
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrifying green synthesis: recent advances in electrochemical annulation reactions
Guilherme M.
Martins
*ab,
Geórgia C.
Zimmer
 c,
Samuel R.
Mendes
d and
Nisar
Ahmed
*a
c,
Samuel R.
Mendes
d and
Nisar
Ahmed
*a
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: guilherme.m.martins@posgrad.ufsc.br; AhmedN14@cardiff.ac.uk
bDepartamento de Química, Universidade Federal de Santa Catarina, Florianópolis, 88040-900 SC, Brazil
cDepartamento de Química, Universidade Federal de Santa Maria, Santa Maria, 97105-900 RS, Brazil
dSINCA – Departamento de Química, Universidade do Estado de Santa Catarina, Joinville, 89219-719 SC, Brazil
First published on 30th June 2020
Abstract
Electricity originating from renewable resources can be used for highly sustainable and economically attractive applications. With electrons as the mass-free reagent, the use of a stoichiometric amount of oxidants in annulation reactions can be avoided, thereby eliminating the production of waste. Considered as a modern reaction configuration, the availability of electrochemical methods is expanding synthetic applications in the field of organic chemistry. Electrochemical transformations possess many benefits over traditional reagent-based methodologies, such as high functional group tolerance, mild conditions, easy scale up setup, high yields and selective transformations. In this review, we targeted electrochemical annulation reactions involving mediators and mediator-free conditions with generation of new C–C, C–heteroatom and heteroatom–heteroatom bonds, their mechanistic insights, as well as the reactivity of substrates. We also explain the recent use of sacrificial electrodes in annulation reactions.
 Guilherme M. Martins | Guilherme M. Martins obtained his Ph.D. in 2019 with the group of Prof. Claudio Silveira (UFSM, Santa Maria – Brazil). He performed an internship in electrosynthesis with Prof. Thomas Wirth and Prof. Nisar Ahmed in 2017 (Cardiff University, Cardiff – UK). In 2019, he conducted postdoctoral research with the group of Prof. Claus Jacob (Universität des Saarlandes, Saarbrücken – Germany). He is currently conducting postdoctoral research on electrosynthesis with the group of Prof. Antônio L. Braga and Prof. Samuel R. Mendes (UFSC, Florianópolis – Brazil). |
 Geórgia C. Zimmer | Geórgia C. Zimmer obtained her Ph.D. in 2019 working on Molecular Machines and Crystal Engineering in the group of Prof. Marcos A. P. Martins (UFSM, Santa Maria – Brazil). In 2019, she developed education activities in the chemistry laboratory at the technical college of Santa Maria (CTISM – Santa Maria – RS). In 2019–2020, she conducted a postdoctoral research with the group of Prof. Vanderlei Folmer (UFRGS, Porto Alegre – Brazil). |
 Samuel R. Mendes | Samuel R. Mendes obtained his Ph.D. in 2011 in Organic Chemistry working in the group of Prof. Claudio Silveira (UFSM, Santa Maria – Brazil). He performed an internship with Prof. Teodoro S. Kaufman in 2009 (Universidad Nacional de Rosario, Rosario – Argentina). In 2011, he conducted postdoctoral research with the group of Prof. Eder J. Lenardao (UFPel, Pelotas – Brazil). He is currently an Associate Professor at the Santa Catarina State University (UDESC, Joinville – Brazil) and a permanent professor at PPGQ-UDESC. |
 Nisar Ahmed | Nisar Ahmed obtained his Ph.D. in 2012 working in the group of Prof. Kwang S. Kim (POSTECH, Korea). Then, he moved to the University of Zurich for a postdoctoral stay with a Novartis Fellowship. In 2015, he became a senior research associate in the University of Bristol. Nisar started his research career in 2017 at Cardiff University, United Kingdom. He is also an Adjunct Professor at the HEJ Research Institute of Chemistry, Pakistan. His research interests are development of green & sustainable technology in organic synthesis using batch & microflow electrochemistry and molecular recognition of biomolecules. |
1. Introduction
Annulation is broadly defined as the construction of a new ring to organic molecules by introducing new bonds between the same or different atoms. In annulation reactions, the new bond-formation via conventional procedures commonly employs stoichiometric chemical oxidants, the use of which reduces the sustainability of the synthesis and carries safety and environmental concerns.1–3 Conventional synthetic methods are generally performed at high temperatures or pressures; however, electrochemical reactions are generally carried out under mild conditions without the addition of external oxidants, and in short reaction times, saving energy. In addition, by changing the operating current, the reaction time can be controlled. By varying the current or voltage, the oxidation or reduction capacity of the electrochemical system can be manipulated, which is another advantage compared to conventional methodologies.4,5 An intriguing discussion on the advantages of electrosynthesis compared to traditional methods was presented by Lei and Yuan, where the authors addressed the difficulties and limitations found in electrochemical reactions. Ways to solve these possible limitations in electrosynthesis, making it cleaner, are discussed.6 Very recently, Gerhard Hilt published an enlightening review addressing the basic strategies and types of applications in organic electrochemistry, containing an overview of the requirements and possibilities in electrosynthesis.7Recent developments in various cyclic reactions have been reported through a range of electrochemical annulations for the development of pharma-, agro- and fine chemicals as well as the synthesis of complex molecules derived from natural products.8–10 Due to the abundance in nature and the respective reactivity of aromatic heterocycles, the direct C–H or C–heteroatom functionalization calls the attention of several scientists, being extensively applied to obtain target compounds. In general, annulation reactions can occur through the intramolecular coupling when the electrogenerated center and another reactive center are in appropriate energies and positions in the molecule, favoring the reaction, or by intermolecular coupling. In the latter case, the annulation reaction involves the formation of two separate or simultaneous bonds.11 Electrochemical conversions include the cleavage of a covalent bond as the main reaction pathway. This way, both indirect electrolysis, which requires electron transfer mediators, and direct electrolysis, which involves the heterogeneous transfer of electrons directly between the electrode and the substrate, play an important role.12 Regarding the radical, it is possible to observe changes in the geometry structure after electron gain or loss, and cleavage or formation of new bonds may occur. With the removal of an electron from the highest occupied molecular orbital (HOMO), a radical cation is formed. On the other hand, if an electron is introduced externally, it will occupy the smallest unoccupied molecular orbital (LUMO), providing an anionic radical. Ionic radicals present a double character, have a charge and therefore are close to the ions, and present an unpaired electron that can react as radicals.13
1.1. Scope of this review
Herein, we present the recent advances in annulation reactions under electrochemical conditions. Over the past few years, researchers in this area have made significant progress, proving that electrolysis is a powerful tool for the hetero- and carboannulations. To aid the reading, this review is divided into five sections: (1) Introduction, (2) Mediator-promoted electrochemical annulation reactions, (3) Mediator-free electrochemical annulation reactions, (4) Sacrificial electrodes for electrochemical annulation reactions and (5) Conclusions and outlook. Electrochemical methodologies related to obtaining new C–C, C–heteroatom and Het–Het bonds are discussed, evaluating the mechanistic pathway, as well as the reactivity of substrates.2. Mediator-promoted electrochemical annulation reactions
Indirect reactions are those in which the exchange of electrons occurs between a mediator and the organic substrate. The mediators allow the transformation of organic compounds at lower oxidation potentials than the standard potentials. They also increase the selectivity and efficiency of the reaction, reducing energy consumption.14 Mediators can be organic derivatives (e.g. triarylamines, TEMPO, and hypervalent compounds), or inorganic derivatives (e.g. transition metal complexes, multivalent metal ions, and halide ions), which can originate from the electrolyte or can be added externally to the system.15 In addition, the use of catalysts is necessary when an organic substrate is not reactive enough or when the reaction leads to parallel reactions, superoxidation, dimerization, or electrode passivation.16 Next, we will discuss the reactivity and the need for the use of mediators in electrochemical annulation reactions.2.1. C–Heteroatom and C–C bond formation
Carbon–heteroatom and carbon–carbon bond forming reactions are constantly applied in the synthesis of medically significant molecules and agrochemicals, and they are of great value to materials science.17 Electrosynthesis catalyzed by metals activates the organic substrate by the active electrogenerated species of the transition metal catalyst, providing an organometallic intermediate that can be reduced or oxidized more easily. The metallic species can be regenerated electrochemically without the addition of external oxidants. Considering this, electrosynthesis catalysed by metals has been explored with several metals, such as Co, Mn, Cu, Ru, Rh, Ir, and Fe, among others.15 Lei and Ackermann group's recently described a C–H/N–H bond functionalization using cobalt as a mediator in electrochemistry.18,19 Similarly, Sundararaju and co-workers reported a cobalt and photoredox catalysis.20 Additionally, Borggraeve and co-workers described an interesting review comparing photoredox catalysis and electrochemistry in organic synthesis.21 Likewise, Ahmed and co-workers summarized some of the latest research efforts in using photoelectrochemical cells.22Protected amides commonly react with alkynes, providing simple isoquinolones, being catalyzed by Ni, Ru, Rh, and Co, among others.23–27 Likewise, Tang and co-workers proposed the first double-electrocatalytic organometallic C−H activation with amides 1 and alkynes 2 for the synthesis of polycyclic isoquinolinones 3 (Scheme 1).28 No external oxidants were applied and after 14 h, using [RuCl2(p-cymene)]2 (5 mol%), NaOPiv (1.0 equiv.), and n-Bu4NClO4 (1.0 equiv.) under a 10 mA current, the products were obtained in yields up to 82%. Substrates with electron-rich and electron-deficient substituents in both alkynes and benzoylamides were well tolerated; however, the increase in the number of methoxy substituents in benzoylamides resulted in decreased reaction efficiency (Scheme 1 – 3b and 3c). The role of the electric current is to anodically oxidize Ru(0) to Ru(II) to regenerate the catalyst in the cycle. Compared to the literature using a strong oxidizer,29 the most notable feature is the effective improvement of product regioselectivity under mild electrical conditions and no use of protective groups.
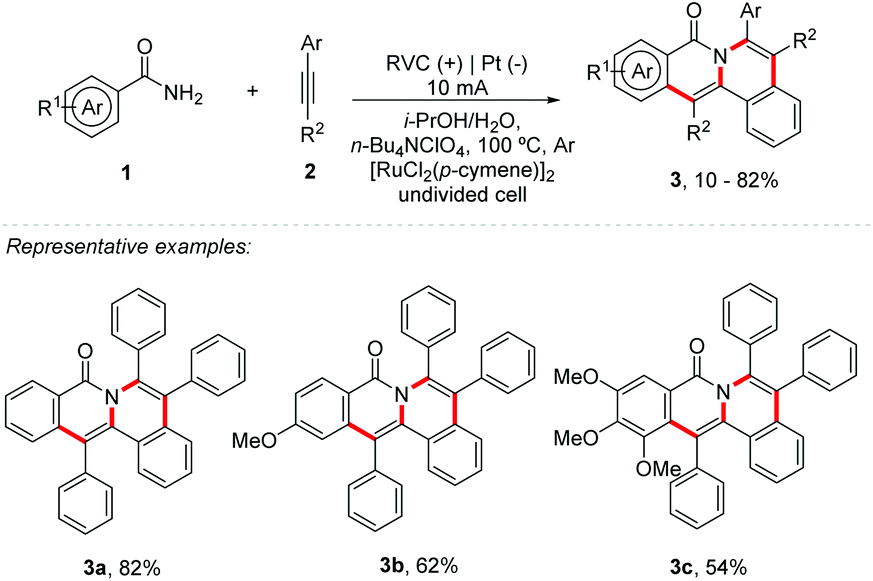 | ||
| Scheme 1 Synthesis of polycyclic isoquinolinones. | ||
Electro-oxidative activation of C–H/N–H carbamate and C–H/O–H 1-naphthols 4 with internal alkynes 5 catalyzed by Ru has been described by Ackermann's group (Scheme 2).30 Good tolerance of functional groups, such as ester, fluorine, chlorine or bromine substituents, was achieved; however, it presented the need to protect the amine group for N–H activation. Asymmetric alkynes provided the desired product 6b with high levels of regio-control (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), favouring the aromatic portion on the nitrogen heteroatom side. The mechanism was proposed through ruthena(II) cycle 7, followed by alkyne insertion.
:1), favouring the aromatic portion on the nitrogen heteroatom side. The mechanism was proposed through ruthena(II) cycle 7, followed by alkyne insertion.
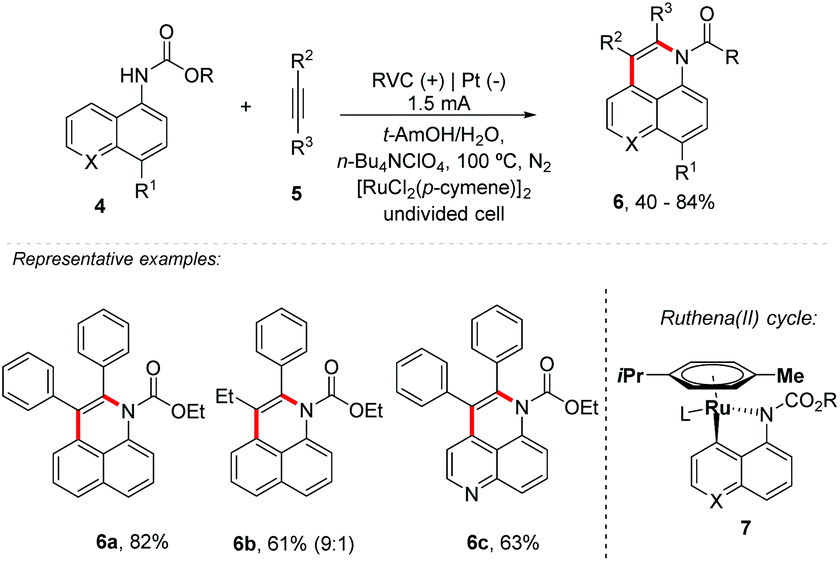 | ||
| Scheme 2 Ruthenium-catalyzed alkyne annulations by C–H/Het–H activation. | ||
Considering the remarkable interest of Ackermann's group in the reactions of C–H/heteroatom–H activation with alkynes,31,32 the group also proposed the synthesis of isochromen-1-one derivatives from alkynes and benzoic acid,33 and good yields (up to 93%) were obtained for both electron-rich and electron-deficient arenes, providing products from symmetric and non-symmetric, aromatic and aliphatic alkynes. Recently, the same group developed a modular electrochemical route for aza-PAHs 10 through a rhodium-catalyzed cascade C–H activation for alkyne annulation (Scheme 3).34 Aryl amidoximes 8 bearing electron-donating and electron-withdrawing groups were well tolerated. Asymmetric substrates were isolated with high levels of regioselectivity and good yields. The authors also isolated two main rodacyclic intermediates (11 and 12, Scheme 3), making it possible to identify the exact order of activation of the C–H bond.
 | ||
| Scheme 3 Rhoda-electrocatalyzed C–H/N–H activation. | ||
In addition, Ackermann's group reported the use of a cobalt catalyst for the activation of C–H/N–H bonds with carbon monoxide and isocyanate derivatives (Scheme 4).35 Both reactions were successful using an RVC anode and a platinum cathode as electrodes with Co(OAc)2 (20 mol%) and NaOPiv (1 equiv.) as additives and benzhydrazide 13 as the starting material common to both methodologies. However, for the reactions with isocyanate the best electrolyte was n-Bu4NBF4 (2 equiv.), DMSO as solvent, and a constant current electrolysis of 4 mA at 120 °C, giving products 14 in up to 82% yield. On the other hand, for the reaction with carbon monoxide, the best electrolyte was n-Bu4NClO4 (0.6 equiv.), with TFE:HFIP (2:1) as a solvent mixture under a constant current electrolysis of 3 mA at room temperature, giving products 15 in up to 89% yield. Competition studies have shown that electron-rich benzhydrazides achieved higher speeds than the electron-poor derivative. According to these results, the authors concluded that the mechanism follows the base-assisted internal electrophilic-type substitution (BIES) C–H metalation.36 In order to understand the role of the cobalt catalyst, the authors performed cyclic voltammetry, concluding that through anodic oxidation a cobalt(III) species was formed.
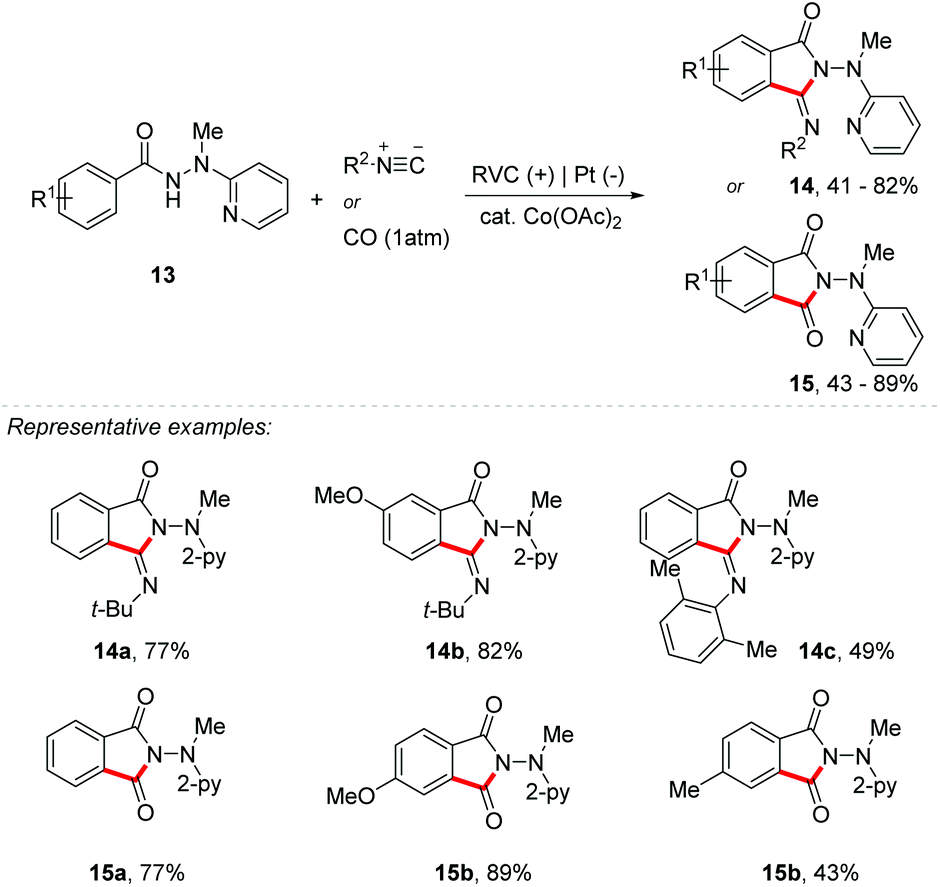 | ||
| Scheme 4 Cobalt catalyst for the activation of C–H/N–H bonds with carbon monoxide and isocyanate derivatives. | ||
Lei and co-workers described a transition metal cobalt catalyzed electrochemical [4 + 2] annulation of sulfonamides 16 with alkynes 17 to obtain sultam derivatives 18 (Scheme 5).37 The reaction was carried out under a constant current electrolysis of 4.0 mA in an undivided cell, with a carbon cloth anode and a stainless steel plate cathode, in the presence of Co(OAc)2·4H2O (20 mol%) and NaOAc (2 equiv.) additives, using a mixture of ethanol and acetic acid as the co-solvent, at 75 °C for 8 h (3.4 Fmol−1). Both electron-deficient and electron-rich acetylene derivatives were efficient in the annulation reaction, affording products in up to 85% yield. Alkyl alkynes were also evaluated, providing products in yields of up to 89%. Following the same modus-operandi, sulfonamides were explored with a wide scope of substituents. Considering a study using cyclic voltammetry, the authors suggest that the mechanism follows via a Co(II) species that initially coordinates with the sulfonamide, providing an intermediate complex 19; through a single electron oxidation, access to the Co(III) complex 20 is achieved. This protocol proved to be practical and scalable, and by scaling up to 5.0 mmol, 86% yield was obtained.
 | ||
| Scheme 5 Substrate scope for the synthesis of sultams via cobalt catalyzed electrochemical [4 + 2] annulation. | ||
It is appropriate here to highlight the advances made in reactions mediated by vitamin B12 derivatives. Through controlled-potential electrolysis, the catalytic cycle is based on a change in the valence of cobalt naturally derived from vitamin B12.38,39 Researchers have already applied cyano-cobalamin and its derivatives as catalysts in organic synthesis, such as double bond addition reactions, dehalogenation, halide coupling, oxidation, ring expansion, and functionalization of C–H bonds, among others.40–43 Important discoveries with a new point of view on the chemistry of a B12 derivative in an organic and aprotic environment were made by Hisaeda and co-workers,44 and these should be considered by the new generation of researchers.
