
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactive extraction of fructose for efficient separation of sucrose-derived glucosides produced by enzymatic glycosylation†‡
Andreas
Kruschitz
a and
Bernd
Nidetzky
 *ab
*ab
aAustrian Centre of Industrial Biotechnology (ACIB), Krenngasse 37, 8010 Graz, Austria
bInstitute of Biotechnology and Biochemical Engineering, Graz University of Technology, NAWI Graz, Petersgasse 12, 8010 Graz, Austria. E-mail: bernd.nidetzky@tugraz.at; Fax: +433168738434; Tel: +433168738400
First published on 30th June 2020
Abstract
The disaccharide sucrose (α-D-glucopyranosyl-1,2-β-D-fructofuranoside) is a highly efficient donor substrate for enzymatic glucosylation reactions. A large number of commercially relevant glucosides are produced via this general route. Every sucrose-based glucosylation involves co-production of D-fructose in amounts corresponding to the target glucoside. A separation technology for selective removal, and efficient recovery, of the D-fructose co-product would be highly desirable as a generic platform for downstream process development. Here, we demonstrate organoboronate-complex reactive extraction of D-fructose into an 1-octanol/hexane (4/1, v/v) solvent using methyltrioctyl ammonium chloride as phase transfer catalyst and extractant. We show separation of the D-fructose co-product (100 mM) from equimolar mixture with 2-α-D-glucosyl-glycerol, a cosmetic ingredient that is manufactured industrially from sucrose and glycerol using sucrose phosphorylase. With the main parameters of extraction, stripping and organic phase recycling identified and their effect on product/co-product yield and purity characterized, we developed a three-step extraction process with alternating extraction and stripping stages that removed the D-fructose efficiently at 50 ml operating scale. 2-α-D-glucosyl-glycerol and D-fructose were cleanly separated and obtained in excellent purity (≥90%) at a recovery of 90% and 83%, respectively. Reactive extraction appears to be faster and more selective and resource-efficient than applicable alternatives for separation, including chromatography and membrane nanofiltration. Thus, the established extraction process is promising for industrial application. It could be generically useful to separate D-fructose from glucosides.
Introduction
With around 200 million tons produced annually worldwide,1 the disaccharide sucrose (α-D-glucopyranosyl-1,2-β-D-fructofuranoside; Fig. 1(a)) represents an important industrial commodity. When compared to other carbohydrates produced at similar scale (e.g., maltodextrins), bulk-grade sucrose offers the advantage of being highly pure (≥95%) and representing a well-defined, single chemical entity. Due to its unique glycoside structure, sucrose exhibits internal energy higher than that of common disaccharides (e.g., maltose).2 Glycoside cleavage liberates the internal energy and can thus promote chemical transformations immediately connected to it. Sucrose is therefore an excellent donor substrate for glycosylation reactions.3,4 Glycosylation involves the D-glucosyl or D-fructosyl moiety of sucrose transferred to an acceptor molecule.5 Our focus here is on the transfer of the D-glucosyl part. Considering reactivity and selectivity of the reaction, glycosylation from sucrose is preferably performed using enzymatic catalysis.4 The relevant enzymes are glycosyltransferases,3–8 glycoside hydrolases and trans-glycosidases (e.g., glucansucrases3,9), as well as glycoside (sucrose) phosphorylases.10,11 Although often specific for the sucrose donor, many of the enzymes mentioned will use a broad range of acceptor substrates. This “acceptor promiscuity” provides the basis for a large number of sucrose-derived glucosides with important uses in the food, cosmetic and pharma sectors11–13 of industry, synthesized via enzymatic (trans)glycosylation. Glucosides of a non-sugar residue (e.g., stevia,12,13 resveratrol,14 arbutin15,16), disaccharides (e.g., nigerose,17 kojibiose18) and oligo/polysaccharides (e.g., glucan,9 dextran19) fall into this diverse category of industrially important products.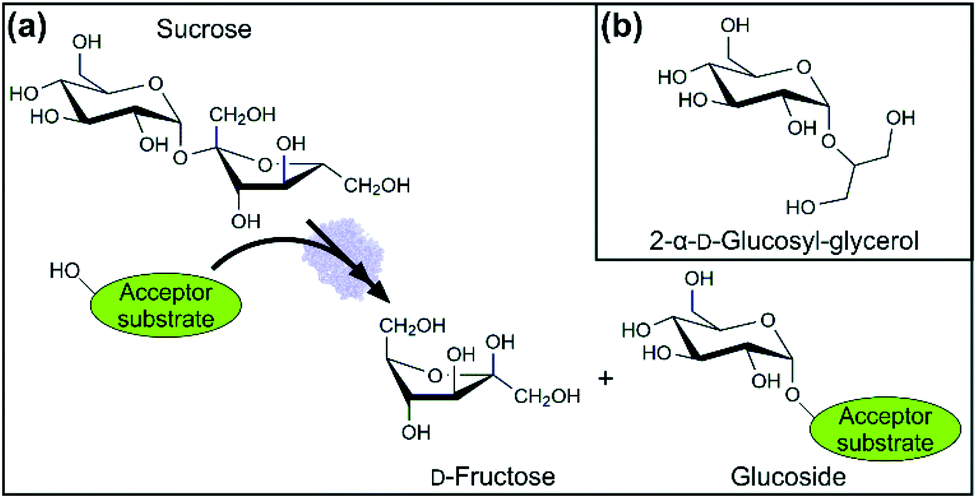 | ||
| Fig. 1 (a) Generic reaction scheme of an enzymatic glycosylation using sucrose as the donor substrate. D-Fructose is formed as a co-product to the target glucoside. (b) 2-α-D-Glucosyl-glycerol, a cosmetic ingredient, is shown as an example of an industrial glucoside synthesized by glycosylation from sucrose. | ||
Defining feature of any sucrose-based glucosylation is the D-fructose (Fru) released as the co-product of the overall reaction. Generally (i.e., side reactions of the Fru excluded), the Fru accumulates in exact correspondence to the sucrose converted. The Fru released is equal to or greater than the target glucoside formed, depending on the actual selectivity of the glucosylation.10 For high-value commercial applications, the Fru must be separated from the product. Strategy for efficient product/co-product separation is thus a central pillar of process development for every sucrose-derived glucoside. A modular separation technology for selective removal, and efficient recovery, of the Fru would be highly desirable. This could serve as a generic platform for the downstream processing in different glucoside production processes.
