
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalysis for versatile polymer degradation†
Paul
McKeown
 *ab,
Muhammed
Kamran
ab,
Matthew G.
Davidson
ab,
Matthew D.
Jones
*ab,
Luis A.
Román-Ramírez
c and
Joseph
Wood
c
*ab,
Muhammed
Kamran
ab,
Matthew G.
Davidson
ab,
Matthew D.
Jones
*ab,
Luis A.
Román-Ramírez
c and
Joseph
Wood
c
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: pm482@bath.ac.uk; mj205@bath.ac.uk
bCentre for Sustainable and Circular Technologies, Bath, BA2 7AY, UK
cSchool of Chemical Engineering, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK
First published on 27th May 2020
Abstract
The use of a simple, cheap and effective organocatalyst, tetramethylammonium methyl carbonate, has been exploited for the transesterification of a range of commercial polymer samples. PLA, PCL, PC and PET have been successfully broken down into useful products, with the repolymerisation of DMT to PET demonstrated, highlighting a truly circular economy approach.
Plastics have had numerous societal benefits, contributing to energy saving and material conservation,1 largely due to their versatile and lightweight properties. However, with demand predicted to increase, there are two urgent challenges that need to be addressed in relation to current usage: (A) Plastics are largely derived from petrochemical feedstocks, which is unsustainable when considering the growing plastic demand and the decreasing reliance placed on these feedstocks.2,3 In response to this there has been a resurgence into research into bio-derived materials including poly(lactic acid) (PLA) and poly(ethylene furanoate) (PEF) among others.4–12 (B) The second issue relates to the extended lifetime of plastics and end-of-life mismanagement causing leaking and persistence in the environment leading to land and sea pollution.13–16 While biodegradable materials are being sought to reduce this issue,17 compostable materials are not compatible with the circular economy as immediate value is lost from the life-cycle and some plastic applications require robust properties.18,19 Therefore, there is a great need for plastic recycling processes in which useful products are the result of the polymer end-of-life.20–22 Mechanical recycling is useful for some materials and can prolong the life time or usage; however, the polymer properties or quality can be impaired and the material downgraded to less demanding uses.22 Chemical recycling offers greater benefits, with polymers being broken down into component parts which could be directly repolymerised, to a material with equivalent physical properties, or to useful chemical platforms.21 For example, by transesterification, PLA can be degraded into lactic acid or alkyl lactate (A-La), the former being a platform chemical and the latter having uses as solvent.23–27 Both degradation products can also be transformed to lactide, which is the first step towards polymer synthesis.28–30 For some polymers, chemical recycling can be achieved thermally by pyrolysis, depolymerising the material directly back to monomers.31 Pyrolysis of PLA can access lactide directly, however this requires high-temperatures and can increase the amount of by-products formed.32–36 Poly(ethylene terephthalate) (PET) can be chemically depolymerised into ethylene glycol (EG) and either bis(2-hydroxyethyl)terephthalate (BHET) or dimethyl terephthalate (DMT) by glycolysis or methanolysis respectively.37–43 The recovery of these monomers can allow for direct repolymerisation to PET of the same quality.39,44 PET can also be degraded by aminolysis yielding a range of amides which can be used as additives to other plastics.45 Chemical recycling of polyesters is typically achieved by a transesterification reaction performed by metal complexes or oxide catalysts.37,46–49 This can lead to toxic residues that may be difficult to remove from the degradation product as well as discolouration. Potentially more benign is the use of organocatalysts which represent a simpler, low cost alternative to metal catalysis which is in alignment with the twelve principles of green chemistry. Examples of organocatalysis applied to polymer degradation has recently been reviewed;50 specific examples of organocatalytic polyester degradation include TBD (for PLA and PET)41,45,51,52 and DMAP (for PLA),53 and ionic liquids (for PLA and PET).40,54–56 Herein, we report the use of a simple onium transesterification catalyst, first reported for monomeric substrates by Ishihara et al.,57 for the mild and selective degradation of a range of polyesters, including PLA and PET.
