
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combined lignin defunctionalisation and synthesis gas formation by acceptorless dehydrogenative decarbonylation†
Zhenlei
Zhang
 ,
Douwe S.
Zijlstra
,
Ciaran W.
Lahive
and
Peter. J.
Deuss
*
,
Douwe S.
Zijlstra
,
Ciaran W.
Lahive
and
Peter. J.
Deuss
*
Department of Chemical Engineering (ENTEG), University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: p.j.deuss@rug.nl
First published on 13th May 2020
Abstract
The valorization of lignin, consisting of various phenylpropanoids building blocks, is hampered by its highly functionalized nature. The absence of the γ-carbinol group in an unnatural C2 β-O-4 motif compared to the native lignin C3 β-O-4 motif provides great opportunities for developing new valorization routes. Thus efficient defunctionalisation approaches that transform the C3 β-O-4 motif into a simplified C2 β-O-4 motif are of interest. Based on a study with a series of model compounds, we established a feasible application of an iridium-catalysed acceptorless dehydrogenative decarbonylation method to efficiently remove the γ-carbinol group in a single step. This defunctionalisation generates valuable synthesis gas, which can be collected as a reaction product. By this direct catalytic transformation, a yield of ∼70% could be achieved for a C3 β-O-4 model compound that was protected from undergoing retro-aldol cleavage by alkoxylation of the benzylic secondary alcohol in the α position. A phenylcoumaran model compound containing a γ-carbinol group as well as a benzylic primary alcohol also proved to be reactive under dehydrogenative decarbonylation conditions, which can further contribute to the reduction of the structural complexity of lignin. Notably, the liberation of synthesis gas was confirmed and the signals for the defunctionalized C2 β-O-4 motif were observed when this dehydrogenative decarbonylation approach was applied on organosolv lignins. This selective defunctionalized lignin in conjunction with the formation of synthesis gas has the potential to enhance the development of profitable and sustainable biorefineries.
 Peter J. Deuss | Peter J. Deuss completed his studies at the University of Amsterdam and thereafter joined the group of Paul Kamer at the University of St. Andrews as a PhD student. He obtained his degree in 2011 and after working at the MRC UK, Laboratory of Molecular Biology Cambridge, he moved to the University of Groningen where, after post-doctoral work in the groups of Katalin Barta and Erik Heeres, he started in 2016 as a tenure-track assistant professor in green and smart biomass processing at the Chemical Engineering Department of the Engineering and Technology Institute Groningen (ENTEG). |
Introduction
Lignin, an amorphous cross-linked aromatic biopolymer, represents a major structural component of lignocellulosic biomass (up to 40%).1,2 Efficient lignin valorisation is of vital importance for the implementation of profitable and sustainable biorefinery schemes for biomass.3–6 Due to its rich aromatic structure, lignin has been identified as a renewable source for a range of key aromatic chemicals that are currently derived from petrochemical routes or as the aromatic component in resins and polymers.7–10 To this end, many elegant selective catalytic chemical reactions have been developed to adjust the chemical structure of lignin or deconstruct it for targeted aromatic chemicals.11–17 As the β-O-4 linking motif accounts for over 50% of the linking motifs that connect the aromatic units in lignin, β-O-4 model compounds are most commonly applied for the development of catalytic methodology in order to avoid the structural complexity of extracted polymeric lignin.18–23 There are two main types of model compounds representing the β-O-4 linking motif currently being applied in such studies: (1) C3 β-O-4 compounds (A-type) that provide a matching chemical representation of the β-O-4 motif most commonly found in native lignin and (2) the C2 β-O-4 compounds (B-type) that are defunctionalized β-O-4 motifs without a γ-carbinol group and easier to access synthetically (Fig. 1a).24–27 The application of the B-type linkage motif was reported to avoid side reactions such as the retro-aldol reaction, which is a typical side reaction for the A-type linkage motif, especially under oxidative depolymerisation conditions.21,28–30 Thus, the use of B-type linkage motif allows for some innovative elegant approaches to valorise lignin that are not accessible for the polymeric lignin that contains A-type β-O-4 motifs.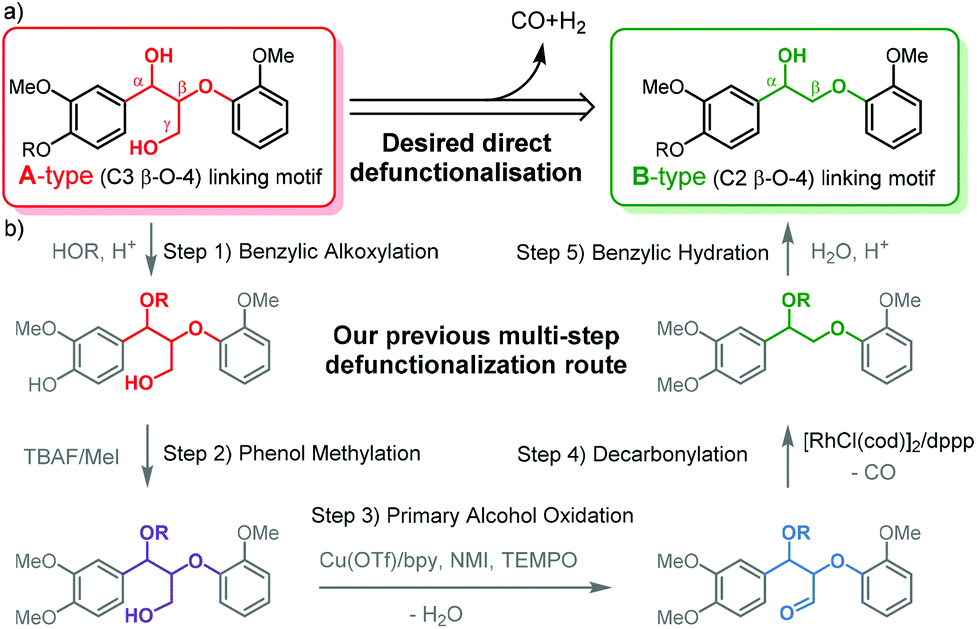 | ||
| Fig. 1 (a) The two types of β-O-4 linkage motifs commonly represented in model compounds used in the chemical methodology development for lignin valorisation and (b) multistep defunctionalisation of alcohol incorporated organosolv lignin via methylation, oxidation and decarbonylation demonstrated in our previous work.31 | ||
For example, Bergman and Ellman et al. demonstrated a hydrogen neutral cleavage method based on B-type dimeric model compounds as well as a B-type synthetic polymer to afford valuable acetophenone.13 However, James et al. reported that this procedure was ineffective for A-type model compounds due to the chelation of the two hydroxyl groups to the catalyst.32 With respect to this discrepancy, the efficient lignin defunctionalisation to remove the γ-carbinol functionality and thus transform an A-type into a B-type structure can allow for the application of such an alternative depolymerisation technology developed for B-type model compounds. Additionally, the defunctionalisation changes other polymer properties such as stability towards condensation18 and solubility in organic solvents due to the removal of hydroxyl groups.33 Previously, we have demonstrated the transformation of A- into B-type structures by stepwise catalytic modification that removed the γ-carbinol functionality over several steps (Fig. 1b).31 This stepwise approach relied on the selective oxidation of the γ-hydroxy group to the aldehyde and a subsequent decarbonylation step. To allow selective oxidation and prevent decomposition via retro-aldol, alkoxylated A-type structures were used as the starting point.29–31 Such alkoxylated structures are readily accessible by organosolv extraction with alcohol under specific conditions.33–35 Nonetheless, the results obtained from the application of this stepwise methodology on ethanol extracted lignin showed a considerable loss of β-O-4 linkages during the catalytic oxidation step, demonstrating that there is significant room for improvement. In addition, the oxidation method did not tolerate the presence of phenol moieties in lignin; this implied that an additional phenol methylation step using environmentally undesirable MeI was necessary.36–38 In order to overcome these drawbacks and provide a shorter route, the application of a single dehydrogenative decarbonylation step that is compatible with free phenols that are inherently present in lignin is attractive.
A recent study by Bruijnincx et al. demonstrated the application of an acceptorless dehydrogenation method to depolymerize lignin allowing for simplified product mixtures (Fig. 2a).30 This methodology relied on the dehydrogenation of the γ-hydroxy group in the β-O-4 structure, subsequent retro-aldol (Cα–Cβ) bond cleavage and finally in situ stabilisation of the formed aldehyde by conversion to the corresponding carboxylic acid or alcohol. Acceptorless decarbonylation itself has also been reported to be an effective approach for in situ stabilization of the lignin acidolysis intermediate aldehydes, removing the aldehyde functionality to avoid undesired repolymerisation.11,39 However, a coupled acceptorless dehydrogenative decarbonylation has not yet been applied for lignin valorisation. For this, we were inspired by the works of the group of Madsen and Sadow40–42 who achieved acceptorless dehydrogenative decarbonylation reactions on a variety of primary alcohols by using iridium or rhodium-based homogeneous catalysts, importantly in conjunction with the release of valuable syngas (Fig. 2b).