He and co-workers developed a new strategy for [4 + 2] annulation of arylglyoxylic acids 21 with internal alkynes 22 using ruthenium catalysis, providing the respective 1H-isochromen-1-one 23 in yields of up to 88% (Scheme 6).45 Symmetric and unsymmetric internal alkynes were suitable, but it was not feasible for terminal alkynes and 1,2-di-o-tolylethyne. Control experiments showed that the reaction under an inert atmosphere was identical to that under air; however with an O2 atmosphere the 23a yield decreased to 43%. The results suggest that the newly formed carboxyl oxygen atoms are not from O2, which was also supported by the 18O-labeled isotope experiment. Interestingly, both oxygen atoms were replaced by 18O isotopes when H218O was used. The authors emphasize that the reaction via electrochemistry occurs through a different pathway from Ru-catalyzed decarboxylative annulations previously reported by Wang and co-workers.46 The Hammett graph analysis for competition experiments is in accordance with the reactivity order of the aryl glyoxylic acids: electron-rich aryl glyoxylic acids > 2-oxo-2-phenylacetic acid > electron-deficient aryl glyoxylic acids.
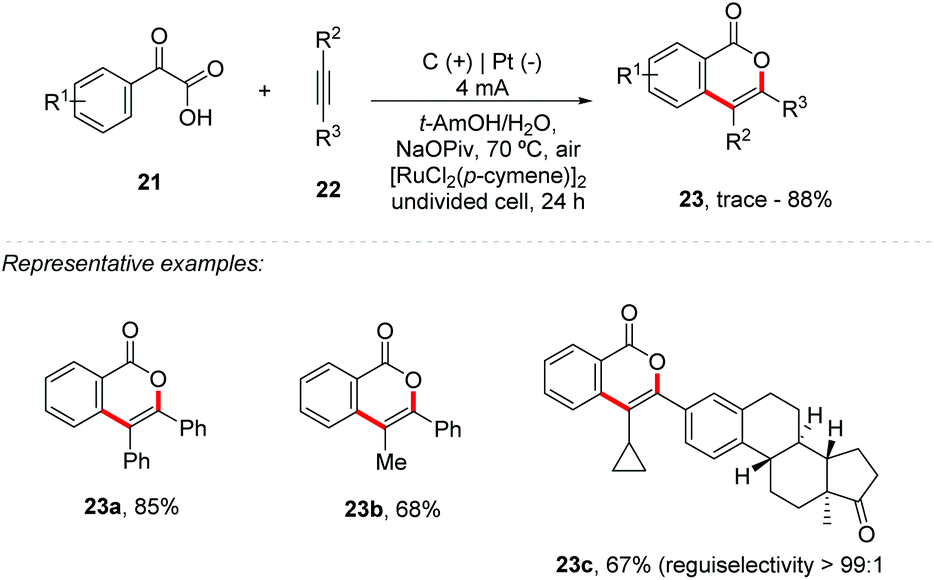 | ||
| Scheme 6 Ruthenium-catalyzed decarboxylative [4 + 2] annulation of arylglyoxylic acids with internal alkynes. | ||
Very recently, Park and co-workers reported an electrochemical double oxidative [3 + 3] cycloaddition for the synthesis of substituted dihydropyrano[4,3-b]indoles 26 (Scheme 7).47 The methodology was extended to the synthesis of 2,3-dihydrofuran derivatives 31 (Scheme 8). Electrolysis was carried out in an undivided cell, equipped with graphite electrodes as the anode and reticulated vitreous carbon (RVC) as the cathode, in acetonitrile as a solvent in the presence of NaI as a mediator and NaBF4 as an electrolyte, under a constant voltage of 3 V. Given the scope of indole 24, the method was efficient with halide, nitro, nitrile, methoxy and N-Boc substituents. N-Alkyl and N-aryl indoles, as well as 7-azaindole, were also compatible in the transformation. Regarding the variation of the active methylene compound (AMC) 25, unsymmetric diketones reacted with complete regioselectivity and several electron donating and withdrawing substituents were efficient in this transformation. According to the authors, the reaction proceeds by radical–radical cross-coupling of the indole/enamine radicals 27 with the radical species derived from the AMCs 28, followed by tandem 6π-electrocyclization.
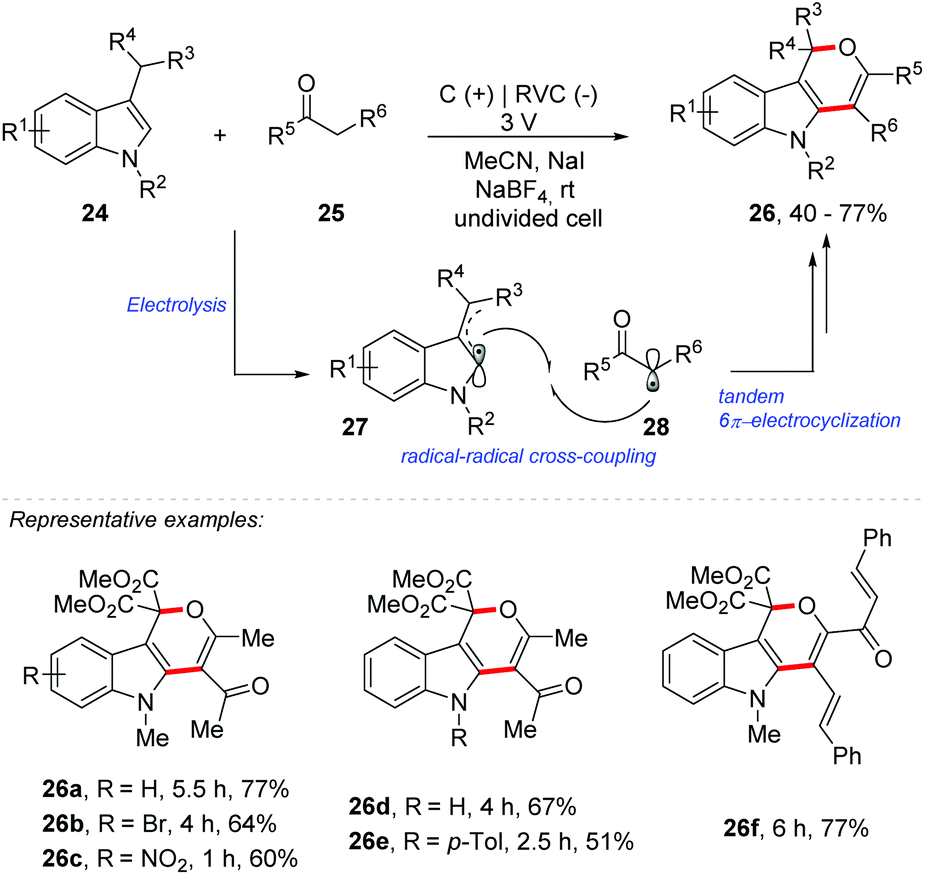 | ||
| Scheme 7 Electrochemical double oxidative [3 + 3] cycloaddition for the synthesis of substituted dihydropyrano[4,3-b]indoles. | ||
 | ||
| Scheme 8 Electrochemical double oxidative [3 + 3] cycloaddition for the synthesis of substituted 2,3-dihydrofuran derivatives. | ||
2.2. C–Heteroatom bond formation
The discovery that catalysts could mediate similar transformations replacing organometallic derivatives was a huge scientific advance for the formation of carbon–heteroatom bonds, being useful for the construction of several heterocycles.48 In addition, mediated reactions are widely used for the formation of C–N bonds, being able to assemble a wide range of heterocycles of interest containing nitrogen, for example, pyrroles, indoles, benzimidazoles, quinolines, among others. Recently, Huang and co-workers developed an efficient approach for the synthesis of quinazolinones 34 from o-aminobenzamides 33 and alcohols 32 by the combination of electrochemistry and a redox-metal catalyst (Scheme 9).49 The authors describe the first Mn catalysed cascade cyclization reaction to afford quinazolinones. Primary benzylic alcohols bearing electron-donating or electron-withdrawing groups were evaluated, and also, sterically hindered substrates. Additionally, o-aminobenzamides bearing electron-donating or electron-withdrawing groups were tested as well. The reaction was performed under a constant current electrolysis of 20 mA in an undivided cell, with Pt foil as electrodes, LiClO4 as the electrolyte, MeCN:H2O (48:2) as the solvent, TFA as an additive, and MnSO4·H2O (10 mol%) as a redox catalyst, under air at room temperature. The authors stated that water had a significant influence on this reaction. The compounds 34 were obtained in good yields, up to 90%. Interestingly, heteroaromatic alcohols with pyridinyl (34c), furanyl and thienyl groups were also suitable coupling partners, with good yields (up to 72%). Halide substituents were well tolerated under the electrochemical conditions. Benzyl methyl ethers were tested affording quinazolinone derivatives in good yields, up to 83%. The reaction mechanism was proposed, and considering the oxidation peak for Mn(II) at 1.87 V (vs. SCE) in the presence of TFA observed by cyclic voltammetry, the authors suggest that the reaction is mediated by Mn(III).
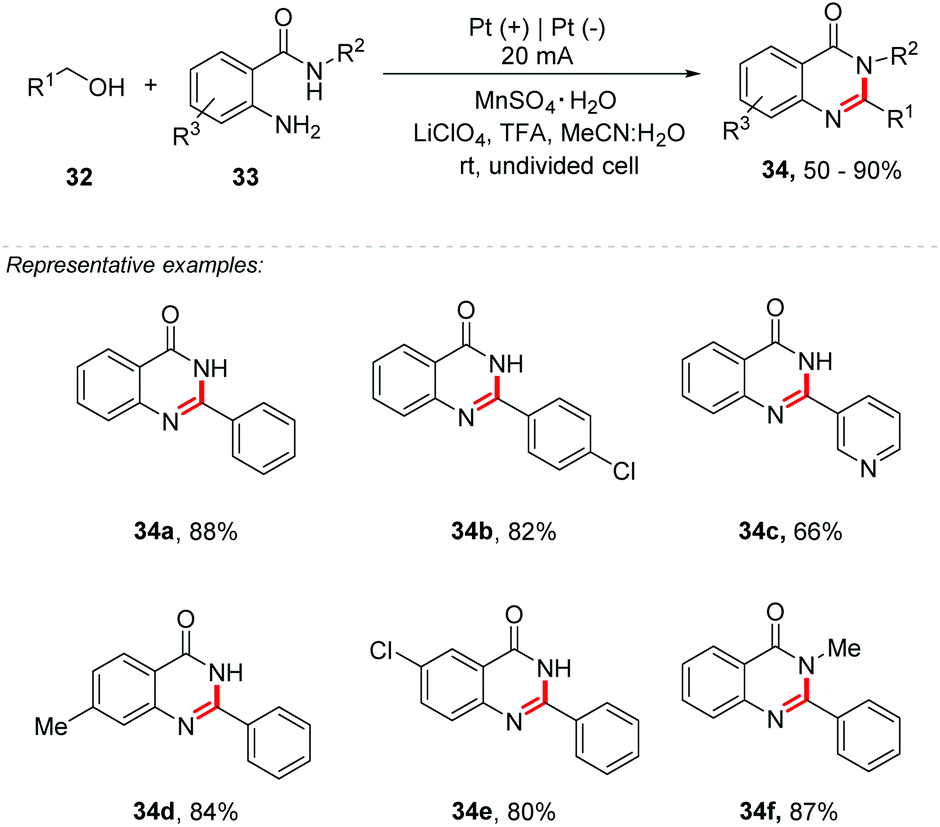 | ||
| Scheme 9 Mn-Catalyzed electrochemical synthesis of quinazolinones from primary alcohols and o-aminobenzamides. | ||
Hypervalent iodine reagents are classified as organic oxidants and have already been explored in various chemical transformations. Due to their great importance, there have already been several revisions and books reporting the recent advances in hypervalent iodine(III)-catalyzed functionalization, and it is still a topic of interest.50–53 Recently, Wirth et al. reported an interesting review on the electrochemical generation of hypervalent iodine reagents through anodic oxidation of iodoarenes, reporting their application as mediators for different chemical transformations.54 Additionally, Powers and co-workers reported electrocatalytic C–N coupling via anodically generated hypervalent iodine oxidants 38 (Scheme 10).55 A series of N-acetylcarbazole 37 were obtained with yields of up to 85% in an undivided cell with glassy carbon anode and platinum cathode electrodes, under a potential of 1.5 V (CPE) to 1.9 V vs. Ag+/Ag reference electrode at room temperature using HFIP as solvent.
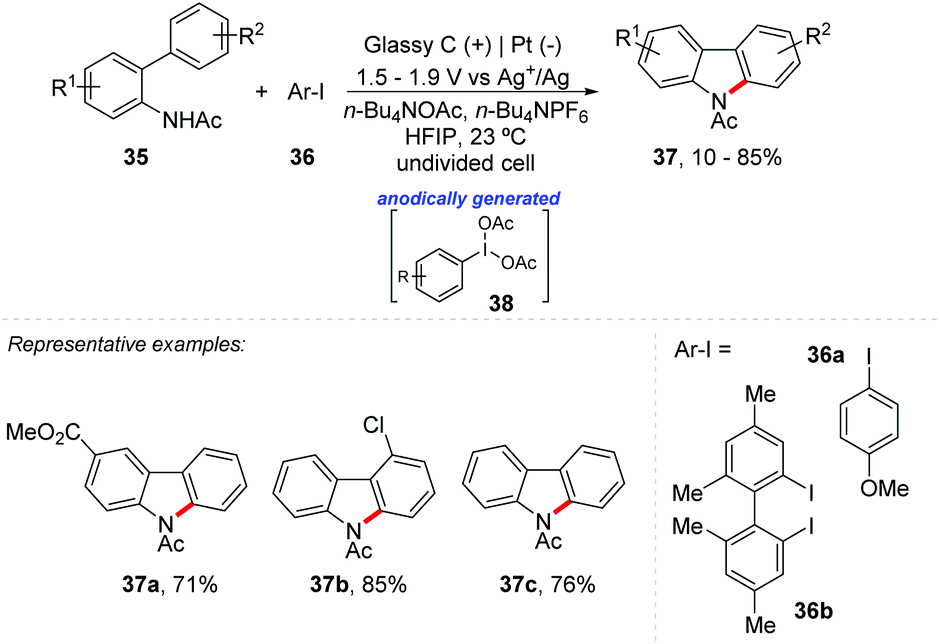 | ||
| Scheme 10 Electrocatalytic C–N coupling via anodically generated hypervalent iodine oxidants. | ||
Lei and co-workers described an intramolecular oxidative electrochemical C–H/N–H cross-coupling methodology to achieve 10H-benzo[4,5]imidazo[1,2-a]-indole derivatives 40 mediated by Cp2Fe, obtaining products with up to 83% yield (Scheme 11).56 An undivided cell was equipped with a graphite electrode at the anode and a nickel electrode at the cathode using a solvent mixture of MeCN:EtOH (1:1) and n-Bu4NBF4 as the electrolyte under a constant current of 8 mA at 70 °C for 4 h (4 Fmol−1). From the anodic oxidation of Cp2Fe to Cp2Fe+, the reaction proceeds to the oxidation of the conjugated base of 39 delivering the radical nitrogen species 41. Intramolecular radical addition, single electron oxidation and deprotonation provide the product 40.
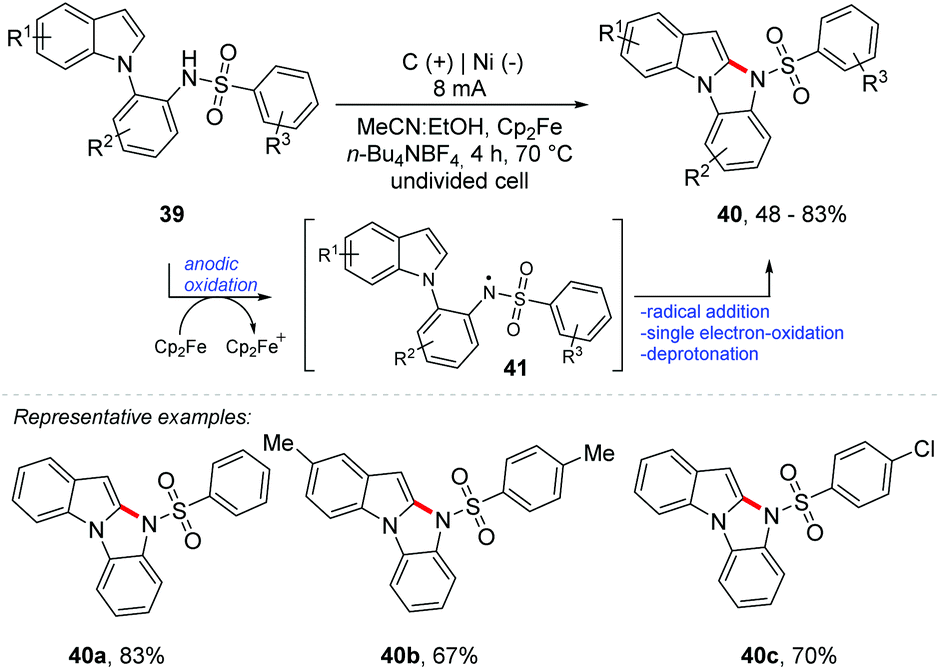 | ||
| Scheme 11 Intramolecular oxidative electrochemical C–H/N–H cross-coupling. | ||
2.3. C–C bond formation
In the last few years, new procedures for obtaining naphthol derivatives by C–H bond activation have been developed by Wang,57 Narender,58 Yu,59 Yamamoto,60 and Larock.61 In the same way, Pan and co-workers proposed a synthesis of 1-naphthols 44 by C-centered radical cyclization of highly functionalized 1,3-dicarbonyl compounds 42 with alkynes 43 by [4 + 2] intermolecular annulation (Scheme 12).62 The reaction was conducted in an undivided cell in the presence of Cp2Fe as a catalyst in a THF:EtOH (1:1) mixed solvent at a constant potential of 1.15 V vs. Ag/AgCl with NaOEt (30 mol%) as the base. Both the electron donating and electron withdrawing substituents were evaluated and all groups showed good yields (up to 84%). However, the electron donating substituents showed better yields than the electron withdrawing groups. From the cyclic voltammetry (Ag/AgCl) experiments, the oxidation potentials of 42a (1.27 V), the conjugate base 45 (0.59 V) and Cp2Fe (0.64 V) were obtained. In addition, the 42a curve together with Cp2Fe had practically no change in the potential compared to the Cp2Fe curve. However, the mixture of 42a, EtO− and Cp2Fe showed a significant increase, indicating that the electron transfer had occurred. In summary, it was observed that the reaction between Cp2Fe and 42a did not occur; however the reaction between the conjugated base 45 and Cp2Fe promotes the transformation efficiently. Based on the control experiments, the reaction mechanism was proposed. It was also possible to detect by HRMS the adducts of intermediate radicals 46 and 47 with TEMPO, proving the presence of these species in the medium (Scheme 13).