Previously, chromatography,20,21 nanofiltration22,23 or selective fermentation24 was applied to remove Fru from reaction mixtures. However, important limitations on the applicability of each method can arise. Chromatography is high in energy and solvent consumption and leads to a diluted product. In addition, it needs to be adapted, at least in its operational details, to each glucoside product anew. For nanofiltration to be usable efficiently, the glucoside product must be larger in mass by about 2.5 to 3-fold than the Fru. Selective fermentation is slow and post-treatment of the fermentation products is necessary. A promising alternative is reactive extraction of the Fru.25–27 Reactive extraction was previously intensively examined for the recovery of carboxylic acids (e.g., lactic and citric acid).28–30 This method can be highly selective,31 generally avoids product dilution32 and has already been used for Fru recovery from different product mixtures.25 Moreover, processes of reactive extraction are usually simple to operate and can be implemented well within existing process lines.31 Reactive extraction was shown for monosaccharide isolation in earlier studies,33–36 but its application to separate Fru from sucrose-derived glucosides was not explored. The goal of the current study was the development of a dedicated separation process based on reactive extraction. The separation should be able to provide reasonably pure streams of the product (glucoside) and the by-product (Fru). It should furthermore be broadly applicable and show suitable scalability.
As shown in Fig. 2, reactive extraction can overcome the low solubility of carbohydrates in organic solvents through complex formation with a carrier that is admixed to the organic phase.37 Hydrophobic organoboronic acids33–35,38–40 are widely used carriers. The anionic boronic acid-carbohydrate complex is stabilized in the organic phase by ion pairing with an organic cation (Fig. 2).33 For the complexation to be efficient, the carbohydrate should exhibit a vicinal diol to interact with the boronic acid.25 Such diol group is present in Fru but not in D-glucose (Glc), with consequent effect on the relative stability of the corresponding complex with boronic acid. Moreover, disaccharides and larger oligosaccharides, apart from some exceptions like lactulose, are generally less well extracted than monosaccharides.25,31,34 This is due to the occurrence of steric hindrance in disaccharides, preventing the complexation with organoboronic acids.41 These considerations lent strong support to the idea that, by exploiting differential complexation with an organoboronic acid carrier, efficient separation of Fru and sucrose-derived glucoside might be achieved. Through selective extraction of the Fru into the organic phase, a purified aqueous solution of the glucoside would be obtained. The Fru co-product might be recovered, now also purified, from the organic phase. Delidovich and Palkovits have carefully studied reactive extraction to separate Fru from Glc.25 The extractable complex of Fru was only stable at a pH above the pKa of the organoboronic acid used. Therefore, to avoid pH drop in the aqueous phase due to proton release during extraction (Fig. 2), pH stabilization (e.g., buffering) was necessary.34 Stripping with acid solution was used for back-extraction of the Fru and concomitantly to recycle the organic phase (Fig. 2).25,32,35 Repeated usage of the organic phase would thus be enabled for a multi-stage extraction effectively removing all of the Fru. Resource consumption and waste formation would so be minimized and the performance of the whole production process could be increased. Fig. 2 depicts the complete process of Fru separation, comprised of reactive extraction and acidic stripping.
 | ||
| Fig. 2 Principle and generic process scheme for reactive extraction to separate and isolate the target glucoside and the D-fructose co-product from enzymatic reaction. Complex formation between organoboronic acid carrier and the vicinal diol group of the carbohydrate is driven by ionization of the organoboronic acid at the aqueous–organic interface. The anionic complex ion pairs with an organic cation (Q+) that is suitably hydrophobic to be dissolved in the organic phase. Q+ serves as a phase transfer catalyst and extractant. Methyltrioctyl ammonium ion (as chloride salt; trade name Aliquat 33632) is often used. Proton release into the aqueous phase requires buffering/pH control.25 Acidic stripping recovers the D-fructose and allows for recycling of the organic phase.26,33–35,62 | ||
Here, we investigated the proposed strategy of reactive extraction for separation of Fru from 2-α-D-glucosyl-glycerol (2-GG) (Fig. 1(b)). The 2-GG is a natural compatible solute that has been commercialized as Glycoin® natural for cosmetic use. Industrial production of 2-GG proceeds according to Fig. 1(a),42–45 using sucrose and glycerol as substrates of a highly efficient trans-glycosylation reaction catalysed by sucrose phosphorylase.46 Beside Fru and 2-GG, there is also glycerol present in the product mixture. It was previously shown that glycerol can be easily removed with diafiltration.47 The remaining separation problem is thus the separation of Fru from 2-GG. Product specifications of Glycoin® natural demand that the Fru is removed to ≤10 wt% of the active ingredient 2-GG. Fru impurity in larger degree can cause a yellowish discoloration of the product mixture during formulation and storage. Ligand exchange column chromatography48 and nanofiltration47 were previously applied with limited success to remove the Fru in requested degree. Both approaches involved rather cumbersome multi-step procedures that resulted in strong dilution of an originally well-concentrated product (≥100 mM) from the enzymatic synthesis. Losses of 2-GG were substantial (≥40%). The Fru got even more diluted, thus generating large volumes of waste from which co-product recovery was not an option. In this study, therefore, we demonstrate reactive extraction to overcome these major limitations and develop an efficient process for the product/co-product separation and recovery of 2-GG and Fru. We show results of a systematic approach in which the main parameters of extraction, stripping and organic phase recycling were identified and their effect on product/co-product yield and purity were characterized. We found operation conditions that might fulfil requirements of process efficiency for industrial use, which was not usually the case in previous studies. 2-GG and Fru were cleanly separated and recovered in excellent purity and yield. The herein developed separation technology can be generically useful to separate, and recover, Fru from product mixtures of enzymatic glucosylation processes across all scales.