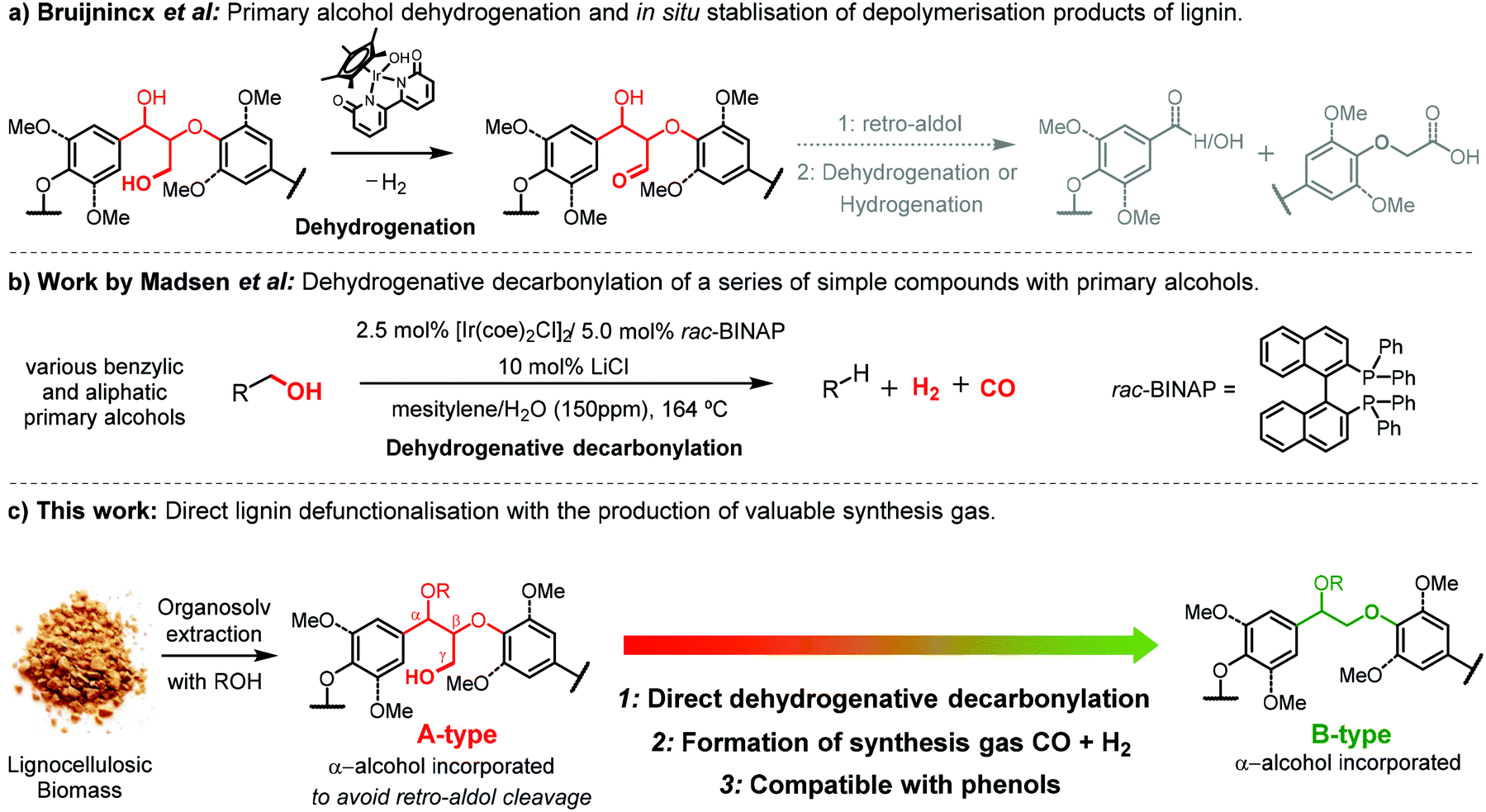 | ||
| Fig. 2 (a) Overview of the primary alcohol dehydrogenation and hydrogen shuttling for in situ stabilisation of depolymerisation products of lignin developed by Bruijnincx et al.30 (b) Dehydrogenative decarbonylation of a series of simple primary alcohols demonstrated by Madsen et al.40 (c) Direct dehydrogenative decarbonylation to defunctionalize a high alcohol incorporated lignin demonstrated in this work. | ||
Syngas or synthesis gas, constituting of CO and H2, is an important feedstock for both the chemical and energy industries; the syngas provides a great opportunity for sustainable energy developments.43 The production of syngas from biomass is considered to be an attractive and sustainable route. Biomass-derived syngas can be typically obtained by gasification of various lignocellulose feedstock or industrial wastes from the pulp and paper industry.44 However, harsh temperatures ranging from 600 to 900 °C are applied in gasification technology with the formation of large amounts of biochar as the side-product.45 In addition, undesirable contaminants, such as nitrogen and sulphur based compounds, exist in the raw biomass gasification derived syngas, combined with the formation of tars impose the necessity for further cleanup.46 In contrast, a much milder temperature is required for the production of syngas by acceptorless dehydrogenative decarbonylation catalytic reaction (around 160 °C). Conceptually, by targeting primary alcohol groups in lignin, much cleaner syngas with a relatively stable syngas composition would also be expected in spite of lignin source variability.
In this contribution, we subsequently evaluated a combination of carefully selected lignin model compounds to show the potential of using the iridium-based acceptorless dehydrogenative decarbonylation40 for lignin defunctionalisation (Fig. 2c). In particular, we aimed for application of this methodology in lignins with high proportions of β-O-4 linkages in order to allow for selective defunctionalisation of an A-type lignin structure to a B-type structure, allowing for further β-O-4 targeted catalytic valorisation. For this purpose, different lignin model compounds were applied to verify that the reaction is compatible with phenols and indeed releases syngas for collection. Then α-alkoxylated A-type model compounds were used to evaluate that the defunctionalisation is possible without degradation via retro-aldol reactions. Finally, α-alkoxylated walnut lignins were used to demonstrate the formation of defunctionalized β-O-4 linkages using this methodology.