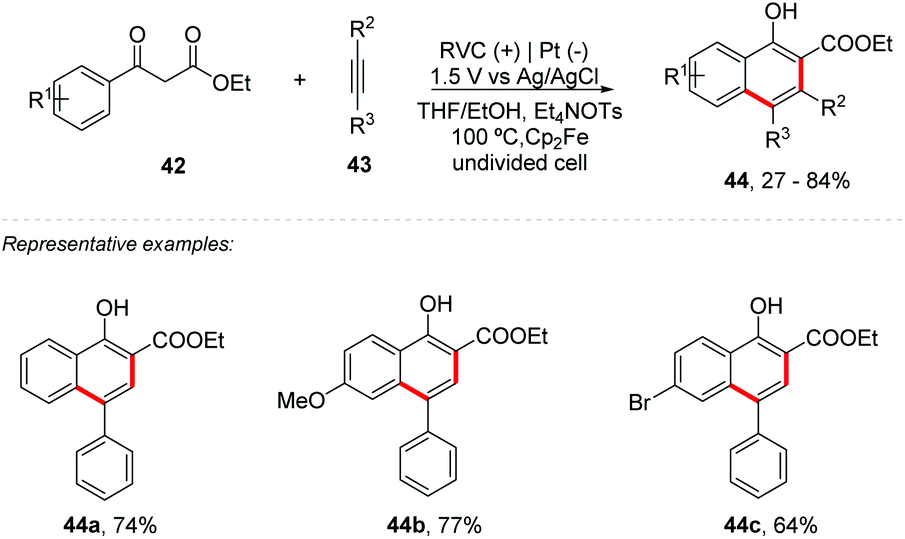 | ||
| Scheme 12 C-Centered radical cyclization of highly functionalized 1,3-dicarbonyl compounds with alkynes. | ||
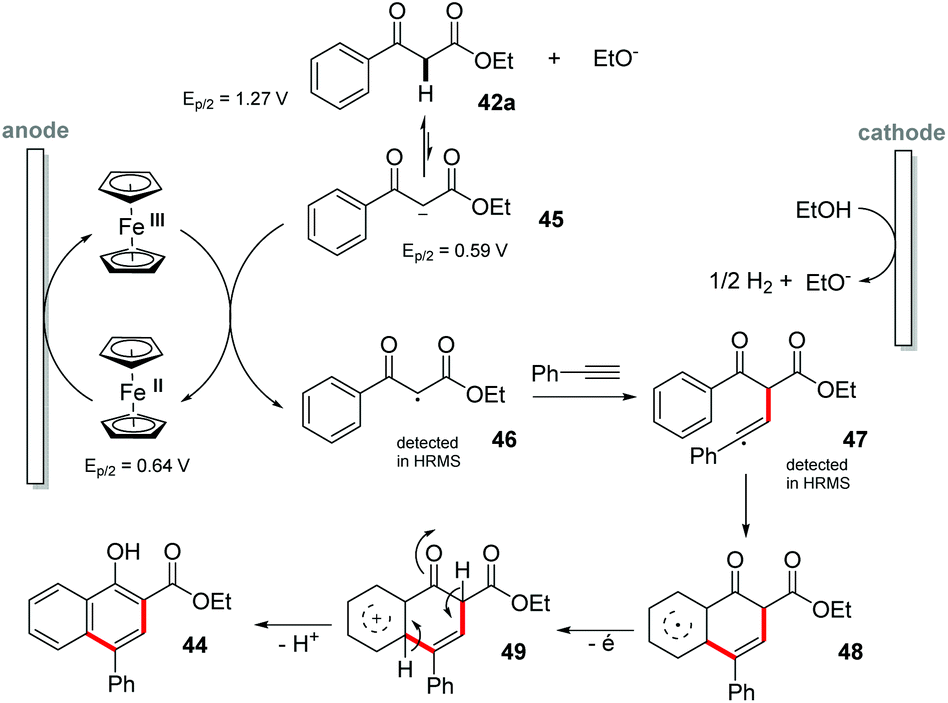 | ||
| Scheme 13 Proposed mechanism for electrochemical synthesis of 1-naphthols. | ||
In recent years, there has been an increasing number of reactions involving iodine as a mediator, often with equivalent or even better efficiency compared to those catalyzed by transition metals. Recently, Lei and co-workers published an overview of the recent development of iodine-mediated electrochemical coupling reactions.63 Zhang and co-workers developed an interesting study of I−, I2, and ICl via cyclic voltammetry.64 Likewise, Lei and co-workers proposed an iodine-promoted intramolecular dehydrogenative annulation of N-aryl enamine 50 for the synthesis of indole derivatives 51 (Scheme 14).65 A constant current of 7 mA in an undivided cell, with a Pt plate as an electrode, KI (0.15 M) as the electrolyte, and DMF:H2O (10:1) as a mixture of solvents at room temperature promoted the formation of products in yields of up to 96%. The use of 10% water showed a reaction improvement. It is noteworthy that KI not only works as an electrolyte, but also plays the role of a redox mediator. Electron-deficient and electron-rich N-phenyl enamines showed excellent yields; however the replacement of the carboxylic ester core by ketone was less efficient (51d). N-Naphthyl and N-pyridyl enamine derivatives were also fruitful, providing the respective products in good yields (51e–51f).
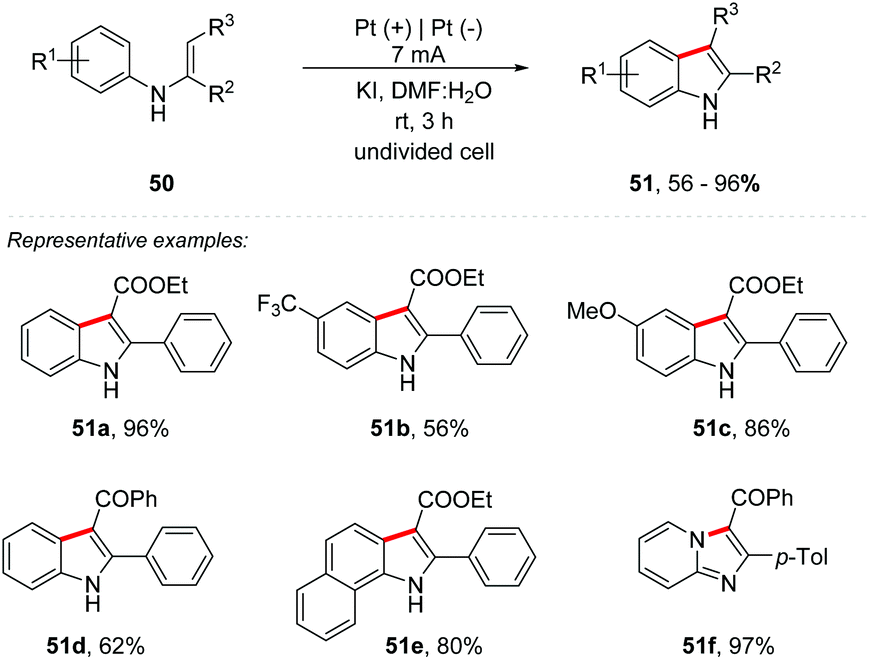 | ||
| Scheme 14 Intramolecular dehydrogenative C–H/C–H cross-coupling for the synthesis of indoles. | ||
Considering the importance of fluorinated molecules, several protocols for fluoroalkylation have been described, such as metallic catalysis66,67 or photoredox catalysis,68,69 and an interesting overview of fluorine in medicinal chemistry has been described by Gouverneur and co-workers.70 Recently, Tian and co-workers reported a tandem trifluoromethylation/cyclization of N-arylacrylamides with TfNHNHBoc to give 3-(β,β,β-trifluoroethyl)-oxindole derivatives.71 Additionally, Xu and co-workers have developed an unexplored ferrocene-mediated electrochemical alkyne difluoromethylarylation, using CF2HSO2NHNHBoc as a CF2H radical source, to provide derivatives of fluorinated dibenzazepines 53 in yields of up to 79% (Scheme 15).72 The reaction was set up in an undivided cell containing an RVC anode and platinum cathode electrodes (7.4 Fmol−1) with Cp2Fe (10 mol%) as a mediator, Na2HPO4 (2 equiv.) as the base, and n-Et4NBF4 (1 equiv.) as the electrolyte in MeOH as the solvent at 70 °C. Enyne 54 was subjected to functionalization and four new C–C bonds and three new rings were obtained, providing the hexacyclic product 55 in 71% yield.
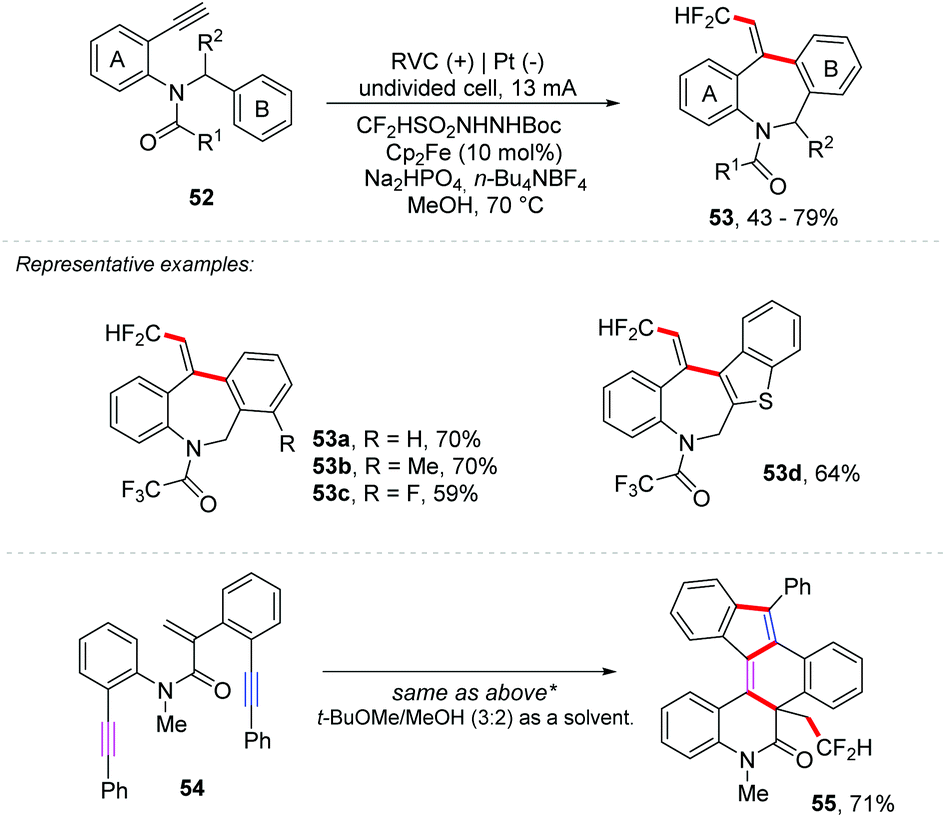 | ||
| Scheme 15 Electrochemical difluoromethylarylation of alkynes. | ||
In addition, Mo and co-workers proposed an Mn-mediated electrochemical strategy for the trifluoromethylation/C(sp2)–H functionalization using sodium trifluoromethanesulfinate (Langlois’ reagent) as the CF3 source (Scheme 16).73 Several oxindoles and related heterocycles containing a trifluoromethyl group, including a series of novel compounds, were obtained with yields of up to 83%. The authors adopted MnBr2 as a mediator and Pt as the electrode with a constant electric current of 10 mA for 6 h. H3PO4 (2 equiv.) was necessary as a sacrificial oxidizer; however the use of AcOH instead of H3PO4 showed practically identical results, and no electrolyte was needed. The Mn(III)–CF3 species is generated by anodic oxidation of Mn(II) in the presence of Langlois’ reagent. The CF3 radical species is added to an olefin and a transitory radical centered 59 is formed, which can be oxidized by the Mn(II) species or at the anode; the elimination of a proton re-aromatizes the ring 60 providing the product 58.
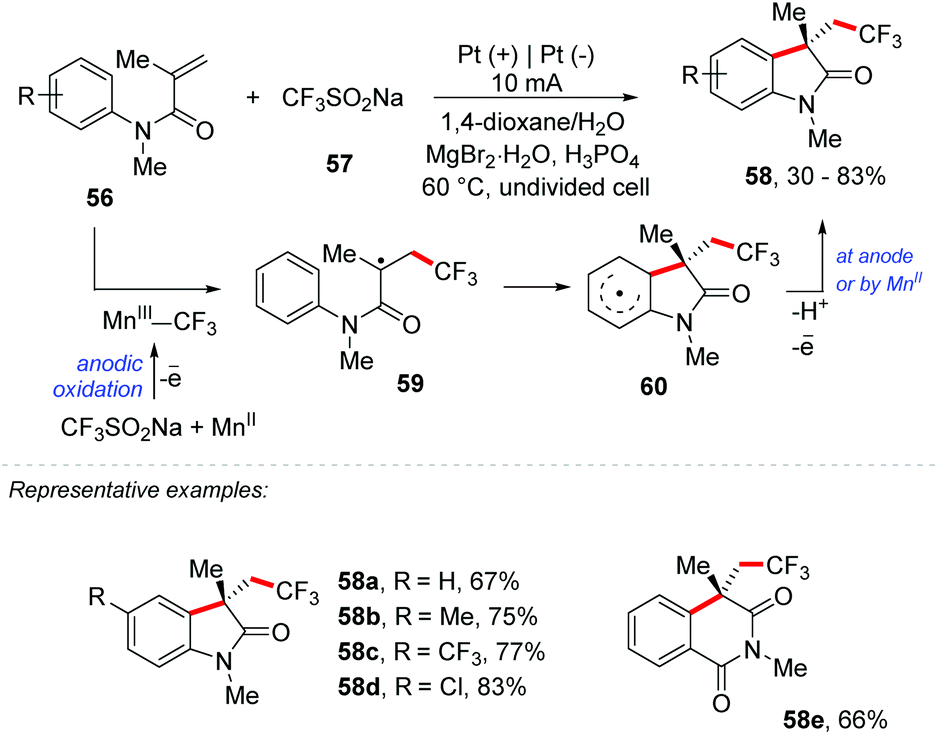 | ||
| Scheme 16 Mn-Mediated electrochemical strategy for the trifluoromethylation/C(sp2)–H functionalization. | ||
3. Mediator-free electrochemical annulation reactions
In the direct reactions, electron transfer occurs directly between the surface of the electrode and the organic substrate; in this context, all the essential properties of electrochemistry are completely applied, without any mediators. The main reaction region is the electrode surface itself, which can also occur close to the surface; however, the surface must have a large electric field, which is the differential of the common redox reactions in heterogeneous catalysts. The reactions on the electrodes occur in specifically exclusive fields, and the adsorption of the reagent/intermediate/product is a very important variable. Another very important factor is the choice of the electrode material, as the material is not only the interface for electron transfer, but also acts as an electrocatalyst.74 The electrode acts as a collector or source of electrons, and the choice of the electrode material is usually dictated by the heterogeneous electron transfer rate, redox potential and propensity to adsorb molecules from the solution. The choice of the electrode material has an influence on the selectivity and reactivity of electrosynthetic reactions, depending also on other experimental parameters, improving the reaction performance.75 In the following section, we will discuss some recent methodologies under conditions free of external mediators for electrochemical annulation reactions.3.1 C–Heteroatom and C–C bond formation
Recently, Li and co-workers proposed a synthesis of isoxazolidine-fused isoquinolin-1(2H)-one derivatives 62 by electrochemical-oxidation-induced intramolecular annulation via amidyl radicals under metal-additive- and external oxidant free conditions (Scheme 17).76 Ethanol as solvent in an undivided cell equipped with carbon felt electrodes afforded products in yields of up to 99%. As the electric current increased, the yield decreased due to the decomposition of the starting material and the final product, with the best-established condition being 2 mA (3 Fmol−1) at reflux temperature under an inert atmosphere. A wide reaction scope was achieved, being efficient for both electron-withdrawing and electron-donating groups; however, meta-substituted benzamides gave product mixtures, being selective for the less hindered position of the aromatic ring. For ortho-substituted benzamides there was no inhibitory effect, and the products were obtained with excellent yields. In the cyclic voltammetry experiments, a 1.5 V vs. SCE oxidation potential for ethanol was observed, as well as a 0.8 V oxidation potential for benzamide 62a. | ||
| Scheme 17 C–H/N–H functionalization. | ||
Based on the control experiments, the authors suggest that the mechanism begins with a cathodic formation of EtO− (Scheme 18); deprotonation of amide 61 provides anion 63, undergoing single-electron-transfer (SET) anodic oxidation to afford the N-centered radical 64. 5-exo-dig annulation delivers radical 65, which proceeds for a second annulation 66. In the end, a re-aromatization affords the product 62.
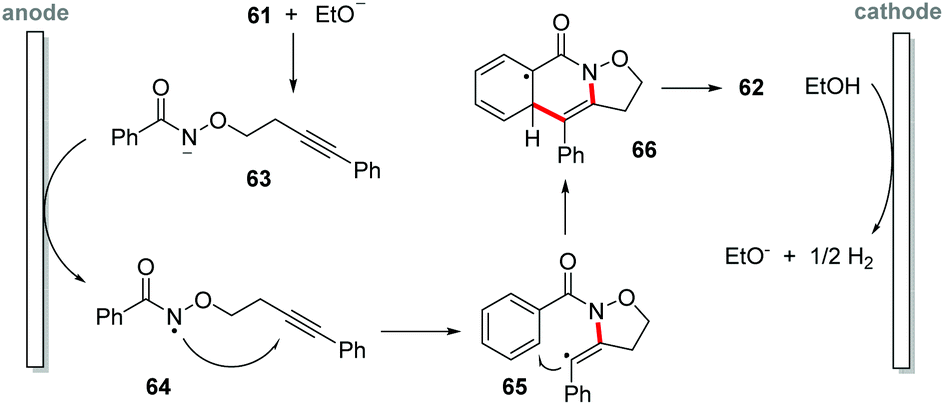 | ||
| Scheme 18 Proposed mechanism for C–H/N–H functionalization. | ||
Compared to C–N bond formation, it is observed that obtaining new C–O bonds has been less explored. However, C–H/O–H functionalization has been efficiently applied in the construction of O-heterocycles, such as benzofurans, benzoxazoles, benzofuranones, oxazolines and others, being of great interest in the medicinal field due to the several biological functions present.77 Recently, Lei and co-workers described an annulation reaction for the preparation of indolines78 and polysubstituted pyrroles.79 Likewise, the author proposed an electrooxidative [3 + 2] annulation using activated phenols 67 with N-acetylindoles 68 under undivided electrolytic conditions with no external oxidant and catalyst, providing the respective benzofuro[3,2]indolines 69 in up to 99% yield (Scheme 19).80 Both 3-substituted N-acetylindoles and 2-substituted N-acetylindoles were suitable for this transformation. 2,3-Disubstituted N-acetylindoles are also efficient in providing excellent yields (69f). With a 6:4 solvent mixture of HFIP:DCM, the product 69a was obtained in 99% yield. Interestingly, using only HFIP as a solvent, the yield decreased from 99 to 78%. Note the importance of HFIP as a co-solvent, probably due to its ability to stabilize aromatic radical cations, as reported by Eberson and co-workers.81–83 Cyclic voltammetry was performed, and the oxidation potential of p-methoxyphenol was close to that of 3-methyl-N-acetylindole; in this way, oxidation of both was possible. A trapping experiment with triethyl phosphite was conducted and no desired products could be observed.
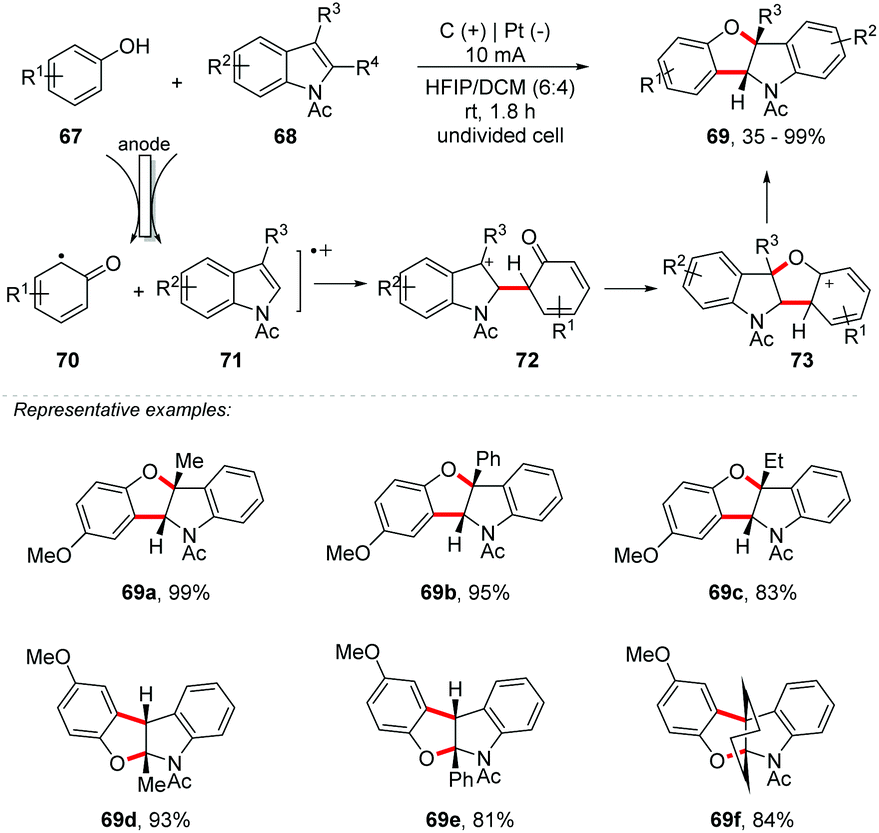 | ||
| Scheme 19 Synthesis of benzofuro[3,2]indolines. | ||
Masson and co-workers developed an electrochemical intramolecular oxytrifluoromethylation of N-tethered alkenyl alcohols to access morpholine derivatives 76 (Scheme 20).84 The authors applied mild reaction conditions with direct anodic oxidation of Langlois’ reagent (CF3SO2Na) 75 as a cheap and easy method to handle the trifluoromethylation reagent. The reaction was performed from alken-6-ol 74 with 75 in an undivided cell, using a graphite carbon anode and a nickel cathode plate as electrodes under a constant current electrolysis of 15 mA, with LiClO4 (0.2 M) as the electrolyte and MeCN as the solvent, 3.2 Fmol−1 at room temperature. Additional experiments have shown that a change of the electrolyte or current intensity (from 15 to 30 mA) leads to a significant decrease in yields. A set of representative N-tethered alken-6-ols with various aryl groups on the alkene proved to be suitable substrates, and morpholine derivatives were obtained in good yields, up to 88%. The N-2-nitrophenylsulfonyl substrates underwent the oxytrifluoromethylation reaction with equal efficiency (76b). Pleasingly, secondary alcohol was well tolerated furnishing the morpholine 76c in good yield as a mixture of diastereoisomers. This condition was found to be efficient in the formation of a seven-membered oxacycle by intramolecular oxytrifluoromethylation of N-tethered alken-7-ol (76d).
