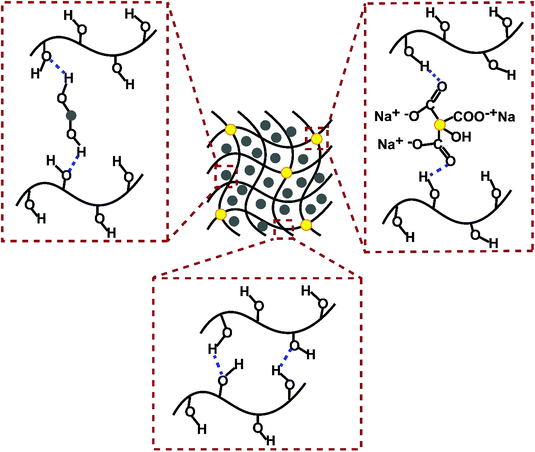 |
| | Scheme 1 Schematic illustration of possible intermolecular interactions involved in CC-PCM formation. Note that one set has been illustrated for simplicity yet all –OH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups are associated with hydrogen bonding. O groups are associated with hydrogen bonding. | |
3.2 Composition
FTIR analysis was used to identify the chemical composition and the nature of reactions between the coupled components in the CC-PCM. Fig. 2a shows the infrared spectra of ERY, PVA and CC-PCM (for different compositions see Table 1). A broad peak at 3600–3000 cm−1 corresponds to the stretching vibration of –OH groups in the molecular structure of the materials. The lower intensity peaks at 2946 and 2900 cm−1 are characteristic of symmetric and anti-symmetric vibrations of –CH2 groups. The peaks at about 1419 and 1064 cm−1 represent the –C–O group.35 Crystallinity in the solid state results in sharper IR spectra35 as evidenced by increasing ERY content (Fig. 2a). Hydrogen bonding largely influences the IR spectra of the compounds. Generally, associated molecules show broader bands at decreased O–H stretching frequency than those of the non-associated state. A shift (60 cm−1) is observed for hydroxyl vibration on the spectrum of CC-PCM compared with that of PVA (Fig. 2c) which indicates intermolecular hydrogen bonding.15,16 Characteristic adsorption peaks of ERY intensify by increasing the ERY content in the CC-PCM. Carboxylate asymmetric stretching vibration at 1580 cm−1 (ref. 35) appears in the spectra of ionically cross-linked CC-PCM (e.g. compositions A1, B2, and C1) originating from the SC (Fig. 2b). No new chemical peak appeared in the spectrum of CC-PCM apart from those of applied compounds, which confirms the physical nature of interactions between PVA and ERY.
Fig. 3 shows the morphological and elemental properties of the CC-PCM (composition C2). The elemental analysis (SEM-EDX) of the CC-PCM (Fig. 3c–f) confirms the presence of the main elements, i.e. carbon (46.5 wt%), oxygen (20.7 wt%) and sodium (2.2 wt%). The EDX mapping and SEM images show a uniform and homogeneous spread of the elements in the sample. Homogeneity and uniformity of the combined components are due to the compatibility of their nature, which prevents phase separation and assists form-stabilization as observed in Fig. 1b. For the case of sodium cation, a honeycomb pattern was observed during EDX mapping measurement. Sodium cations originate from the ionic agent and their homogeneous spread within the sample suggests an even cross-linking of the PVA chains and better stabilization of the PCM, which is consistent with the following DSC analysis. SEM images of CC-PCM (in nano- and micro-scales) are presented in Fig. 3a and b. The CC-PCM possesses a three-dimensional fibrillar network morphology, consisting of a continuum between partially coalesced rounded pores and fibrillar structures. Similar morphological structuring was observed previously for PVA hydrogels.36,37 The structural morphology of the CC-PCM confirms the homogeneous spread of the polymeric network in the CC-PCM.
 |
| | Fig. 3 SEM images of CC-PCM (C2) in microscale (a) and nanoscale (b) bars. EDX analysis of CC-PCM (C2) including carbon in purple (c), oxygen in red (d), sodium in green (e), and quantitative plot of the elements (f). Note that the detected Au (23.4 wt%) and Pd (7.2 wt%) originate from the coating, and the dark spots in the EDX images are related to the voids and grooves present on the sample surface. | |
3.3. Thermal energy storage
3.3.1 Cold-crystallization versus crystallization.
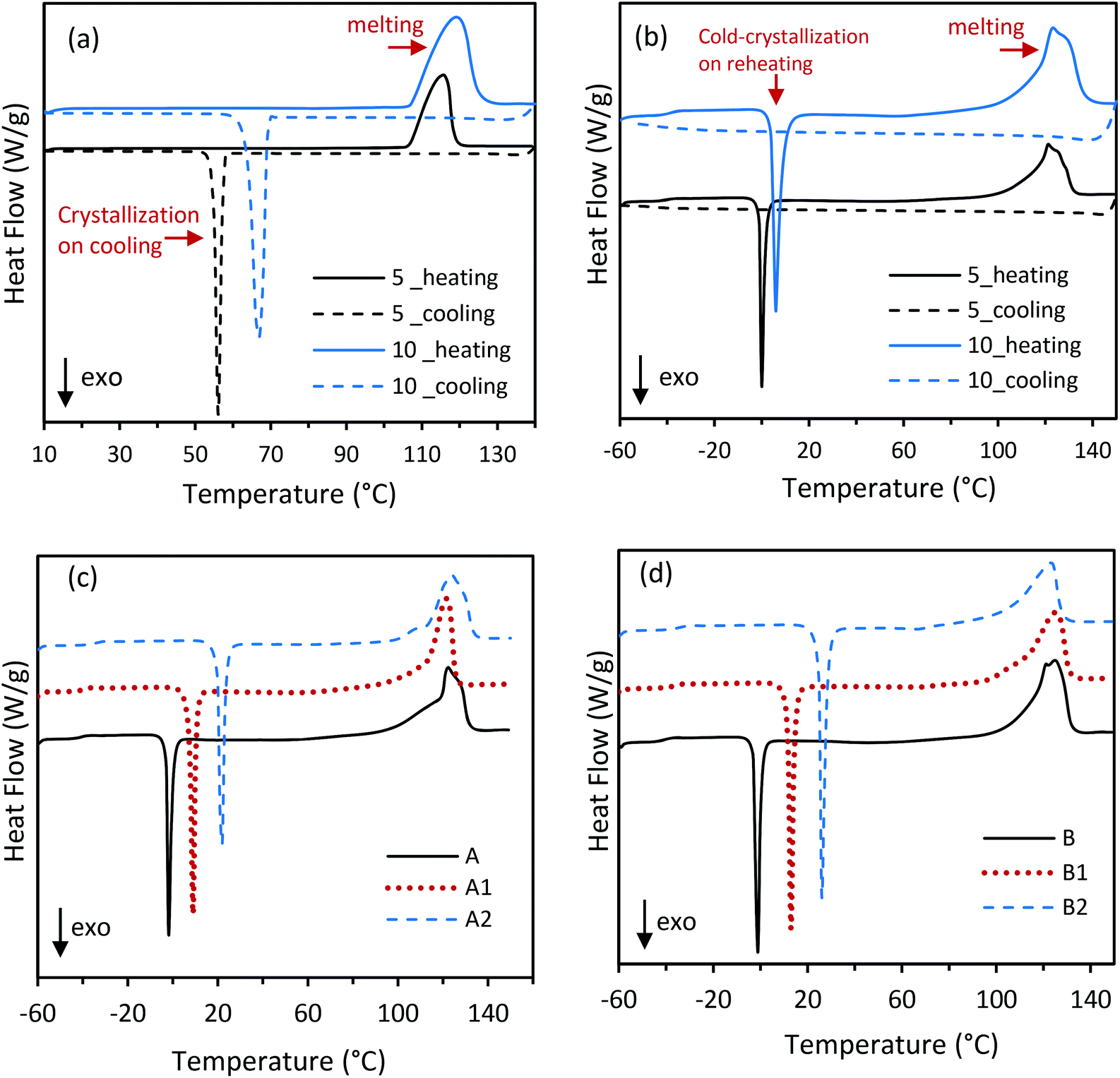
Although ERY shows a high extent of supercooling, it crystallizes upon cooling within the temperature range −5 to 80 °C, depending on the cooling conditions e.g. scan rate,13,38 as also has been shown in the DSC curves in Fig. 4a. For instance, the crystallization of ERY starts at 57.5 °C on 5 K min−1 cooling rate. Supercooled ERY spontaneously relaxes from a thermodynamically metastable liquid into a crystalline state; that is, it releases the latent heat at an undesired time, which hinders the long term heat storage.24 However, ERY within PVA matrices shows different thermal behavior under cooling and heating conditions. As showed by DSC measurement in Fig. 4b, CC-PCM (composition C) did not crystallize upon cooling but supercooled until below −20 °C when vitrification into a glass occurred. Relaxation time for the dynamic properties of polyol molecules in the PVA matrices is prolonged due to strong intermolecular interactions with the polymer chains;38 for example, numerous hydrogen bonds between hydroxyl groups on PVA and ERY (Scheme 1) confirmed by FTIR spectra (Fig. 2). Fig. 4b shows that after the glass transition upon reheating, cold-crystallization occurred when the latent heat was released. Similar to the crystallization on cooling, a dependency to the heating conditions, such as the scan rate, was observed for