Results and discussion
Dehydrogenative decarbonylation of 4-methoxybenzenepropanol (BP1)
Targeting the defunctionalisation of lignin, we previously reported the sequential oxidation of the γ-cabinol group followed by decarbonylation to release CO. We aimed to simplify this process by performing these two key transformations in sequence in one step (steps 3 and 4, Fig. 1b). Additionally, we saw the opportunity for the release of syngas by acceptorless dehydrogenation and decarbonylation in conjunction with the targeted defunctionalisation of the native β-O-4 motif at the γ position. Of the reported catalytic systems that can perform both dehydrogenation and decarbonylation of primary alcohols without being deactivated by the formation of syngas, we found that the one reported by Madsen et al. consisting of a commercial iridium precursor and BINAP the most convenient to use.40 In this catalytic system, [Ir(coe)2Cl]2 or [Ir(cod)Cl]2 serves as a metal precursor with rac-BINAP as a ligand and LiCl as an additive in mesitylene. In addition, the catalytic solution is saturated with water (150 ppm), which leads to overall improved performance although the mechanism for this remains uncertain. This catalyst was reported to achieve good product yields for both benzylic and non-benzylic primary alcohols.40 Initially, we tested this reported iridium-catalysed dehydrogenative decarbonylation procedure on 4-methoxybenzenepropanol (BP1), which mimics the presence of a γ-carbinol group in lignin (Fig. 3). However, the reaction with the reported conditions proceeded sluggishly. The reaction with BP1 in mesitylene at 165 °C only yielded 23% of 1-ethyl-4-methoxybenzene (BE1) after 36 hours, although with a promising 81% selectivity (Fig. 3a).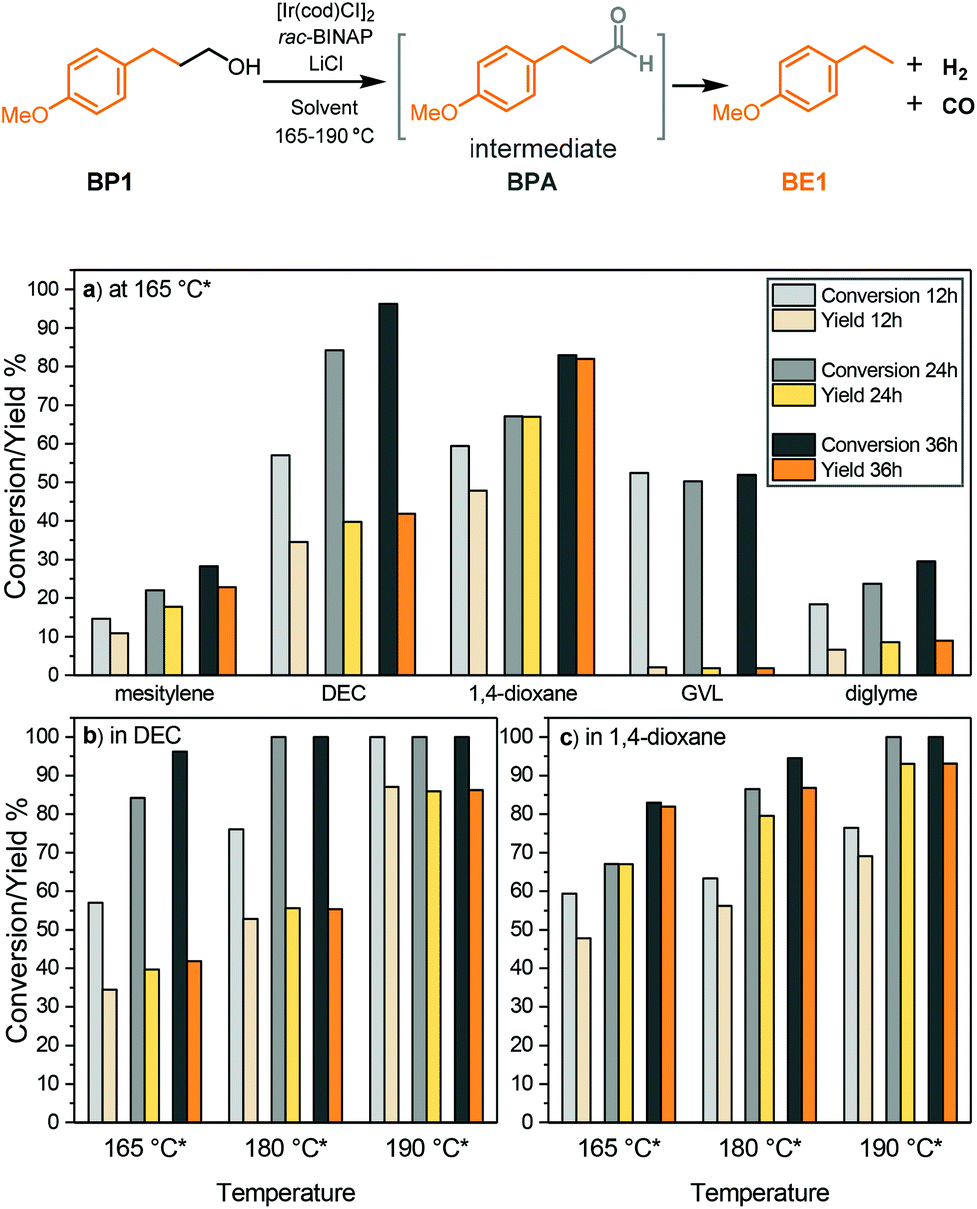 | ||
Fig. 3 Overview of the iridium catalysed dehydrogenative decarbonylation of 4-methoxybenzenepropanol (BP1) to 1-methoxy-4-ethylbenzene (BE1). (a) Solvent screening at 165 °C*, (b) temperature screening in diethyl carbonate, and (c) temperature screening in 1,4-dioxane. Reaction conditions: 15 mg substrate, with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:4 ratio of [Ir(cod)Cl]2:rac-BINAP:LiCl (2.5 mol% [Ir(cod)Cl]2, other components were adjusted to the [Ir(cod)Cl]2 amount to maintain the ratio), performed under a N2 atmosphere in 2 mL solvent. Yield and conversion were determined by GC-FID using n-octadecane as an internal standard. *Set temperature of the heating block; the measured corresponding inside temperature is shown in Table S1.† :2:4 ratio of [Ir(cod)Cl]2:rac-BINAP:LiCl (2.5 mol% [Ir(cod)Cl]2, other components were adjusted to the [Ir(cod)Cl]2 amount to maintain the ratio), performed under a N2 atmosphere in 2 mL solvent. Yield and conversion were determined by GC-FID using n-octadecane as an internal standard. *Set temperature of the heating block; the measured corresponding inside temperature is shown in Table S1.† | ||
We were keen to move away from using mesitylene as it is not a suitable solvent for dissolving most types of lignin. Therefore, we screened a set of alternative solvents that are more polar and are known to have a higher capability for dissolution of lignin (Fig. 3a). To our delight the use of diethyl carbonate, considered to be a green organic solvent,47,48 had a significant positive effect on the reaction rate, reaching 35% yield with 60% selectivity after 12 hours at 165 °C (Fig. 3a). Notably, substantially cleaner GCMS spectra were obtained by using diethyl carbonate instead of mesitylene (Fig. S1 and S2a†). The dehydrogenation intermediate 4-methoxybenzenepropanal (BPA) was also observed in the spectra, though the amount was very low (Fig. S1†); therefore we suspected the presence of some higher molecular weight species that were not visible by GC to explain the poor carbon balance. The yield of BE1 then could be increased to 42% by leaving the reaction for 36 hours although the selectivity decreased to 44%. At the same time, it was noteworthy that the inside temperature of the reaction was lower than 165 °C that was used as a set temperature for the heating block, which corresponds to the boiling point of mesitylene that was used in the reported procedure. Therefore, the inside temperature was measured and found to be only 139 °C. One reason for this difference was the partial refluxing of the solvent (boiling point of diethyl carbonate = 126 °C), as the reaction vessel was only heated at the bottom. Thus, the differences between the actual inside temperature and set temperature are listed for solvents with a relatively low boiling point (Table S1†). Another such solvent that was tested was 1,4-dioxane. Although 1,4-dioxane may impose potential risks for human health and the environment,47,49 it is still one of the best lignin extraction media due to its ability to dissolve many types of lignin as well as its stability during the lignin solvolysis process.33,50 By changing the solvent to 1,4-dioxane, the yield of BE1 was greatly improved to 82%, along with a satisfactory conversion of 83% at 165 °C (inside 130 °C) after 36 hours (Fig. 3a). Some polar solvents with significantly higher boiling points were also tested. However, in diglyme also the selectivity remained below 40% at all times and in GVL tremendous side reactions were observed, leading to very poor selectivity (Fig. S2d†).
Based on the preliminary solvent screening at 165 °C, diethyl carbonate and 1,4-dioxane were selected for further investigation at increased temperature. Diethyl carbonate as a solvent at 180 °C (inside 145 °C) leads to a 14% increase in the yield at 36 hours compared to the reaction performed at 165 °C; however, the selectivity was only 55%, with still no apparent identifiable side products (Fig. 3b). A further increase in the reaction temperature to 190 °C (inside 148 °C) led to full conversion of BP1 as well as remarkably improved selectivity. Within 12 hours, the yield of BE1 reached 86% and maintained relatively constant despite an increase in time, indicating that the product was stable in solution. In 1,4-dioxane, the yield after 36 hours was enhanced from 82% to 93% by increasing the temperature from 180 °C to 190 °C (inside temperatures of 138 and 142 °C, respectively). (Fig. 3c). Overall, these stepwise screenings show the possibility of applying the dehydrogenative decarbonylation of primary alcohols in 1,4-dioxane and diethyl carbonate, with elevated temperatures being beneficial for both yield and selectivity.
Dehydrogenative decarbonylation of 4-hydroxy-3-methoxybenzenepropanol (BP2)
The presence of phenol groups in lignin has proven to be detrimental to many strategies that have taken an oxidative approach to the γ-cabinol group in lignin as it frequently results in an insoluble and intractable material.31,38 To prevent this problem, an additional methylation step using TBAF/MeI was required to convert phenols prior to selective primary alcohol oxidation (step 2, Fig. 1b).31,38 Nevertheless, this environmentally unfriendly methylation step can be avoided when the lignin defunctionalisation catalyst can tolerate phenol moieties. Therefore, 4-hydroxy-3-methoxybenzenepropanol (BP2), containing a phenol, was used as a model compound for the dehydrogenative decarbonylation catalyst in both 1,4-dioxane and diethyl carbonate.The reactions were performed at 165, 180 and 195 °C and sampled at 24 and 36 hours. Compared with the non-phenolic model compound BP1, much better conversion of BP2 and yield of 4-ethylphenol (EP2) were already observed at 165 °C in both diethyl carbonate and 1,4-dioxane (Fig. 4, entries 1–4). On increasing the temperature to 180 °C and 190 °C, full conversion and >98% selectivity were achieved for both solvents within 24 hours (Fig. 4, entries 5–12). The results clearly show that this phenolic model BP2 is actually more selectively converted at higher rates compared to its non-phenolic counterpart. The catalysts might benefit from an increase in the acidity of the solution, which might be one of the reasons for the requirement of the addition of H2O as an additive. This defunctionalisation approach thus allows for the combination of the dehydrogenation and decarbonylation steps, as well as the omission of the toxic phenol methylation step.