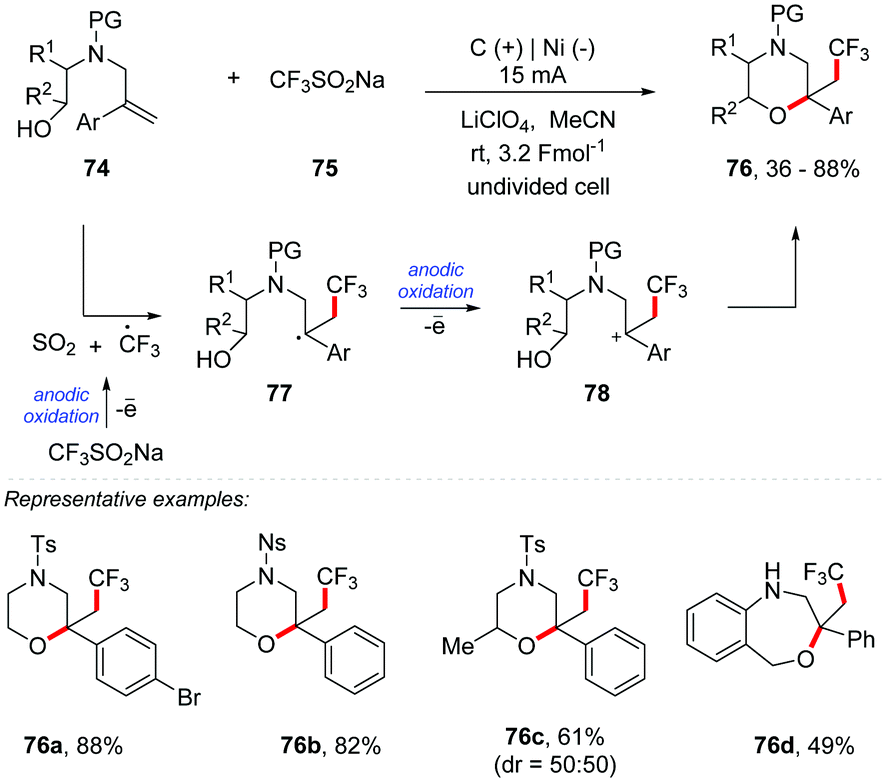 | ||
| Scheme 20 Intramolecular trifluoromethylation of N-tethered alken-6-ols and alken-7-ols. | ||
Liao and co-workers developed an electrochemical strategy for a trifluoromethylation/SO2 insertion/cyclization process with Langlois’ reagent as a source of CF3 and SO2, promoting the synthesis of trifluoromethylated cyclic N-sulfonylimines 81 (Scheme 21). Two new bonds (C–S/S–N), two new C–C bonds and two new rings were formed in a single step. Cyanamides substituted in ortho and para positions were well tolerated (yields of up to 62%); however the meta-substituted analogue 81b provided a mixture of regioisomers (1.2:1) with yields of up to 47%. Heterocyclic scaffolds such as indoles and naphthyls were fruitful with high regioselectivity and yields of up to 47%. N-Aryl-cyanamides with disubstituted alkenes were also evaluated, providing products with moderate yields (32–40%).
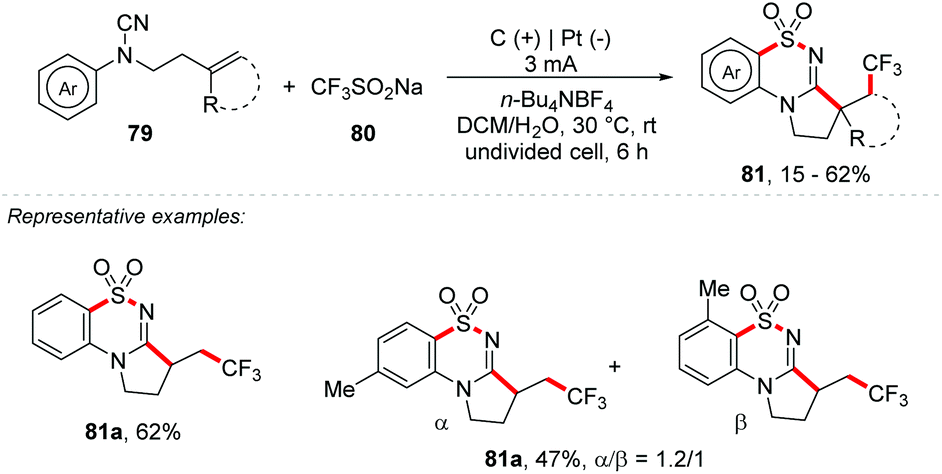 | ||
| Scheme 21 Trifluoromethylation/SO2 insertion/cyclization process with Langlois’ reagent as a source of CF3 and SO2. | ||
Xu and co-workers developed an electrochemical fluoromethylation triggered lactonization of alkenes 82 using CF2HSO2Na and CF3SO2Na as the reagents (Scheme 22).85 The reaction was performed under a constant current electrolysis of 6.7 mA in an undivided cell with a Pt plate as an electrode, MeCN:H2O (7:1) as a mixture of solvents, and HOAC (3 equiv.) as an additive at room temperature for 3 h. Both CF2H- and CF3-containing lactone products (84) were obtained in yields up to 93%. The mechanism was studied by cyclic voltammetry experiments. CF2HSO2Na and CF3SO2Na showed oxidation potentials of 0.72 V and 1.06 V (vs. Ag/AgCl), respectively. The oxidation potential of alkenes was 1.58 V (vs. Ag/AgCl). These results indicated that CF2HSO2Na and CF3SO2Na are much easier to be electrochemically oxidized to generate fluoromethyl radicals than the alkene moiety.
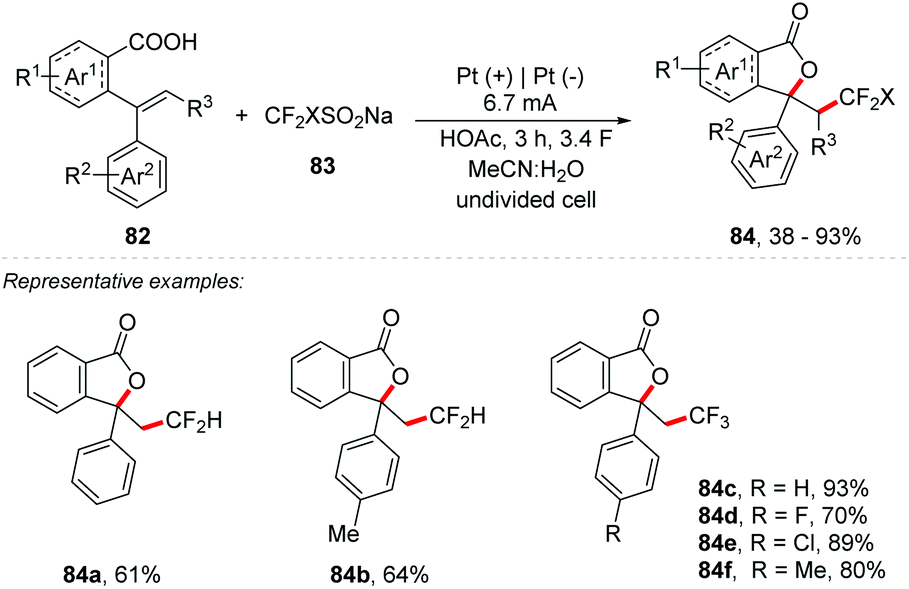 | ||
| Scheme 22 Electrochemical carboxydifluoromethylation. | ||
Shi and co-workers proposed a synthesis of benzofuran derivatives 87via electrochemical cross-dehydrogenative coupling between phenols 85 and β-dicarbonyl compounds 86 (Scheme 23).86 The reaction proved to be chemoselective, without by-products from homocoupling reactions. Interestingly, HFIP played a crucial role in the reaction, and according to the authors it has a fundamental importance in the coupling stage, also facilitating the anodic current, which helps in the oxidation of phenol. A crucial step is the anodic oxidation of phenol, which provides the phenoxonium cation, being isomerized to the oxonium ion, a reactive Michael acceptor, which reacts with the nucleophilic species β-dicarbonyl compounds 86. Intermolecular condensation delivers benzofurans 87. It is worth mentioning that according to the cyclic voltammetry results, the authors determined that the phenol must be more easily oxidized than the enol from diketone.
 | ||
| Scheme 23 Synthesis of benzofuran derivatives via electrochemical cross-dehydrogenative coupling. | ||
An interesting approach proposed by Guo and co-workers describes an electrochemical oxidative cyclization of activated alkynes to access functionalized coumarins and quinolinones with selenium or sulphur, reaching products 90 in yields of up to 86% (Scheme 24).87 The reaction was completely inhibited with the addition of TEMPO, verifying that the mechanistic route follows a radical pathway. The oxidation peak of diphenyl diselenide and diphenyl disulfide was determined by cyclic voltammetry with values of 1.47 V and 1.76 V, respectively (vs. Ag/AgCl). The phenyl-3-phenylpropiolate 88a showed a peak of oxidation at 2.43 V (vs. Ag/AgCl). It should be noted that the reactions with disulfides must be carried out under an inert atmosphere, to minimize oxidation. As part of the group's interest, the same authors developed an intriguing electrochemical route for the synthesis of spiro[4.5]trienone derivatives through radical-initiated dearomative spirocyclization of alkynes with diselenides (Scheme 25).88 The products were obtained in an undivided cell equipped with graphite at the anode and platinum at the cathode, with a solvent mixture of MeCN:HFIP (3:1) using n-Bu4NPF6 as the electrolyte under a constant current of 15 mA at room temperature. The scope was evaluated by varying the electron donating and withdrawing substituents, as well as different alkyl diselenides, with diphenyl ditellurides also being efficient, delivering the products 94 in yields of up to 86%. It is important to use the derivative of 92 with a methoxy group at position 4, which will be responsible for oxidizing the ring and forming the product with a cyclohexa-2,5-dien-1-one scaffold. Additionally, in 2013, Wang and co-workers proposed a route to obtain a tricyclic spiro dihydrofuran scaffold via multi-component electrochemical reaction of dimedone with aryl aldehyde.89
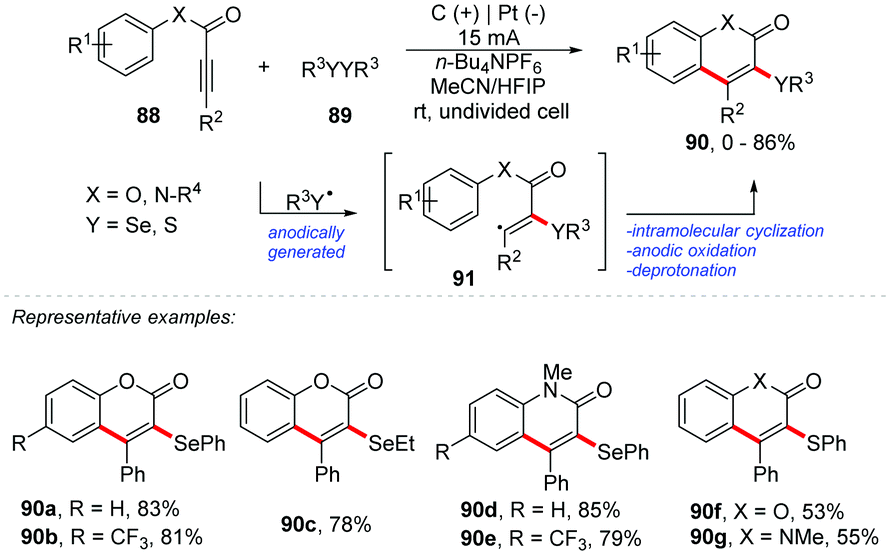 | ||
| Scheme 24 Cyclization of activated alkynes with diselenides or disulfides. | ||
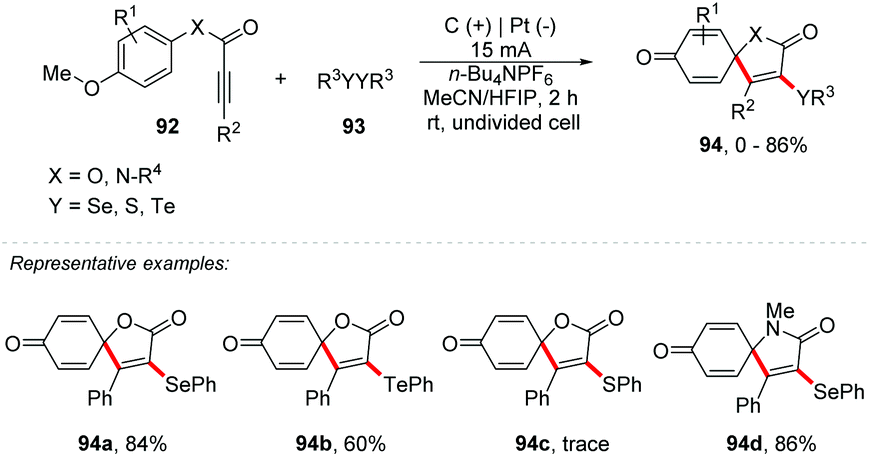 | ||
| Scheme 25 Electrochemical synthesis of spiro[4.5]trienones. | ||
Very recently, Martins and co-workers published an overview with recent advances in electrochemical chalcogen (S/Se)-functionalization of organic molecules, and more information about reactions involving chalcogens in electrochemistry can be found.90
3.2 C–Heteroatom bond formation
The oxidative functionalization of C–heteroatom represents one of the most powerful strategies to achieve organic compounds and access target molecular structures.91 Lei and Stahl groups proposed an Hofmann–Löffler–Freytag procedure for C(sp3)–H amination.92,93 Harran and co-workers developed an electrochemical and a hypervalent iodine(III) route for the direct synthesis of benzofuro[2,3-b]indoline through the oxidative coupling of indoles and phenols.94,95 In addition, Vincent and co-workers performed the synthesis of benzofuro[2,3-b]indoline derivatives by [3 + 2] oxidative coupling between nucleophilic phenols and indoles with NIS as an oxidant.96 There are many reports on cross-coupling and difunctionalization of alkenes by electrochemical methods, and good reviews have recently been published.97,98Lei and co-workers proposed an exciting method for a dearomative annulation of indoles and benzofurans under electrochemical conditions, providing several highly functionalized five to eight-membered heterocycle-2,3-fused indolines and dihydrobenzofurans 97 (Scheme 26).99 Desired [3 + 2], [4 + 2], [5 + 2] and [6 + 2] annulation occurred regioselectively with nucleophilic species of N, O, and S in yields up to 88%. The mechanism was evaluated through cyclic voltammetry, electron paramagnetic resonance (EPR), radical capture experiments and kinetic studies. Considering these results, the authors proposed that the reaction proceeds through the cross-coupling between the radical of the nucleophilic species and the indole radical cation 98 at the C-3 position, considering that the radical species is stabilized by the aromatic ring. The second nucleophilic attack would take place in the imine C-2 formed in the five-membered indole ring 99, delivering the final product 97.
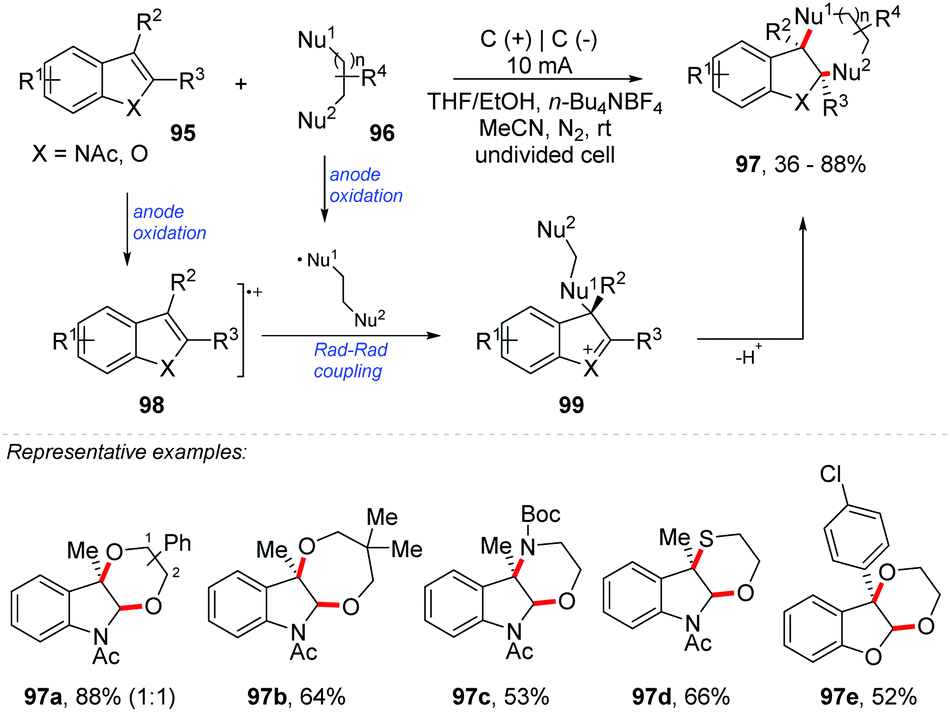 | ||
| Scheme 26 Dearomative annulation of indoles and benzofurans. | ||
Based on their experience, the same authors also developed an electrochemical intermolecular dearomative [4 + 2] annulation between 1-alkylindole 100 and indole-1H-carboxamide 101 to obtain pyrimido[5,4-b]indole derivatives 102, with many biological and pharmaceutical applications (Scheme 27).100
 | ||
| Scheme 27 Intermolecular dearomative [4 + 2] annulation between indole-1H-carboxamide and 1-alkylindole. | ||
An important protocol was developed by Tong and co-workers, using KX/oxone for halo-oxidative cyclization (Br/Cl) of tryptamine/tryptophan derivatives (78–95% yield), also proposing a concise total synthesis of cyclo-tryptamine alkaloid protubonines A and B, which was efficiently completed in 7 steps with 29% overall yield.101 Likewise, Vincent and co-workers reported a clean and simple approach via electrosynthesis, being environmentally friendly and efficient for bromination of tryptophan, tryptamine and tryptophol derivatives 103 (Scheme 28).102 The electrochemical reactions were performed in the ElectraSyn 2.0 package (IKA) with a platinum electrode at the cathode and a graphite electrode at the anode. The electrophilic bromine reagent was generated in situ by the electrochemical oxidation of MgBr2, and no toxic by-products were obtained, avoiding an additional electrolyte. Replacing MgBr2 with MgCl2 delivered the equivalent of 104b in just 37% yield. It is worth mentioning that the same authors developed an electrochemical approach to perform a general oxidative and dearomative difunctionalization of indoles with two C–O or two C–N new bonds.103
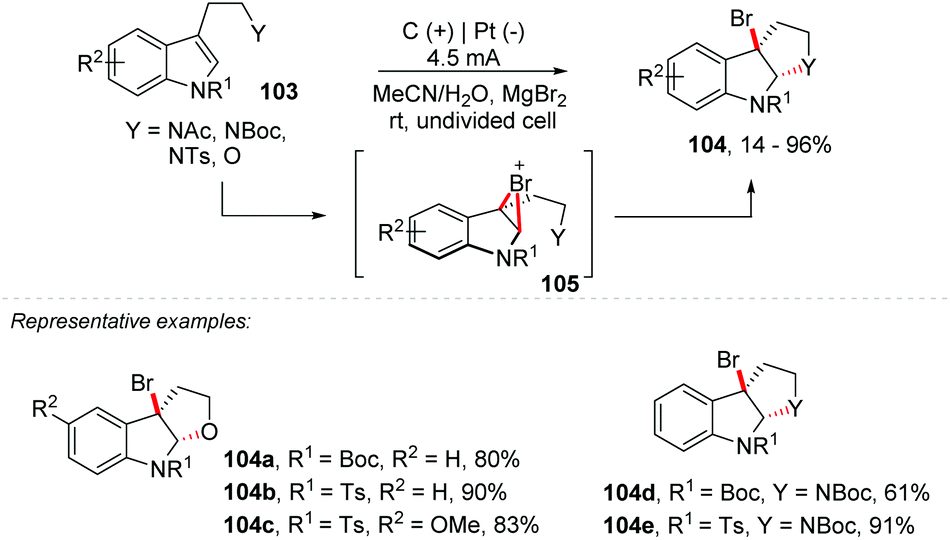 | ||
| Scheme 28 Dearomative annulation of indoles. | ||
Recently, Ruan and co-workers proposed an elegant electrolytic route for the synthesis of medium-sized lactams 107 (8–11-membered rings) (Scheme 29).104 The reaction followed a rare migration of amidyl radicals by C–C bond cleavage, delivering products in up to 98% yield. The reaction was carried out in an undivided cell equipped with a graphite electrode at the anode and a platinum electrode at the cathode, in a mixture of MeCN:H2O (9:1) as the solvent and n-Bu4NBF4 (1 equiv.) as the electrolyte, under a current of 8 mA at room temperature in an open system. The reaction scope was evaluated and the electron donating and withdrawing groups proved to be efficient for the transformation; however some substrates containing azacycles showed less efficiency (107d). The mechanism was suggested to be anodic oxidation of 106, leading to the radical amidyl 108, in which intramolecular cyclization could proceed with the ring to form N–C bonds. The selective cleavage of the C–C bond followed by single-electron oxidation would deliver products 107. From the cyclic voltammetry experiments, the authors observed that the potential of 106 (2.2 V vs. Ag/AgCl) was below that of 107 (2.5 V vs. Ag/Cl), protecting the possible degradation.
 | ||
| Scheme 29 Synthesis of medium-sized lactams (5–11-membered rings). | ||
Zhang and co-workers reported a dehydrogenerative C–N cross-coupling reaction under electrochemical conditions for the synthesis of 1,2,4-triazole-fused heterocycles 111 (Scheme 30).105 The method proved to be efficient for a wide variety of substrates with yields of up to 94%, tolerating groups such as halide, CF3, CO2Me, nitrile, amine, and alkyl groups, and is also efficient for heterocyclic portions such as furans, indoles, pyridines and thiazoles. The undivided cell was equipped with a graphite electrode at the anode and a platinum electrode at the cathode with a mixture of MeCN:H2O (9:1) as the solvent and n-Bu4NBF4 as the electrolyte, under a constant current of 10 mA.