The organocatalyst, [NMe4]+[OCO2Me]−, 1, was prepared according to literature methods from tetramethyl ammonium hydroxide and dimethyl carbonate (DMC).57 The synthesis of this catalyst (Fig. 1) was also scaled up from the reported method. This catalyst was then applied to the degradation of various polymers. The initial step of the transesterification is the in situ generation of the active catalyst by breakdown of the initial carbonate anion, to form CO2 and methanol (MeOH). With the addition of alcohol, the carbonate anion is replaced by an alkoxide which can then perform transesterification with the assistance of the methyl hydrogen atoms acting as Brønsted acids to activate the carbonyl group (Scheme S1†).57 The ability of these hydrogen atoms to hydrogen bond has previously been demonstrated, and is achieved through delocalisation of the positive charge.58–60 For PLA, degradation was initially performed at 50 °C in line with previous studies,46,61 using 4 wt% of 1 (2.0 mol% with respect to ester linkages; Table 1, entry 1). The reaction was monitored by 1H NMR spectroscopy through consideration of the methine units whereby internal methine units ([Int]) react with alcohol to form chain end methine units ([CE]) which can react further to form alkyl lactate ([A-La]) (Scheme S2†). Selectivity (SA-La) and yield (YA-La) relate to amount of alkyl lactate relative to [CE] and the conversion of [Int] (see ESI† for equations). Using MeOH/THF, a commercial PLA sample (PLA cup) was degraded into methyl lactate (Me-La) exclusively within 1 h (Xint = YMe-La = 100%). The reaction mixture remained colourless throughout. Due to the rapidity of the reaction under these conditions, the catalyst loading could be reduced to 1 wt% (0.5 mol%; Table 1, entry 2) with reasonable activity and selectivity still being achieved (Xint = 100%, YMe-La = 83%). This result is competitive with degradation via Zn(II) complexes previously reported.61 Kinetic analysis of the degradation under these conditions was performed in accordance with previous methods.46,61–63 The reaction profile conforms to a two-step consecutive reaction model with kinetic constants (k1 = 0.103 min−1, k2 = 0.037 min−1) evaluated based on this mechanism (Fig. 2 and Scheme S2†). For the degradation of PLA towards Me-La, excess MeOH (7 equivalents per ester unit) was initially added to drive the reaction towards the product. However, the degradation was also successful on addition of 1 equivalent of alcohol per ester unit, with only a slight decrease in selectivity (Table 1, entry 3). The addition of a stoichiometric amount of MeOH can be beneficial as this limits the reformation of the linear dimer on removal of the excess alcohol. The addition of 3 equivalents of MeOH relative to polymer chains, rather than ester linkages, was performed to reduce the molecular weight by a factor of four in a predictable manner. A reasonable agreement is achieved between predicted and measured molecular weight (Mn,theo = 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200; Mn = 8450, Đ = 1.61; Fig. S5†). Despite the use of 4 wt% catalyst loading, the polymer degradation was much slower (∼24 h) compared to previous experiments with ester-stoichiometric or – excess MeOH amounts. This is likely related to the lower concentration of transesterification alcohol functional groups, which are now secondary alcohol groups of polymeric chains, which would necessarily be hindered towards reaction. Furthermore, as chain scission is suggested to be random, extensive transesterification reactions would be required to reduce the polymer to a consistent weight. Compared to other degradation reactions, with sub-stoichiometric amounts of alcohol, the solution appears turbid, likely due to the formation of [NMe4]+[O-Poly]−. The apparent insolubility of this species could also contribute to the extended reaction time. In comparison, Hedrick and co-workers have demonstrated the use of DMAP and 4-pyrrolidinopyridine for the controlled reduction of PLA.64 With relatively high loading (7.6 wt%), reactions required 3–4 days at 38 °C.
200; Mn = 8450, Đ = 1.61; Fig. S5†). Despite the use of 4 wt% catalyst loading, the polymer degradation was much slower (∼24 h) compared to previous experiments with ester-stoichiometric or – excess MeOH amounts. This is likely related to the lower concentration of transesterification alcohol functional groups, which are now secondary alcohol groups of polymeric chains, which would necessarily be hindered towards reaction. Furthermore, as chain scission is suggested to be random, extensive transesterification reactions would be required to reduce the polymer to a consistent weight. Compared to other degradation reactions, with sub-stoichiometric amounts of alcohol, the solution appears turbid, likely due to the formation of [NMe4]+[O-Poly]−. The apparent insolubility of this species could also contribute to the extended reaction time. In comparison, Hedrick and co-workers have demonstrated the use of DMAP and 4-pyrrolidinopyridine for the controlled reduction of PLA.64 With relatively high loading (7.6 wt%), reactions required 3–4 days at 38 °C.