cold-crystallization on reheating. Indeed, after the CC-PCM is charged with heat by melting, a stable supercooling allows the charged material to be stored at lower temperatures, while the vitrification tendency prevents the risk of undesired crystallization during the storage time and solves the metastability issue. The stored heat then can be recovered through cold-crystallization on reheating, which is a controllable way to exploit the subcooled PCM for long-term heat storage without the risk of unexpected heat release.13,24 The required heat pulse for trigging the cold-crystallization on reheating depends on the temperature deference (ΔT) between the storage temperature (Ts) and Tcc. The relationship Q = m·Cp·ΔT can be used to determine the amount of the trigging heat (Q), where m is the mass and Cp is the average specific heat capacity of the material within the ΔT range. An example for Q calculation is provided in the section 3.3.3.
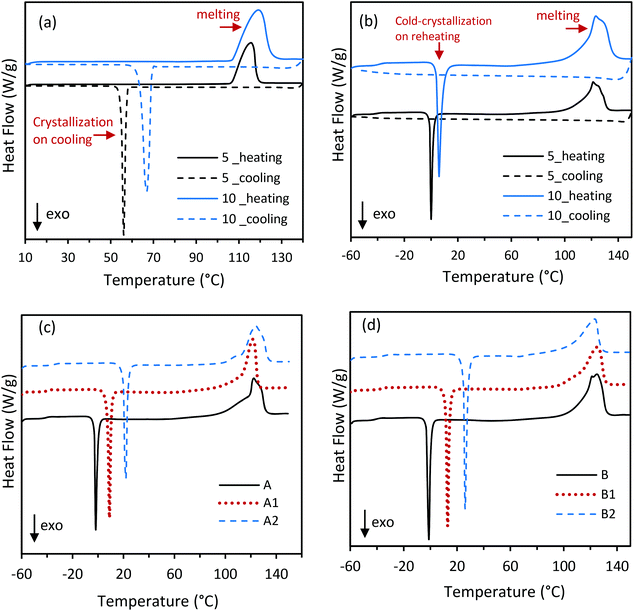 |
| | Fig. 4 DSC curves of (a) ERY (crystallization) and (b) CC-PCM (cold-crystallization of composition C) under 5 and 10 K min−1 scan rates: heating (lines)-cooling (dashes), (c) compositions A, A1, and A2 and (d) compositions B, B1, and B2 under 5 K min−1 heating scan rate (see Fig. S2† for C-compositions). See calculated latent heat values by DSC in Table 2. | |
Fig. 5a shows OM images of ERY upon cooling from the melt state at 120 °C. A microscopic video (1) of the crystallization of ERY is provided in the ESI.† It was observed that crystallization first starts on cooling (∼45 °C) and proceeds quickly. Fig. 5b presents the OM images of CC-PCM (A1) from the melt (120 °C) to a deep supercooled state at −20 °C and cold-crystallization on subsequent reheating. A microscopic video (2) of the phase transition of CC-PCM is also provided in the ESI.† Similarly, micrographs of composition A (without ionic agent) confirmed the glass-transition on cooling and cold-crystallization on reheating, as illustrated in Fig. S3 in the ESI.† The microscopic observation demonstrated that CC-PCM does not crystalize on cooling instead it supercools steadily until −20 °C. On reheating, cold-crystallization started first by the formation of several nuclei, which grew into spherulites on further heating as seen in Fig. 5b until the crystal grains were completed. Unlike bulk PCM, the CC-PCM shows a spherulitic morphology. Spherulites, as spherical crystalline regions, are associated with linear polymers such as PVA. The formation and morphology of spherulites are influenced by different factors e.g. temperature, nuclei number, and polymer structure.39 Spherulization can result in, for example, increased density, hardness and tensile strength possibly due to intermolecular interactions in highly ordered and dense molecular packs. Further isothermal analysis can be conducted to determine the nuclei formation and growth rate of the cold-crystallization in future research.
 |
| | Fig. 5 Micrographs of (a) crystallization of ERY on cooling from the melt (120 °C) and (b) supercooling of CC-PCM (A1) on cooling from the melt (120 °C) and cold-crystallization on reheating from the deeply supercooled state (−20 °C). Microscopic videos of both phenomena are provided in the ESI.† Micrographs of composition A (without ionic agent) are provided in Fig. S3 in the ESI.† Note that due to the high scan rate on the heating stage, the temperatures showed by the microscope may not be very exact. | |
3.3.2. Compound ratio.
The thermal energy storage characteristics, e.g. phase transition temperatures and the corresponding latent heat release and absorption, depend on several parameters including the polymer/PCM ratio in the CC-PCM. Table 2 is a compilation of these thermal properties of the CC-PCM, which were determined by DSC, including the glass-transition (Tg), cold-crystallization (Tcc), and melting (Tm) temperatures, and the latent heat of cold-crystallization (ΔHcc) and melting (ΔHm). The ESI† includes an example DSC curve with the heat storage values shown in Fig. S1.† As seen in Table 2, increasing the PVA content from 20 to 30 percentage (composition A, B, C, excluding those with the ionic agent) results in a small increase of the glass transition temperature (∼1 °C) and cold-crystallization temperature (∼4 °C) and a reduced melting temperature (∼2 °C). Whereas, decreasing the ERY content from 80 to 70 percentage led to a decrease of the latent heat of melting (ΔHm) from 266 J g−1 to 236 J g−1 and the latent heat of cold-crystallization (ΔHcc) ranging from −102 J g−1 to −118 J g−1, which is consistent with their fraction of thermally active compound (ERY, 340 J g−1 enthalpy of melting). The differences between the solidification and melting heats originate from their dependency on the temperature and specific heat capacity, as the following relationship shows:| | 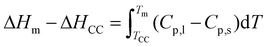 | (1) |
where Cp,l and Cp,s are the specific heat capacity of liquid and solid states, respectively, and T is the temperature.