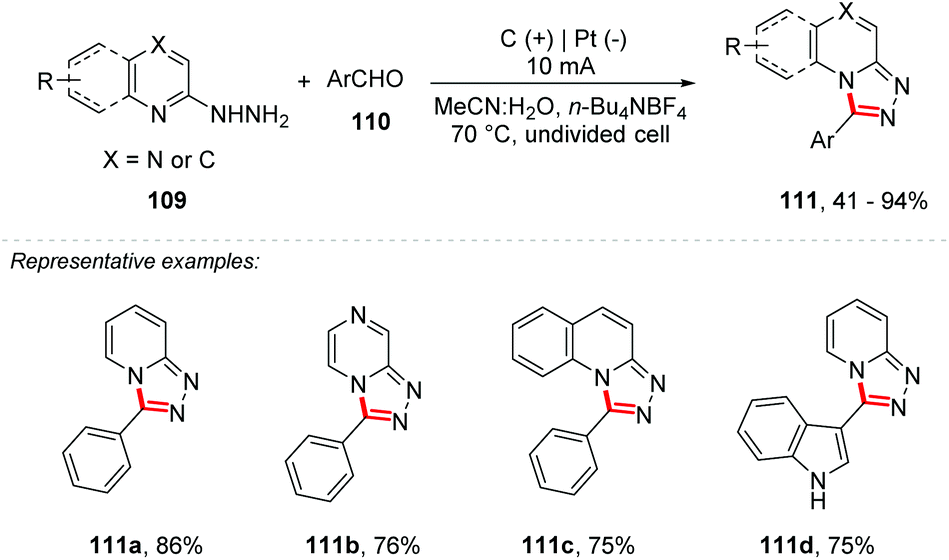 | ||
| Scheme 30 Cyclization of activated alkynes with diselenides or disulfides. | ||
Among different approaches, the direct C–H/O–H oxidative coupling reaction gained special attention due to its high atom economy. Recently, Prabhu and co-workers reported a cascade reaction involving the activation of C–H/O–H bonds for regioselective oxidative ringing [4 + 2] and concomitant lactonization with carboxylic acids.106 Similarly, Yatham and co-workers developed the first photocatalyzed dehydrogenative lactonization of 2-arylbenzoic acids by CeCl3.107 Palladium and copper catalysts have also been developed for C–H/O–H activation.108,109 However, these methods make industrial use unfeasible due to their high cost. Additionally, the treatment of carboxylic acids with a base generates carboxylate anions, which can undergo anodic oxidation to provide carboxylate radicals, which could be added to alkenes to produce lactones. This suggests that lactonization may occur if the C–O coupling is fast enough to overcome Kolbe's decarboxylation.110,111 If 1,5-hydrogen-atom-transfer (HAT) makes remote C–H homolytic cleavage possible, the lactonization would be achieved. Considering this, Xu and co-workers described an electrochemical dehydrogenative lactonization of carboxylic acids 112 with C(sp2/sp3)–H bonds (Scheme 31).112 The procedure required n-Bu4NOAc as the electrolyte in a mixture of MeCN:MeOH (9:1) with platinum electrodes under a current of 13.3 mA cm−2. It exhibited a broad substrate scope, and high regioselectivity under conditions free of external oxidants. Scalability was assessed with 40 g of 112a, demonstrating its potential in industrial applications (113a – 84% yield). From cyclic voltammetry experiments, it was observed that the carboxylate moiety has an oxidation potential of 1.4 V vs. Ag/AgCl, while the aromatic moiety has an oxidation potential above 2.0 V vs. Ag/AgCl, indicating that the carboxylate moiety is oxidized preferentially, being consistent with radical trapping experiments.
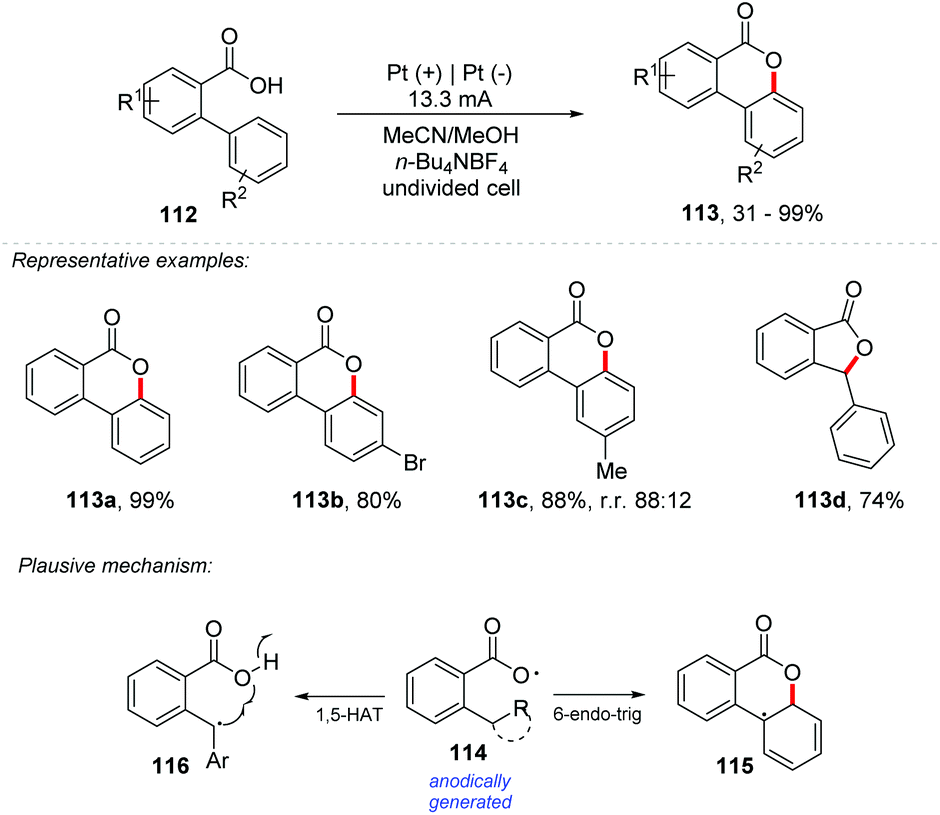 | ||
| Scheme 31 Lactonization of C(sp2/sp3)–H bonds. | ||
Lei and co-workers reported an electrochemical oxidative cascade cyclization of olefinic carbonyls with diselenides for C–O and C–Se bond formation (Scheme 32).113 A series of functionalized dihydrofurans and oxazolines 119 were synthesized. Several diaryl diselenides were employed, providing products in good yields (up to 93%). It is important to mention that the reaction was well tolerated with dimethyl and dibenzyl diselenides. Olefinic carbonyls containing γ,δ-substituted groups were also efficient. The steric effect had a great influence on the reaction (Scheme 32 – 119c). To explore the mechanism, control experiments were conducted under different conditions, and only traces of 119a were detected when 2 equivalents of TEMPO were added. According to cyclic voltammetry analysis, oxidation and reduction peaks of diphenyl diselenide were observed at 1.8 V and −1.5 V vs. Ag/AgCl, respectively. Thus, considering the control reactions and the results reported in the literature,114,115 the authors suggest a mechanism route via a radical intermediate. Diphenyl diselenide is reduced at the cathode to give an anion radical as an intermediate, which is further decomposed to give the phenyl-selenium radical and phenyl-selenium anion.
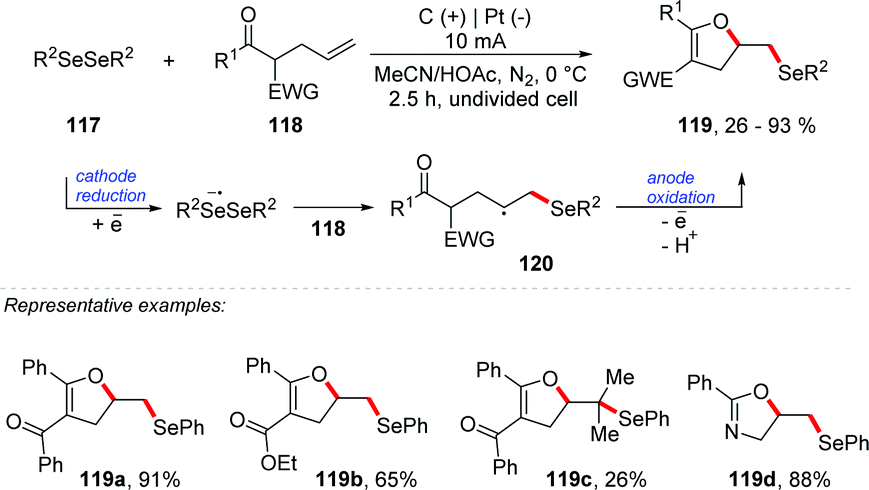 | ||
| Scheme 32 Cyclization of olefinic carbonyls with diselenides. | ||
In addition, an electrochemical synthesis of selenyl-dihydrofurans via anodic selenofunctionalization of allyl-naphthol/phenol derivatives was proposed by Braga and co-workers.116 Siewert and co-workers developed an anodic amination and esterification reaction of non-activated alkenes catalyzed by diselenides.117 Torii and co-workers described the first procedure involving selenium-based catalysts in electrosynthesis.118 Wirth and co-workers re-studied Torri's reaction, applying the electrochemical sequence of selenenylation/elimination reactions using catalytic amounts of diphenyl diselenide.119 It is worth mentioning that in the review paper by Breder and co-workers, the authors describe the recent oxidative alkene functionalization via selenium-π-acid catalysis.120
3.3 C–C bond formation
In the last century, we have seen huge advances in the construction of new C–C bonds by radical reactions. Due to its high reactivity and practical tolerance, it provided motivation for the development of new synthetic methodologies.121 Currently, several methods for obtaining radical carbons are known, e.g. using halides, carboxylic acids, borates, alcohols, and silicates, metal catalysis, photoredox catalysis, ultrasound, microwaves, and electrochemistry, among others.21,122–130 In the same way, the construction of functionalized ring systems through an oxidative annulation process is possible. An example of this was proposed by Wright and co-workers, who developed a two-stage electrochemical annulation for fused furan products 124 (Scheme 33).131 After the conjugated addition of a furyethyl cuprate and the capture of the enolate as the corresponding silyl enol ether, the annulation step involves the anodic coupling of furan and silyl enol ether (123), providing the six-membered ring. High current densities may promote the formation of oligomers by providing other electroactive species.132 With relatively low current densities (0.5–1 mA cm−2) the amount of charge required for complete consumption of the enol ether was almost the same as the theoretical amount (2 Fmol−1). However, with a high current density (>1 mA cm−2), the amount of charge consumed was greater than the theoretical amount. | ||
| Scheme 33 Preparation of fused furans. | ||
Lei and co-workers proposed an electrochemical oxidative [4 + 2] annulation between alkenes and tertiary anilines for the synthesis of tetrahydroquinoline derivatives 128 (yields of up to 72%) (Scheme 34).133 The undivided cell was equipped with platinum electrodes, n-Bu4NBF4 as the electrolyte, and MeCN:AcOH (4:1) as a solvent mixture under a constant current (3–5 mA) at room temperature. The methodology presented a wide reaction scope, both for electron donating groups and for electron withdrawing groups. Additionally, heterocyclic enamines were well tolerated (naphthyl, thiophene, tetrahydrofuran, pyridine, and pyrrolidinone). The authors propose that the mechanism involves anodic oxidation of 126 to generate the radical cation stabilized by acetic acid, which after deprotonation would deliver the tertiary α-amino carbon radical 129. This radical would react with 127via radical addition providing 130, followed by intramolecular cyclization. Anodic oxidation of 131 delivers product 128.
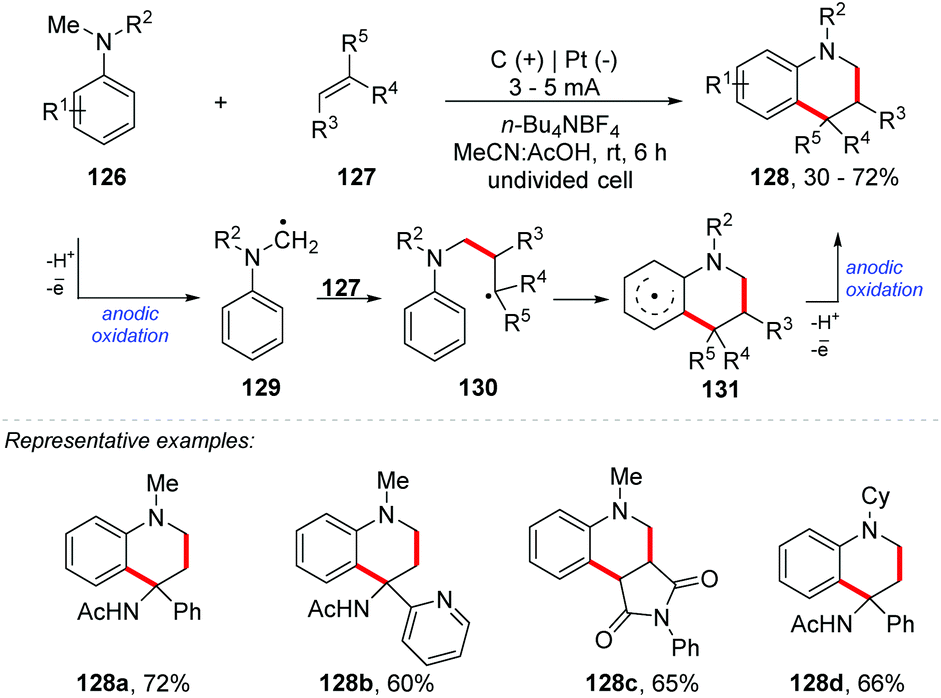 | ||
| Scheme 34 Electrochemical oxidative [4 + 2] annulation between alkenes and tertiary anilines. | ||
3.4 Heteroatom–heteroatom bond formation
There are several methods for reactions for the formation of new C–C and C–Het bonds; however the formation of new Het–Het bonds remains statistically less explored. This topic was recently discussed by Antonchick and co-workers in the book Patai's Chemistry of Functional Groups, highlighting reactions mediated by hypervalent iodine.134 Compounds containing Het–Het bonds (Het = N, S, O, P) have already been found and isolated from a variety of natural products, and their biological activities and reactivities have attracted the attention of chemists and chemical biologists.135Xu and co-workers report an oxidizing reagent- and transition metal-free synthesis of [1,2,3]triazolo[1,5-a] pyridines 133 through electrochemical dehydrogenative cyclization of pyridyl hydrazones 134 (Scheme 35).136 The reaction was carried out in two stages: first the hydrazine 132 was obtained by refluxing the respective ketone with hydrazine in MeOH in the presence of AcOH (0.1 equiv.). After complete conversion, the n-Et4NPF6 electrolyte, water, and K2CO3 (1.0 equiv.) were added, equipping the undivided cell with an RVC electrode at the anode and Pt at the cathode.
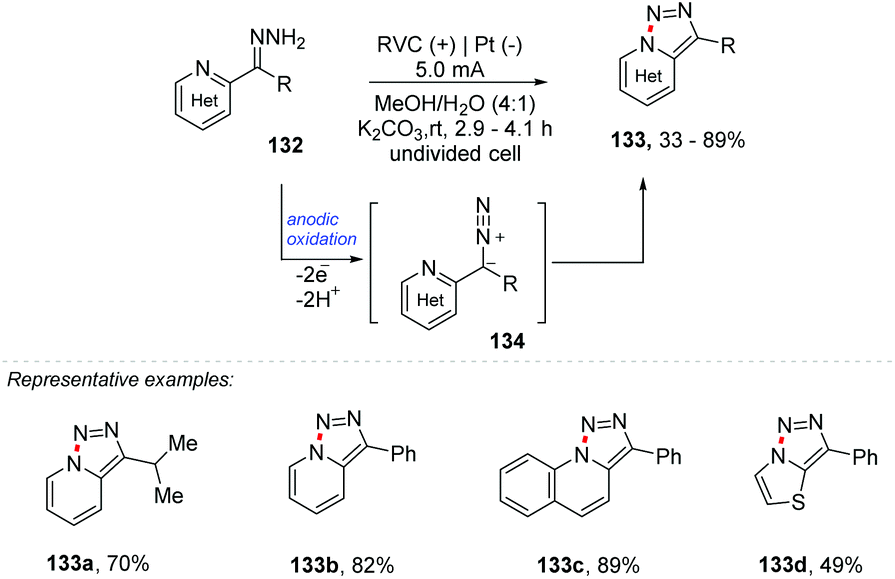 | ||
| Scheme 35 Cyclization of pyridyl hydrazones. | ||
Another important heterocycle is the phthalazin-1,4-dione derivatives. This skeleton is part of a range of drugs with anti-bacterial, anti-inflammatory, anti-convulsant and analgesic properties, and has been applied as an anesthetic in the past.137–141 So far, methods for the synthesis of these derivatives involved the use of metals, such as Ni, Rh, Pd, Cu, and others.142–145 Electrosynthetic methods for the formation of N–N bonds have recently been explored by the Waldvogel and Baran group.146–151 Considering this, Waldvogel and co-workers proposed the synthesis of phthalazin-1,4-diones 136 from phthaldianilides 135via dehydrogenative N–N bond formation (Scheme 36).152 The undivided cell was equipped with graphite electrodes at the anode and platinum at the cathode with HFIP as the solvent and only 0.01 M n-Bu4NPF6, under a constant current of 3 mA. The method proved to be efficient for a wide variety of substrates with yields of up to 89%, tolerating substituent groups such as halide, triflate, nitrile, and alkyl groups, and being also efficient for heterocyclic portions such as pyridine 136c. The authors consider the formation of amidyl radicals as intermediates, which would be stabilized by the HFIP solvent.