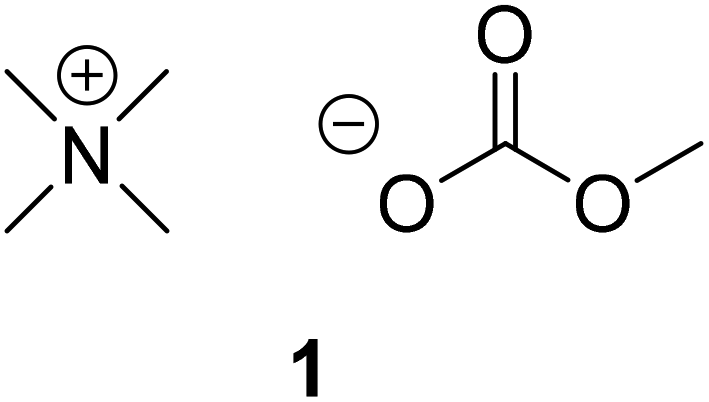 | ||
| Fig. 1 Structure of organocatalyst, 1. | ||
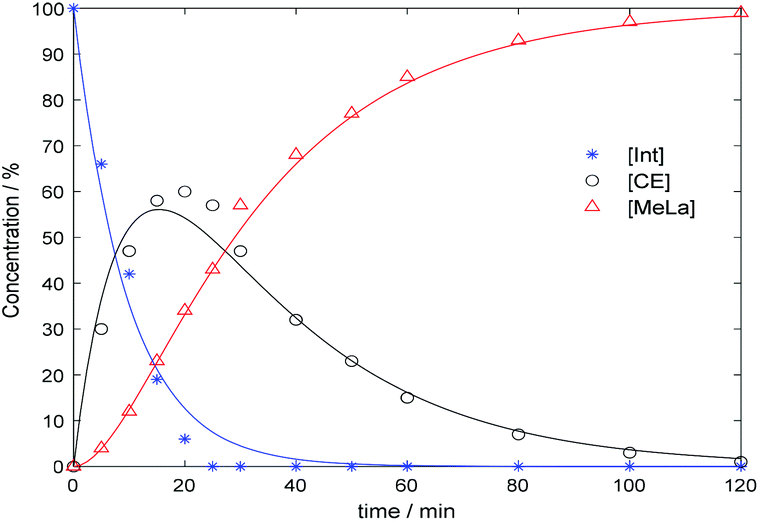 | ||
| Fig. 2 Kinetic profile for PLA degradation with 1 and MeOH at 50 °C. Lines represent kinetic model. | ||
| Entry | Solvent | Time/h | X Int | S A–La | Y A–La |
|---|---|---|---|---|---|
| Conditions: 50 °C, solvent/MeOH = 4:1, nMeOH:nester = 7:1; internal methine group conversion (XInt), A-La selectivity (SA-La) and A-La yield (YA-La) measured by 1H NMR spectroscopy.a 4 wt% loading of 1, 2.0 mol% relative to ester linkages.b 1 wt% loading of 1, 0.5 mol% relative to ester linkages.c MeOH, nMeOH:nester = 1:1.d EtOH, nEtOH:nester = 7:1.e 100 °C solvent-free.f MeOH, nMeOH:nester = 14:1.g EtOH, nEtOH:nester = 14:1. |
|||||
| 1a | THF | 1 | 100 | 100 | 100 |
| 2b | THF | 1 | 100 | 83 | 83 |
| 3b,c | THF | 1 | 99 | 70 | 67 |
| 4b,d | THF | 1 | 85 | 24 | 21 |
| 5b | EtOAc | 1 | 100 | 75 | 75 |
| 6b | 2-MeTHF | 1 | 93 | 69 | 64 |
| 7b | DMC | 1 | 88 | 41 | 36 |
| 8b | Acetone | 1 | 62 | 19 | 12 |
| 9b | ACN | 1 | 24 | 13 | 3 |
| 10b,e,f | — | 0.17 | 100 | 100 | 100 |
| 11b,e,g | — | 0.5 | 100 | 100 | 100 |
The organocatalyst, 1, is also active with ethanol (EtOH) rather than MeOH as transesterification agent. After 1 h, reasonable conversion of Int groups are observed but selectivity towards ethyl lactate (Et-La) remains low (Table 1, entry 4). Increased selectivity is achieved with extended reaction time (3 h, SEt-La = 75%) suggesting EtOH to be less active in the transesterification, in agreement with previous results.46,61,65
The solvent scope was expanded to include more benign solvents to further improve the green credentials of the process, at the reduced catalyst loading (Fig. 3, Table 1, entries 5–9). Under these conditions, the choice of solvent is limited by the need to dissolve the polymer, however the high activity of 1 reduces this limitation. Ethyl acetate (EtOAc), 2-methyl tetrahydrofuran (2-MeTHF), acetone, acetonitrile (ACN), DMC and cyclopentyl methyl ether (CPME) were selected as “greener” alternatives to THF.66–70
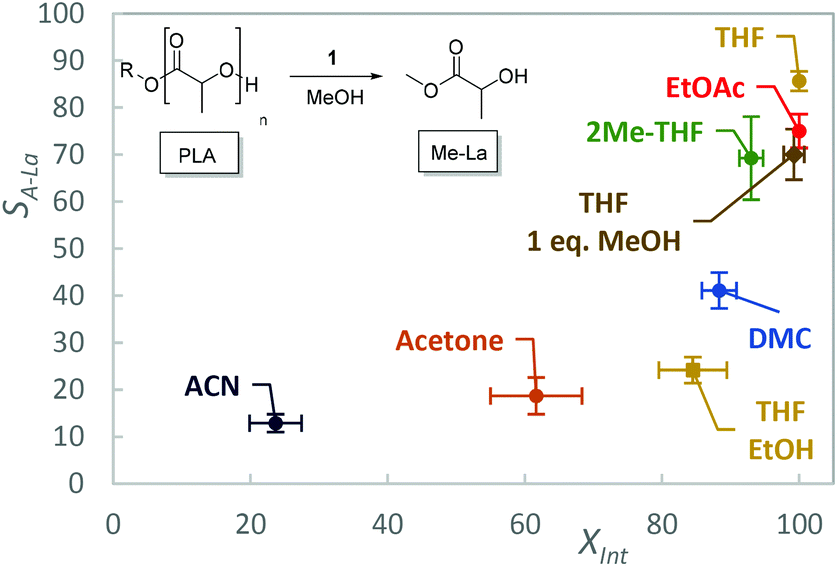 | ||
| Fig. 3 Degradation of PLA with 1, conversion of Int (XInt) and selectivity towards alkyl lactate (SA-La) with various solvents. Error bars represent one standard deviation based on three repeats. | ||
With the exception of DMC, 2-MeTHF and toluene, solvents were not treated or stored under inert anhydrous conditions, demonstrating the tolerance of this catalyst to residual moisture. Except for CPME, for which the PLA did not dissolve within the 1 h reaction time, 1 was active in the additional solvents. While THF afforded the highest selectivity, potentially due to the excellent polymer solubility and ion stabilisation, EtOAc and 2-MeTHF are shown to be credible replacements. ACN was less selective, and this is in contrast to the solvent performance when used with Zn(II) complexes for which a comparable selectivity to THF was observed.61 This could be an indication of an incompatibility between the organocatalyst and ACN. It is noted that ACN has the highest dielectric constant and dipole moment of the solvents used and we tentatively suggest there is extensive hydrogen bonding between this solvent and the cation, inhibiting the activation of the carbonyl group. This hypothesis is potentially supported by computational literature studies, in which this interaction is suppressed by water but remains competitive with less polar solvents such as THF and toluene.60 While these initial results are promising for the application of 1 for PLA degradation under mild conditions, they are currently inferior to what is achievable with TBD, which shows exceptional activity at room temperature (1 mol%, 2 min).51