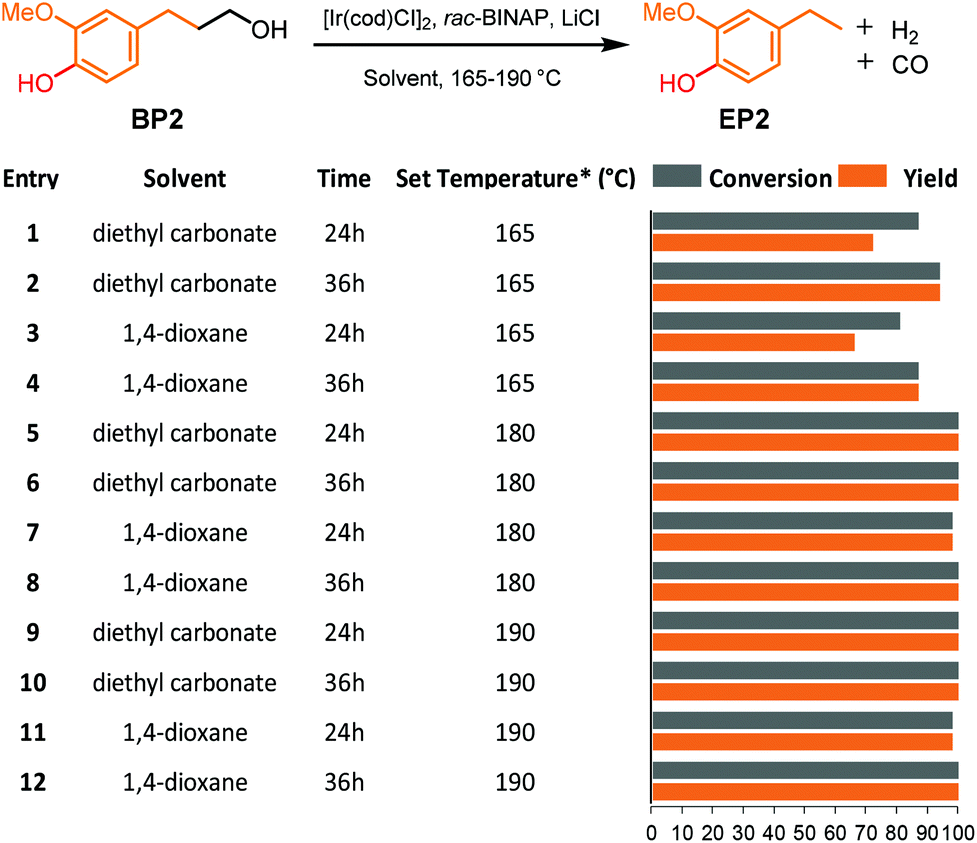 | ||
| Fig. 4 Overview of the dehydrogenative decarbonylation of 4-hydroxy-3-methoxybenzenepropanol (BP2) to 2-methoxy-4-ethylphenol (EP2). Reaction conditions: 15 mg substrate, [Ir(cod)Cl]2 2.5 mol%, 4:2:1 LiCl:rac-BINAP:[Ir(cod)Cl]2, 165-190 °C in 2 mL solvent. Yield and conversion were determined by GC-FID using n-ocatadecane as an internal standard. *Set temperature of the heating block; the measured corresponding inside temperature is shown in Table S1.† | ||
Next, we aimed to confirm the release of valuable syngas. For this purpose, gas evolution was monitored for the reaction with BP2 in diethyl carbonate at 190 °C. 1 mmol starting material was reacted in a flask with condensers, connected with a water-filled burette for the quantification of gas formation monitored over time. As shown in Fig 5, the volume of syngas steadily increased as a function of time. Finally, 38.3 mL syngas was produced at about 30 hours, which is equivalent to 1.55 mmol syngas calculated using the ideal gas equation of state. This equates to an 82% yield of syngas being produced with 100% selectivity for EP2 as detected by GC-FID analysis, with theoretically maximum yields being calculated as 40.7 mL of syngas. Furthermore, the CO:H2 ratio analysed by GC-TCD was 1.1:1, indicating the successful implementation of acceptorless dehydrogenation and decarbonylation without the requirement of a sacrificial agent. Interestingly, a small amount of CO2 was also detected in the gas phase, which could derive from the water–gas shift reaction between the liberated CO and the added water from the catalyst preparation. Additionally, the steady continued gas formation indicated that the catalyst appears to be relatively stable.
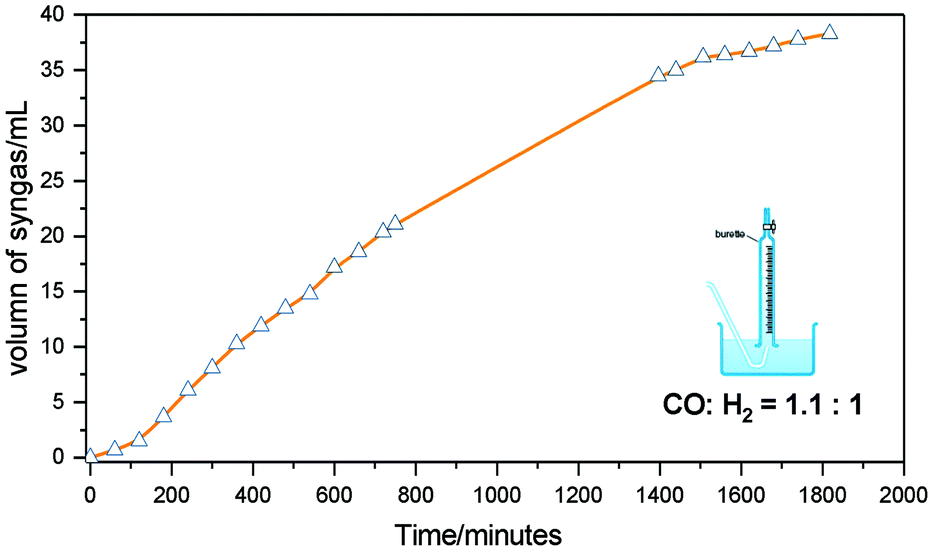 | ||
| Fig. 5 Syngas formation over time using 4-hydroxy-3-methoxybenzenepropanol (BP2) as a model compound. Reaction conditions: 1 mmol substrate, [Ir(cod)Cl]2 2.5 mol%, rac-BINAP 5 mol%, LiCl 10 mol%, 190 °C (setting temperature of the oil bath) in 2 mL solvent. Yield and conversion were determined by GC-FID using n-ocatadecane as external standard. | ||
Catalytic defunctionalisation of A-OEt and A-OBuvia dehydrogenative decarbonylation
With the successful demonstration of acceptorless catalytic dehydrogenative decarbonylation on benzenepropanol model compounds, the α-ethoxylated β-O-4 motif A-OEt was tested as the starting material in the same catalytic system. Compared with the native β-O-4 motif A-OH, the α-ethoxylated A-OEt motif avoids decomposition via retro-aldol reaction29,30 under these catalytic conditions and thus allows for selective modification on the γ position of the motif followed by defunctionalisation.31 Furthermore, this motif is readily accessible by the extraction of lignin with ethanol.31,33 When A-OEt in 1,4-dioxane was heated to 190 °C in the presence of 5 mol% of [Ir(cod)Cl]2, only 27% yield was observed with full conversion of A-OEt (Fig. 6, entry 1). When the reaction time was doubled to 48 hours, the yield increased only by 8% reaching 35% (Fig. 6, entry 2). An increase in the temperature to 200 °C (inside 146 °C) led to further improvement, achieving 46% yield at 24 hours and 59% yield at 48 hours (Fig. 6, entries 3 and 4). Yet, there was no further improvement in the product yield at 210 °C (inside 150 °C, Fig. 6, entry 5). In these reactions, the main side products were identified as veratrylglycol-β-guaiacyl ether (SP1), 1,2-dimethoxy-4-ethylbenzene (SP2) and guaicol (SP3), see Scheme 1 and Fig. S3.† There were two possible routes for the formation of these side products (Scheme 1). Either the desired defunctionalized β-O-4 motif B-OEt eliminated ethanol affording SP, which can undergo hydrogenation resulting in SP1 and further hydrogenolysis to SP2 and SP3 or the starting β-O-4 motif A-OEt was directly cleaved by hydrogenolysis to obtain guaiacol (SP3) and SP4, which was converted by dehydrogenative decarbonylation to SP2. SP4 was not detected by GC-MS, but the route through SP4 cannot be excluded as its formation might be obscured due its fast consumption by the dehydrogenative decarbonylation catalyst. Both routes require the consumption of hydrogen, which is formed in the catalytic dehydrogenation reaction.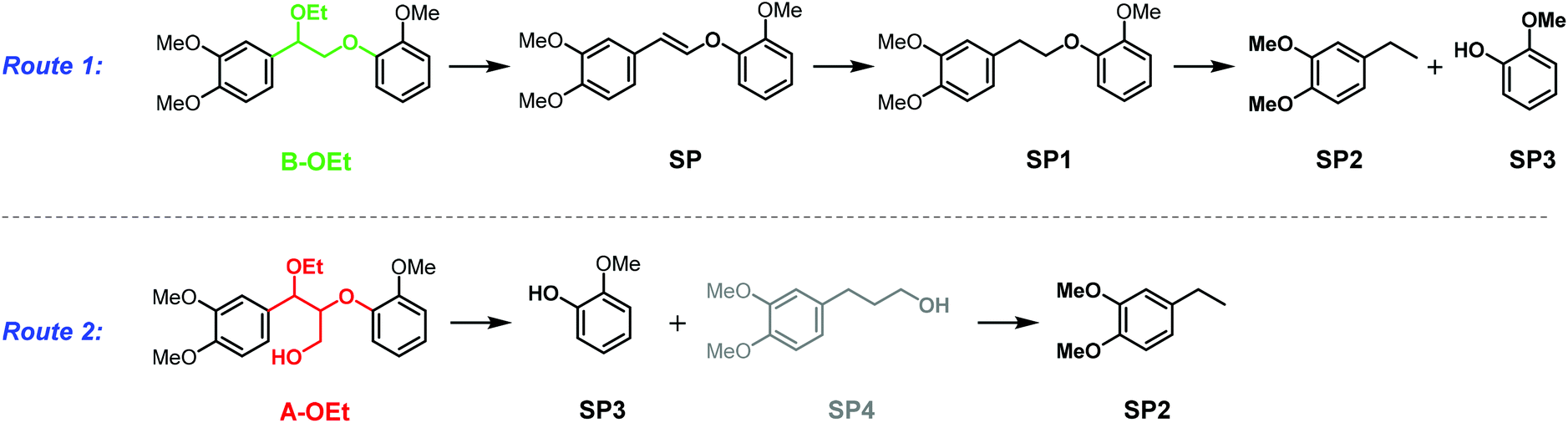 | ||
| Scheme 1 Possible side reactions during dehydrogenative decarbonylation of A-OEt or A-OBu. | ||
 | ||
| Fig. 6 Catalytic dehydrogenative decarbonylation A-OEt or A-OBu to B-OEt or B-OBu. Reaction conditions: 15 mg substrate, [Ir(cod)Cl]2 5 mol%, 4:2:1 LiCl:rac-BINAP:[Ir(cod)Cl]2, as catalyst; 190–210 °C in 1 mL solvent. Yield and conversion were determined by GC-FID using n-decane as an external standard; *Set temperature of the heating block; the measured corresponding inside temperature is shown in Table S1.† | ||