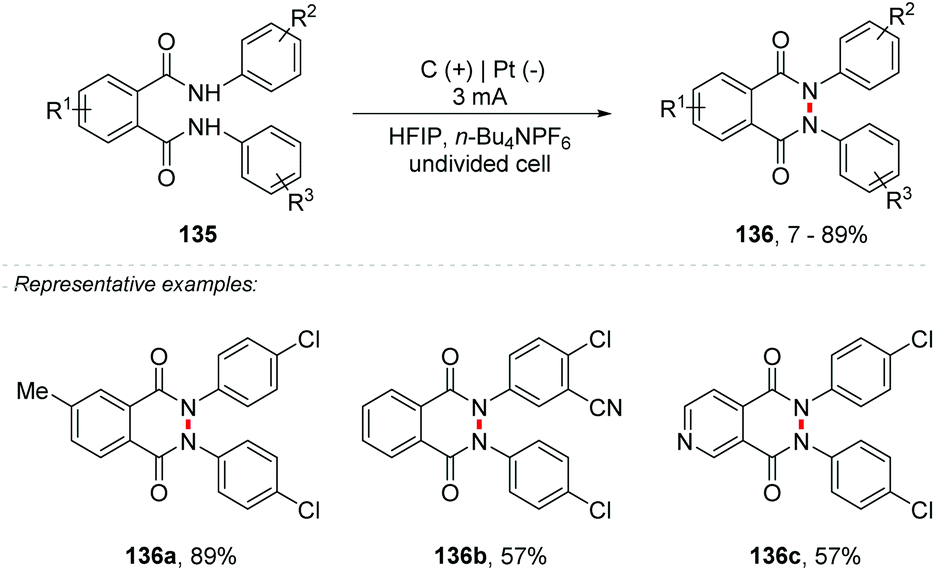 | ||
| Scheme 36 Synthesis of phthalazin-1,4-diones from phthaldianilides via dehydrogenative N–N bond formation. | ||
Waldvogel and co-workers also reported an electrochemical approach for S–S coupling of dithioanilines 137, providing the cyclic 3,5-diimido-1,2-dithiolane derivatives 138 with yields of up to 89%, tolerating various groups of substituents (Scheme 37).153 In 2016, the same authors proposed a method for access to pyrazolidin-3,5-diones through anodic N–N bond formation.150 Additionally, in 2018, the group developed an electrochemical conversion of phthaldianilides to phthalazin-1,4-diones by dehydrogenative N–N bond formation.152
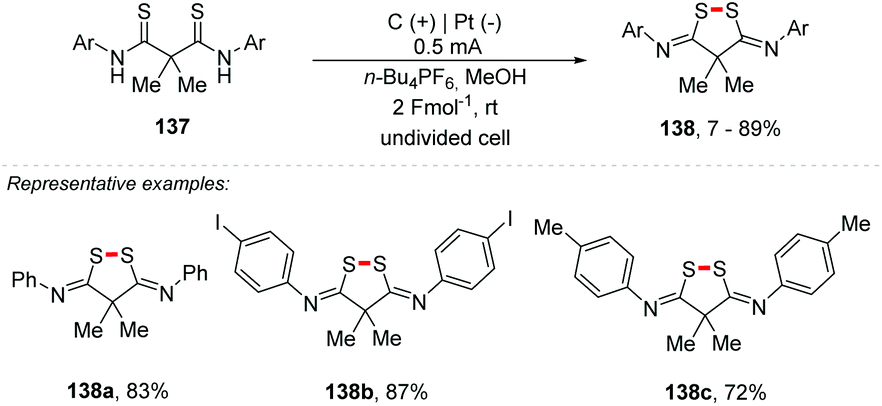 | ||
| Scheme 37 S–S coupling of dithioanilines. | ||
Additionally, Waldvogel and co-workers considering their experience, as well as recent publications proposing the electrochemical reduction of aryl-nitro derivatives,154 nitrones155 and the synthesis of heterocycles containing nitrogen,148,152,156 proposed the synthesis of 2,1-benzisoxazoles (140a–c) and quinoline N-oxides (141a–b) through cathodic reduction of the nitro portion of o-nitrobenzaldehyde and o-nitrocinnamaldehyde, with subsequent intramolecular cyclization (Scheme 38).157 Using glassy carbon at the anode and Boron Doped Diamond (BDD) at the cathode as electrodes in a mixture of HFIP:H2O (1:1) under a current of 2.4 mA it was possible to obtain the products in good yields (29–70%). The use of HFIP is crucial for the total conversion, avoiding the need for additives, and can be recovered at the end. It highlights that the addition of 4% (v/v) acetone facilitated the dissolution of the substrate. Using the theoretical amount of charge (4 Fmol−1) the yield was low, requiring up to 7 Fmol−1. The authors explain this restriction due to the less favored condensation between the hydroxylamine and the carbonyl moiety of the enal, because of the alkene conjugation, and if the condensation is not fast enough, there may be a re-oxidation of the hydroxylamine to nitrous species. Other electrosynthetic methodologies for obtaining 2,1-benzisoxazoles have been reported.158–160
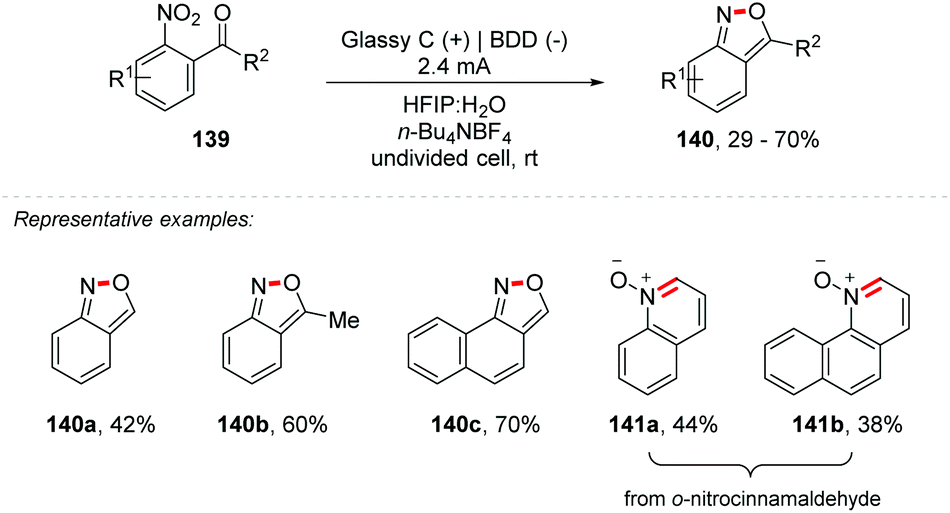 | ||
| Scheme 38 Synthesis of 2,1-benzisoxazoles and quinoline N-oxides. | ||
Recently, oxidative N–S bond formation has been established with catalysis by copper,161 I2,162,163 hypervalent iodine,164 and DCC in the presence of KF/Al2O3 with thiourea, etc.165 It is free of catalysts via the use of elemental sulfur with amidines166 and hydrazones,167 and dimerization of thioamides168 or thiobenzamides with methyl bromocyanoacetate.169 Yang and co-workers describe a facile and efficient protocol for the synthesis of 3-substituted 5-amino-1,2,4-thiadiazoles 143 that avoids the use of metal catalysts and stoichiometric oxidants (Scheme 39).170 The thiadiazoles were obtained via electrochemical oxidative intramolecular dehydrogenative N–S bond formation with imidoyl thioureas 142, in yields of up to 92%. The reaction was carried out using various imidoyl thioureas in the presence of MeCN at room temperature, and an undivided cell was equipped with a carbon rod anode and a Pt plate cathode under a 10 mA constant current using 0.03 M n-Bu4NBF4 as the electrolyte. Importantly, alkyls of imidoyl thioureas were tolerated in this reaction and the corresponding products were obtained in good yields (143c and 143f). The plausible mechanism was proposed and the formation of the radical cation 144 was supported by cyclic voltammetry (vs. SCE) results. To evaluate the scalability, a gram-scale reaction was performed, delivering 143a in 74% yield.
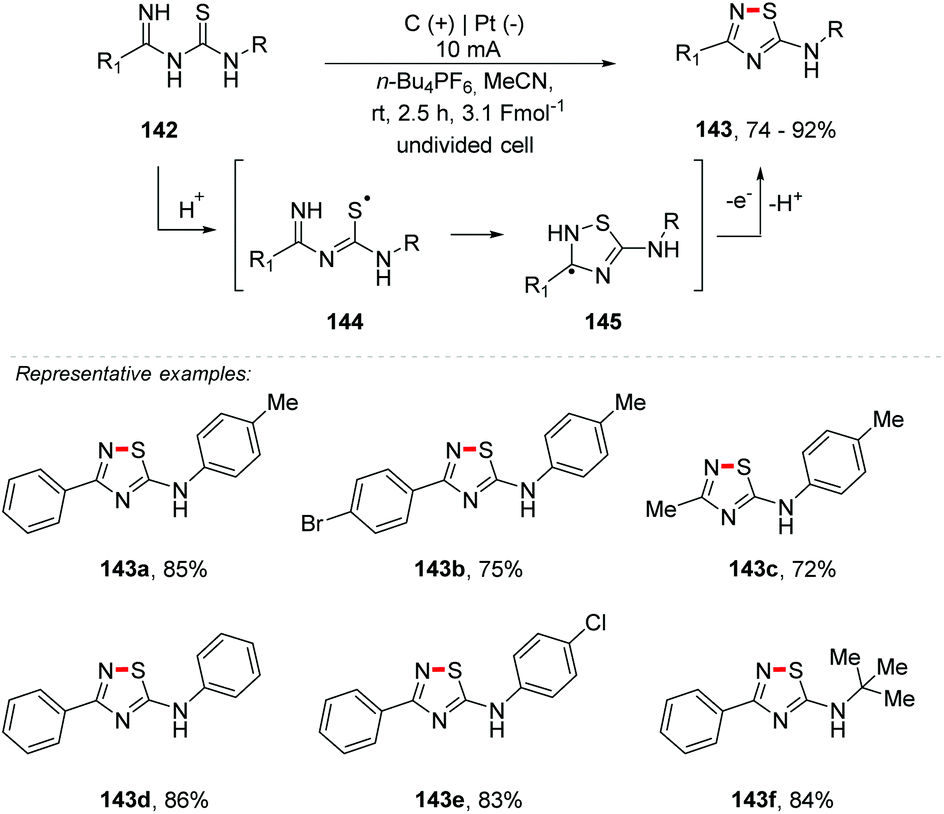 | ||
| Scheme 39 N–S bond formation of thiadiazoles. | ||
3.5 Carboannulations involving fullerenes
Electrolysis has been shown to be an important ally in fullerene chemistry. The reversion of electrophilic-to-nucleophilic reactivity of fullerenes has great potential, being a new field for chemists.171,172 Electrochemical generation of C60n− anions was first employed by Kadish and co-workers in 1993 for the controlled functionalization of fullerenes.173 Currently, researchers are improving these studies; Suzuki and co-workers conducted the reaction of C60˙− with alkyl dihalides and alkyl halides. Cycloaddition and 1,4-dialkylation products were obtained (Scheme 40).174 The authors suggest that the reaction proceeds via electron transfer from C60˙− to the alkyl halide (148 or 149), and the alkyl radical attacks C60. Then, the addition of the alkyl radical to the alkylated C60 radical intermediates provides the cyclization products. Thus, the high recovery of C60 (39–63%) at the end of the reaction would be justified.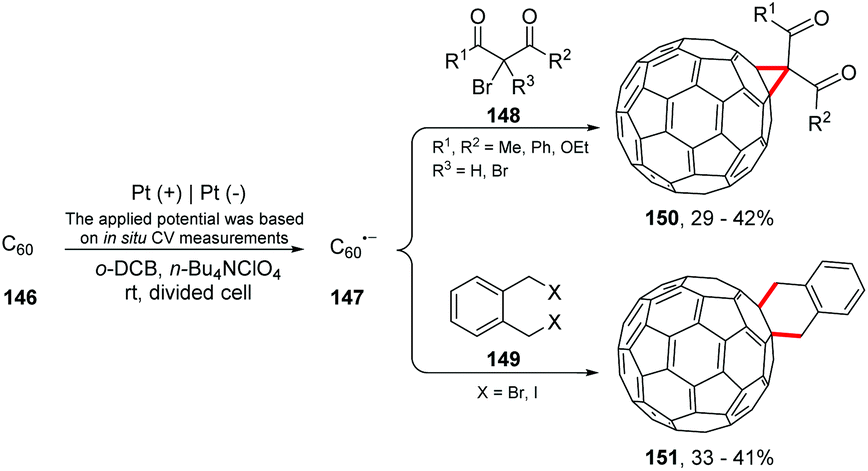 | ||
| Scheme 40 Reaction of C60˙− with alkyl dihalides and alkyl halides. | ||
Gao and co-workers proposed an electrosynthetic approach for the preparation of C60o-quinodimethane bisadducts [C60(QM)2] (Scheme 41).175 The authors emphasized the importance of these products in the study of organic solar cells (OSC);176 however, the synthetic methodologies previously described resulted in up to 8 regioisomers.177 Electrolysis showed high regiocontrol, providing cis-2, trans-3 and C60(QM)2 products.
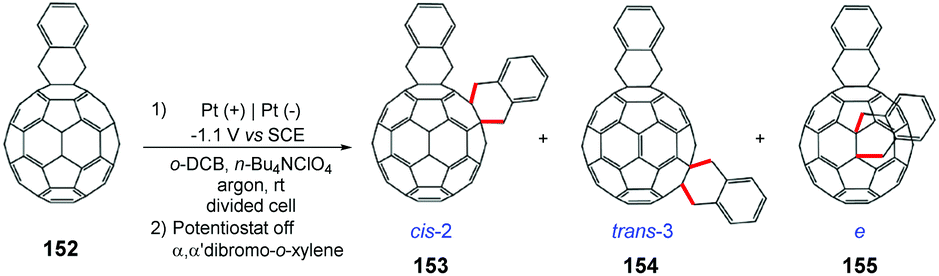 | ||
| Scheme 41 Regiocontrolled electrosynthesis of [60]fullerene bisadducts [C60(QM)2]. | ||
Wang and co-workers proposed a reaction with electrochemically generated dianions of [60]fullerene 158 with bulky secondary alkyl bromides (Scheme 42).178 In a divided cell equipped with platinum electrodes under an argon atmosphere, the reaction was controlled until it reached the theoretical number of coulombs necessary for the complete conversion of C60 to C602− (−1.2 V vs. SCE); the potentiostat was turned off and then the respective alkyl bromide was added. Using BrCH(CO2Et)2 it was possible to obtain the carboannulation product 157. The authors emphasize that the reaction of C602− with of BrCHPh2 and TFA led to the monoalkylation product. In the absence of TFA the reaction proceeded to form the di-alkylation products.
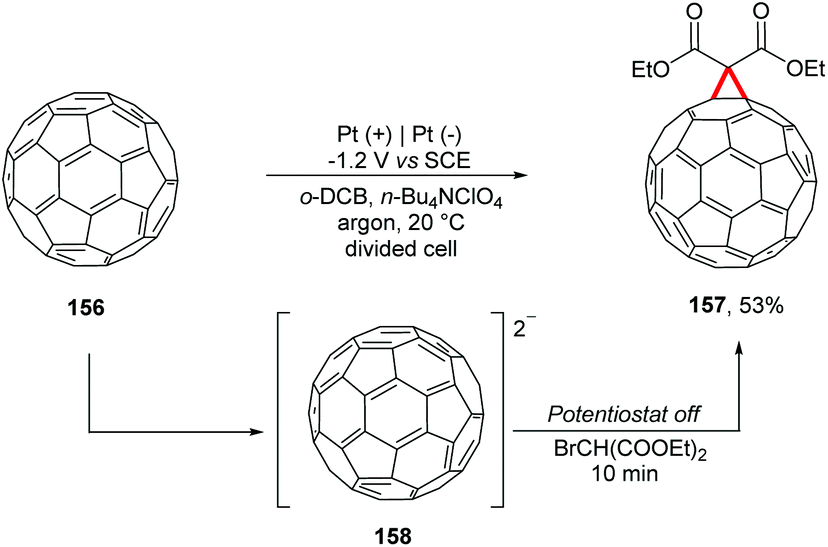 | ||
| Scheme 42 Reactions with electrochemically generated dianions of [60]fullerene. | ||
The same group described a cyclopropanation reaction of electrochemically generated dianionic [60]fullerobenzofurans 163 with diethyl dibromomalonate (Scheme 43). The sterically favored bisadducts e160 were obtained as the major product, and trans-3 bisadducts 161 and 162 were obtained as minor products.179 Considering the previous work, the group emphasized that the fused benzofuran heterocycle derivatives did not reorganize from [6,6]-bonds to [5,6]-bonds.180–184 For the electrophilic reactions of fullerene dianionic derivatives, these results provided information about the control factors, such as electronic and steric factors.
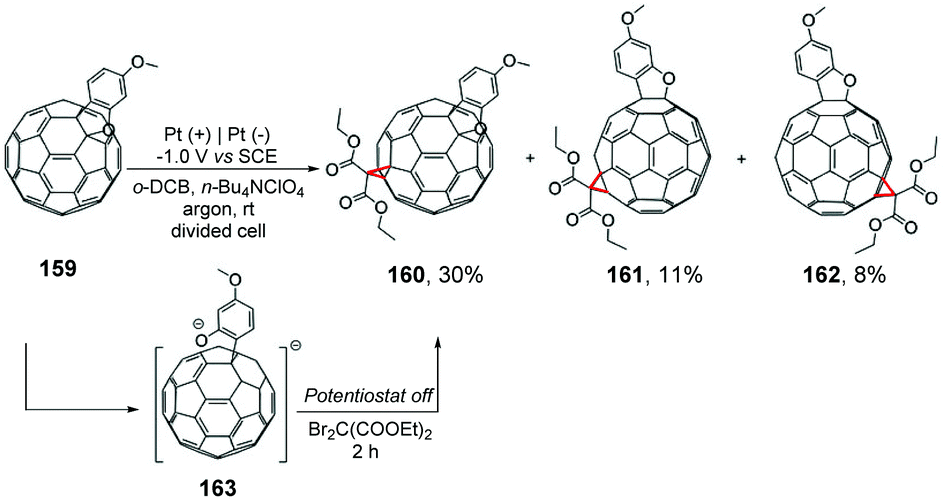 | ||
| Scheme 43 Reactions of electrochemically generated dianions of [60]fullerene with diethyl dibromomalonate. | ||
Wang and co-workers also reported an interesting reaction between the electrochemically generated dianionic [60]fulleroindoline and 1,2-bis(bromomethyl)benzene for the regioselective synthesis of the tetra-functionalized 1,2,4,17-adduct (cis-3′) and 1,2,3,4,9,10-adduct (cis-1) of C60 (Scheme 44).185 The experimental procedure followed the same modus operandi previously discussed by the authors; however when the reaction was carried out at 0 °C, product 166 (cis-3′) was obtained in 49% yield. By carrying out the reaction at a temperature of 25 °C, product 167 (cis-1) was obtained in 33% yield. The authors observed that at room temperature isomer 166 (cis-3′) has a tendency to decompose to isomer 167 (cis-1). Considering that the potential energy surface (PES) scan enabled a reasonable prediction of the ring-closure position, theoretical calculations were performed, showing that cis-3′ isomer 166 was less stable than cis-1 isomer 167 by 14.2 kcal mol−1 at the B3LYP/6-31G(d) level. The authors also observed that protonation of dianionic 168 with 2 equivalents of TFA at 0 °C for 5 min provides 1,2,3,4,9,10-adduct 169 (“S”) in 59% yield.
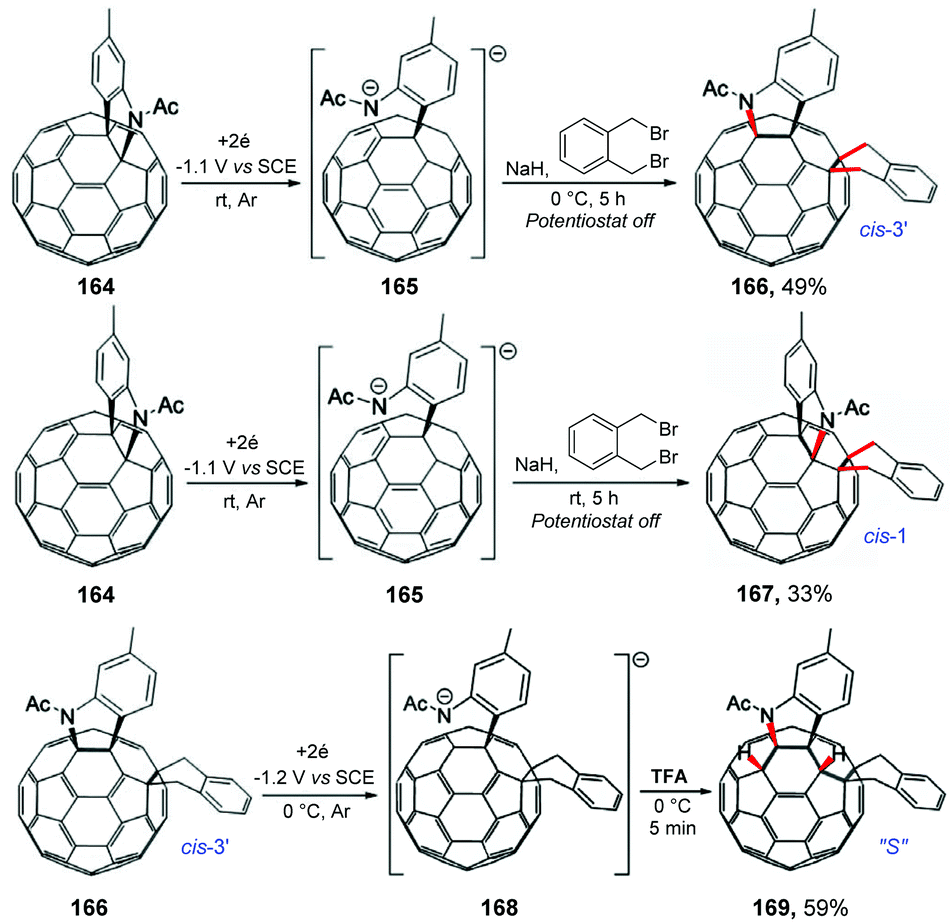 | ||
| Scheme 44 Regiocontrolled electrochemical reaction between the electrochemically generated dianionic [60]fulleroindoline and 1,2-bis(bromomethyl)benzene. | ||
In the same year, Wang and co-workers performed the carboannulation of the electrochemically generated dianionic [60]fulleroindoline 170 with phthaloyl chloride 171, providing the products 1,2,4,17-functionalized [60]fullerene derivatives 172 (cis-3′) (Scheme 45).172 The method presented a unique addition pattern, with a hererocyclic rearrangement to a [5,6]-junction and the carbocycle was fused to an adjacent [6,6]-junction, being in agreement with the biscycloadducts discussed previously. It is worth mentioning that the 1,2,4,17-adducts 172 can also be protonated with TFA to provide hexafunctionalized fullerene products with an “S”-shaped addition pattern with yields of up to 40%.