Experimental
Chemicals
Naphthalene-2-boronic acid (97%) was from Matrix Scientific (Columbia, South Carolina, USA) and Aliquat 336 was from Thermo Fisher Scientific (Kandel, Germany). Other chemicals were of reagent grade from Carl Roth (Karlsruhe, Germany), Merck (Darmstadt, Germany) or Honeywell (Charlotte, North Carolina, US). Raw and purified Glycoin® natural were from bitop AG (Dortmund, Germany). The raw Glycoin® natural is a solution containing around 1885 mM glycerol, 325 mM Fru and 320 mM 2-GG. The purified Glycoin® natural contains 52.8 wt% 2-GG, 1.4 wt% Glc, 0.7 wt% Fru, 6.6 wt% 1-α-D-glucosyl-glycerol and 38.5 wt% water.Extraction of Fru
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 3 min, the two phases were separated with a pipette.
000 rpm for 3 min, the two phases were separated with a pipette.
The extraction was then tested at higher carbohydrate concentrations. The aqueous phase contained around 110 mM Fru and 110 mM 2-GG and was made of raw Glycoin® diluted 3-fold with 0.3 M Na2CO3-NaHCO3 buffer (pH ∼ 10.5). Raw Glycoin® is a reaction mixture that is directly obtained from biosynthesis. The organic phase contained 300 mM organoboronic acid and 200 mM Aliquat 336. As negative control, extraction was carried out with an organic phase containing either 300 mM organoboronic acid, 200 mM Aliquat 336 or none of both compounds. Mixing and phase separation was performed as described above. The aqueous phases were analysed by HPLC.
Recovery of Fru and the organic phase
First, extraction was performed with an organic phase, containing 100 mM organoboronic acid and 200 mM Aliquat 336. For the aqueous phase, raw Glycoin® was diluted 3-fold with 0.3 M Na2CO3-NaHCO3 buffer (pH ∼ 10.5). Second, the organic phase was stripped with HCl solutions. The stripping solutions contained 0, 50, 100, 150, 500 or 1000 mM of HCl, respectively. The ratio of stripping to organic phase was either 1/1 or 2/1. Extraction and stripping were carried out for 5 min each. Mixing and phase separation of both extraction and stripping were carried out as described above. Extraction and stripping trials were performed in duplicate. The aqueous and stripping phases were analysed by HPLC.Recycling of the organic phase
An organic phase, compromising 100 mM organoboronic acid and 200 mM Aliquat 336, was mixed with raw Glycoin® diluted 3-fold with 0.3 M Na2CO3-NaHCO3 buffer (pH ∼ 10.5). The organic phase was then stripped with 150 mM HCl. Of this recovered organic phase, 0.9 mL were mixed with a new fraction of the diluted Glycoin® solution and stripped again with 150 mM HCl. Extraction and stripping were performed for 5 min each. The ratios of aqueous to organic phase and stripping to organic phase were 1/1, respectively. Mixing and phase separation were done as described above. Extraction and stripping trials were performed in duplicate. Aqueous and stripping phases were analysed by HPLC.Extraction process
Separation of Fru from 2-GG was performed with a three-step extraction cascade at a scale of 50 ml (25 ml per phase). The experimental set-up was that of a mixer-settler. Mixing was performed in a glass beaker on a magnetic stirrer (MR 3001K, Heidolph Instruments) at maximum speed for 5 min. Phase separation was achieved by gravity in a 100 ml separation funnel. The aqueous to organic and stripping to organic phase ratios were 1/1. In a first step, the aqueous phase made of raw Glycoin® diluted 3-fold with Na2CO3-NaHCO3 buffer (pH ∼ 10.5) was mixed with an organic phase, containing 100 mM organoboronic acid and 200 mM Aliquat 336. The organic phase was then stripped with 150 mM HCl. The recovered organic phase was recycled and again mixed with the already extracted aqueous phase. The organic phase was then again stripped with 150 mM HCl. The same was conducted for a third time to have three extraction and three stripping steps in total. In-between the extraction steps, the pH of the aqueous phase was adjusted to around 10.5 with 5 M NaOH. The whole process is depicted in Fig. 3. Aqueous and stripping phases were analysed by HPLC. | ||
| Fig. 3 Flow sheet of the three-step extraction process, including the three extraction steps and the three stripping steps to recover D-fructose and to recycle the organic phase. | ||
Extraction parameters
The relative amount of Fru extracted (EFru) from the aqueous into the organic phase was calculated with eqn (1): | (1) |
The relative amount of Fru stripped (SFru) from the organic into the stripping phase was calculated with eqn (2):
 | (2) |
As general performance parameters, the 2-GG recovery Y2-GG (eqn (3)) and the purity Pu (eqn (4) & (5)) of 2-GG and Fru were used.