Degradation was also viable in the absence of solvent at higher temperatures (100 °C). In MeOH (2 mL, nMeOH:nester = 14:1; Table 1, entry 10), complete conversion and high selectivity were achievable within 10 min. The production of Et-La at the same temperature is also feasible (30 min, nEtOH:nester = 14:1; Table 1, entry 11). For comparison, Liu and co-workers have demonstrated high temperature methanolysis mediated by ionic liquids.54 At 100 °C, [Bmim][OAc] (5 wt%) required more than 3 h to achieve a high yield of Me-La, however, it is worth noting less equivalents of methanol were used (nMeOH:nester = 6:1). Liu and co-workers have also demonstrated DBU based ionic liquids, with which improved, more competitive, results could be achieved (1 h, 70 °C).71,72 Enthaler and co-workers have recently demonstrated the methanolysis of PLA by DMAP, DBU and TBD at high temperatures, achieving high yields within 10 min.53 However, reactions were performed at 180 °C, with high catalyst loading (5 mol%) and equivalents of MeOH (23.1), highlighting the mild conditions for the results achieved with 1.
The organocatalyst was also able to facilitate the ring-opening polymerisation of lactide (LA). For the polymerisation of rac-LA, while reasonable conversion is achieved within 2 h (LA]:[1]:[BnOH] = 100:1:1, 79%, toluene, 80 °C), there is evidence of backbiting, with meso-LA being observed in the crude 1H NMR spectrum (see ESI†). Accordingly, atactic PLA was observed (Pr = 0.50). The molecular weight of the polymer was lower than anticipated based on the amount of co-initiator, benzyl alcohol (Mn,theo = 11500 g mol−1, Mn, = 7400 g mol−1, Đ = 1.17). This reduced molecular weight can be attributed to the activation of the catalyst, which produces a stoichiometric amount of MeOH which can act as a chain transfer agent, halving the expected molecular weight. Indeed, MeOH and potential methoxy end group resonances are visible in the crude NMR spectrum. MALDI-ToF analysis of polymer derived from 1 shows a polymer distribution centred on 5900 g mol−1 and this molecular weight is consistent with methoxy and benzoxy initiation. This distribution is asymmetric with a tail towards low molecular weight. Peak spacing is observed to be 72 g mol−1 indicative of extensive transesterification of the growing chain. End group analysis is inconclusive, with MeOH or BnOH fitting reasonably well with the residual mass. For the polymerisation of L-LA under the same conditions (81%, Mn,theo = 11750 g mol−1, Mn, = 7550 g mol−1, Đ = 1.05), there is also evidence of epimerisation, with meso-LA again being observed as well as tetrads other than the expected [iii]. Therefore 1 is not suitable for polymerisation and is also much less active compared to literature organocatalysts.73,74 However, 1 is more relevant to chemical recycling where less control is required. The polymerisation of ε-CL under these conditions was unsuccessful.
Poly(caprolactone) (PCL) is a non-renewable polyester that has seen a resurgence in recent times, having biomedical applications (Scheme 1).75 The degradation of PCL (beads, Sigma Aldrich, Mn = 45000 g mol−1) was unsuccessful under the same solvent conditions (THF, 50 °C) applicable to PLA, despite complete solubilisation of the polymer. This may be related to an increased crystallinity and hydrophobicity making PCL more difficult to degrade. To achieve PCL degradation, it was necessary to increase the reaction temperature to 100 °C for which toluene is a more appropriate solvent. With 4 wt% of 1 (3.2 mol% relative to polymer repeat unit) high conversion towards methyl 6-hydroxyhexanoate was observed after 16 hours at this temperature. The degradation of PCL shows the versatility of this catalyst towards other polyester materials. While the product is not directly amenable to polymer reformation, it is still a value-added material with a relatively high market value (Fluorochem, 1 g, £315.00).