In addition, we tested this catalyst on the α-butoxylated model compound A-OBu, which represents the β-O-4 motif obtained from lignin from n-butanol extraction instead of ethanol extraction.34,51 The reaction of A-OBu at 200 °C for 24 hours in 1,4-dioxane gave 57% yield of B-OBu, which was comparable to 59% yield obtained for A-OEt (Fig. 6, entry 6). An even higher yield of 70% was obtained by doubling the reaction time to 48 hours (Fig. 6, entry 7). In order to show its compatibility for a greener solvent, this reaction was also performed in diethyl carbonate at 200 °C (inside 154 °C); however, the yield dropped dramatically to only 22% even after 48 hours (Fig. 6, entry 8). The GC spectra of this mixture showed two new signals, which were identified to correspond to two diastereomers of an A-OBu analogue on which ethyl carbonate got attached on the γ position in conjunction with ethanol liberation (Fig. S4 and S5†). This side reaction accounted for the low yield of the B-OBu in diethyl carbonate. However, due to the reversible nature of this side reaction, this could be avoided to a certain extent by increasing the reaction temperature. At 210 °C (inside 159 °C) a yield of 61% of B-OBu was achieved at 24 hours and 65% yield was obtained at 48 hours (Fig. 6, entries 9 and 10). In summary, this dehydrogenative decarbonylation catalytic system could work satisfactorily for α-ethoxylated and butoxylated β-O-4 motifs at 200–210 °C which corresponds to an inside temperature of 146–150 °C in 1,4-dioxane and 154–159 °C in diethyl carbonate.
Dehydrogenative decarbonylation of other primary alcohol containing lignin structures
Considering the presence of other structures in lignin that contain primary alcohols, we further examined the effectiveness of dehydrogenative decarbonylation on a phenylcoumaran model compound (PC) shown in Fig. 7 that contains a γ-carbinol as found in the lignin β-5 motif as well as a benzylic primary alcohol. This model compound was synthesized via the direct reduction of a previously reported intermediate structure reported for the synthesis of β-5 models (see ESI S3.0† for details).18 After conducting the dehydrogenative decarbonylation reaction with 5 mol% [Ir(cod)Cl]2 for 48 hours, most of PC was converted (Fig. 7). However, the expected compound PC0 was not identified by GC-MS. Instead, PC2 was detected by GC-MS to be the main product, with PC1 as the minor product (Fig. 7 and S6†). PC1 is thought to form via a ring-opening reaction that forms a phenol and a stilbene. The stilbene then can afford PC2 by hydrogenation (Fig. 7). Similar to the β-O-4 model reaction, the exchange reaction between diethyl carbonate and the γ-carbinol group was identified as well as shown by the identification of PC3 (Fig. 7 and S7†). Therefore, under the dehydrogenative decarbonylation reaction conditions, other structural motifs in lignin such as the γ-carbinol of β-5 linkage motifs and possible benzylic primary alcohols are verified to be able to contribute to the formation of syngas as well via the removal of these primary alcohol groups, leading to even further defunctionalisation of lignin structures.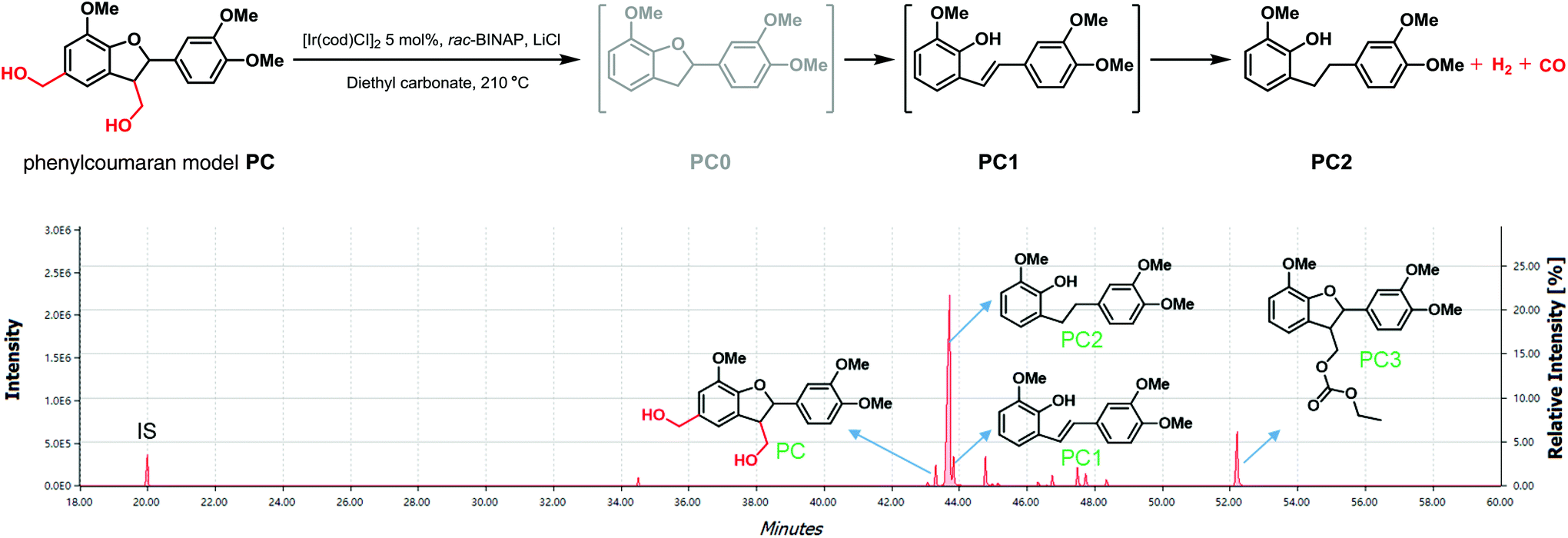 | ||
| Fig. 7 Dehydrogenative decarbonylation of the phenylcoumaran model compound (PC). Reaction conditions: 10 mg substrate, [Ir(cod)Cl]2 5 mol%, 4:2:1 LiCl:rac-BINAP:[Ir(cod)Cl]2, heating block set at 210 °C in 1 mL solvent. | ||
Dehydrogenative decarbonylation of walnut organosolv lignins (LA-OEt and LA-OBu)
Based on the successful application of this catalytic methodology on a series of representative model compounds, we investigated the effectiveness of the defunctionalisation on isolated lignin. For this purpose, organosolv lignins with high levels of β-O-4 motifs as well as with high levels of α-ethoxy or α-butoxy incorporation were used as starting materials. According to the 2D-HSQC NMR spectra shown in Fig. 8a and c, the obtained α-protected lignins (LA-OR) consisted of mainly the β-O-4 α-OR structure, R = Et, Bu (see ESI S4.0 and Table S2† for details). No further phenol methylation step, as required in our previous procedure,31,51 was performed as the dehydrogenative decarbonylation catalyst was confirmed to be compatible with phenol groups in model studies. When dehydrogenative decarbonylation was performed on α-ethoxylated lignin (LA-OEt), at the set temperature of 205 °C in diethyl carbonate for 48 hours, an obvious product signal was observed in the 2D-HSQC NMR spectra, and there was a good agreement of the product spectra with those of the defunctionalized β-O-4 model compound. The β-signal of the desired defunctionalized lignin (LB-OEt) was clearly identified (Fig. 8b). To have a better understanding on the effectiveness of the acceptorless dehydrogenative decarbonylation on lignin, a tentative semi-quantification was carried out based on the 2D-HSQC spectra of lignin (Table S2,† for detailed calculation, see ESI† section 4.4). The defunctionalized linkage, LB-OEt, was obtained in about 15% yield and 16% selectivity based on the conversion of the starting β-O-4 α-OEt. In accordance with our model compound dehydrogenative decarbonylation study, the