 | (3) |
 | (4) |
 | (5) |
HPLC analysis
Samples were analysed by HPLC on a Shimadzu LC-20AD (Kyoto, Japan) equipped with an autosampler (SIL-20AC HT) and a RI-detector (RID-20A). A YMC-Pack Polyamine II/S-5 μm/12 nm column (250 × 4.6 mm) (YMC, Kyoto, Japan)49 equipped with a guard column (20 × 4.0 mm) or a Bio-Rad Aminex HPX-87C column (300 × 7.8 mm) (Bio Rad, Hercules, US)50 equipped with a guard column (30 × 4.6 mm) was used. Elution was performed isocratically with 75/25 acetonitrile/water (YMC column) or water (Aminex column). The sample injection volume was 20 μl. The YMC column was operated at 1 ml min−1, ambient temperature and the run time was 20 min. The Aminex column was operated at 0.5 ml min−1, 80 °C and the run time was 25 min.Results and discussion
Reactive extraction of the Fru from mixture with 2-GG
To examine whether reactive extraction is suitable in principle to separate Fru from 2-GG, we performed a set of extraction experiments in which complexing agent (55 mM) was present in large excess over carbohydrate substrate (10 mM). Phase catalyst/extractant (170 mM) was added further in excess, to ensure that all organoboronic acid, complexed and free, could undergo ion pairing in the organic phase. Solutions of individual compounds (Fru, 2-GG) and their mixture were used. Fig. 4 shows HPLC chromatograms from the aqueous feed phase before and after extraction. Using single-compound solutions, Fru was extracted almost completely (≥95%) whereas 2-GG was not extracted within limits of accuracy in mass balance (≤3%). Using the binary compound mixture, the extraction behaviour of the individual compounds was exactly the same within limits of error. The negative controls, lacking the organoboronic acid and/or Aliquat 336, showed no extraction for both Fru and 2-GG. These results reveal a high degree of selectivity in reactive extraction of Fru and 2-GG under the conditions used. Clean separation of the two compounds should thus be feasible with the method in principle. The molecular reason underlying the effect arguably is that 2-GG shows weak/no complex formation with the organoboronic acid, in contrast to Fru that is known to be complexed strongly. To evaluate the performance of the reactive extraction at industrially relevant substrate loadings, we increased the concentrations of Fru and 2-GG to 110 mM, respectively. The organoboronic acid (300 mM) and Aliquat 336 (200 mM) were still used in excess. Around 42% of the Fru was extracted into the organic phase. A partition coefficient Po/a ( = cFru,o/cFru,a) of ∼0.73 can be calculated from the data. Previous studies of Fru extraction used as carrier primary amines that were dissolved in organic phase or ionic liquid. Such carrier-facilitated extractions gave lower Po/a values, typically 0.5 or smaller.37,51 In the conditions used here, 2-GG was not extracted in detectable amounts. This result implies importantly that the reactive extraction retained its near-perfect selectivity at elevated substrate concentrations. Clean removal of Fru from 2-GG was thus achieved. The negative controls showed that both organoboronic acid and Aliquat 336 had to be present in the organic phase for any extraction of Fru to happen. This observation is in line with a previous study of reactive extraction of Glc, D-xylose and L-arabinose.26 | ||
| Fig. 4 HPLC chromatograms from reactive extraction of Fru, 2-GG or mixture of Fru and 2-GG. (a) 10 mM Fru, (b) 10 mM 2-GG or (c) 10 mM Fru and 10 mM 2-GG. The black line is the aqueous feed solution before extraction, the grey line is the aqueous phase after extraction. The retention time of Fru was around 12.3 min and of 2-GG around 13.6 min. | ||
Extraction was also performed with organic phases made of 1-octanol/heptane and 1-octanol/cyclohexane. Both organic phases showed a similar extraction potential of Fru as 1-octanol/hexane (see Table S1 in the ESI‡). 2-GG was not extracted in detectable amounts with any of the tested solvents. Organic phases of pure heptane and cyclohexane were also investigated. Yet, the solubility of organoboronic acid in these two solvents is much worse than in their mixtures with 1-octanol, making them unsuitable for reactive extraction.
Optimization of the Fru extraction
Having demonstrated the suitability of reactive extraction to separate Fru from 2-GG in principle, we moved on to challenge the efficiency of the method. We in particular considered dependence of the extraction yield (% Fru removed into the organic phase) on main operational parameters of the extraction, namely time and concentrations of complexing agent and phase transfer catalyst/extractant. Previous studies showed that the reactive extraction may require more than one hour to complete, and organoboronic acid and Aliquat 336 were often used in high excess.25,32,35 Long extraction time and requirement for high loadings of complexing agent and extractant detract from practicability and economic feasibility of the separation method. The herein used system was systematically explored in search of optimum conditions of operation.We first showed that extraction time in the range 5–60 min had no effect on the Fru yield (42.6 ± 2.3%). Therefore, the reactive extraction of Fru is achieved considerably faster than in previous studies.25,34 This is a major improvement in view of a possible industrial application. Then, we analysed the effect of concentration change for the organoboronic acid (100–500 mM) and the Aliquat 336 (100–300 mM). Note that the organoboronic acid was dissolved completely in the organic phase under all the conditions used. This was inconsistent with previous studies, where organoboronic acids only dissolved when Aliquat 336 was used in a molar ratio ≥1.34,38 The aqueous to organic phase ratio used was 1/1. The extraction yields dependent on compound concentration are summarized in Table 1.
| OCa | Boronic acid [mM] | Aliquat 336 [mM] | Phase ratiob | E Fru [%] |
|---|---|---|---|---|
| a Operating condition. b Phase ratio of aqueous to organic phase. The aqueous phase contained ∼123 mM D-fructose. | ||||
| 1 | 100 | 100 | 1/1 | 27.2 ± 0.4 |
| 2 | 100 | 200 | 1/1 | 41.9 ± 0.6 |
| 3 | 100 | 300 | 1/1 | 43.5 ± 0.6 |
| 4 | 300 | 100 | 1/1 | 21.9 ± 0.3 |
| 5 | 300 | 200 | 1/1 | 38.1 ± 0.5 |
| 6 | 300 | 300 | 1/1 | 38.8 ± 0.5 |
| 7 | 500 | 100 | 1/1 | 13.6 ± 0.2 |
| 8 | 500 | 200 | 1/1 | 39.7 ± 0.6 |
| 9 | 500 | 300 | 1/1 | 49.0 ± 0.7 |
| 10 | 100 | 300 | 1/2 | 59.0 ± 0.8 |
| 11 | 300 | 300 | 1/2 | 63.2 ± 0.9 |
| 12 | 500 | 300 | 1/2 | 61.4 ± 0.9 |
When Aliquat 336 was used about equimolar to Fru (123 mM), the extraction yield decreased as the concentration of organoboronic acid increased (operation conditions 1,4 & 7 in Table 1). Similar effect was noted in a study of Griffin and Shu,34 who suggested competition between complexed and non-complexed anionic species of organoboronic acid for ion-pairing with Aliquat 336 as the possible reason. Here, the extraction yield increased considerably (1.54-fold) upon increase in the Aliquat 336 concentration from 100 to 200 mM. Further increase in Aliquat 336 to 300 mM was no longer effective, however. Interestingly, when Aliquat 336 and organoboronic acid were increased simultaneously, the extraction yield was hardly affected. Operating condition 9 in Table 1 was the exception, giving a slight increase in yield by 5.5% at maximum loading of organoboronic acid. 2-GG was not extracted under any of the conditions shown.