Table 2 Latent heat storage properties of CC-PCM with varying contents of ERY, PVA and SC (see Table 1 for compositions) determined by DSC under 5 K min−1 scan rate; tuning the thermal properties by composition. Tg is the end of the glass transition region on the heating DSC curve; Tcc and Tm are the onset temperatures of phase transition measured by line fitting. ΔHm and ΔHcc are the corresponding area under DSC peaks. The average values of three measurements on three separate samples are reported along with their standard deviation
| Composition |
T
g
|
T
cc
|
ΔHcc |
T
m
|
ΔHm |
| (°C) |
(°C) |
(J g−1) |
(°C) |
(J g−1) |
| A |
−39.2 ± 0.9 |
−5.2 ± 2.7 |
−102.2 ± 2.3 |
116.2 ± 2.2 |
266.0 ± 2.5 |
| A1 |
−33.7 ± 0.2 |
10.1 ± 0.1 |
−142.0 ± 1.9 |
112.8 ± 0.8 |
263.1 ± 0.6 |
| A2 |
−30.5 ± 0.2 |
22.0 ± 1.7 |
−154.8 ± 2.5 |
112.1 ± 0.8 |
255.9 ± 0.5 |
| B |
−38.2 ± 0.4 |
−2.9 ± 0.6 |
−118.0 ± 0.9 |
114.4 ± 0.1 |
257.6 ± 3.4 |
| B1 |
−33.3 ± 0.2 |
11.9 ± 0.1 |
−133.0 ± 2.0 |
109.8 ± 0.5 |
239.4 ± 2.8 |
| B2 |
−31.2 ± 0.7 |
25.4 ± 0.2 |
−156.9 ± 0.5 |
105.4 ± 0.5 |
232.8 ± 2.2 |
| C |
−38.1 ± 0.7 |
−1.5 ± 0.6 |
−105.5 ± 0.9 |
116.5 ± 0.4 |
235.9 ± 12.5 |
| C1 |
−32.8 ± 1.1 |
13.5 ± 0.9 |
−133.9 ± 0.5 |
114.6 ± 0.3 |
231.1 ± 9.2 |
| C2 |
−28.5 ± 0.2 |
28.6 ± 0.3 |
−152.5 ± 0.7 |
106.3 ± 1.0 |
218.5 ± 0.5 |
3.3.3. Ionic cross-linker.
PVA matrices can provide functional properties such as super-absorbency, fluid retention capacity, and structural and thermal stability for the CC-PCM. Several factors such as polymer ratio, hydrophilicity, cross-linking density, and ionic strength influence the functional properties, such as swelling capacity, of a super-adsorbent.32 However, covalent cross-linking agents pose the risk of toxicity and other drawbacks.40 Therefore, trisodium citrate (SC), a food grade product consisting of three sodium cations and a citrate anion with three carboxylic groups (Fig. 1), was incorporated into the system to act as an ionic cross-linker to provide ionic strength. The ionic crosslinking agent bridges between the polymer chains through reversible electrovalent (ionic) bonds.41 It gives the advantage of easy reconfiguration leading to tunable physical properties and self-healing from physical damage, unlike chemical crosslinking through irreversible covalent bonds. The effect of SC addition and concentration on the latent heat storage properties measured by DSC can be seen in Fig. 4c, d and Fig. S2.†Table 2 is a tabulation of the glass transition and phase change properties of CC-PCM with increasing amount of the ionic agent. Interestingly, increasing the ionic agent content in all three compositions i.e. A, B, C, results in a significant rise of glass transition and cold-crystallization temperatures, while reducing the melting temperature. Tunable thermal properties can further provide the advantage of adjusting the desired thermal features according to the working temperature required in specific applications. The latent heat of cold-crystallization (ΔHcc) was improved up to −157 J g−1 (for composition B2 with 75 wt% ERY, 18.75 wt% PVA and 6.25 wt% SC), while the latent heat of melting (ΔHm) was observed to be reduced down to 233 J g−1 (B2). As mentioned in the previous section, the amount of trigging heat (Q) for cold-crystallization can be estimated by Q = m·Cp·ΔT. For example, if the melted CC-PCM (B2) is stored at Ts ∼ 5 °C and the cold-crystallization starts at 25 °C (Tcc in Table 2) with a Cp of 2.2 J g−1 K−1 (Fig. S4†), Q = 44 J g−1 is consumed to initiate the cold-crystallization, while, ΔHcc = −157 J g−1 (Table 2) is recovered during cold-crystallization. This storage approach can be also considered as an upgrading of the heat quality, by which low temperature heat is transformed to medium/high temperature heat. There exist plenty of low temperature industrial and domestic waste heat sources that can be sustainably utilized to trigger cold-crystallization.
In the phase transition zone between the PVA super-adsorbent32,42 and the surrounding environment, polar molecules of ERY can be present in different sorption states including non-freezing bound (NFB), freezing bound (FB) and free molecules.43 The free fraction, if it exists, is expected to behave similar to pristine ERY as presented in Fig. 4a. Since CC-PCM behaved differently, shown in Fig. 4b along with Fig. 4c, d and Fig. S2,† the free ERY molecules do not exist. The FB fraction is moderately attracted by the polymeric chains, while NFB are strongly attached molecules. Therefore, the FB fraction in the CC-PCM can undergo phase transition, i.e. cold-crystallization and melting, whereas, the NFB fraction is incapable of phase transition due to the strong adsorptive forces from the super-adsorbent.13 The mass fraction (W) of the FB and NFB correlates with the melting enthalpy (ΔHm) (ref. 43) according to:
| | 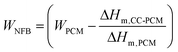 | (2) |
where
WPCM is the mass fraction of the PCM (ERY) in the CC-PCM and Δ
Hm,CC–PCM and Δ
Hm,PCM are the latent heat of melting for CC-PCM and the pristine PCM, respectively. Interestingly, the FB fractions in the CC-PCM without ionic cross-linking correlate very well with the total mass fraction of ERY in the material (
WPCM = 0.7–0.8 ≈
WFB = 0.7–0.8) suggesting that all the molecules of ERY are capable of phase transition due to a moderate involvement with the polymeric matrices. By increasing the ionic cross-linker content, the FB fraction ranged within
WFB = 0.64–0.76 for the composition containing
WPCM = 0.7–0.8. This suggests that incorporation of the SC will cause stronger involvement of the PCM with the polymer matrices leading to NFB fractions in the material. The high FB fractions for ERY by PVA are in agreement with the FB fractions of ERY and water involved with other polymers reported previously.
13,44 The latent heat of fusion obtained with our CC-PCM (218–266 J g
−1) can compete with commonly used PCMs such as paraffin (100–200 J g
−1) (
ref. 45) and fatty acids (140–260 J g
−1).
46 Of note, these common PCMs are able to store heat only for short-term, from a day up to a week,
10 whereas, CC-PCM is intended for longer time heat storage (from weeks to seasonal).
7,8,11 Recently, we have demonstrated that heat can be reliably stored for at least a three-month period without any noticeable decrease in the recovered cold-crystallization heat by scaling up the erythritol dispersed in covalently crosslinked polyacrylate.
27 In the scaling-up process, bulk samples (160 g) were charged with the heat during melting, supercooled and stored at a constant temperature within 0–10 °C for up to three months, and finally cold-crystallized for heat recovery
via an applied thermal chamber set-up. After the storage period of three months, the cold-crystallization heat of the samples was up to 70% of the heat charged during melting, confirming the laboratory scale DSC measurements.