derivatives of veratrylglycol-β-guaiacyl ether (SP1) and 1,2-dimethoxy-4-ethylbenzene (SP2) seem to be part of the cause (Scheme 1). Accordingly, we checked the existence of the LD structure in defunctionalized lignin by overlaying the product 2D-HSQC NMR spectra with 4-ethylguaicol, the derivative of 1,2-dimethoxy-4-ethylbenzene (SP2), which showed good agreement in the aliphatic region (Fig. S18†). Furthermore, in order to confirm the presence of the LC structure, the reported NMR data of veratrylglycol-β-guaiacyl ether (SP1)25 were compared with the 2D-HSQC NMR spectra of the defunctionalized lignin. The comparison indicated that the β signal in the LC structure actually corresponded to Hα–Cβ (δH/δC 3.99–4.29/73.19–74.62 ppm) in the 2D-HSQC NMR spectra (Fig. 8b). In addition, there were some unknown impurities originating from the starting lignin which overlapped with the signals in this region. Based on the linkages calculated by 2D-HSQC NMR (Table S2†), the LC structure, another type of defunctionalized β-O-4 linkage, was obtained in a yield of about 38% with 40% selectivity. Thus for the α-ethoxylated lignin, an overall 53% yield of the defunctionalized lignin was achieved by taking both the LB-OEt and LC structures into account. Likewise, α-butoxylated lignin underwent the dehydrogenative decarbonylation reaction under the same conditions. There was an obvious shift of the β signal in the LB-OBu structure as well (Fig. 8d). Similarly, LC and LD structures were found in the product. In particular, this dehydrogenative decarbonylation methodology remained effective when the starting lignin was extended to a pentanolsolv lignin (LA-OPn), showing the possibility of using different alcohols to protect the α-hydroxy group during the extraction stage (Fig. S14 and S15†) prior to the application in iridium-based dehydrogenative decarbonylation.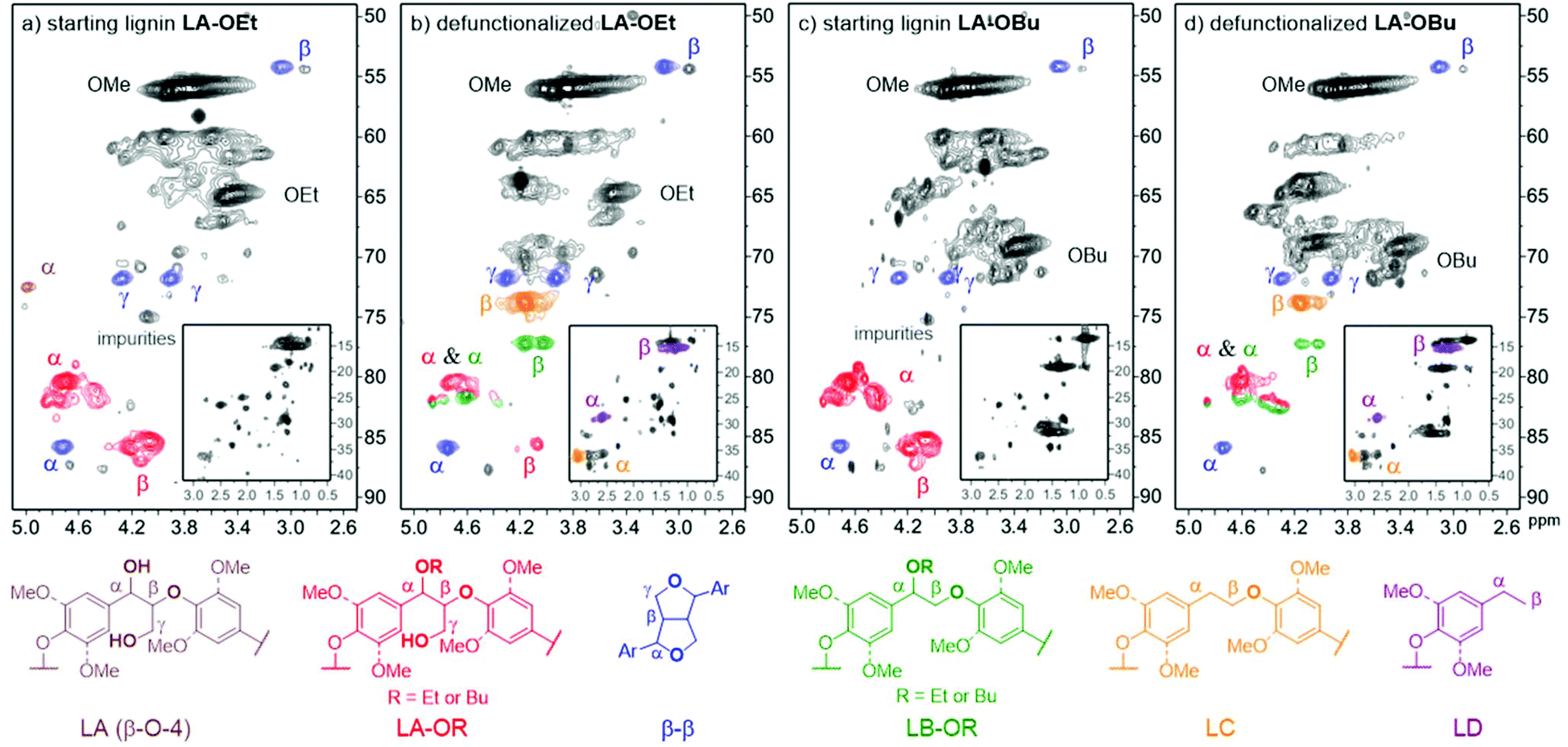 | ||
| Fig. 8 2D-HSQC NMR spectra (600 MHz, CDCl3) of the linkage region of (a) α-ethoxylated lignin (starting lignin), (b) defunctionalized α-ethoxylated lignin, (c) α-butoxylated lignin (starting lignin), and (d) defunctionalized α-butoxylated lignin; defunctionalized lignins were obtained by dehydrogenative decarbonylation; reaction conditions: 200 mg LA-OEt or LA-OBu with [Ir(cod)Cl]2 13.4 wt%, rac-BINAP 24.9 wt%, LiCl 3.4 wt% and a drop of water dissolved in 2.5 mL diethyl carbonate reacting at a set temperature of 205 °C for 48 hours. | ||
The release of syngas from lignins by dehydrogenative decarbonylation
The volume and composition of the gas released were analysed to confirm the formation of valuable syngas during defunctionalization of lignin by dehydrogenative decarbonylation. 200 mg of ethanosolv lignin generated 5.4 mL syngas with a CO:H2 ratio of 4.2:1 after reacting in 1,4-dioxane at 205 °C for 48 hours. We suspect that the higher content of CO than that of H2 was due to the consumption of H2 for the side reactions, analogous to those shown in Scheme 1. Thus some of the liberated H2 was likely consumed by hydrogenation or hydrogenolysis of the β-O-4 linkage to afford LC or LD structures (Fig. 8), which was consistent with the model compound study. To exclude that demethoxylation of the aromatic rings contributes to synthesis gas formation we calculated the S/G and β–β:-OMe ratio in the 2D-HSQC NMR spectra and found that the ratios were not significantly changed (Table S3†). Interestingly, by changing the solvent to diethyl carbonate, significantly higher amount of syngas, 18.9 mL, was generated. However, in this case also 11.6 mL methane, the product of dehydrogenative decarbonylation of ethanol, was observed. This could be from ethanol released from the lignin structure but the decomposition of diethyl carbonate could not be excluded. As mentioned previously in the model compound studies, the diethyl carbonate could react with alcohols resulting in ethanol formation. Additionally, methane was observed in the butanosolv lignin reaction in diethyl carbonate, however, in a much smaller amount of 9% compared to 17% for ethanosolv lignin. This suggested that methane was generated from the ethanol released from ethanol elimination from the β-O-4 motif as well as from exchange reactions between alcohols and diethyl carbonate. No methane was detected in the reactions with BP2 showing that diethyl carbonate decomposition only occurs at the more elevated reaction temperatures used on A-type model compounds and lignin. Analogously, 4% propane was found in the gas phase in the reaction using butanosolv in diethylcarbonate. No propane was found in the gas released from the dehydrogenative decarbonylation of ethanosolv or pentanosolv lignins, confirming also that alcohols released by elimination from the lignin structure are in part dehydrogenative decarbonylated to give syngas and the corresponding alkanes. Nevertheless, these results show the possibility to apply dehydrogenative decarbonylation for lignin defunctionalisation with a joint production of syngas.