The here obtained extraction yields for Fru can be compared to Delidovich and Palkovits25 who used phenylboronic acid and hydroxymethyl-phenylboronic acid as complexing agents. They applied similar Fru concentrations (∼166 mM) and used a high excess of boronic acid (400 mM) and Aliquat 336 (400 mM),25 somewhat comparable to our operating condition 9 (Table 1). Their yields were in the range 20–72%.25
In a last step, we analysed effect of the aqueous to organic phase ratio (Table 1, operating conditions 10–12). Doubling the volume of the organic phase caused the Fru yield to increase ∼1.5-fold to around 60%. However, balancing this increase in Fru yield against the increase in operating costs resulting from the double amount of organoboronic acid and Aliquat 336 used in the process, we decided to keep the aqueous to organic phase ratio at 1/1. From Table 1, we selected the condition best suited for reactive extraction as 100 mM organoboronic acid and 200 mM Aliquat 336 (entry 2). This represents a notable improvement compared to previous studies, where sugar extraction was carried out with a high excess of boronic acid and Aliquat 336.25,31,32 This condition was used further to develop the extraction process.
Recovery of Fru
The Fru is a valuable co-product of the glucoside synthesis from sucrose. Therefore, a purification technology that allows for full recovery of the Fru separated from the glucoside product would be highly desirable. The previously used approaches of 2-GG isolation did not include the realistic option of Fru recovery.47,48 There are interesting chemical (non-food) uses for Fru “waste streams” obtained by downstream processing of the product mixtures from sucrose-based enzymatic glycosylations.52–54 One route that appears promising in particular is chemical conversion of the Fru into the platform chemical hydroxymethylfurfural.55–57 To support this conversion, a technical-grade purity of the Fru should be achieved (≥90%) and other carbohydrates should not be present.58Stripping of the organic phase post-extraction serves the dual purpose of recovery of the Fru and re-use of the organic phase. In comparable studies, the stripping was performed with acid concentrations much higher than the carbohydrate concentration in the organic phase.34,35 Considering the possibility of acid-catalysed degradation of the Fru, we were alert to the proper selection of the stripping conditions, in view of further processing of the Fru thus obtained. Generally, the acidity of the stripping solution should be as low as possible. The organic phase applied to the stripping was previously used to extract the Fru (∼129 mM) from diluted raw Glycoin®. Around 54% of the Fru was extracted into the organic phase which showed a final Fru concentration of 69.5 ± 2.7 mM. We examined effect of the acid concentration from HCl on the relative amount of Fru stripped from the organic phase. Results are shown in Fig. 5. There was a steep, effectively linear increase in the Fru yield upon increase in the HCl concentration from 0 to 100 mM. Further increase in HCl to 150 mM caused the Fru yield to increase only slightly by 5%, thus reaching its maximum value of around 90%. The yield was unchanged when the HCl concentration was even further ramped up to 1.0 M. Collectively, these results serve to demonstrate that the Fru can be recovered efficiently from its stabilized organoboronic acid complex in the organic phase. This is potentially quite relevant because a usable/processable Fru co-product of relatively high purity can add significant value to the enzymatic production of sucrose-derived glucosides overall.
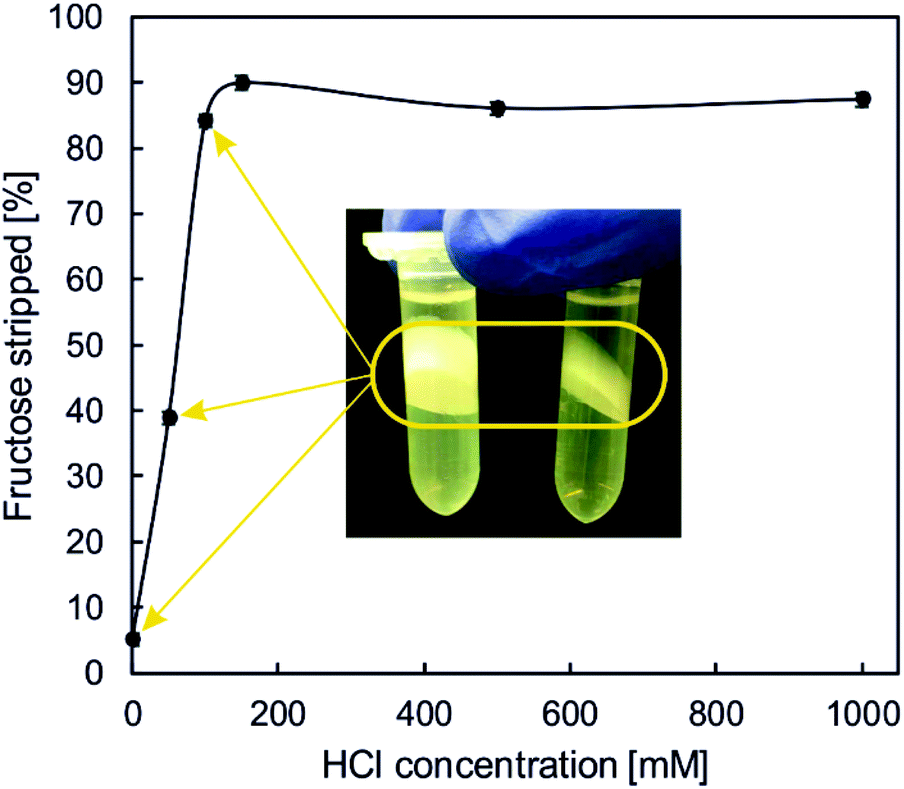 | ||
| Fig. 5 Relative amount of D-fructose stripped from the organic phase depending on the HCl concentration used in the stripping phase. Crud formation (highlighted in the picture with the two microtubes) occurred at HCl concentrations of 0, 50 and 100 mM (indicated by the three arrows). | ||
When performing the stripping at HCl concentrations ≤100 mM, we noticed formation of a whitish gel that accumulated as a crud phase between the aqueous stripping phase and organic phase (Fig. 5). The appearance of a crud phase is not uncommon in aqueous–organic extraction of carbohydrates.59 Salt precipitation could be the reason for crud phase formation.60 The formed crud severely exacerbated the phase separation and the recovery of the individual phases. Its formation needs to be absolutely avoided during the extraction process. Solution of 150 mM HCl was thus used further.