27 Of note, the scaling-up studies on the cold-crystallization of erythritol dispersed in ionically crosslinked polyvinyl alcohol introduced herein are on-going and will be reported in our future work.
3.4. Glass transition
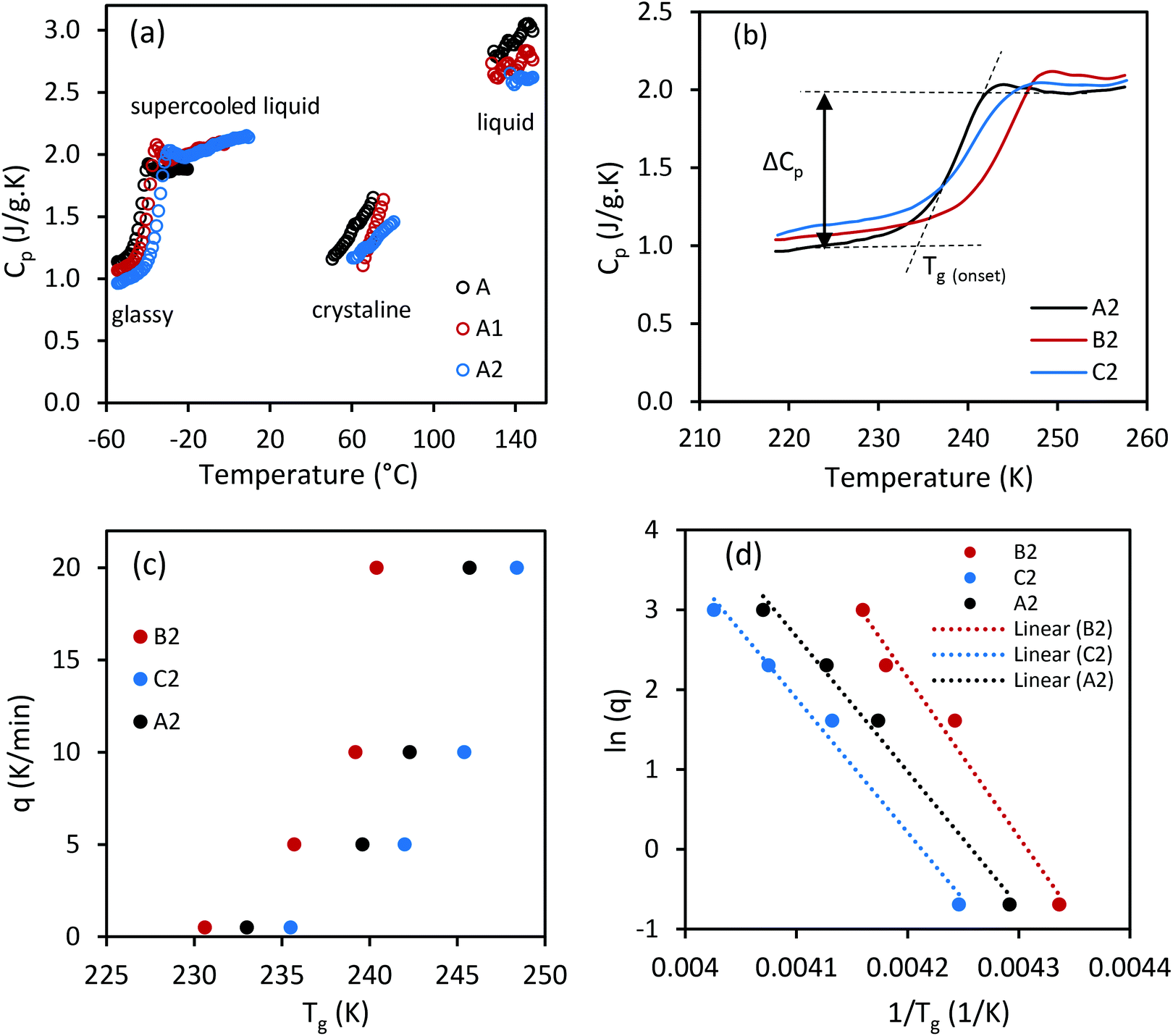
Good glass-formers normally maintain a liquid state over a wide temperature range. On reheating, yet below the melting temperature, the glass-former abruptly turns from the amorphous solid into a supercooled liquid instead of directly transforming to a crystalline form,26 which is also observed in Fig. 6a and Fig. S4† for the CC-PCM. A profound vitrification on cooling indicates a readily formed glassy state even with a slow cooling rate (see Fig. S2 and S5†).47 The glass transition temperature rises by the temperature scan rate (q) and ionic cross-linker, as observed in Fig. 6c and Table 2. The structural relaxation time of a liquid highly depends on the temperature. At sufficiently low temperatures, a rapid change of temperature results in vitrification of the liquid due to significantly prolonged structural relaxation time, i.e. formation of glass from a supercooled liquid.48 However, with a slower scan rate, the liquid PCM may have more time to relax into crystal form on cooling. The CC-PCM (compositions including the ionic crosslinker) is unable to crystallize on cooling even with a slow scan rate (0.5 K min−1), see Fig. S2.† The dependence of glass transition on the temperature scan rate (q) is given by the following equation:48| | 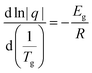 | (3) |
where Eg is the activation energy for structural relaxation (enthalpy or volume) and R is the gas constant. The plot of ln (q) versus 1/Tg is given in Fig. 6d.
 |
| | Fig. 6 (a) Specific heat capacity (Cp) of CC-PCM (A-compositions) (see Fig. S4† for the Cp of other CC-PCM compositions) determined by DSC. (b) Cp change in the glass transition region on heating under 5 K min−1 DSC scan rate. (c) Dependence of glass transition temperature on the DSC scan rate (q) and (d) Ln (q) versus 1/Tg based on eqn (3). | |
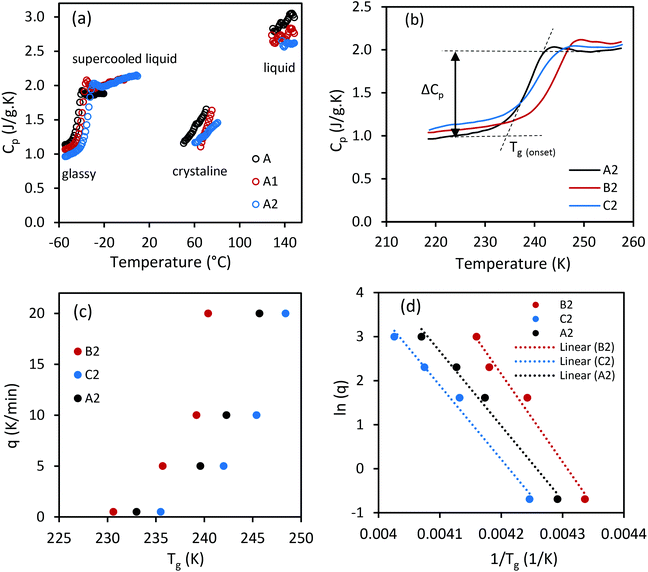
The fragility concept of glass forming liquids assists in understanding the relaxation process and glass transition of supercooled liquids.49 The steepness index (m-fragility) can be determined according to:
| | 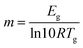 | (4) |
Liquids are classified from strong to fragile based on their behavior on continuous cooling (viscous slowdown).25,50 The strong-fragile classification of liquids explains the sensitivity of their structure to temperature changes. Fragile liquids demonstrate non-Arrhenius behavior with large ΔCP (60–80%) at Tg, including substances without directional intermolecular bonds such as molten salts.51 Hydrogen bonding specially contributes to ΔCP.25 Whereas, strong liquids (small m) follow Arrhenius behavior with small heat capacity changes (Cp,l/Cp,g ≈ 1.1) at Tg, including network liquids and covalent glass formers.51 Strong liquids have a smaller number of potential energy hypersurfaces, hence a lower quantity of energy minima compared to that of fragile liquids. The glass transition properties of CC-PCM including activation energy, m-fragility, and thermodynamic fragility are given in Table 3. As observed in Table 3 and Fig. 6, CC-PCM shows small values for m fragility (kinetic strength) and large values for ΔCP (thermodynamic fragility) at Tg, which is typical of hydrogen bonded organic liquids. Angell25,50 explained that thermodynamically fragile and kinetically strong liquids have a similar density of energy minima to that of fragile liquids. However, the potential energy barrier between the hypersurfaces is larger likely due to the need for breaking specific hydrogen bonds for the rearrangement of molecules.