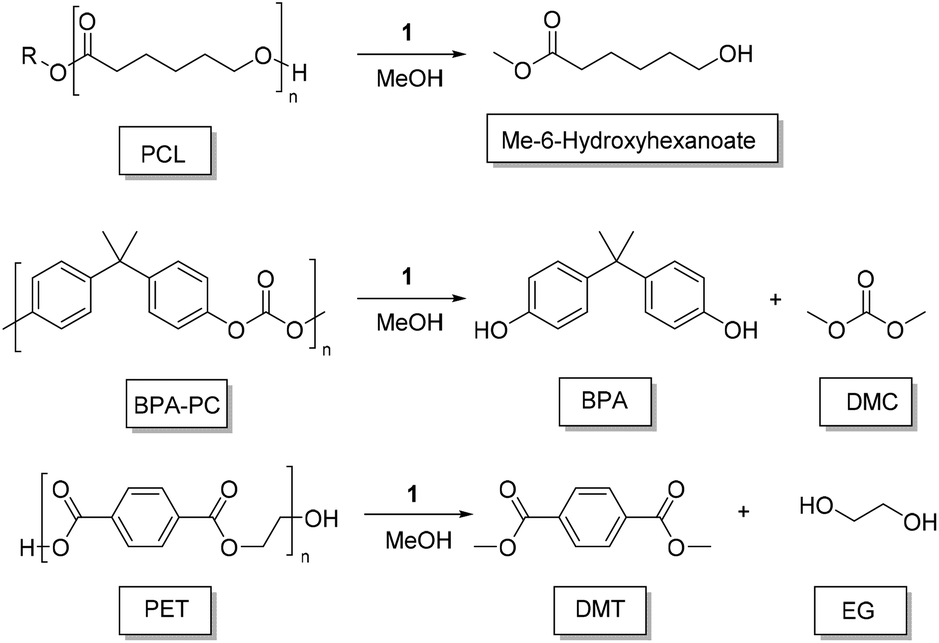 | ||
| Scheme 1 Further polymers degraded in this study. | ||
Polycarbonates (PC) represent a widely used class of thermoplastics with commodity and engineering applications. Poly(bisphenol A carbonate) (BPA-PC) is the most widely used PC, and is therefore an important material to be able to recycle efficiently.50,76 BPA-PC (Sigma Aldrich, Mn = 45000 gmol−1) was degraded in either 2-MeTHF or DMC at 75 °C,77 with 1 wt% of 1 (1.4 mol% relative to polymer repeat unit). Complete conversion of the polymer was observed after 3 h. Prior to the formation of BPA, mono- and di-carbonate BPA are formed. With 2Me-THF, selectivity is high towards BPA after 3 h (SBPA = 83%) whereas for DMC the BPA carbonate intermediates were more pronounced (SBPA = 42%). After 20 h, BPA-PC had been fully converted to BPA and DMC (see ESI†). While promising, and amenable to further optimisation, these results fall short compared to the application of literature examples (eg, TBD: 2 mol%, 2 h, 75 °C).72,77,78
For poly(ethylene terephthalate) (PET) (Bottle, Mn ∼ 40000 g mol−1),79 degradation is complicated by the general insolubility of the polymer. However, degradation could be performed as low as 100 °C with 4 wt% of 1 (5.4 mol%, relative to polymer repeat unit), relatively mild conditions for PET methanolysis, in toluene. Despite an extended reaction time (16 h), PET could be disassembled into dimethyl terephthalate (DMT) and ethylene glycol, with the diester being isolated by solvent removal and washing with MeOH (72% yield). PET degradation is typically performed, via glycolysis, at high temperatures (≥120 °C), with high conversion to BHET generally achieved within 1–12 h,40,41,55,72,80 and in exceptional cases within minutes.52,81 Relative to previous studies, we report milder conditions and, to the best of our knowledge, the first example of organocatalyzed PET methanolysis. To further demonstrate the applicability of this catalyst to PET recycling, the recovered DMT monomer was repolymerised {with Ti(IV) catalyst, see ESI†}. Under these conditions, recovered DMT gave PET with equivalent thermal properties (measured by DSC, Fig. 4) to that prepared using commercially available DMT (Sigma Aldrich, 99%). With a melt temperature of 253 °C, the recycled monomer gave a material with competitive properties that of the original PET bottle (Tm = 247 °C).