Conclusion
In this study, we report the first example of combined targeted lignin defunctionalisation and biomass-derived synthesis gas formation. This is achieved by iridium catalysed acceptorless dehydrogenative decarbonylation. Its effectiveness relies on the high activity as well as the high selectivity towards primary alcohol groups in the lignin structure. Here, our key innovation is the extension of the application of this catalytic methodology for the first time to lignin model compounds with different primary alcohol groups. For the model compounds with a representative β-O-4 motif, 70% selectivity towards the removal of the γ-carbinol group to obtain the desired defunctionalized lignin structure was achieved. The reactions could be performed in 1,4-dioxane which is highly compatible with many lignins. Although diethyl carbonate was shown to be a significantly greener alternative, it also contributes to synthesis gas formation at more elevated temperatures. Furthermore, the dehydrogenative decarbonylation reaction was demonstrated on organosolv lignin. The same defunctionalised β-O-4 motif was clearly visible by 2D-HSQC NMR for α-ethoxylated, α-butoxylated and α-pentoxylated lignins, although the high temperatures required to obtain efficient conversion lead to the formation of some side-products that could be identified using the data from the model compound reactions. Nevertheless, this defunctionalisation route is promising. Compared with our previous defunctionalisation strategy that required firstly oxidation followed by a decarbonylation step, this dehydrogenative decarbonylation methodology can not only combine these transformations into a single step but also tolerate phenol groups, which lead to the omission of the toxic methylation step on lignin. Meanwhile, this direct dehydrogenative decarbonylation defunctionalisation method is atom-economical and waste-free by producing synthesis gas as another valuable product, offering great opportunity for further releasing the valorisation potential of lignin yet with a well preserved and less complex β-O-4 linkage structure. Additionally, this study shows that the dehydrogenative decarbonylation methodology can serve as a powerful tool to sustainably valorise alcohol containing substrates which are widely accessible from renewable biomass resources. Studies on the further development of the catalyst and reaction set-up are required and are ongoing to achieve more efficient and selective modification of lignin, particularly to obtain selectively defunctionalized lignin structures.Experimental
Chemicals
1,5-Cyclooctadiene-iridium(I) chloride dimer ([Ir(cod)Cl]2) and rac-BINAP were purchased from Fluorochem. Other chemicals obtained from Acros, Sigma-Aldrich or Fluorochem were used as received. Both 1,4-dioxane and diethyl carbonate utilised in this work were obtained anhydrous under argon and used using standard Schlenk techniques. The α-ethoxylated lignin C3 β-O-4 model compound A-OEt and α-butoxylated lignin C3 β-O-4 model compound A-OBu were synthesized following the published procedure.31,51 The C2 model compounds 4-(1-ethoxy-2-(2-methoxyphenoxy)ethyl)-1,2-dimethoxybenzene (B-OEt) and 4-(1-butoxy-2-(2-methoxyphenoxy)ethyl)-1,2-dimethoxybenzene (B-OBu) were synthesized as previously reported.31 The synthesis of the phenylcoumaran model compound was provided in the ESI (ESI S3.0† for details). α-ethoxylated lignin (LA-OEt), α-butoxylated lignin (LA-OBu) and α-pentoxylated lignin (LA-OPn) were obtained following a similar procedure provided in the ESI (ESI S4.0† for details).Dehydrogenative decarbonylation of model compounds
A typical procedure for dehydrogenative decarbonylation reaction is as follows: catalyst stock solution was premade by dissolving 8.75 μmol [Ir(cod)Cl]2, 17.5 μmol rac-BINAP, 35 μmol LiCl, a drop of water and 0.15 mmol n-octadecane in a 10 mL volumetric flask, and was then sonicated for 10 minutes. The model compound (e.g. 4-methoxybenzenepropanol or 4-hydroxy-3-methoxybenzenepropanol, 0.07 mmol) was weighed out in a 20 mL pressure vial (Biotage) equipped with a magnetic stirring bar. 2 mL catalyst stock solution was added to the vial, and the vial was then sealed with a Biotage airtight cap, which allowed for the reaction to be run under the pressure caused by heating the reaction above the boiling point of the respective solvent used. The vial was flushed with nitrogen for two minutes via a needle connection to ensure a nitrogen atmosphere. The mixture was then heated to the desired temperature in a heating block. Before sampling, the vial was cooled in a cold water bath. A 0.2 mL sample was taken through the septum of the vial cap using a syringe equipped with a thin needle. The sample was diluted in 0.8 mL 1,4-dioxane and analysed by GC-MS and GC-FID. After sampling, the vial was flushed with nitrogen for 2 minutes and reheated to the appropriate temperature. For the starting materials A-OEt and B-OEt, 0.04 mmol was used and n-decane was applied as an external standard while the other relative quantities and procedures were kept similar. Regarding the β-5 model compound, 29 μmol was used for each reaction with 1,4-dinitrobenzene as an internal standard. After the reaction, it was analysed by both NMR and GC-MS.Collection and quantification of syngas from the model compound
To collect gas released from the reaction, 25 μmol [Ir(cod)Cl]2, 50 μmol rac-BINAP, 100 μmol LiCl and a drop of water were dissolved in 2 mL diethyl carbonate in a 15 mL Schlenk tube equipped with a magnetic stirring bar. After stirring for 10 minutes, 1 mmol 4-hydroxy-3-methoxybenzenepropanol was added. A short condenser was then installed on the Schlenk tube, and the condenser was further connected to a tube fitted with a long needle, allowing the gas to escape. Prior to heating the oil bath to 190 °C, the whole set-up was flushed with nitrogen for 2 minutes. Once the flushing was completed, the liberated gas volume was measured via an inverted burette placed inside a water bath. Similarly, a blank reaction was performed without the addition of the starting material, and the volume increase due to heating was recorded when it became constant. The actual gas volume was corrected by deducting the volume increase generated by the blank reaction.Dehydrogenative decarbonylation of lignin and the collection of the formed syngas
In a typical reaction, 26.9 mg [Ir(cod)Cl]2, 49.8 mg rac-BINAP, 6.8 mg LiCl and a drop of water were dissolved in 2.5 mL solvent in a 20 mL pressure vial (Biotage) equipped with a magnetic stirring bar. After premixing for 10 minutes, 200 mg lignin (e.g. α-ethoxylated lignin LA-OEt) was added into the vial and the vial was sealed with an airtight cap bought form Biotage. Prior to heating the vial to the desired temperature, the vial was flushed with nitrogen for 2 minutes through needles. After the reaction mixture was cooled to room temperature, the cap was pierced with the needle of a syringe which was fitted with a valve, such that the gas formed could penetrate into the syringe allowing the reading of the volume of the gas until no change was observed. Then the collected gas was subjected to GC-TCD analysis. Finally, the lignin was recovered by precipitation via the addition of 150 mL of water to the mixture. The lignin was collected as a solid by vacuum filtration and dried at 50 °C overnight to yield the lignin product as a brown powder.Analytical procedures
Analytical procedures for NMR, GC-FID, GC-MS, GC-TCD, etc. are described along with analysis results relevant to the manuscript in the ESI (see ESI S1.0† for details).Abbreviations
| bpy | 2,2′-Bipyridine |
| Cod | 1,5-Cyclooctadiene |
| dppp | 1,3-Bis(diphenylphosphino)propane |
| ES | External standard |
| GC-FID | Gas chromatography with a flame ionization detector |
| GC-MS | Gas chromatography with a mass spectrometry detector |
| GC-TCD | Gas chromatography with a thermal conductivity detector |
| HSQC | Heteronuclear single quantum coherence spectroscopy |
| IS | Internal standard |
| [Ir(cod)Cl]2 | Bis(1,5-cyclooctadiene)diiridium(I) dichloride |
| [Ir(coe)2Cl]2 | Chlorobis(cyclooctene)iridium(I) dimer |
| NMI | 1-methylimidazole |
| [RhCl(cod)]2 | Chloro(1,5-cyclooctadiene)rhodium(I) dimer |
| rac-BINAP | [2,2’-Bis(diphenylphosphino)-1,1’-binaphthyl] |
| TBAF | Tetrabutylammonium fluoride |
| TEMPO | 2,2,6,6-Tetramethylpiperidine 1-oxyl |
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Z. Z. acknowledges the China Scholarship Council for funding (grant number 201704910922). The work performed by C. W. L. has partly been conducted within the framework of the Dutch TKI-BBEI project “CALIBRA”, reference TEBE117014. The financial support of the Dutch Ministry of Economic Affairs and Climate for this project is gratefully acknowledged. Additionally, the authors would like to thank Johanna H. L. Kaelen and Lambert J. Deuss for providing walnut shells used for lignin extraction. Analytical support was provided by Leon Rohrbach.References
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Marita, R. D. Hatfield, S. A. Ralph, J. H. Christensen and W. Boerjan, Phytochem. Rev., 2004, 3, 29–60 CrossRef CAS.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- S. Gillet, M. Aguedo, L. Petitjean, A. R. C. Morais, A. M. da Costa Lopes, R. M. Lukasik and P. T. Anastas, Green Chem., 2017, 19, 4200–4233 RSC.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed. Engl., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- Y. Jing, Y. Guo, Q. Xia, X. Liu and Y. Wang, Chem, 2019, 5, 2520–2546 CAS.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, J. Am. Chem. Soc., 2015, 137, 7456–7467 CrossRef CAS PubMed.