Table 3 Glass transition properties of CC-PCM calculated viaeqn (2) and (3). Cp values were measured by DSC at Tg(onset) under 5 K min−1 scan rate
| Composition |
E
g (kJ mol−1) |
R
2
|
m
|
ΔCp J g−1 K−1 |
C
p,l/Cp,g |
| A2 |
140.51 |
0.99 |
14.19 |
0.91 |
1.73 |
| B2 |
165.97 |
0.98 |
16.78 |
0.94 |
1.72 |
| C2 |
139.38 |
0.98 |
14.06 |
0.80 |
1.60 |
3.5. Crystallinity
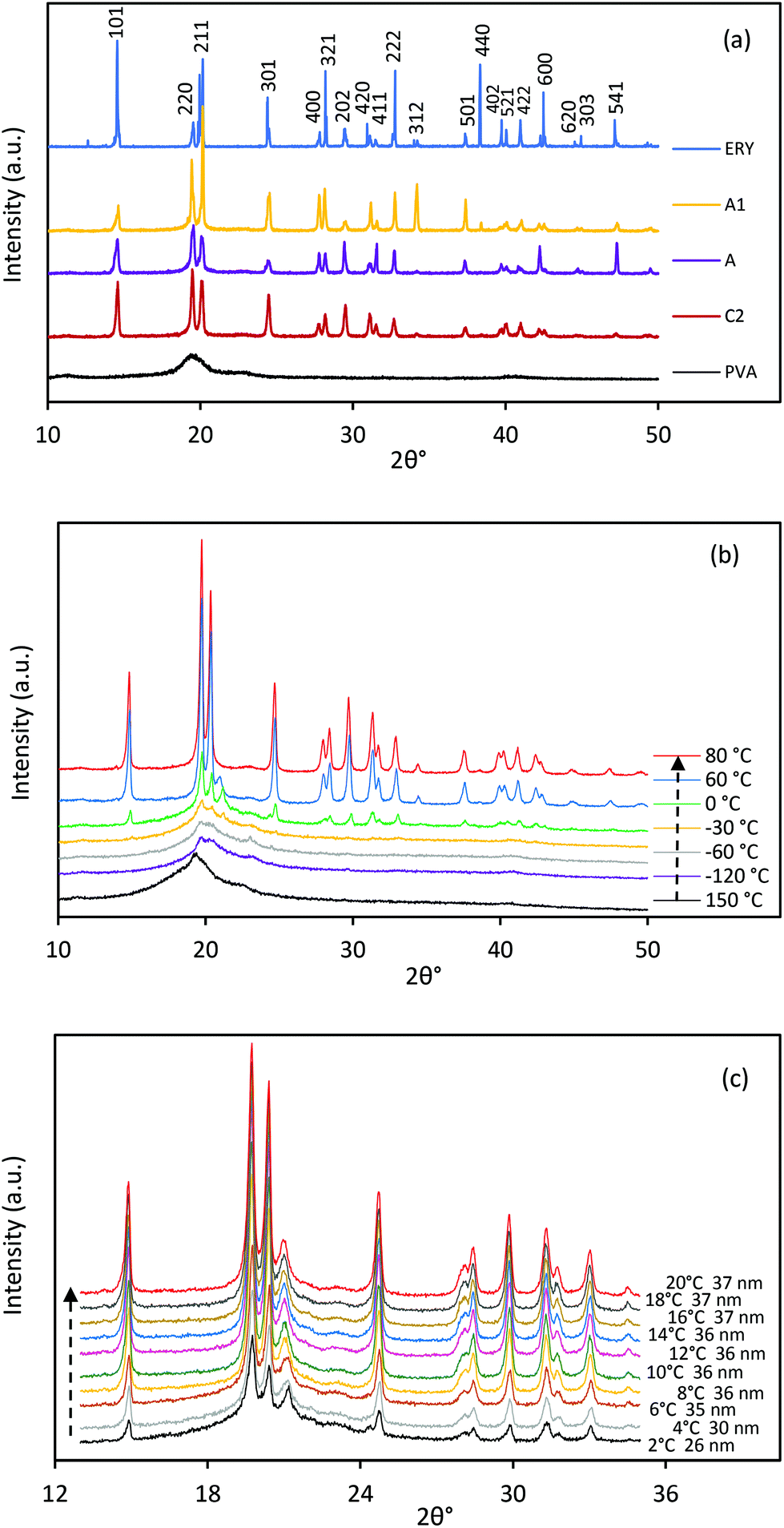
Fig. 7a shows the XRD pattern of PVA, ERY and CC-PCM (crystalline) at room temperature. A broad, shallow peak at 2θ = 19.4° was detected for PVA, which indicates a low degree of molecular orientation as well as intermolecular hydrogen bonding between the PVA chains. ERY showed three well-defined peaks at 2θ = 14.5°, 19.5°, and 20.1° and several lower intensity peaks at higher angles. All the characteristic peaks of ERY are observed in the XRD pattern of crystalline CC-PCM. In situ temperature XRD was performed to reveal the structural changes during cooling and heating cycles. In situ temperature XRD profiles of CC-PCM are depicted in Fig. 7b. Crystalline CC-PCM showed a series of sharp peaks due to reflected beams by lattice planes, while in the amorphous state, one broad peak centered in the range, where the strong peak was detected for the crystalline state of the same composition. This occurs due to the long-range order of crystal versus the short-range order of glass. The diffraction pattern of molten and glassy state of the CC-PCM, at ∼+150 °C and −60 °C respectively, exhibits a broad peak with a maximum at 2θ = 19.4° related to PVA52 and amorphous ERY,53 as seen in Fig. 7b. With the temperature approaching 0 °C, the XRD patterns show negligible differences. At ∼0 °C, small peaks start to appear at 2θ = 19.7°, 20.3° and 21.5°. Fig. 7c shows the fast scan XRD profile with the temperature under 1 K min−1 rate within the 0–20 °C range. As the temperature approaches 20 °C, small peaks are replaced by numerous narrow peaks with the most intense ones located at 2θ = 14.8°, 19.7°, 20.3°, and 24.5°, indicating the rapid transformation of the amorphous state into a crystalline phase (rapid cold-crystallization). A low intensity peak was observed at 2θ = 21.5° which diminished when the temperature was raised to 60 °C and disappeared at 80 °C, suggesting the presence of a metastable crystal form that is transformed to the stable crystal form by time and temperature.53In situ temperature XRD confirmed the prevention of undesired crystallization on cooling and a fast cold-crystallization on reheating.