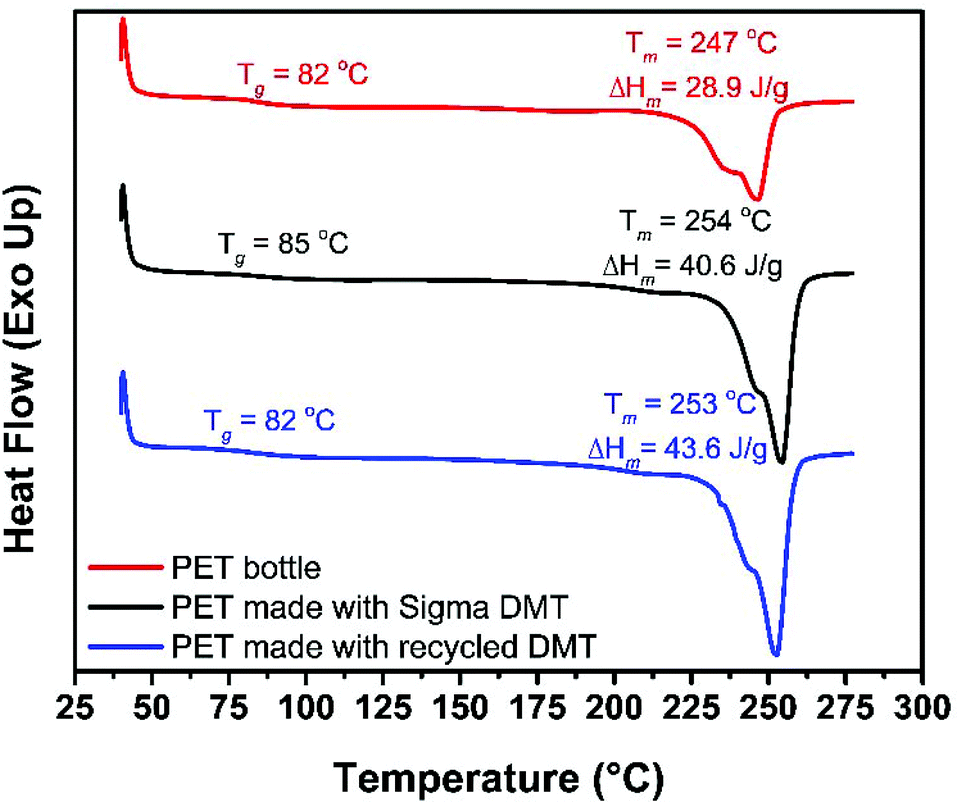 | ||
| Fig. 4 DSC trace of commercial and prepared PET samples. | ||
Conclusions
The chemical recycling of plastics is important to mitigate the pollution caused by these materials and to shift towards a more sustainable circular economy. We have reported the use of a simple organocatalyst capable of degrading a range of polyesters to their component parts, which represent value-added chemicals or monomers for repolymerisation. For PLA, we have demonstrated the use of a range of solvents in conjunction with the organocatalyst and the production of methyl- and ethyl-lactate. Kinetic data revealed a consecutive reaction pathway. PCL and PC have also been degraded to their components requiring higher temperatures to achieve this. PET has also been degraded by 1, with the di-ester being recovered and used to reform polymers with comparable thermal properties to the original material. Further work is ongoing to optimise the degradation of plastics in the presence of 1.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PM and LARR gratefully acknowledge the financial support of ESPRC (Grant No. EP/P016405/1). MK acknowledged the support of the EU Horizon 2020 research and innovation programme under grant agreement H2020-MSCA-CO-FUND, #665992 and Corbion.References
- A. L. Andrady and M. A. Neal, Philos. Trans. R. Soc., B, 2009, 364, 1977–1984 CrossRef CAS PubMed.
- S. Shafiee and E. Topal, Energy Policy, 2009, 37, 181–189 CrossRef.
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737–4753 RSC.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G. J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- M. R. Thomsett, J. C. Moore, A. Buchard, R. A. Stockman and S. M. Howdle, Green Chem., 2019, 21, 149–156 RSC.
- A. Bhaumik, G. I. Peterson, C. Kang and T.-L. Choi, J. Am. Chem. Soc., 2019, 141, 12207–12211 CrossRef CAS PubMed.
- R. C. Thompson, C. J. Moore, F. S. vom Saal and S. H. Swan, Philos. Trans. R. Soc., B, 2009, 364, 2153–2166 CrossRef CAS PubMed.
- R. Hatti-Kaul, L. J. Nilsson, B. Zhang, N. Rehnberg and S. Lundmark, Trends Biotechnol., 2019, 1–18 Search PubMed.
- M. R. Thomsett, T. E. Storr, O. R. Monaghan, R. A. Stockman and S. M. Howdle, Green Mater., 2016, 4, 115–134 CrossRef.
- H. C. Quilter, M. Hutchby, M. G. Davidson and M. D. Jones, Polym. Chem., 2017, 8, 833–837 RSC.
- P. VanWouwe, M. Dusselier, E. Vanleeuw and B. Sels, ChemSusChem, 2016, 9, 907–921 CrossRef CAS PubMed.