Increase in the phase ratio of organic to stripping phase from 1/1 to 1/2 was considered, but it did not improve the Fru recovery. Overall, about 88% of the extracted Fru could be stripped from the organic phase. With these results, as shown later, a multi-stage process of Fru extraction was developed (Fig. 3). The stripping phase had the same volume as the organic phase. The pH of the resulting strip solution was around 1.5 and thus by 0.7 pH units higher than the pH of the 150 mM HCl solution. The pH change during stripping was probably caused by both the transfer of HCl into the organic phase and the transfer of OH− ions into the aqueous phase due to the cleavage of the complex.32 The concentration ratio of HCl and Fru during the stripping used here was ∼2.16. This is in line with literature,32 showing that stripping of xylulose/xylose from an organic phase was readily achieved for a concentration ratio of acid to carbohydrate of around 2. However, the carbohydrate concentrations used in that earlier study (≤10 mM) were much lower than the ones used here.
Recycling of the organic phase
We have shown (see Table 1) that complete removal of the Fru was not achievable in single-stage extraction. Considering extraction over multiple stages to that end, the aspect of recycling of the organic phase becomes of particular importance. We therefore examined repeated extraction of Fru with an already used and recovered organic phase. Operating condition 2 from Table 1 was used. Using the fresh organic phase, the Fru yield was 52.5 ± 0.3%. Using the recycled organic phase, the Fru yield was almost identical (48.3 ± 0.7%; N = 2). Capacity of the organic phase for Fru extraction appears to not have changed due to recycling. Our findings are in accordance with Sánchez-Bastardo et al.,25 who recycled the organic phase for Glc, D-xylose and L-arabinose extraction. The authors observed no loss of boronic acid into the aqueous phase.25 A toxic contamination of the aqueous phase is thus avoided. Here, the amount of Fru recovered by stripping was similar for fresh (88.5 ± 0.7%) and recycled (90.9 ± 1.4%) organic phase. The recyclability of the organic phase was thus confirmed. Organic phase recycling is indispensable for a sustainable and cost-effective multi-step reactive extraction process.Three-stage extraction process
Considering the efficiencies of single-stage extraction and stripping, we devised reactive extraction in three stages with intermediate stripping and recycling of the organic phase (Fig. 3). We estimated from mass balance that removal of Fru to at least 90% should thus be achievable. The extraction was performed at 50 ml scale, using an aqueous to organic and an organic to stripping phase ratio of 1/1, respectively (operating condition 2, Table 1). The aqueous feed phase contained 123 mM Fru and 114 mM 2-GG. Phase separation by gravity was complete within 3 min. The pH of the aqueous phase was dropped from its initial value of 10.5 to around 9.6 after the extraction. The pH drop can be explained by the release of protons from water during Fru complex formation and extraction (see Fig. 2).25 The pH was thus only slightly above the pKa of the organoboronic acid which is around 9.35 The pH was therefore re-adjusted in between the extraction steps. To illustrate the importance of pH control, we performed the extraction without a pH adjustment. The extraction of Fru at the second stage was 20% lower as compared to extraction with pH adjustment. Decreased efficiency of carbohydrate extraction at lowered pH was noted previously33 and the requirement for pH control in between the extraction stages was pointed out.34Table 2 presents a summary of extraction at each stage. The relative amount of Fru extracted was constant, consistent with the notion that the extraction capacity of the organic phase did not decrease. Of the total Fru, 89% could be removed by the three-stage process. The total recovery of the extracted Fru in all three stripping streams was 93%. In stage 2, the relative extraction of Fru was even larger than 100%, implying that Fru not stripped in stage 1 could be recovered in stage 2. The overall Fru recovery was 83%. The Fru purity in the combined stripping streams from stage 1–3 (∼75 ml) was 92%. The Fru concentration was 33 mM. The 2-GG remaining in the aqueous solution after stage 3 meets the market requirements in terms of Fru content below 10 wt%. The aqueous solution recovered from the process had a volume of around 24 ml and contained 14 mM Fru and 107 mM 2-GG. The purity of 2-GG was increased from 57% to 92% in process (Fig. 6). Around 90% of the 2-GG were recovered. A selective separation of Fru and 2-GG was thus achieved.
 | ||
| Fig. 6 Course of the 2-GG purity (●) and the ratio of Fru to 2-GG in the aqueous phase (♦) throughout the three-stage extraction process. | ||
| Step | Fru extracted [mmol] | E Fru [%] | S Fru [%] |
|---|---|---|---|
| 1 | 1.58 ± 0.01 | 51.5 ± 0.9 | 86.7 ± 1.3 |
| 2 | 0.83 ± 0.01 | 55.4 ± 1.0 | 105.5 ± 1.6 |
| 3 | 0.34 ± 0.00 | 51.0 ± 0.9 | 90.4 ± 1.4 |
| Total | 2.75 ± 0.02 | 89.4 ± 1.6 | 92.8 ± 1.4 |
It is the first time that a multi-step reactive extraction process was developed that is capable of separating Fru from the sucrose-derived glucoside 2-GG. The whole process can be considered as very efficient and sustainable. Waste formation is reduced to a minimum, since the organic phase, including the organoboronic acid and Aliquat 336, can be effectively recycled. If one is concerned about the usage of hexane in the organic phase, even though it is recycled, it can be replaced by less harmful cyclohexane and heptane (see Table S1 in the ESI‡). The amount of applied organoboronic acid and Aliquat 336 appears to be economically acceptable. They were considerably lower than in comparable studies.25,31,32 26 g of product could be thus purified with one litre of organic phase in one cycle. This is a major improvement to chromatography, where around one litre of solvent is necessary to purify 1 g of product.48 Dilution of the Fru co-product is not large (3-fold), this is another advantage over chromatography and nanofiltration. In diafiltration, for example, Fru is multiple diluted (≥10-fold), since many diafiltration steps are necessary to remove Fru.47 Besides its acid content, equivalent to salt load if the pH was changed to a more neutral value, the Fru stream is highly pure. Considering literature, its composition seems to meet the demand for further conversions, in particular dehydration into hydroxymethylfurfural.56,58
The process is also low in energy consumption. Heating energy is not required, since operation is at ambient temperature. Mixing is the only energy input required. However, since extraction and stripping occur fast, the energy demand for mixing is also small. The production of pure 2-GG and Fru solutions is achieved within about an hour. This is an important improvement to previously proposed reactive extraction processes for sugar separation, wherein already one extraction step took at least 30 min25,34,35 Moreover, reactive extraction works much faster than nanofiltration23,61 or selective fermentation,24 which usually need several hours to remove Fru. The established process can be considered as a new and promising downstream process strategy to separate Fru from sucrose-derived glucosides across all scales.
Conclusions
A reactive extraction process for efficient separation of Fru from 2-GG was developed. Selective extraction of Fru was possible due to complex formation with naphthalene-2-boronic acid dissolved in the organic phase. In contrast, 2-GG was hardly extracted by this principle. A selective separation of the two compounds could thus be achieved. Reactive extraction parameters, including reaction time, carrier and counter-ion concentration in the organic phase and the phase ratio were optimised. Economically feasible parameters were found, providing a fast and selective separation of Fru and 2-GG. Recovery of the extracted Fru by stripping with an acidic solution was shown to be well feasible. The overall efficiency of the enzymatic production of glucoside using sucrose as donor is thereby increased. The recyclability of the organic phase was also confirmed. Resource consumption and waste formation could be thereby minimized. This is important since the organic phase itself, the carrier and the counter-ion are environmentally rather unfriendly. Based on these findings a three-step extraction process was established. It enabled the production of both a pure (90%) product (2-GG) and a by-product (Fru) stream, which is a considerable benefit for the enzymatic glycosylation process. The recoveries of both compounds were >80%. The established process could be highly interesting for industrial application. It is very selective and works much faster than other separation techniques (e.g., chromatography or nanofiltration). It is resource- and energy-efficient and can be considered as sustainable. The biggest asset of the developed extraction process is that it is not only restricted to the separation of Fru from 2-GG. The principle could be broadly applied in the downstream processing of sucrose-derived glucoside solutions obtained by enzymatic transglycosylation reactions, taking into account that the glucoside does not form a complex with the applied carrier. This is a big advantage compared to chromatography or nanofiltration. Since for those techniques, the operation concept and equipment (e.g., resin or membrane) have to be adjusted individually for each separation problem.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 761030 (CARBAFIN).Notes and references
- O. Sahu, Ann. Agrar. Sci., 2018, 16, 389–395 CrossRef.
- K. Buchholz and J. Seibel, Carbohydr. Res., 2008, 343, 1966–1979 CrossRef CAS PubMed.
- P. Monsan, M. Remaud-Siméon and I. André, Curr. Opin. Microbiol., 2010, 13, 293–300 CrossRef CAS PubMed.
- J. Seibel, H.-J. Jördening and K. Buchholz, Biocatal. Biotransform., 2006, 24, 311–342 CrossRef CAS.
- C. A. G. M. Weijers, M. C. R. Franssen and G. M. Visser, Biotechnol. Adv., 2008, 26, 436–456 CrossRef CAS PubMed.
- F. J. Plou, M. T. Martín, A. Gomez De Segura, M. Alcalde and A. Ballesteros, Can. J. Chem., 2002, 80, 743–752 CrossRef CAS.
- B. Nidetzky, A. Gutmann and C. Zhong, ACS Catal., 2018, 8, 6283–6300 CrossRef CAS.
- K. Schmölzer, M. Lemmerer, A. Gutmann and B. Nidetzky, Biotechnol. Bioeng., 2017, 114, 924–928 CrossRef PubMed.
- M. Remaud-Simeon, R. M. Willemot, P. Sarçabal, G. Potocki De Montalk and P. Monsan, J. Mol. Catal. B: Enzym., 2000, 10, 117–128 CrossRef CAS.
- V. Puchart, Biotechnol. Adv., 2015, 33, 261–276 CrossRef CAS PubMed.
- C. Luley-Goedl and B. Nidetzky, Biotechnol. J., 2010, 5, 1324–1338 CrossRef CAS PubMed.
- K. Olsson, S. Carlsen, A. Semmler, E. Simón, M. D. Mikkelsen and B. L. Møller, Microb. Cell Fact., 2016, 15, 1–14 CrossRef PubMed.
- V. Chatsudthipong and C. Muanprasat, Pharmacol. Ther., 2009, 121, 41–54 CrossRef CAS PubMed.
- P. Jeandet, E. Sobarzo-Sánchez, A. S. Silva, C. Clément, S. F. Nabavi, M. Battino, M. Rasekhian, T. Belwal, S. Habtemariam, M. Koffas and S. M. Nabavi, Biotechnol. Adv., 2020, 39, 107461 CrossRef CAS PubMed.
- Y. H. Moon, S. H. Nam, J. Kang, Y.-M. Kim, J.-H. Lee, H.-K. Kang, V. Breton, W.-J. Jun, K.-D. Park, A. Kimura and D. Kim, Appl. Microbiol. Biotechnol., 2007, 77, 559–567 CrossRef CAS PubMed.
- S. Kitao and H. Sekine, Biosci. Biotechnol. Biochem., 1994, 58, 38–42 CrossRef CAS PubMed.
- M. Kraus, J. Görl, M. Timm and J. Seibel, Chem. Commun., 2016, 52, 4625–4627 RSC.
- K. Beerens, K. De Winter, D. Van De Walle, C. Grootaert, S. Kamiloglu, L. Miclotte, T. Van De Wiele, J. Van Camp, K. Dewettinck and T. Desmet, J. Agric. Food Chem., 2017, 65, 6030–6041 CrossRef CAS PubMed.
- P. Monsan and F. Paul, FEMS Microbiol. Rev., 1995, 16, 187–192 CrossRef CAS.
- C. Nobre, P. Suvarov and G. De Weireld, N. Biotechnol., 2014, 31, 55–63 CrossRef CAS PubMed.
- D. Campos, L. Mescua, A. Aguilar-Galvez, R. Chirinos and R. Pedreschi, Int. J. Food Sci. Technol., 2017, 52, 2637–2646 CrossRef CAS.
- L. Moreno-Vilet, J. Bonnin-Paris, S. Bostyn, M. A. Ruiz-Cabrera and M. Moscosa-Santillán, Sep. Purif. Technol., 2014, 131, 84–93 CrossRef CAS.
- W. Li, J. Li, T. Chen and C. Chen, J. Membr. Sci., 2004, 245, 123–129 CrossRef CAS.