 |
| | Fig. 7 (a) High resolution XRD pattern of PVA, ERY and CC-PCM (A, A1, and C2) at room temperature, (b) in situ temperature XRD profile of CC-PCM (A1), (c) fast X-ray scans within 0–20 °C under 1 K min−1 heating rate (temperature and (220) peak crystallite size are denoted next to each scan); demonstration of crystallization prevention on cooling and a fast crystal growth by cold-crystallization during reheating. | |
The high resolution XRD profiles of CC-PCM compositions in Fig. 7a correspond well to the reference structure of erythritol (PDF 00-033-166554) and the calculated pattern of single crystals by Ceccarelli et al.55 (the difference plot is provided in Fig. S6†). The crystal structure belongs to the tetragonal category (space group I41/a) including eight molecules in the unit cell. The refinement of unit cell parameters was carried out based on the XRD pattern of C2 as a = b = 12.879 Å, c = 6.867 Å. Two types of hydrogen bonding participate in the crystal structure forming a three-dimensional network of hydrogen bonds.55,56 The outer hydroxyl groups (CH2OH) of the adjacent ERY molecules form a helical chain arrangement of hydrogen bonds, while, the inner hydroxyl groups (CHOH) of the adjacent molecules form a quadrilateral arrangement of hydrogen bonds around the molecular symmetry.55,56 The crystallite size (L) was calculated according to the Scherrer formula:
| | 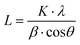 | (5) |
where
K is the Scherrer constant,
λ is the wavelength of radiation,
β is the full width at half maximum of the peak (FWHM), and
θ is the diffraction angle of the peak. According to the high-resolution XRD profiles shown in
Fig. 7a, the average crystallite sizes for compositions C2, A, and A1 were determined as 34.51 nm (35 nm for (220) peak), 45.21 nm (26 nm for (220) peak), and 46.87 nm (37 nm for (220) peak), respectively. Their crystallinity percentages were calculated as 71%, 70%, and 69%, respectively. Furthermore, the crystallite size of the (220) peak (A1) denoted next to each scan in
Fig. 7c confirms a trend of rapid crystallization occurring at around and after 2 °C. No significant crystallite growth is observed after 8–10 °C,
i.e. no peak sharpening or amorphous background intensity reduction.
3.6. Thermal stability
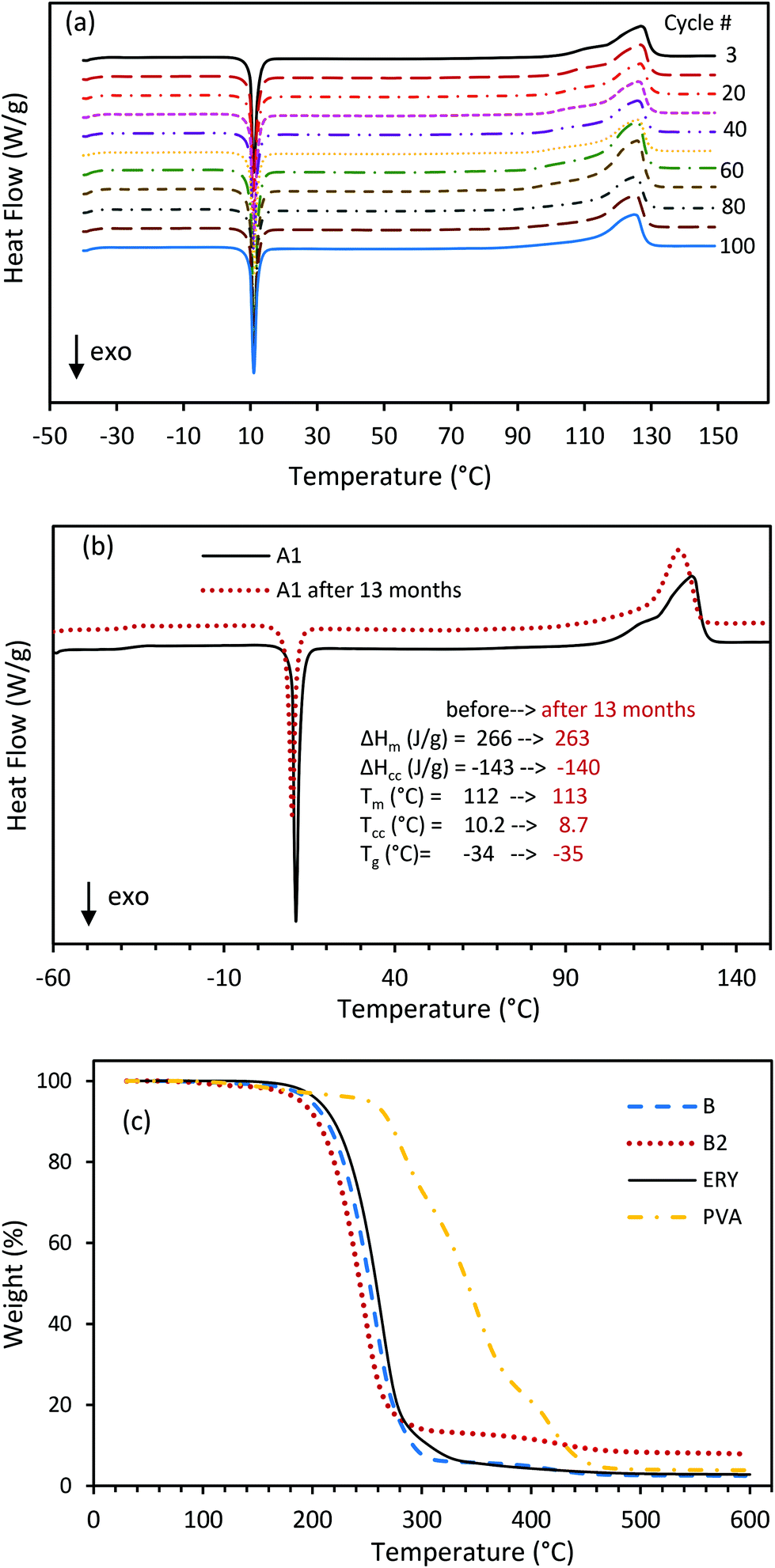
The consistency in the phase change behavior of the CC-PCM was examined over 100 successive DSC heating–cooling cycles (Fig. 8a). CC-PCM demonstrated remarkable repeatability and accuracy of cold-crystallization and melting over cycling, with slightly increased latent heat and no observable change in fusion temperatures. The increase in the latent heat after storage and cycling originates from the increase in homogeneity and consolidation of the PCM over time and cycling.30 The material showed excellent structural and mechanical stability without any rupture or leakage of the PCM during prolonged and repeated heating–cooling cycles. The improving effect of cycling has been previously reported for hydrated PVA gels.36,52 Thermal cycling enhances gelation and induces the growth of highly crystalline regions.36 Consecutive melting-cooling cycles homogenize the polyol swollen PVA organogel through crystal connection. Analogous to a model proposed for PVA hydrogels,36,37 CC-PCM can be considered as a fibrillar polymeric network providing connectivity throughout the organogel structure where the polyol molecules are adsorbed within the pores, as schematically presented in Fig. 1c and showed by SEM (Fig. 3). CC-PCM with the ionic agent showed high thermal durability (aging) over time. Fig. 8b illustrates the aging effect on latent heat storage properties of composition A1 after 13 months of storage at room temperature, confirming no observable changes in temperatures and amount of heat absorption and release. However, for the compositions without an ionic cross-linker, random crystallization on cooling was observed at long time points (e.g. 12 months) or very slow scan rates (e.g. 0.5 K min−1), which suggests the importance of the ionic agent for long-term stability.