- E. T. H. Vink, S. Davies and J. J. Kolstad, Ind. Biotechnol., 2010, 6, 212–224 CrossRef CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- J. G. B. Derraik, Mar. Pollut. Bull., 2002, 44, 842–852 CrossRef CAS PubMed.
- L. Lebreton, B. Slat, F. Ferrari, B. Sainte-Rose, J. Aitken, R. Marthouse, S. Hajbane, S. Cunsolo, A. Schwarz, A. Levivier, K. Noble, P. Debeljak, H. Maral, R. Schoeneich-Argent, R. Brambini and J. Reisser, Sci. Rep., 2018, 8, 1–15 CrossRef CAS PubMed.
- T. Narancic, S. Verstichel, S. Reddy Chaganti, L. Morales-Gamez, S. T. Kenny, B. De Wilde, R. Babu Padamati and K. E. O'Connor, Environ. Sci. Technol., 2018, 52, 10441–10452 CrossRef CAS PubMed.
- M. Hong and E. X.-Y. Chen, Green Chem., 2017, 9, 3692–3706 RSC.
- J. Payne, P. Mckeown and M. D. Jones, Polym. Degrad. Stab., 2019, 165, 170–181 CrossRef CAS.
- H. Sardon and A. P. Dove, Science, 2018, 360, 380–381 CrossRef CAS PubMed.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- S. Aparicio and R. Alcalde, Green Chem., 2009, 11, 65–78 RSC.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
- R. Mülhaupt, Macromol. Chem. Phys., 2013, 214, 159–174 CrossRef.
- F. Gironi, S. Frattari and V. Piemonte, J. Polym. Environ., 2016, 24, 328–333 CrossRef CAS.
- S. Planer, A. Jana and K. Grela, ChemSusChem, 2019, 12, 4655–4661 CrossRef CAS PubMed.
- R. De Clercq, M. Dusselier, C. Poleunis, D. P. Debecker, L. Giebeler, S. Oswald, E. Makshina and B. F. Sels, ACS Catal., 2018, 8, 8130–8139 CrossRef CAS.
- M. Ghadamyari, S. Chaemchuen, K. Zhou, M. Dusselier, B. F. Sels, B. Mousavi and F. Verpoort, Catal. Commun., 2018, 114, 33–36 CrossRef CAS.
- M. Dusselier, P. Van Wouwe, A. Dewaele, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 78–80 CrossRef CAS PubMed.
- S. Da Ros, R. S. Braido, N. L. de Souza e Castro, A. L. T. Brandão, M. Schwaab and J. C. Pinto, J. Anal. Appl. Pyrolysis, 2019, 144, 104706 CrossRef CAS.
- F. Kopinke, M. Remmler, K. Mackenzie, M. Moder and O. Wachsen, Polym. Degrad. Stab., 1996, 53, 329–342 CrossRef CAS.
- M. Noda and H. Okuyama, Chem. Pharm. Bull., 1999, 47, 467–471 CrossRef CAS.
- Y. Fan, H. Nishida, Y. Shirai, Y. Tokiwa and T. Endo, Polym. Degrad. Stab., 2004, 86, 197–208 CrossRef CAS.
- L. Feng, S. Feng, X. Bian, G. Li and X. Chen, Polym. Degrad. Stab., 2018, 157, 212–223 CrossRef CAS.
- A. Undri, L. Rosi, M. Frediani and P. Frediani, J. Anal. Appl. Pyrolysis, 2014, 110, 55–65 CrossRef CAS.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022–1028 CrossRef.
- A. B. Raheem, Z. Z. Noor, A. Hassan, M. K. Abd Hamid, S. A. Samsudin and A. H. Sabeen, J. Cleaner Prod., 2019, 225, 1052–1064 CrossRef CAS.
- S. Wang, C. Wang, H. Wang, X. Chen and S. Wang, Polym. Degrad. Stab., 2015, 114, 105–114 CrossRef CAS.
- A. M. Al-Sabagh, F. Z. Yehia, A. M. M. F. Eissa, M. E. Moustafa, G. Eshaq, A. R. M. Rabie and A. E. Elmetwally, Ind. Eng. Chem. Res., 2014, 53, 18443–18451 CrossRef CAS.
- C. Jehanno, I. Flores, A. P. Dove, A. J. Müller, F. Ruipérez and H. Sardon, Green Chem., 2018, 20, 1205–1212 RSC.
- V. Sinha, M. R. Patel and J. V. Patel, J. Polym. Environ., 2010, 18, 8–25 CrossRef CAS.
- K. R. Delle Chiaie, F. R. McMahon, E. J. Williams, M. J. Price and A. P. Dove, Polym. Chem., 2020, 11, 1450–1453 RSC.
- J. Hopewell, R. Dvorak and E. Kosior, Philos. Trans. R. Soc., A, 2009, 364, 2115–2126 CrossRef CAS PubMed.
- K. Fukushima, J. M. Lecuyer, D. S. Wei, H. W. Horn, G. O. Jones, H. A. Al-Megren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, J. E. Rice and J. L. Hedrick, Polym. Chem., 2013, 4, 1610–1616 RSC.