- C. Guerrero, C. Vera and A. Illanes, Int. Dairy J., 2018, 81, 131–137 CrossRef CAS.
- I. Delidovich and R. Palkovits, Green Chem., 2016, 18, 5822–5830 RSC.
- N. Sánchez-Bastardo, I. Delidovich and E. Alonso, ACS Sustainable Chem. Eng., 2018, 6, 11930–11938 CrossRef.
- I. Delidovich, M. S. Gyngazova, N. Sánchez-Bastardo, J. P. Wohland, C. Hoppe and P. Drabo, Green Chem., 2018, 20, 724–734 RSC.
- K. L. Wasewar, A. B. M. Heesink, G. F. Versteeg and V. G. Pangarkar, J. Biotechnol., 2002, 97, 59–68 CrossRef CAS PubMed.
- K. L. Wasewar, A. A. Yawalkar, J. A. Moulijn and V. G. Pangarkar, Ind. Eng. Chem. Res., 2004, 43, 5969–5982 CrossRef CAS.
- Y. K. Hong, W. H. Hong and D. H. Han, Biotechnol. Bioprocess Eng., 2001, 6, 386–394 CrossRef CAS.
- M. Wang, F. Ye, H. Wang, R. Yang and X. Hua, ACS Sustainable Chem. Eng., 2020, 8, 3465–3476 CrossRef CAS.
- B. Li, P. Relue and S. Varanasi, Green Chem., 2012, 14, 2436–2444 RSC.
- M. Matsumoto, K. Ueba and K. Kondo, Sep. Purif. Technol., 2005, 43, 269–274 CrossRef CAS.
- G. J. Griffin and L. Shu, J. Chem. Technol. Biotechnol., 2004, 79, 505–511 CrossRef CAS.
- T. C. R. Brennan, S. Datta, H. W. Blanch, B. A. Simmons and B. M. Holmes, BioEnergy Res., 2010, 3, 123–133 CrossRef CAS.
- B. Li, S. Varanasi and P. Relue, Green Chem., 2013, 15, 2149–2157 RSC.
- D. Hameister, S. Illner, C. Vogel, D. Michalik and U. Kragl, Sep. Purif. Technol., 2014, 132, 438–445 CrossRef CAS.
- H. A. Aziz, A. H. Kamaruddin and M. Z. A. Bakar, Sep. Purif. Technol., 2008, 60, 190–197 CrossRef CAS.
- S. Shinkai, K. Tsukagoshi, Y. Ishikawa and T. Kunitake, J. Chem. Soc., Chem. Commun., 1991, 1039–1041 RSC.
- G. Schroer, J. Deischter, T. Zensen, J. Kraus, A. C. Pöppler, L. Qi, S. Scott and I. Delidovich, Green Chem., 2020, 22, 550–562 RSC.
- M. Wang, F. Ye, H. Wang, H. Admassu, M. A. A. Gasmalla, X. Hua and R. Yang, Chem. Eng. J., 2019, 370, 1274–1285 CrossRef CAS.
- D. K. Hincha and M. Hagemann, Biochem. J., 2004, 383, 277–283 CrossRef CAS PubMed.
- K. Yoshida, A. Takenaka, T. Nitta and M. Iki, 2007137862, 2007.
- U. Breitenbach, V. Kallmayer, T. Raschke, C. Scherner, W. Siefken and S. Viala, WO2006/122669, 2006.
- J. A. Novejarque Conde, US2012/0308621, 2012.
- C. Luley-Goedl, T. Sawangwan, M. Mueller, A. Schwarz and B. Nidetzky, Food Technol. Biotechnol., 2010, 48, 276–283 CAS.
- A. Kruschitz and B. Nidetzky, Sep. Purif. Technol., 2020, 241, 116749 CrossRef CAS.
- C. Goedl, T. Sawangwan, M. Mueller, A. Schwarz and B. Nidetzky, Angew. Chem., Int. Ed., 2008, 47, 10086–10089 CrossRef CAS PubMed.
- M. Holtkamp, F. A. Erhardt, H. J. Jördening and S. Scholl, Chem. Eng. Process., 2009, 48, 852–858 CrossRef CAS.
- A. K. Goulas, P. G. Kapasakalidis, H. R. Sinclair, R. A. Rastall and A. S. Grandison, J. Membr. Sci., 2002, 209, 321–335 CrossRef CAS.
- D. Hameister and U. Kragl, Eng. Life Sci., 2006, 6, 187–192 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- N. do Amaral Sampaio Neta, J. C. S. dos Santos, S. de Oliveira Sancho, S. Rodrigues, L. R. B. Gonçalves, L. R. Rodrigues and J. A. Teixeira, Food Hydrocolloids, 2012, 27, 324–331 CrossRef.
- I. K. M. Yu and D. C. W. Tsang, Bioresour. Technol., 2017, 238, 716–732 CrossRef CAS PubMed.
- Y. Miyagawa, M. Yoshino, T. Kobayashi and S. Adachi, J. Appl. Glycosci., 2015, 62, 143–147 CrossRef CAS.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- F. Menegazzo, E. Ghedini and M. Signoretto, Molecules, 2018, 23, 1–18 CrossRef PubMed.
- I. K. M. Yu, K. L. Ong, D. C. W. Tsang, M. A. Haque, T. H. Kwan, S. S. Chen, K. Uisan, S. Kulkarni and C. S. K. Lin, Catal. Today, 2018, 314, 70–77 CrossRef CAS.
- N. Mungma, M. Kienberger and M. Siebenhofer, ChemEngineering, 2019, 3, 43 CrossRef CAS.
- G. M. Ritcey, Hydrometallurgy, 1980, 5, 97–107 CrossRef CAS.
- R. C. Kuhn, F. Maugeri Filho, V. Silva, L. Palacio, A. Hernández and P. Prádanos, J. Membr. Sci., 2010, 365, 356–365 CrossRef CAS.
- M. Takeuchi, K. Koumoto, M. Goto and S. Shinkai, Tetrahedron, 1996, 52, 12931–12940 CrossRef CAS.
Footnotes |
| † Data accessibility statement: Data obtained in the current study are available from the DOI: 10.5281/zenodo.3927374. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc01408g |
| This journal is © The Royal Society of Chemistry 2020 |