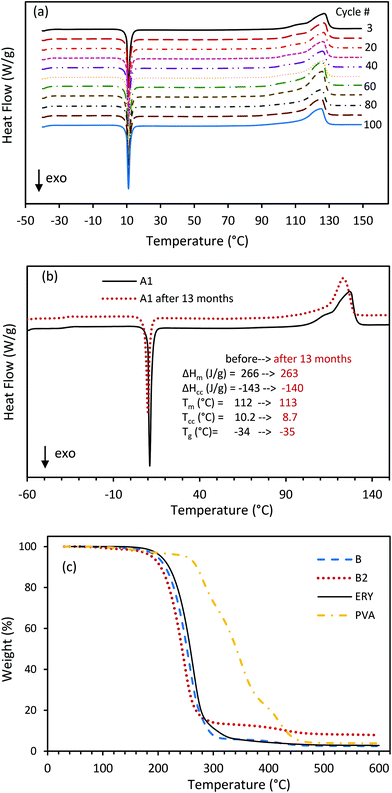 |
| | Fig. 8 (a) Repeatability of phase change behavior of the CC-PCM (A1) over 100 DSC heating–cooling cycles (5 K min−1 scan rate); demonstration of high cycling repeatability by DSC. (b) Thermal energy storage properties of CC-PCM (A1) before and after 13 months (in red) i.e. aging effect measured by DSC. (c) TGA profile of the ERY, PVA and CC-PCM (compositions B and B2). | |
The stability of the deeply supercooled state (between −40 °C and 15 °C) and the effect of cooling end temperature on cold-crystallization are also shown in Fig. S7.† Unlike other existing PCMs, CC-PCM provides better stability against undesired crystallization by cooling or storage time. The charged material is stable against spontaneous crystallization as long as it is kept below the cold-crystallization temperature (Tcc), as shown in the DSC graphs in Fig. S7a† for composition B2. Similar results have been reported previously for cold-crystallization of an erythritol-polyacrylate system.13,27 The end temperature of cooling for cold-crystallization to occur on the subsequent reheating was also analyzed with DSC, which is shown in Fig. S7b.† It was observed that the material can cold-crystalize on reheating if it has been cooled down below the Tcc, e.g. 0 °C for the composition B2. Glass transition (the end temperature of the glass transition region in the DSC heating curve [K]) is important when selecting a storage temperature (Ts) for the charged CC-PCM. The storage temperature is set above Tg and below the heat release temperature (Tcc). A possible approach is to select the storage temperature between the range 1.1 Tg and 1.2 Tg. Preceding the cooling to a deeply supercooled state T ≤ 1.1 Tg may provide a higher stability for a long storage period, due to the effect of energy minima (larger potential energy barriers) on dynamics and decoupling of translational diffusion and viscosity.13,57
The thermal degradability of ERY, PVA and CC-PCM was also analyzed by thermogravimetric analysis (TGA) from room temperature to 600 °C, as illustrated in Fig. 8c. CC-PCM was stable within the operation temperature range (up to 150 °C). A one-step thermal degradation within the 200–300 °C temperature range was observed for both ERY and CC-PCM. PVA showed a two-step thermal degradation process. The weight loss that started at around 250 °C is related to the dissociation of strong intermolecular hydrogen bonding and structural decomposition, which was followed by decomposition of polyene residues to carbon within the 350–450 °C temperature range.
4. Conclusions
We demonstrated an innovative and sustainable approach for storing heat for long-term and releasing it on demand through a latent heat process. Via a simple fabrication method, we developed a cold-crystalizing phase change material (CC-PCM) for long-term latent heat storage (LLHS). The material is composed of high-latent-heat erythritol PCM dispersed within the ionic cross-linked matrices of polyvinyl alcohol (PVA: a water-soluble and biocompatible polymer). We studied the material for the compositional, morphological and thermal energy storage properties via common standard methods such as FTIR, SEM-EDX, in situ temperature XRD, OM, DSC, and TGA. The unique property of CC-PCM was the ability of the polymeric matrices to inhibit undesired crystallization of the PCM during cooling by providing strong intermolecular interactions (hydrogen bonding), leading to vitrification instead of crystallization. The material was able to release the latent heat through cold-crystallization (CC) on reheating, which was determined by DSC and visualized though optical microscopy. Vitrification tendency of CC-PCM mitigates the risk of unexpected discharge of the charged heat during storage time and cold-crystallization provides controllability on the heat release. The addition of an ionic citrate cross-linker further tunes the vitrification and cold-crystallization properties of the CC-PCM. The homogeneity and intermolecular interactions of CC-PCM were confirmed by SEM-EDX, XRD and FTIR. Thermal stability was confirmed by TGA along with 100 consecutive DSC heating–cooling cycles. The CC-PCM demonstrated high latent heat of melting (up to 266 J g−1) and cold-crystallization (up to 157 J g−1 depending on the composition of CC-PCM) determined by DSC. Furthermore, the PVA super-adsorbent was able to swell and adsorb liquid PCM resulting in shape-stability and leakage-preventive properties above the melting point. The high latent heat, thermal stability against undesired crystallization at low temperatures, and inhibited fluidity above the melting temperature make this new class of PCMs promising for LLHS, especially for colder climates.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This article is an enhanced extension of work originally presented as a conference presentation in Eurotherm Seminar #112 Advances in Thermal Energy Storage (2019, Lleida Spain). Finnish Cultural Foundation (Suomen Kulttuurirahasto, 00191186 Central Fund) and Business Finland (HeatStock Project) are acknowledged for financial support of this research. We would like to thank Polymer Technology Research Group at School of Chemical Technology, Aalto University, for providing the Infrared Spectroscopy and Thermogravimetric equipment, and the Nanomicroscopy Center (OtaNano), Aalto University, for providing the electron microscopy and X-ray diffraction equipment. The authors thank Senior Scientist Yangling Ge, Aalto University, for her advice with XRD analysis. We would also like to acknowledge ZAE BAYERN-Garching, for providing the optical microscope equipped with a heating–cooling stage. We thank Christoph Rathgeber for hosting the first author's research visit at ZAE BAYERN and Dovile Rudaleviciene for assisting in the OM measurement.
References
- T. Kousksou, P. Bruel, A. Jamil, T. El Rhafiki and Y. Zeraouli, Sol. Energy Mater. Sol. Cells, 2014, 120, 59–80 CrossRef CAS.
- X. Huang, X. Chen, A. Li, D. Atinafu, H. Gao, W. Dong and G. Wang, Chem. Eng. J., 2019, 356, 641–661 CrossRef CAS.
- M. K. Rathod and J. Banerjee, Renewable Sustainable Energy Rev., 2013, 18, 246–258 CrossRef CAS.
- M. M. Farid, A. M. Khudhair, S. A. K. Razack and S. Al-Hallaj, Energy Convers. Manage., 2004, 45, 1597–1615 CrossRef CAS.
- S. N. Gunasekara, R. Pan, J. N. Chiu and V. Martin, Appl. Energy, 2016, 162, 1439–1452 CrossRef CAS.
- G. Diarce, I. Gandarias, A. Campos-Celador, A. García-Romero and U. J. Griesser, Sol. Energy Mater. Sol. Cells, 2015, 134, 215–226 CrossRef CAS.
- E. P. Del Barrio, R. Cadoret, J. Daranlot and F. Achchaq, Sol. Energy Mater. Sol. Cells, 2016, 155, 454–468 CrossRef.
- J. Xu, R. Z. Wang and Y. Li, Sol. Energy, 2014, 103, 610–638 CrossRef CAS.