- L. A. Román-Ramírez, P. Mckeown, M. D. Jones and J. Wood, ACS Catal., 2019, 9, 409–416 CrossRef.
- R. Petrus, D. Bykowski and P. Sobota, ACS Catal., 2016, 6, 5222–5235 CrossRef CAS.
- C. Fliedel, D. Vila-Viçosa, M. J. Calhorda, S. Dagorne and T. Avilés, ChemCatChem, 2014, 6, 1357–1367 CrossRef CAS.
- P. McKeown and M. D. Jones, Sustainable Chem., 2020, 1, 1–22 CrossRef.
- C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172–186 RSC.
- F. A. Leibfarth, N. Moreno, A. P. Hawker and J. D. Shand, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4814–4822 CrossRef CAS.
- K. Fukushima, O. Coulembier, J. M. Lecuyer, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. Mcneil, P. Dubois, R. M. Waymouth, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1273–1281 CrossRef CAS.
- C. Alberti, N. Damps, R. R. R. Meißner and S. Enthaler, ChemistrySelect, 2019, 4, 6845–6848 CrossRef CAS.
- X. Song, X. Zhang, H. Wang, F. Liu, S. Yu and S. Liu, Polym. Degrad. Stab., 2013, 98, 2760–2764 CrossRef CAS.
- Q. Wang, X. Yao, S. Tang, X. Lu, X. Zhang and S. Zhang, Green Chem., 2012, 14, 2559–2566 RSC.
- H. Wang, Z. Li, Y. Liu, X. Zhang and S. Zhang, Green Chem., 2009, 11, 1568–1575 RSC.
- M. Hatano, Y. Tabata, Y. Yoshida, K. Toh, K. Yamashita, Y. Ogura and K. Ishihara, Green Chem., 2018, 20, 1193–1198 RSC.
- S. Shirakawa, S. Liu, S. Kaneko, Y. Kumatabara, A. Fukuda, Y. Omagari and K. Maruoka, Angew. Chem., Int. Ed., 2015, 54, 15767–15770 CrossRef CAS PubMed.
- M. T. Reetz, S. Hütte and R. Goddard, J. Am. Chem. Soc., 1993, 115, 9339–9340 CrossRef CAS.
- C. E. Cannizzaro and K. N. Houk, J. Am. Chem. Soc., 2002, 124, 7163–7169 CrossRef CAS PubMed.
- P. McKeown, L. A. Román-Ramírez, S. Bates, J. Wood and M. D. Jones, ChemSusChem, 2019, 5233–5238 CrossRef CAS PubMed.
- L. A. Román-Ramírez, P. McKeown, M. D. Jones and J. Wood, ACS Omega, 2020, 5, 5556–5564 CrossRef PubMed.
- L. A. Roman Ramirez, P. McKeown, C. Shah, J. Abraham, M. D. Jones and J. Wood, Ind. Eng. Chem. Res., 2020, acs.iecr.0c01122 CrossRef.
- T. G. F. Nederberg, E. F. Connor and J. L. Hedrick, Chem. Commun., 2011, 0, 2066–2067 Search PubMed.
- F. Liu, J. Guo, P. Zhao, Y. Gu, J. Gao and M. Liu, Polym. Degrad. Stab., 2019, 167, 124–129 CrossRef CAS.
- K. Watanabe, N. Yamagiwa and Y. Torisawa, Org. Process Res. Dev., 2007, 11, 251–258 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed.
- D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156–159 CrossRef CAS.
- Y. E. Jad, G. A. Acosta, T. Govender, H. G. Kruger, A. El-Faham, B. G. De La Torre and F. Albericio, ACS Sustainable Chem. Eng., 2016, 4, 6809–6814 CrossRef CAS.
- F. Liu, J. Guo, P. Zhao, Y. Gu, J. Gao and M. Liu, Polym. Degrad. Stab., 2019, 167, 124–129 CrossRef CAS.
- M. Liu, J. Guo, Y. Gu, J. Gao and F. Liu, ACS Sustainable Chem. Eng., 2018, 6, 15127–15134 CrossRef CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- J. G. Kim, Polym. Chem. 10.1039/C9PY01927H.
- T. Do, E. R. Baral and J. G. Kim, Polymer, 2018, 143, 106–114 CrossRef CAS.
- E. Quaranta, D. Sgherza and G. Tartaro, Green Chem., 2017, 19, 5422–5434 RSC.
- S. Farah, K. R. Kunduru, A. Basu and A. J. Domb, in Poly(Ethylene Terephthalate) Based Blends, Composites and Nanocomposites, eds. P. M. Visakh and M. Liang, Elsevier, Oxford, 2015, pp. 143–165 Search PubMed.
- J. Sun, D. Liu, R. P. Young, A. G. Cruz and N. G. Isern, ChemSusChem, 2018, 781–792 CrossRef PubMed.
- K. Fukushima, D. J. Coady, G. O. Jones, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2013, 1606–1611 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc01252a |
| This journal is © The Royal Society of Chemistry 2020 |