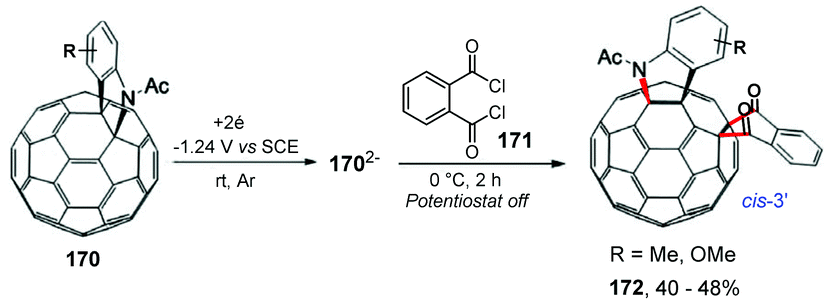 | ||
| Scheme 45 Carboannulation of the electrochemically generated dianionic [60]fulleroindoline with phthaloyl chloride. | ||
4. Sacrificial electrodes for electrochemical annulation reactions
A wide literature search has shown that in the last 20 years there have been few methodologies for organic electrosynthesis using sacrificial electrodes. The application of sacrificial metals has been studied in the past for carboxylation reactions, functionalization of organic halides, formation of metal complexes, etc.186,187 In 1986, Le Guillanton and co-workers used an efficient sulfur/carbon mix cathode for the synthesis of thiophenes and related compounds from acetylenes.188 The same authors reported an overview of the synthesis of thioorganic compounds derived from the electrogenerated species Sx2− and Sy2+.189 The direct synthesis of diaryl diselenides and ditellurides using Se and Te electrodes has also been explored.190Over 30 years ago, Pahil and Banait proposed the synthesis of copper(II) alkoxides (e.g. Cu(OR): R = methyl, ethyl, propyl, butyl, amyl, phenyl and quinoline groups), by direct electrolysis of alcohols in DMF using a copper sacrificial anode and n-Bu4NCl as a supporting electrolyte.191 Recently, Wilden and co-workers proposed an electrochemical synthesis of copper(I) acetylides 175 (Scheme 46) using simultaneous oxidation of copper foil and Hofmann elimination of quaternary ammonium salts.192,193 The authors evaluated its importance through a click reaction (CuAAC reaction), successfully obtaining two conditions in a one-pot electrochemical process, which is a promising approach for these reactions. According to control reactions, they propose that the active Cu species is Cu(MeCN)4X (X = PF6 or CH3C6H4SO3).
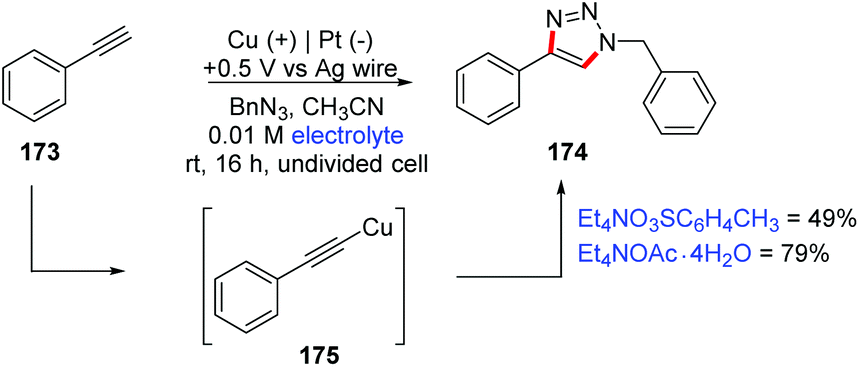 | ||
| Scheme 46 One-pot electrochemical CuAAC reaction by electrochemically mediated cyclization of 2-allylphenols using the Cu sacrificial electrode. | ||
In addition, the same group reported an electrochemical method to generate Zn(II) in situ from a zinc-coated graphite electrode, achieving iodocyclization of 2-allylphenols 176 to obtain dihydrobenzofuran derivatives 177 (Scheme 47).194 Considering that the standard reduction potential of Zn(II) is lower than that of I2 (−0.76 V/+0.54 V vs. SHE), and the first oxidation of I− to form the reactive oxidative species I3− occurs at +1.0 V (vs. Ag quasi-reference electrode, QRE), the authors set the potential at +1.2 V to a zinc-coated graphite electrode (40 mol% Zn0) in a divided ‘H cell’.
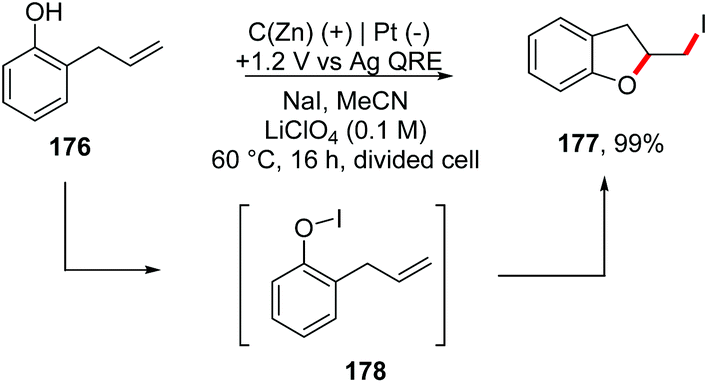 | ||
| Scheme 47 Electrochemically mediated cyclization of 2-allylphenols. | ||
Hara and co-workers described the synthesis of succinic acid derivatives 180 through a cyclization reaction of 2-(2-propynyloxy)bromobenzenes 179, followed by CO2 carboxylation mediated by methyl 4-tert-butylbenzoate using Mg electrodes at the anode and Pt at the cathode under a current of 5 mA cm−2 (Scheme 48).195 Derivatives of indoline, indane, dihydrobenzothiophene, dihydrobenzofuran and tetrahydropyran were obtained with yields of up to 93%. According to the authors, the role of Mg(II) in the solution is to form a stable enolate and also to prevent the oxidized species at the anode. The same group also reported an electrochemical carboxylation of flavones in the presence of carbon dioxide by using an undivided cell equipped with a Pt cathode and Mg anode at 0 °C.196 Further clarification on reactions involving carbon dioxide and sacrificial electrodes can be obtained in the review paper by Filardo and co-workers.197
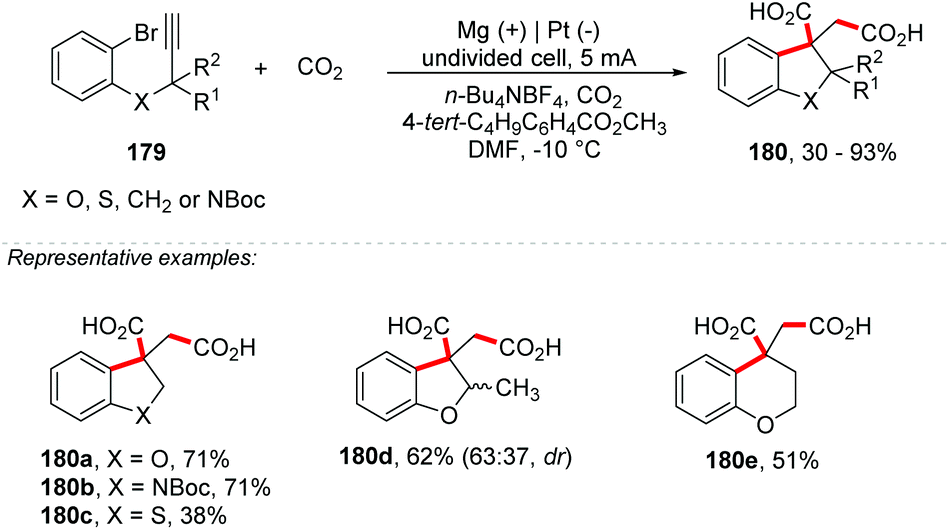 | ||
| Scheme 48 Cyclization of 2-(2-propynyloxy)bromobenzenes followed by tandem carboxylation. | ||
5. Conclusions and outlook
Recent advances in electrochemical annulation reactions considering methodologies for obtaining new C–C, C–heteroatom and heteroatom–heteroatom bonds were discussed, evaluating the mechanistic pathway, as well as the reactivity of substrates. Additionally, it has been shown that this approach provides new synthetic routes, yielding products with excellent atom economy, following green chemistry principles. However, there is a lot to be done in the development of this technology, mainly with regard to hetero- and carboannulations. The substrate scope is still narrow, limiting control in synthesis, especially when compared to the various natural heterocycles present in nature. That is why the continuous study and applications of new electrosynthetic routes are so important, as it allows us to better understand this technology and to have control of these reaction conditions. Despite advances in the application of electrochemistry for the functionalization of organic compounds, most synthetic protocols require a complicated catalytic system or adverse conditions, which need to be improved. The use of new electrode materials must be implemented, such as sacrificial electrodes, which have been explored in the past in simpler molecules. We believe that the ongoing efforts to develop efficient reaction methods in this field, as well as scientific advancement, should provide more access to diverse heterocycles, which may facilitate synthetic control of new molecules. Despite all the benefits offered by electrosynthesis, this is still not a common method in research laboratories. Considering this, our objective is to motivate and demystify electrochemistry in organic synthesis, disseminating this technology so that, in the future, we will have more research groups performing research in this field.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The Marie Skłodowska-Curie Actions COFUND Early Career Fellowship (Grant 663830) to Dr Nisar Ahmed and EPSRC funding (EP/T019719/1) are gratefully acknowledged. We also thank CNPq for their funding support (G. M. M, G. C. Z and S. R. M.).References
- D. Mal, Anionic Annulations in Organic Synthesis, Elsevier, 2019 Search PubMed.
- G. S. Singh, M. D′hooghe and N. De Kimpe, in Comprehensive Heterocyclic Chemistry III, Elsevier, 2008, pp. 1–110 Search PubMed.
- J. Feng and B. Liu, Tetrahedron Lett., 2015, 56, 1474–1485 CrossRef CAS.
- Encyclopedia of Electrochemistry, ed. A. J. Bard, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007 Search PubMed.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC.
- Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed.
- G. Hilt, ChemElectroChem, 2020, 7, 395–405 CrossRef CAS.
- A. Shatskiy, H. Lundberg and M. D. Kärkäs, ChemElectroChem, 2019, 6, 4067–4092 CrossRef CAS.
- R. Francke, Beilstein J. Org. Chem., 2014, 10, 2858–2873 CrossRef PubMed.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020 DOI:10.10621/acs.chemrev.9b00482.
- B. Speiser, Choice Rev. Online, 2005, 42, 42 Search PubMed.
- Organic Electrochemistry, ed. O. Hammerich and B. Speiser, CRC Press, 5th edn, 2015 Search PubMed.
- M. A. Bohn, A. Paul and G. Hilt, in Encyclopedia of Radicals in Chemistry, Biology and Materials, John Wiley & Sons, Ltd, Chichester, UK, 2012 Search PubMed.
- Y. N. Ogibin, M. N. Elinson and G. I. Nikishin, Russ. Chem. Rev., 2009, 78, 89–140 CrossRef CAS.
- A. V. Listratova, N. Sbei and L. G. Voskressensky, Eur. J. Org. Chem., 2020, 2020, 2012–2027 CrossRef CAS.
- Fundamentals and Applications of Organic Electrochemistry, ed. T. Fuchigami, S. Inagi and M. Atobe, John Wiley & Sons Ltd, Chichester, United Kingdom, 2014 Search PubMed.
- E. J. E. Caro-Diaz, M. Urbano, D. J. Buzard and R. M. Jones, Bioorg. Med. Chem. Lett., 2016, 26, 5378–5383 CrossRef CAS PubMed.
- S. Tang, D. Wang, Y. Liu, L. Zeng and A. Lei, Nat. Commun., 2018, 9, 798 CrossRef PubMed.
- C. Tian, L. Massignan, T. H. Meyer and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 2383–2387 CrossRef CAS PubMed.
- D. Kalsi, S. Dutta, N. Barsu, M. Rueping and B. Sundararaju, ACS Catal., 2018, 8, 8115–8120 CrossRef CAS.
- R. H. Verschueren and W. M. De Borggraeve, Molecules, 2019, 24, 2122 CrossRef PubMed.
- T. Hardwick, A. Qurashi, B. Shirinfar and N. Ahmed, ChemSusChem, 2020, 13, 1967–1973 CrossRef CAS PubMed.
- W.-Z. Weng, J. Xie and B. Zhang, Org. Biomol. Chem., 2018, 16, 3983–3988 RSC.
- N. S. Upadhyay, V. H. Thorat, R. Sato, P. Annamalai, S.-C. Chuang and C.-H. Cheng, Green Chem., 2017, 19, 3219–3224 RSC.
- S. L. Yedage and B. M. Bhanage, Green Chem., 2016, 18, 5635–5642 RSC.
- X. Yu, K. Chen, S. Guo, P. Shi, C. Song and J. Zhu, Org. Lett., 2017, 19, 5348–5351 CrossRef CAS PubMed.
- B. Li, H. Feng, S. Xu and B. Wang, Chem. – Eur. J., 2011, 17, 12573–12577 CrossRef CAS PubMed.
- Z.-Q. Wang, C. Hou, Y.-F. Zhong, Y.-X. Lu, Z.-Y. Mo, Y.-M. Pan and H.-T. Tang, Org. Lett., 2019, 21, 9841–9845 CrossRef CAS PubMed.
- G. Song, D. Chen, C.-L. Pan, R. H. Crabtree and X. Li, J. Org. Chem., 2010, 75, 7487–7490 CrossRef CAS PubMed.
- R. Mei, J. Koeller and L. Ackermann, Chem. Commun., 2018, 54, 12879–12882 RSC.
- R. Mei, N. Sauermann, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2018, 140, 7913–7921 CrossRef CAS PubMed.
- L. Yang, R. Steinbock, A. Scheremetjew, R. Kuniyil, L. H. Finger, A. M. Messinis and L. Ackermann, Angew. Chem., Int. Ed. Engl., 2020, 27, 11130–11135 CrossRef PubMed.
- Y. Qiu, C. Tian, L. Massignan, T. Rogge and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 5818–5822 CrossRef CAS PubMed.
- W.-J. Kong, Z. Shen, L. H. Finger and L. Ackermann, Angew. Chem., 2020, 59, 5551–5556 CrossRef CAS PubMed.
- S. C. Sau, R. Mei, J. Struwe and L. Ackermann, ChemSusChem, 2019, 12, 3023–3027 CrossRef CAS PubMed.
- D. Gallego and E. A. Baquero, Open Chem., 2018, 16, 1001–1058 CAS.
- Y. Cao, Y. Yuan, Y. Lin, X. Jiang, Y. Weng, T. Wang, F. Bu, L. Zeng and A. Lei, Green Chem., 2020, 22, 1548–1552 RSC.
- M. J. Hossain, T. Ono, Y. Yano and Y. Hisaeda, ChemElectroChem, 2019, 6, 4199–4203 CrossRef CAS.
- D. Lexa and J. M. Saveant, Acc. Chem. Res., 1983, 16, 235–243 CrossRef CAS.
- M. Giedyk, K. Goliszewska and D. Gryko, Chem. Soc. Rev., 2015, 44, 3391–3404 RSC.
- H. Shimakoshi and Y. Hisaeda, Curr. Opin. Electrochem., 2018, 8, 24–30 CrossRef CAS.
- I. A. Dereven'kov, D. S. Salnikov, R. Silaghi-Dumitrescu, S. V. Makarov and O. I. Koifman, Coord. Chem. Rev., 2016, 309, 68–83 CrossRef.
- H. Shimakoshi, Z. Luo, T. Inaba and Y. Hisaeda, Dalton Trans., 2016, 45, 10173–10180 RSC.
- M. Giedyk, H. Shimakoshi, K. Goliszewska, D. Gryko and Y. Hisaeda, Dalton Trans., 2016, 45, 8340–8346 RSC.
- M.-J. Luo, T.-T. Zhang, F.-J. Cai, J.-H. Li and D.-L. He, Chem. Commun., 2019, 55, 7251–7254 RSC.
- H. Tan, H. Li, J. Wang and L. Wang, Chem. – Eur. J., 2015, 21, 1904–1907 CrossRef CAS PubMed.
- S. Choi, J. Park, E. Yu, J. Sim and C. Park, Angew. Chem., Int. Ed., 2020, 59, 11886–11891 CrossRef CAS PubMed.
- Catalyzed Carbon-Heteroatom Bond Formation, ed. A. K. Yudin, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2010 Search PubMed.
- D. Z. Lin, Y. L. Lai and J. M. Huang, ChemElectroChem, 2019, 6, 4188–4193 CrossRef CAS.
- Hypervalent Iodine Chemistry, ed. T. Wirth, Springer International Publishing, Cham, 2016, vol. 373 Search PubMed.
- I. F. D. Hyatt, L. Dave, N. David, K. Kaur, M. Medard and C. Mowdawalla, Org. Biomol. Chem., 2019, 17, 7822–7848 RSC.
- X. Li, P. Chen and G. Liu, Beilstein J. Org. Chem., 2018, 14, 1813–1825 CrossRef CAS PubMed.
- S. R. Kandimalla, S. P. Parvathaneni, G. Sabitha and B. V. S. Reddy, Eur. J. Org. Chem., 2019, 2019, 1687–1714 CrossRef.
- M. Elsherbini and T. Wirth, Chem. – Eur. J., 2018, 24, 13399–13407 CrossRef CAS PubMed.
- A. Maity, B. L. Frey, N. D. Hoskinson and D. C. Powers, J. Am. Chem. Soc., 2020, 142, 4990–4995 CrossRef CAS PubMed.
- Y. Yu, Y. Yuan, H. Liu, M. He, M. Yang, P. Liu, B. Yu, X. Dong and A. Lei, Chem. Commun., 2019, 55, 1809–1812 RSC.
- T. Lu, Y.-T. Jiang, F.-P. Ma, Z.-J. Tang, L. Kuang, Y.-X. Wang and B. Wang, Org. Lett., 2017, 19, 6344–6347 CrossRef CAS PubMed.
- G. Naresh, R. Kant and T. Narender, Org. Lett., 2015, 17, 3446–3449 CrossRef CAS PubMed.
- H. Jiang, Y. Cheng, Y. Zhang and S. Yu, Org. Lett., 2013, 15, 4884–4887 CrossRef CAS PubMed.
- N. Asao, K. Takahashi, S. Lee, T. Kasahara and Y. Yamamoto, J. Am. Chem. Soc., 2002, 124, 12650–12651 CrossRef CAS PubMed.
- X. Zhang, S. Sarkar and R. C. Larock, J. Org. Chem., 2006, 71, 236–243 CrossRef CAS PubMed.
- M.-X. He, Z.-Y. Mo, Z.-Q. Wang, S.-Y. Cheng, R.-R. Xie, H.-T. Tang and Y.-M. Pan, Org. Lett., 2020, 22, 724–728 CrossRef CAS PubMed.
- K. Liu, C. Song and A. Lei, Org. Biomol. Chem., 2018, 16, 2375–2387 RSC.
- C. L. Bentley, A. M. Bond, A. F. Hollenkamp, P. J. Mahon and J. Zhang, Anal. Chem., 2016, 88, 1915–1921 CrossRef CAS PubMed.
- S. Tang, X. Gao and A. Lei, Chem. Commun., 2017, 53, 3354–3356 RSC.
- G. Li, C. Zhang, C. Song and Y. Ma, Beilstein J. Org. Chem., 2018, 14, 155–181 CrossRef CAS PubMed.
- Z.-Y. Li, L. Li, Q.-L. Li, K. Jing, H. Xu and G.-W. Wang, Chem. – Eur. J., 2017, 23, 3285–3290 CrossRef CAS PubMed.
- X. Pan, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 1163–1185 RSC.
- T. Koike and M. Akita, Chem, 2018, 4, 409–437 CAS.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- J.-Y. Guo, R.-X. Wu, J.-K. Jin and S.-K. Tian, Org. Lett., 2016, 18, 3850–3853 CrossRef CAS PubMed.