- L. Scapino, H. A. Zondag, J. Van Bael, J. Diriken and C. C. M. Rindt, Renewable Sustainable Energy Rev., 2017, 76, 1314–1331 CrossRef CAS.
- B. Sandnes and J. Rekstad, Sol. Energy, 2006, 80, 616–625 CrossRef CAS.
- E. P. Del Barrio, A. Godin, M. Duquesne, J. Daranlot, J. Jolly, W. Alshaer, T. Kouadio and A. Sommier, Sol. Energy Mater. Sol. Cells, 2017, 159, 560–569 CrossRef CAS.
- P. G. Debenedetti and F. H. Stillinger, Nature, 2001, 410, 259 CrossRef CAS PubMed.
- S. Puupponen and A. Seppälä, Sol. Energy Mater. Sol. Cells, 2018, 180, 59–66 CrossRef CAS.
- S. Puupponen, V. Mikkola, T. Ala-Nissila and A. Seppälä, Appl. Energy, 2016, 172, 96–106 CrossRef CAS.
- P. Karimineghlani, E. Emmons, M. J. Green, P. Shamberger and S. A. Sukhishvili, J. Mater. Chem. A, 2017, 5, 12474–12481 RSC.
- P. Karimineghlani, A. Palanisamy and S. A. Sukhishvili, ACS Appl. Mater. Interfaces, 2018, 10, 14786–14795 CrossRef CAS PubMed.
- Y. Wei, J. Li, F. Sun, J. Wu and L. Zhao, Green Chem., 2018, 20, 1858–1865 RSC.
- Z. Liu, J. Lyu, D. Fang and X. Zhang, ACS Nano, 2019, 13, 5703–5711 CrossRef CAS PubMed.
- G. Li, X. Zhang, J. Wang and J. Fang, J. Mater. Chem. A, 2016, 4, 17042–17049 RSC.
- X. Wang, G. Li, G. Hong, Q. Guo and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 41323–41331 CrossRef CAS PubMed.
- G. Li, G. Hong, D. Dong, W. Song and X. Zhang, Adv. Mater., 2018, 30, 1801754 CrossRef PubMed.
- J. Lyu, G. Li, M. Liu and X. Zhang, Langmuir, 2019, 35, 943–949 CrossRef CAS PubMed.
- P. Li, J.-Y. Hwang and Y.-K. Sun, ACS Nano, 2019, 13, 2624–2633 CAS.
- K. Iwase, Y. Nagano, I. Yoshikawa, H. Houjou, Y. Yamamura and A. K. Saito, J. Phys. Chem. C, 2014, 118, 27664–27671 CrossRef CAS.
- C. A. Angell, Science, 1995, 267, 1924 CrossRef CAS PubMed.
- Y. Yue and C. A. Angell, Nature, 2004, 427, 717–720 CrossRef CAS PubMed.
- K. Turunen, M. R. Yazdani, S. Puupponen, A. Santasalo-Aarnio and A. Seppälä, Appl. Energy, 2020, 266, 114890 CrossRef CAS.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397 CrossRef CAS PubMed.
- D. G. Atinafu, W. Dong, C. Wang and G. Wang, J. Mater. Chem. A, 2018, 6, 8969–8977 RSC.
- P. Lu, W. Chen, J. Fan, R. Ghaban and M. Zhu, ACS Sustainable Chem. Eng., 2018, 6, 2656–2666 CrossRef CAS.
-
J. D. Menczel and R. B. Prime, Thermal Analysis of Polymers: Fundamentals and Applications, John Wiley & Sons, 2014 Search PubMed.
- Y. Zhao, H. Su, L. Fang and T. Tan, Polymer, 2005, 46, 5368–5376 CrossRef CAS.
- M. M. Ibrahim, S. A. Hafez and M. M. Mahdy, Asian J. Pharm. Sci., 2013, 8, 48–57 CrossRef CAS.
- M. Chaouat, C. Le Visage, W. E. Baille, B. Escoubet, F. Chaubet, M. A. Mateescu and D. Letourneur, Adv. Funct. Mater., 2008, 18, 2855–2861 CrossRef CAS.
-
P. Larkin, IR and Raman Spectroscopy : Principles and Spectral Interpretation, Elsevier, Saint Louis, US, 2011 Search PubMed.
- P. J. Willcox, D. W. Howie Jr., K. Schmidt-Rohr, D. A. Hoagland, S. P. Gido, S. Pudjijanto, L. W. Kleiner and S. Venkatraman, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 3438–3454 CrossRef CAS.
- F. Yokoyama, I. Masada, K. Shimamura, T. Ikawa and K. Monobe, Colloid Polym. Sci., 1986, 264, 595–601 CrossRef CAS.
- K. Nakano, Y. Masuda and H. Daiguji, J. Phys. Chem. C, 2015, 119, 4769–4777 CrossRef CAS.
- M. L. Di Lorenzo, Prog. Polym. Sci., 2003, 28, 663–689 CrossRef CAS.
- X. Z. Shu, K. J. Zhu and W. Song, Int. J. Pharm., 2001, 212, 19–28 CrossRef CAS PubMed.
- T. Jóźwiak, U. Filipkowska, P. Szymczyk, J. Rodziewicz and A. Mielcarek, React. Funct. Polym., 2017, 114, 58–74 CrossRef.
- D. Dhara, C. K. Nisha and P. R. Chatterji, J. Macromol. Sci., Part A: Pure Appl.Chem., 1999, 36, 197–210 CrossRef.
- T. C. Maloney, H. Paulapuro and P. Stenius, Nord. Pulp Pap. Res. J., 1998, 13(1), 31–36 CAS.
- K. Nakamur, Y. Minagaw, T. Hatakeyam and H. Hatakeyama, Thermochim. Acta, 2004, 416, 135–140 CrossRef CAS.
- R. Baetens, B. P. Jelle and A. Gustavsen, Energy Build., 2010, 42, 1361–1368 CrossRef.
- Y. Yuan, N. Zhang, W. Tao, X. Cao and Y. He, Renewable Sustainable Energy Rev., 2014, 29, 482–498 CrossRef CAS.
- T. Hatakeyma, H. Kasuga, M. Tanaka and H. Hatakeyama, Thermochim. Acta, 2007, 465, 59–66 CrossRef CAS.
- C. T. Moynihan, A. J. Easteal, J. Wilder and J. Tucker, J. Phys. Chem., 1974, 78, 2673–2677 CrossRef CAS.
- L.-M. Wang, V. Velikov and C. A. Angell, J. Chem. Phys., 2002, 117, 10184–10192 CrossRef CAS.
- C. A. Angell, J. Non-Cryst. Solids, 1991, 131–133, 13–31 CrossRef CAS.
- R. Böhmer, K. L. Ngai, C. A. Angell and D. J. Plazek, J. Chem. Phys., 1993, 99, 4201–4209 CrossRef.
- R. Ricciardi, F. Auriemma, C. De Rosa and F. Lauprêtre, Macromolecules, 2004, 37, 1921–1927 CrossRef CAS.
- A. J. L. Jesus, S. C. C. Nunes, M. R. Silva, A. M. Beja and J. S. Redinha, Int. J. Pharm., 2010, 388, 129–135 CrossRef PubMed.
- ICDD, PDF-4+/Organics 00-033-1665, USA, 2020.
- C. Ceccarelli, G. A. Jeffrey and R. K. McMullan, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 3079–3083 CrossRef.
- A. Shimada, Acta Crystallogr., 1958, 11, 748–749 CrossRef CAS.
- P. G. Debenedetti and F. H. Stillinger, Nature, 2001, 410, 259–267 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc01427c |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Jarkko
Etula
*a,
Jarkko
Etula