- Z. Sun, G. Bottari, A. Afanasenko, M. C. A. Stuart, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
- J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554–12555 CrossRef CAS PubMed.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef CAS PubMed.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- . Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen, H. Matsushima, J. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, eaau1567, DOI:10.1126/science.aau1567.
- C. W. Lahive, P. J. Deuss, C. S. Lancefield, Z. Sun, D. B. Cordes, C. M. Young, F. Tran, A. M. Slawin, J. G. de Vries, P. C. Kamer, N. J. Westwood and K. Barta, J. Am. Chem. Soc., 2016, 138, 8900–8911 CrossRef CAS PubMed.
- S. Liu, L. Bai, A. P. van Muyden, Z. Huang, X. Cui, Z. Fei, X. Li, X. Hu and P. J. Dyson, Green Chem., 2019, 21, 1974–1981 RSC.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed. Engl., 2015, 54, 258–262 CrossRef CAS PubMed.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- S. Rautiainen, D. Di Francesco, S. N. Katea, G. Westin, D. N. Tungasmita and J. S. M. Samec, ChemSusChem, 2019, 12, 404–408 CrossRef CAS PubMed.
- I. Bosque, G. Magallanes, M. Rigoulet, M. D. Karkas and C. R. J. Stephenson, ACS Cent. Sci., 2017, 3, 621–628 CrossRef CAS PubMed.
- J. Buendia, J. Mottweiler and C. Bolm, Chemistry, 2011, 17, 13877–13882 CrossRef CAS PubMed.
- D. Alam, M. Y. Lui, A. Yuen, T. Maschmeyer, B. S. Haynes and A. Montoya, Ind. Eng. Chem. Res., 2018, 57, 2014–2022 CrossRef CAS.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- Y. Ren, M. Yan, J. Wang, Z. C. Zhang and K. Yao, Angew. Chem., Int. Ed. Engl., 2013, 52, 12674–12678 CrossRef CAS PubMed.
- V. E. Tarabanko, N. A. Fomova, B. N. Kuznetsov, N. M. Ivanchenko and A. V. Kudryashev, React. Kinet. Catal. Lett., 1995, 55, 161–170 CrossRef CAS.
- S. Dabral, J. G. Hernandez, P. C. J. Kamer and C. Bolm, ChemSusChem, 2017, 10, 2707–2713 CrossRef CAS PubMed.
- C. S. Lancefield, L. W. Teunissen, B. M. Weckhuysen and P. C. A. Bruijnincx, Green Chem., 2018, 20, 3214–3221 RSC.
- Z. Zhang, C. W. Lahive, D. S. Zijlstra, Z. Wang and P. J. Deuss, ACS Sustainable Chem. Eng., 2019, 7, 12105–12116 CAS.
- A. Wu, B. O. Patrick, E. Chung and B. R. James, Dalton Trans., 2012, 41, 11093–11106 RSC.
- D. S. Zijlstra, C. W. Lahive, C. A. Analbers, M. B. Figueirêdo, Z. Wang, C. S. Lancefield and P. J. Deuss, ACS Sustainable Chem. Eng., 2020, 8, 5119–5131 CrossRef CAS.
- C. S. Lancefield, I. Panovic, P. J. Deuss, K. Barta and N. J. Westwood, Green Chem., 2017, 19, 202–214 RSC.
- S. Bauer, H. Sorek, V. D. Mitchell, A. B. Ibáñez and D. E. Wemmer, J. Agric. Food Chem., 2012, 60, 8203–8212 CrossRef CAS PubMed.
- F. Lu and J. Ralph, Plant J., 2003, 35, 535–544 CrossRef CAS PubMed.
- D. M. Miles-Barrett, J. R. D. Montgomery, C. S. Lancefield, D. B. Cordes, A. M. Z. Slawin, T. Lebl, R. Carr and N. J. Westwood, ACS Sustainable Chem. Eng., 2017, 5, 1831–1839 CrossRef CAS.
- I. Panovic, D. M. Miles-Barrett, C. S. Lancefield and N. J. Westwood, ACS Sustainable Chem. Eng., 2019, 7, 12098–12104 CAS.
- R. Jastrzebski, S. Constant, C. S. Lancefield, N. J. Westwood, B. M. Weckhuysen and P. C. Bruijnincx, ChemSusChem, 2016, 9, 2074–2079 CrossRef CAS PubMed.
- E. P. K. Olsen and R. Madsen, Chem. – Eur. J., 2012, 18, 16023–16029 CrossRef CAS PubMed.
- A. Mazziotta and R. Madsen, Eur. J. Org. Chem., 2017, 2017, 5417–5420 CrossRef CAS.
- H. A. Ho, K. Manna and A. D. Sadow, Angew. Chem., Int. Ed. Engl., 2012, 51, 8607–8610 CrossRef CAS PubMed.
- J. R. Rostrup-Nielsen and L. J. Christiansen, Concepts in Syngas Manufacture, Imperial College Press, Singapore, 2011 Search PubMed.
- V. S. Sikarwar, M. Zhao, P. Clough, J. Yao, X. Zhong, M. Z. Memon, N. Shah, E. J. Anthony and P. S. Fennell, Energy Environ. Sci., 2016, 9, 2939–2977 RSC.
- M. M. Yung, W. S. Jablonski and K. A. Magrini-Bair, Energy Fuels, 2009, 23, 1874–1887 CrossRef CAS.
- P. J. Woolcock and R. C. Brown, Biomass Bioenergy, 2013, 52, 54–84 CrossRef CAS.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13 Search PubMed.
- J. A. Stickney, S. L. Sager, J. R. Clarkson, L. A. Smith, B. J. Locey, M. J. Bock, R. Hartung and S. F. Olp, Regul. Toxicol. Pharmacol., 2003, 38, 183–195 CrossRef CAS PubMed.
- M. T. Amiri, G. R. Dick, Y. M. Questell-Santiago and J. S. Luterbacher, Nat. Protoc., 2019, 14, 921–954 CrossRef PubMed.
- I. Panovic, C. S. Lancefield, D. Phillips, M. J. Gronnow and N. J. Westwood, ChemSusChem, 2019, 12, 542–548 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details on analytical procedures, data on set vs. the measured T of reactions, the synthetic procedure for PC, procedures for the extraction of lignin and analytic GCMS and NMR data. See DOI: 10.1039/d0gc01209b |
| This journal is © The Royal Society of Chemistry 2020 |