- P. Xiong, H.-H. Xu, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2018, 140, 2460–2464 CrossRef CAS PubMed.
- Z. Zhang, L. Zhang, Y. Cao, F. Li, G. Bai, G. Liu, Y. Yang and F. Mo, Org. Lett., 2019, 21, 762–766 CrossRef CAS PubMed.
- T. Fuchigami, M. Atobe and S. Inagi, Fundamentals and Applications of Organic Electrochemistry, John Wiley & Sons Ltd, Chichester, United Kingdom, 2014 Search PubMed.
- A. M. Couper, D. Pletcher and F. C. Walsh, Chem. Rev., 1990, 90, 837–865 CrossRef CAS.
- L.-B. Zhang, R.-S. Geng, Z.-C. Wang, G.-Y. Ren, L.-R. Wen and M. Li, Green Chem., 2020, 22, 16–21 RSC.
- P. K. Singh and O. Silakari, ChemMedChem, 2018, 13, 1071–1087 CrossRef CAS PubMed.
- Q. Wang, P. Wang, X. Gao, D. Wang, S. Wang, X. Liang, L. Wang, H. Zhang and A. Lei, Chem. Sci., 2020, 11, 2181–2186 RSC.
- X. Gao, P. Wang, Q. Wang, J. Chen and A. Lei, Green Chem., 2019, 21, 4941–4945 RSC.
- K. Liu, S. Tang, P. Huang and A. Lei, Nat. Commun., 2017, 8, 775 CrossRef PubMed.
- L. Eberson, M. P. Hartshorn and O. Persson, J. Chem. Soc., Perkin Trans. 2, 1995, 1735 RSC.
- L. Eberson, O. Persson and M. P. Hartshorn, Angew. Chem., Int. Ed. Engl., 1995, 34, 2268–2269 CrossRef.
- L. Eberson, M. P. Hartshorn and O. Persson, J. Chem. Soc., Chem. Commun., 1995, 1131 RSC.
- A. Claraz, T. Courant and G. Masson, Org. Lett., 2020, 22, 1580–1584 CrossRef CAS PubMed.
- S. Zhang, L. Li, J. Zhang, J. Zhang, M. Xue and K. Xu, Chem. Sci., 2019, 10, 3181–3185 RSC.
- Y. Wang, B. Tian, M. Ding and Z. Shi, Chem. – Eur. J., 2020, 26, 4297–4303 CrossRef CAS PubMed.
- J. Hua, Z. Fang, J. Xu, M. Bian, C. K. Liu, W. He, N. Zhu, Z. Yang and K. Guo, Green Chem., 2019, 21, 4706–4711 RSC.
- J. Hua, Z. Fang, M. Bian, T. Ma, M. Yang, J. Xu, C. Liu, W. He, N. Zhu, Z. Yang and K. Guo, ChemSusChem, 2020, 13, 2053–2059 CrossRef CAS PubMed.
- C. Yao, Y. Wang, T. Li, C. Yu, L. Li and C. Wang, Tetrahedron, 2013, 69, 10593–10597 CrossRef CAS.
- G. M. Martins, A. G. Meirinho, N. Ahmed, A. L. Braga and S. R. Mendes, ChemElectroChem, 2019, 6, 5928–5940 CrossRef CAS.
- Y. Liu, J. Kim and J. Chae, Curr. Org. Chem., 2014, 18, 2049–2071 CrossRef CAS.
- X. Hu, G. Zhang, F. Bu, L. Nie and A. Lei, ACS Catal., 2018, 8, 9370–9375 CrossRef CAS.
- F. Wang and S. S. Stahl, Angew. Chem., Int. Ed., 2019, 58, 6385–6390 CrossRef CAS PubMed.
- H. Ding, P. L. DeRoy, C. Perreault, A. Larivée, A. Siddiqui, C. G. Caldwell, S. Harran and P. G. Harran, Angew. Chem., Int. Ed., 2015, 54, 4818–4822 CrossRef CAS PubMed.
- A. W. G. Burgett, Q. Li, Q. Wei and P. G. Harran, Angew. Chem., Int. Ed., 2003, 42, 4961–4966 CrossRef CAS PubMed.
- N. Denizot, A. Pouilhès, M. Cucca, R. Beaud, R. Guillot, C. Kouklovsky and G. Vincent, Org. Lett., 2014, 16, 5752–5755 CrossRef CAS PubMed.
- G. M. Martins, B. Shirinfar, T. Hardwick and N. Ahmed, ChemElectroChem, 2019, 6, 1300–1315 CrossRef CAS.
- H. Mei, Z. Yin, J. Liu, H. Sun and J. Han, Chin. J. Chem., 2019, 37, 292–301 CAS.
- K. Liu, W. Song, Y. Deng, H. Yang, C. Song, T. Abdelilah, S. Wang, H. Cong, S. Tang and A. Lei, Nat. Commun., 2020, 11, 3 CrossRef PubMed.
- C. Song, K. Liu, X. Jiang, X. Dong, Y. Weng, C. Chiang and A. Lei, Angew. Chem., 2020, 59, 7193–7197 CrossRef CAS PubMed.
- J. Xu and R. Tong, Green Chem., 2017, 19, 2952–2956 RSC.
- J. Wu, H. Abou-Hamdan, R. Guillot, C. Kouklovsky and G. Vincent, Chem. Commun., 2020, 56, 1713–1716 RSC.
- J. Wu, Y. Dou, R. Guillot, C. Kouklovsky and G. Vincent, J. Am. Chem. Soc., 2019, 141, 2832–2837 CrossRef CAS PubMed.
- Z. Xu, Z. Huang, Y. Li, R. Kuniyil, C. Zhang, L. Ackermann and Z. Ruan, Green Chem., 2020, 22, 1099–1104 RSC.
- Z. Ye, M. Ding, Y. Wu, Y. Li, W. Hua and F. Zhang, Green Chem., 2018, 20, 1732–1737 RSC.
- A. Kumar and K. R. Prabhu, J. Org. Chem., 2020, 85, 3548–3559 CrossRef CAS PubMed.
- K. Wadekar, S. Aswale and V. R. Yatham, Org. Biomol. Chem., 2020, 18, 983–987 RSC.
- J. Gallardo-Donaire and R. Martin, J. Am. Chem. Soc., 2013, 135, 9350–9353 CrossRef CAS PubMed.
- Y. Li, Y.-J. Ding, J.-Y. Wang, Y.-M. Su and X.-S. Wang, Org. Lett., 2013, 15, 2574–2577 CrossRef CAS PubMed.
- R. J. Perkins, H.-C. Xu, J. M. Campbell and K. D. Moeller, Beilstein J. Org. Chem., 2013, 9, 1630–1636 CrossRef PubMed.
- H. Kolbe, Ann. Chem. Pharm., 1849, 69, 257–294 CrossRef.
- S. Zhang, L. Li, H. Wang, Q. Li, W. Liu, K. Xu and C. Zeng, Org. Lett., 2018, 20, 252–255 CrossRef CAS PubMed.
- Z. Guan, Y. Wang, H. Wang, Y. Huang, S. Wang, H. Tang, H. Zhang and A. Lei, Green Chem., 2019, 21, 4976–4980 RSC.
- L. Sun, Y. Yuan, M. Yao, H. Wang, D. Wang, M. Gao, Y.-H. Chen and A. Lei, Org. Lett., 2019, 21, 1297–1300 CrossRef CAS PubMed.
- Q.-B. Zhang, P.-F. Yuan, L.-L. Kai, K. Liu, Y.-L. Ban, X.-Y. Wang, L.-Z. Wu and Q. Liu, Org. Lett., 2019, 21, 885–889 CrossRef CAS PubMed.
- M. R. Scheide, A. R. Schneider, G. A. M. Jardim, G. M. Martins, D. C. Durigon, S. Saba, J. Rafique and A. L. Braga, Org. Biomol. Chem., 2020, 18, 4916–4921 RSC.
- M. Wilken, S. Ortgies, A. Breder and I. Siewert, ACS Catal., 2018, 8, 10901–10912 CrossRef CAS.
- S. Torii, K. Uneyama and M. Ono, Tetrahedron Lett., 1980, 21, 2653–2654 CrossRef CAS.
- O. Niyomura, M. Cox and T. Wirth, Synlett, 2006, 251–254 CAS.
- S. Ortgies and A. Breder, ACS Catal., 2017, 7, 5828–5840 CrossRef CAS.
- U. Wille, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2012, 108, 228 RSC.
- X. Zeng, W. Yan, S. B. Zacate, T.-H. Chao, X. Sun, Z. Cao, K. G. E. Bradford, M. Paeth, S. B. Tyndall, K. Yang, T.-C. Kuo, M.-J. Cheng and W. Liu, J. Am. Chem. Soc., 2019, 141, 11398–11403 CrossRef CAS PubMed.
- S. P. Pitre, N. A. Weires and L. E. Overman, J. Am. Chem. Soc., 2019, 141, 2800–2813 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- J. C. Lo, D. Kim, C.-M. Pan, J. T. Edwards, Y. Yabe, J. Gui, T. Qin, S. Gutiérrez, J. Giacoboni, M. W. Smith, P. L. Holland and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 2484–2503 CrossRef CAS PubMed.
- S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS PubMed.
- Á. Díaz-Ortiz, P. Prieto and A. de la Hoz, Chem. Rec., 2019, 19, 85–97 CrossRef PubMed.
- R. T. McBurney, F. Portela-Cubillo and J. C. Walton, RSC Adv., 2012, 2, 1264–1274 RSC.
- T. G. McKenzie, F. Karimi, M. Ashokkumar and G. G. Qiao, Chem. – Eur. J., 2019, 25, 5372–5388 CrossRef CAS PubMed.
- Z. Chen, M.-Y. Rong, J. Nie, X.-F. Zhu, B.-F. Shi and J.-A. Ma, Chem. Soc. Rev., 2019, 48, 4921–4942 RSC.
- C. R. Whitehead, E. H. Sessions, I. Ghiviriga and D. L. Wright, Org. Lett., 2002, 4, 3763–3765 CrossRef CAS PubMed.
- J. P. Coleman, R. Lines, J. H. P. Utley and B. C. L. Weedon, J. Chem. Soc., Perkin Trans. 2, 1974, 1064 RSC.
- P. Huang, P. Wang, S. Wang, S. Tang and A. Lei, Green Chem., 2018, 20, 4870–4874 RSC.
- L. Bering and A. P. Antonchick, in PATAI'S Chemistry of Functional Groups, John Wiley & Sons, Ltd, Chichester, UK, 2018, pp. 1–57 Search PubMed.
- A. J. Waldman, T. L. Ng, P. Wang and E. P. Balskus, Chem. Rev., 2017, 117, 5784–5863 CrossRef CAS PubMed.
- P. Xu and H. Xu, ChemElectroChem, 2019, 6, 4177–4179 CrossRef CAS.
- A. M. Khalil, M. A. Berghot, M. A. Gouda and S. A. El Bialy, Monatsh. Chem., 2010, 141, 1353–1360 CrossRef CAS.
- V. S. Rao Chunduru and V. R. Rao, Synth. Commun., 2013, 43, 923–929 CrossRef CAS.
- R. P. Jain and J. C. Vederas, Bioorg. Med. Chem. Lett., 2004, 14, 3655–3658 CrossRef CAS PubMed.
- R. W. Carling, K. W. Moore, L. J. Street, D. Wild, C. Isted, P. D. Leeson, S. Thomas, D. O'Connor, R. M. McKernan, K. Quirk, S. M. Cook, J. R. Atack, K. A. Wafford, S. A. Thompson, G. R. Dawson, P. Ferris and J. L. Castro, J. Med. Chem., 2004, 47, 1807–1822 CrossRef CAS PubMed.
- H. P. Kaufmann, Z. Angew. Chem., 1927, 40, 69–79 CrossRef CAS.
- J. B. Diccianni, C. Hu and T. Diao, Angew. Chem., 2016, 128, 7660–7664 CrossRef.
- D.-G. Yu, M. Suri and F. Glorius, J. Am. Chem. Soc., 2013, 135, 8802–8805 CrossRef CAS PubMed.
- Q.-Z. Zheng, P. Feng, Y.-F. Liang and N. Jiao, Org. Lett., 2013, 15, 4262–4265 CrossRef CAS PubMed.
- C. Chen, G. Tang, F. He, Z. Wang, H. Jing and R. Faessler, Org. Lett., 2016, 18, 1690–1693 CrossRef CAS PubMed.
- T. Gieshoff, D. Schollmeyer and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 9437–9440 CrossRef CAS PubMed.
- S. R. Waldvogel and B. Janza, Angew. Chem., 2014, 126, 7248–7249 CrossRef.
- T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, J. Am. Chem. Soc., 2017, 139, 12317–12324 CrossRef CAS PubMed.
- T. Gieshoff, D. Schollmeyer and S. R. Waldvogel, Angew. Chem., 2016, 128, 9587–9590 CrossRef.
- T. Gieshoff, D. Schollmeyer and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 9437–9440 CrossRef CAS PubMed.
- B. R. Rosen, E. W. Werner, A. G. O'Brien and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5571–5574 CrossRef CAS PubMed.
- A. Kehl, T. Gieshoff, D. Schollmeyer and S. R. Waldvogel, Chem. – Eur. J., 2018, 24, 590–593 CrossRef CAS PubMed.
- V. M. Breising, T. Gieshoff, A. Kehl, V. Kilian, D. Schollmeyer and S. R. Waldvogel, Org. Lett., 2018, 20, 6785–6788 CrossRef CAS PubMed.
- E. Rodrigo and S. R. Waldvogel, Green Chem., 2018, 20, 2013–2017 RSC.
- E. Rodrigo and S. R. Waldvogel, Chem. Sci., 2019, 10, 2044–2047 RSC.
- A. Kehl, V. M. Breising, D. Schollmeyer and S. R. Waldvogel, Chem. – Eur. J., 2018, 24, 17230–17233 CrossRef CAS PubMed.
- E. Rodrigo, H. Baunis, E. Suna and S. R. Waldvogel, Chem. Commun., 2019, 55, 12255–12258 RSC.
- S. Hosseini, S. A. Bawel, M. S. Mubarak and D. G. Peters, ChemElectroChem, 2019, 6, 4318–4324 CrossRef CAS.
- R. Han, K. I. Son, G. H. Ahn, Y. M. Jun, B. M. Lee, Y. Park and B. H. Kim, Tetrahedron Lett., 2006, 47, 7295–7299 CrossRef CAS.
- B. H. Kim, Y. M. Jun, Y. R. Choi, D. B. Lee and W. Baik, Heterocycles, 1998, 48, 749–754 CrossRef CAS.
- H.-Y. Kim, S. H. Kwak, G.-H. Lee and Y.-D. Gong, Tetrahedron, 2014, 70, 8737–8743 CrossRef CAS.
- N. Tumula, N. Jatangi, R. K. Palakodety, S. Balasubramanian and M. Nakka, J. Org. Chem., 2017, 82, 5310–5316 CrossRef CAS PubMed.
- B. Wang, Y. Meng, Y. Zhou, L. Ren, J. Wu, W. Yu and J. Chang, J. Org. Chem., 2017, 82, 5898–5903 CrossRef CAS PubMed.
- A. Mariappan, K. Rajaguru, N. M. Chola, S. Muthusubramanian and N. Bhuvanesh, J. Org. Chem., 2016, 81, 6573–6579 CrossRef CAS PubMed.
- Y. Dürüst, M. Yıldırım and A. Aycan, J. Chem. Res., 2008, 2008, 235–239 CrossRef.
- H. Xie, J. Cai, Z. Wang, H. Huang and G.-J. Deng, Org. Lett., 2016, 18, 2196–2199 CrossRef CAS PubMed.
- S. Mo, Q. Teng, Y. Pan and H. Tang, Adv. Synth. Catal., 2019, 361, 1756–1760 CrossRef CAS.
- Z. Wang, X. Meng, Q. Li, H. Tang, H. Wang and Y. Pan, Adv. Synth. Catal., 2018, 360, 4043–4048 CrossRef CAS.
- A. S. Mayhoub, E. Kiselev and M. Cushman, Tetrahedron Lett., 2011, 52, 4941–4943 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, L. Hu, L. Li, K. Liu, T. Yang and C. Zhou, J. Org. Chem., 2020, 85, 3358–3363 CrossRef CAS PubMed.
- L. Echegoyen and L. E. Echegoyen, Acc. Chem. Res., 1998, 31, 593–601 CrossRef CAS.
- X.-X. Yan, B. Li, H.-S. Lin, F. Jin, C. Niu, K.-Q. Liu, G.-W. Wang and S. Yang, Research, 2020, 2020, 1–9 CrossRef PubMed.
- C. Caron, R. Subramanian, F. D'Souza, J. Kim, W. Kutner, M. T. Jones and K. M. Kadish, J. Am. Chem. Soc., 1993, 115, 8505–8506 CrossRef CAS.
- Y. Maeda, M. Sanno, T. Morishita, K. Sakamoto, E. Sugiyama, S. Akita, M. Yamada and M. Suzuki, New J. Chem., 2019, 43, 6457–6460 RSC.
- Z.-J. Li, S. Wang, S.-H. Li, T. Sun, W.-W. Yang, K. Shoyama, T. Nakagawa, I. Jeon, X. Yang, Y. Matsuo and X. Gao, J. Org. Chem., 2017, 82, 8676–8685 CrossRef CAS PubMed.
- H. Li, L. Guo, C.-N. Li, C. Wang, G. Wang, S. Wen, J. Wu, W. Dong, Z.-J. Li and S. Ruan, ACS Sustainable Chem. Eng., 2019, 7, 8579–8586 CrossRef CAS.
- K. Kordatos, S. Bosi, T. Da Ros, A. Zambon, V. Lucchini and M. Prato, J. Org. Chem., 2001, 66, 2802–2808 CrossRef CAS PubMed.
- K.-Q. Liu and G.-W. Wang, Tetrahedron Lett., 2019, 60, 1049–1052 CrossRef CAS.
- J.-J. Wang, H.-S. Lin, C. Niu and G.-W. Wang, Org. Biomol. Chem., 2017, 15, 3248–3254 RSC.
- R. Liu, F. Li, Y. Xiao, D.-D. Li, C.-L. He, W.-W. Yang, X. Gao and G.-W. Wang, J. Org. Chem., 2013, 78, 7093–7099 CrossRef CAS PubMed.
- H.-L. Hou, Z.-J. Li, Y. Wang and X. Gao, J. Org. Chem., 2014, 79, 8865–8870 CrossRef CAS PubMed.
- H.-L. Hou, Z.-J. Li and X. Gao, Org. Lett., 2014, 16, 712–715 CrossRef CAS PubMed.
- Y. Xiao, S.-E. Zhu, D.-J. Liu, M. Suzuki, X. Lu and G.-W. Wang, Angew. Chem., Int. Ed., 2014, 53, 3006–3010 CrossRef CAS PubMed.
- F. Li, J.-J. Wang and G.-W. Wang, Chem. Commun., 2017, 53, 1852–1855 RSC.
- K.-Q. Liu, J.-J. Wang, X.-X. Yan, C. Niu and G.-W. Wang, Chem. Sci., 2020, 11, 384–388 RSC.
- C. Saboureau, M. Troupel and J. Perichon, J. Appl. Electrochem., 1990, 20, 97–101 CrossRef CAS.
- A. Rodríguez and J. A. García-Vázquez, Coord. Chem. Rev., 2015, 303, 42–85 CrossRef.
- G. Le Guillanton, Q. T. Do and J. Simonet, Tetrahedron Lett., 1986, 27, 2261–2262 CrossRef CAS.
- G. Le Guillanton, Sulfur Rep., 1992, 12, 405–431 CrossRef CAS.
- C. Degrand and R. Prest, J. Electroanal. Chem., 1990, 282, 281–286 CrossRef CAS.
- J. S. Banait and P. K. Pahil, Synth. React. Inorg. Met.-Org. Chem., 1986, 16, 1217–1224 CrossRef CAS.
- P. W. Seavill, K. B. Holt and J. D. Wilden, RSC Adv., 2019, 9, 29300–29304 RSC.
- P. W. Seavill, K. B. Holt and J. D. Wilden, Green Chem., 2018, 20, 5474–5478 RSC.
- D. Li, P. W. Seavill and J. D. Wilden, ChemElectroChem, 2019, 6, 5829–5835 CrossRef CAS.
- A. Katayama, H. Senboku and S. Hara, Tetrahedron, 2016, 72, 4626–4636 CrossRef CAS.
- H. Senboku, Y. Yamauchi, N. Kobayashi, A. Fukui and S. Hara, Electrochim. Acta, 2012, 82, 450–456 CrossRef CAS.
- G. Silvestri, S. Gambino and G. Filardo, Acta Chem. Scand., 1991, 45, 987–992 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |