
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of the molecular motifs of mannan and xylan populations on their recalcitrance and organization in spruce softwoods†
Antonio
Martínez-Abad‡
 a,
Amparo
Jiménez-Quero‡
a,
Jakob
Wohlert
bc and
Francisco
Vilaplana
*ac
a,
Amparo
Jiménez-Quero‡
a,
Jakob
Wohlert
bc and
Francisco
Vilaplana
*ac
aDivision of Glycoscience, Department of Chemistry, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, Stockholm, Sweden. E-mail: franvila@kth.se
bDivision of Biocomposites, Department of Fibre and Polymer Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, Stockholm, Sweden
cWallenberg Wood Science Center, KTH Royal Institute of Technology, Stockholm, Sweden
First published on 19th May 2020
Abstract
Softwood from conifers constitutes one of the main terrestrial renewable resources for the production of bio-based materials and platform chemicals. Lignocellulose from softwoods has a distinct molecular composition compared to other plant biomass sources, where acetylated galactoglucomannan is the main hemicellulose with minor amounts of arabinoglucuronoxylan. Here, we reveal the presence of mannan and xylan populations in spruce softwoods with distinct molecular features based on their extractability using sequential hydrothermal treatment by subcritical water without previous delignification. An accessible acetylated mannan population has been identified with simple profiles of glucosyl and galactosyl motifs and without the existence of a regular acetylation pattern. The xylan populations are extracted at intermediate times, and they exhibit the presence of major and minor regular intramolecular domains with different relative abundances based on extractability. Finally, a recalcitrant mannan population with complex glucosylation and galactosylation profiles was identified at longer extraction times. Molecular dynamics simulations revealed that the presence of consecutive mannose units in the backbone prevents the tight association with cellulose surfaces, which may explain the different extractabilities of the two isolated mannan populations. The combination of sequential hydrothermal treatment, comprehensive carbohydrate sequencing and molecular dynamics simulations offers new insights into the distinct features of the mannan and xylan populations in softwoods, and their putative organization in the lignocellulosic matrix.
Introduction
Humankind has exploited plant lignocellulose biomass for centuries for its use as an energy source and for materials in a myriad of applications. In the current situation of climate change due to carbon dioxide emissions caused by fossil resources, lignocellulose biomass emerges as the most abundant renewable resource on the biosphere1 for the production of advanced materials, platform chemicals and bioenergy in future cascade biorefineries2,3 contributing to a balanced carbon cycle. Lignocellulosic biomass consists of a dense polymeric network of cellulose microfibrils embedded in a tightly connected matrix of hemicelluloses and lignin, with well-defined ordering from the nano- to the macroscale.4,5 The composition and molecular structure of the cellulose, hemicellulose and lignin components in lignocellulosic biomass differ not only for the different plant species, but also in the different tissues and developmental stages.6 The complex and recalcitrant molecular structure and supramolecular architecture of lignocellulosic biomass, fundamental to its biological role in plant secondary cell walls, hampers its efficient fractionation and exploitation for a wide portfolio of potential bio-based products.7,8 Lignocellulose recalcitrance is attributed to its structural features at the nano- and macroscales, and is determined by the composition, organization and supramolecular interactions between the cellulose, hemicellulose, and lignin components.5,7,9–12 However, conflicting information has been reported about the occurrence and nature of the intermolecular forces between cell wall components and the origin of this recalcitrance.9,10Softwoods from conifers constitute one of the main terrestrial renewable resources in the Northern hemisphere, especially in the Nordic countries, for the production of bio-based materials and platform chemicals. In softwoods, acetylated galactoglucomannan (acGGM) is the most abundant hemicellulose accounting for 20–25% of the total dry mass, with a minor content (10–15% dry weight) of arabinoglucuronoxylan (AGX) (Fig. 1A).13 Recently, the presence of regular motifs in the molecular structure of softwood AGX has been reported by combined enzymatic deconstruction using a selective β-glucuronoxylanase and oligosaccharide sequencing by mass spectrometry.14,15 These studies have revealed the presence of major intramolecular domains in AGX with even spacing of the mGlcA and Araf substitutions, and minor domains of clustered and consecutive mGlcA spacing. These precise substitution patterns are compatible with softwood xylan binding to the hydrophilic and hydrophobic surfaces of cellulose in a 2-fold screw xylan conformation. However, the presence of regular molecular motifs, in terms of backbone organization and patterns of acetylation and galactose substitution, has not been reported yet for softwood glucomannans.16 We are only now starting to understand how the fine and heterogeneous molecular structure of hemicelluloses modulates the interactions with cellulose microfibrils through hydrogen bonding and non-polar interactions,14,15,17–19 and with lignin through covalent linkages.11,20,21 The spatial organization of the cellulose, hemicellulose and lignin components in lignocellulosic biomass is largely unknown, although recent studies by advanced imaging and solid scattering techniques are starting to reveal the exquisite supramolecular architecture of microfibrillar and macrofibrillar domains.22–25 Indeed, the combined presence of rigid mannan and xylan domains closely interacting with cellulose microfibrils and unbound matrix mannan and xylan populations has been recently demonstrated.23 However, the distinct molecular nature of the rigid and flexible hemicellulose populations is not known.
 | ||
| Fig. 1 Description of the hydrothermal process, composition and molecular structure of the polysaccharide fractions from spruce softwood. (A) Polysaccharides in spruce wood. Cellulose consists of a linear backbone of β-(1→4)-linked glucopyranosyl (Glc) units. Acetylated galactoglucomannan (acGGM) consists of a backbone of β-(1→4)-linked mannopyranosyl (Man) and glucopyranosyl (Glc) units, with α-(1→6) galactopyranosyl (Gal) and acetylated in the O-2 and/or O-3 positions of the Man units.33,34 Arabinoglucuronoxylan (AGX) consists of a backbone of β-(1→4)-linked xylopyranosyl (Xyl) units, substituted by α-(1→2) 4-O-methyl glucuronic acid (mGlcA) units and arabinofuranose units (Araf) at the α-(1→3) position.35,36 (B). Sequential extraction of spruce hemicelluloses using subcritical water. (C) Monosaccharide composition of the spruce chips, the consecutive extracts and the residue. (D) Molar mass distributions of the extracts. | ||
In this work, we have used mild acid hydrothermal treatment by sequential subcritical water extraction (SWE) to isolate the different hemicellulose populations based on their extractability and recalcitrance in the lignocellulosic network, minimizing the occurrence of deacetylation and autohydrolysis.11,26,27 SWE is considered a green extraction method using pressurized water as a solvent, offering a safe, sustainable and cost-effective alternative to current biomass extraction methods.28 The dielectric potential and ionization of water at high temperatures and high hydrostatic pressures decreases polarity, contributing to the extraction and dissolution of less polar, less soluble high molecular weight biopolymers. An important advantage of using SWE for hemicellulose extraction is the capacity to release intact decorated biopolymers, as opposed to conventional acid or basic methods that cause extensive depolymerisation and loss of valuable functionalities (e.g. acetylation, phenolic acids).27,29 This ‘hemicellulose-first’ approach contributes to an improved material sustainability of future biorefineries, since it isolates the hemicelluloses in polymeric form for their potential use in films, hydrogels, and food additives,30–32 and it also enables the potential further valorization of the native lignin and cellulose components. This approach also maintains the polymeric functionality of the lignocellulosic components without the need to deconstruct them into a sugar or phenolic platform for further processing into chemicals or bioenergy. In our work, we have comprehensively analysed the isolated spruce softwood hemicelluloses by mass spectrometry (MS) based glycomic analysis, in order to correlate the molecular structure of the mannan and xylan populations with their extractability. We have evaluated the influence of distinct mannan molecular features on their interactions with cellulose surfaces using molecular dynamics simulations. The fundamental understanding provided by the combination of sequential subcritical water extraction, advanced carbohydrate sequencing and molecular modelling provides detailed molecular insights about the different mannan and xylan domains in softwoods, their contribution to their supramolecular organization in the lignocellulosic matrix and their recalcitrance to hydrothermal processing.
Materials and methods
Materials, extraction, and purification
Norway spruce chips (Picea abies), a kind gift from the Wallenberg Wood Science Centre (Stockholm, Sweden), were milled to a particle size <1 mm, defatted, freeze-dried and kept at −20 °C before use. The biomass was subjected to SWE at pH 5 (formate buffer 0.2 M), 170 °C and 100 bars in a pressurized liquid extraction equipment (ASE-300, Dionex, USA). Extraction was carried out over 7 consecutive cycles of 5, 15, 20, 20, 40, 80 and 120 min, corresponding to a total residential time of 5 hours for the biomass. The extracts were subjected to dialysis (3 days) using 6–8 kDa Spectra/Por 3 membranes (Spectrum, USA) to remove salts and low molecular weight molecules. The high molecular weight fractions (S1–S7) and the residual insoluble biomass (R) were freeze-dried for further analysis.Compositional analysis of the wood fractions
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 analytical columns (Polymer Standards Services, Mainz, Germany) as previously reported.29 Calibration was performed using pullulan standards provided by Polymer Standards Services (PSS, Mainz, Germany).
000 analytical columns (Polymer Standards Services, Mainz, Germany) as previously reported.29 Calibration was performed using pullulan standards provided by Polymer Standards Services (PSS, Mainz, Germany).
Enzymatic profiling and sequencing of hemicellulose fractions from spruce wood
Molecular dynamics simulations
Results and discussion
Distinct mannan and xylan populations are sequentially extracted from spruce softwood using subcritical water
Hemicellulose extraction from spruce softwood chips was performed using sequential subcritical water under buffered conditions without previous delignification, in order to monitor the extractability of different hemicellulose populations based on their molecular structure (Fig. 1A). The use of buffered mild acidic conditions during subcritical water extraction minimizes the occurrence of potential deacetylation and autohydrolysis, resulting in extracted hemicelluloses with molecular structures as intact as possible.11 The conditions of extraction were fixed at 170 °C and pH 5 using 7 sequential cycles between 5–300 min, based on initial trials on defatted spruce chips (results not shown) and in agreement with similar procedures reported for the extraction of acetylated galactoglucomannan from spruce softwoods.52,53 The mass balances, average composition and molar mass of the different fractions obtained during the sequential extraction process are presented in Table 1. From the mass balances, only 8% of the total solids were extracted in the form of high molar mass polysaccharides using subcritical water after 5 h of extractions, whereas 15% of the total solids were lost corresponding to low molar mass compounds washed out during dialysis. These yields are significantly lower than those obtained in our previous study using birch wood (a typical hardwood species from the Nordic countries),11 which indicates the denser structure and higher recalcitrance of softwoods to processing. Hemicelluloses were mainly extracted throughout the sequential process, reaching purities between 700–800 mg g−1. The remaining solid content of the extracts can be assigned to non-carbohydrate components, mainly lignin, with a general content of 20–25% and increase in the extracts at a longer residence time. Cellulose was not primarily extracted in the subcritical water process and was mainly left in the residue.| Spruce | E1 | E2 | E3 | E4 | E5 | E6 | E7 | ∑Si | R | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Yields determined gravimetrically and referred to the original birch chips. b Determined from the complete monosaccharide composition (ESI Table S1†). c Determined from the Klason lignin. d Determined after saponification and HPLC-UV analysis. e Determined by SEC-DRI. n.a: not applicable; n.d.: not determined. | ||||||||||
| Extraction times (min) | n.a. | 5 | 15 | 20 | 20 | 40 | 80 | 120 | n.a. | n.a. |
| Total yielda (%) | 100 | 2.1 | 1.5 | 0.6 | 0.5 | 1.8 | 0.9 | 0.3 | 7.7 | 76.6 |
| Carbohydrate contentb (mg g−1) | 636.5 | 752.3 | 779.0 | 731.1 | 740.7 | 762.6 | 719.8 | 702.0 | n.a. | 675.2 |
| Celluloseb (%) | 60.5 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | 89.0 |
| Xylanb (%) | 14.0 | 9.3 | 27.2 | 37.6 | 42.8 | 50.1 | 33.1 | 18.7 | n.d. | 0.3 |
| Mannanb (%) | 24.1 | 78.5 | 68.8 | 58.3 | 52.8 | 45.4 | 64.6 | 80.3 | 10.7 | |
| Pectinb (%) | 1.3 | 12.0 | 3.9 | 4.1 | 4.4 | 4.5 | 2.0 | 0.6 | n.d. | 0.0 |
| Lignin contentc (mg g−1) | 363.5 | 247.7 | 221.0 | 268.9 | 259.3 | 237.4 | 280.2 | 298.0 | n.d. | 324.8 |
| Acetyl contentd (%) | n.a. | 5.97 (0.08) | 3.67 (0.83) | 3.31 (0.10) | 3.46 (0.20) | 3.15 (0.05) | 1.61 (0.85) | 1.14 (0.04) | n.a | n.a |
| Degree of acetylation (DAcGGM) | n.a. | 0.42 (0.01) | 0.28 (0.07) | 0.32 (0.01) | 0.36 (0.02) | 0.38 (0.01) | 0.14 (0.07) | 0.08 (0.00) | n.a | n.a |
|
M
ne (kDa) |
n.a. | 3.5 | 4.0 | 3.9 | 3.6 | 2.7 | 2.1 | 1.5 | n.a | n.a |
|
M
we (kDa) |
17.6 | 15.5 | 12.6 | 10.9 | 19.4 | 4.94 | 3.93 | |||
The time evolution of the monosaccharide composition of the extracts shows very interesting profiles (Fig. 1C, Table 1 and ESI Table S1†). At short residence times (5 min, fraction S1), a population of glucomannans was easily extracted together with the pectic components rich in galacturonic acid. This first extracted glucomannan population shows a relatively high degree of acetylation (DAc of 0.30–0.42) and a Man:Glc:Gal ratio of 3.7:1:0.3. This initial population corresponds well with the previously reported water-soluble acetylated galactoglucomannans as not yet affected by deacetylation induced by alkaline conditions or by harsher hydrolytic tratments.34,54,55 As extraction progresses, arabinoglucuronoxylan (AGX) becomes particularly enriched in the extracts between 20–90 min of extraction. This AGX shows an average ratio of Ara:mGlcA:Xyl of 1:2:6, in the ranges previously reported for xylan extracted by both alkaline35 and hydrothermal15 processes. The substitution of the spruce AGX is also modified with the extraction time, with an apparent increase of glucuronidation and a reduction of arabinosylation with extraction times. Interestingly, at much longer extraction times (from 100–300 min in residence times, extracts S6 and S7) a second population of glucomannan becomes predominantly extracted, with a much lower degree of acetylation (DAc of 0.10–0.15) and a Man:Glc:Gal of 3:1:1.2 with higher Glc and Gal content. The low acetylation content in this second glucomannan population could be both due to the fact that natively it has lower acetylation, or could be caused by deacetylation during the prolonged extraction times. The near absence of galactose in the residue could also hint towards possible random cleavage of these decorations at longer residence times, underestimating the native ratio of this recalcitrant mannan population. The molar mass distributions of the hemicellulose populations show a progressive decrease with extraction time, with the first mannan and xylan populations showing typical weight-average molar mass (Mw) values of 20 kDa, and the more recalcitrant populations in the range of 4–5 kDa. This indicates the extent of hydrolysis caused by the prolonged exposure of the hemicelluloses under the subcritical water conditions of extraction between 100–300 min. However, the time evolution shows that specific mannan and xylan hemicellulose populations with a distinct molecular structure can be extracted during sequential subcritical water extraction, indicating their distinct recalcitrance and potential different interconnectivities in the softwood lignocellulosic network. These differences will be studied in detail by combining mass spectrometric glycomic analyses and molecular dynamics simulations.
Oligomeric mass profiling (OLIMP) of mannan and xylan in spruce softwoods
Enzymatic digestions and subsequent mass spectrometric analysis by ESI-MS were performed on the extracts to identify the oligomeric patterns for spruce mannans and xylans during the sequential extraction (ESI Fig. S1†). A GH5 β-mannanase was used to digest the acetylated glucomannans; this enzyme requires the presence of a mannose residue in the −1 and the +2 positions of enzymatic cleavage, but can tolerate a Glc residue in the backbone in the +1 and −2 positions (Fig. 2A). The oligomeric mass profiling of acetylated GGM shows a ladder of hexose (H) oligosaccharides (Fig. 2B) where it is possible to distinguish the number of acetyl substitutions (AcnHm), but unfortunately does not enable the identification of the Man, Glc and Gal constituents, as they all are hexose isomers. As can be observed, in the initial extracts the presence of acetylated oligosaccharides (H3Ac2 and H4Ac2) is quite high, but this decreases progressively for the latter extracts, where the non-acetylated oligosaccharides prevail. Despite extensive chemical depolymerisation at later stages of extraction, the mannan populations (S6 and S7) exhibit longer oligomeric profiles with lower acetylation compared to the initial accessible mannans (S1 or S2). This hints towards the presence of more complex motifs in the mannan populations extracted at longer times, with glucose or galactose units in the backbone and as decorations at “intolerable positions” for the selected mannanase. In parallel, a GH30 β-glucuronoxylanase was used specifically to digest AGX; this enzyme recognizes the presence of a (m)GlcA in the −2 position and releases acidic xylo-oligosaccharides (Fig. 2A).15,56,57 The OLIMP of AGX by ESI-MS enables the determination of the spacing and number of glucuronic acid substituents in the aldouronic acid oligosaccharides (UXOs) from spruce AGX (PnUm), but unfortunately does not distinguish the isobaric Ara and Xyl pentose units (Fig. 2C). The evolution of the UXOs with the extraction time indicates the prevalence of P6U and P7U oligosaccharides for the shorter extraction times as previously reported. Indeed, the OLIMP for the extracts S1 to S4 are fairly similar, indicating the presence of a rather homogeneous AGX population. On the other hand, the increase of the relative abundance of more closely-spaced UXOs (P3U and P4U) can be observed for longer extraction times, which reveals the presence of more recalcitrant AGX motifs with tighter glucuronidation.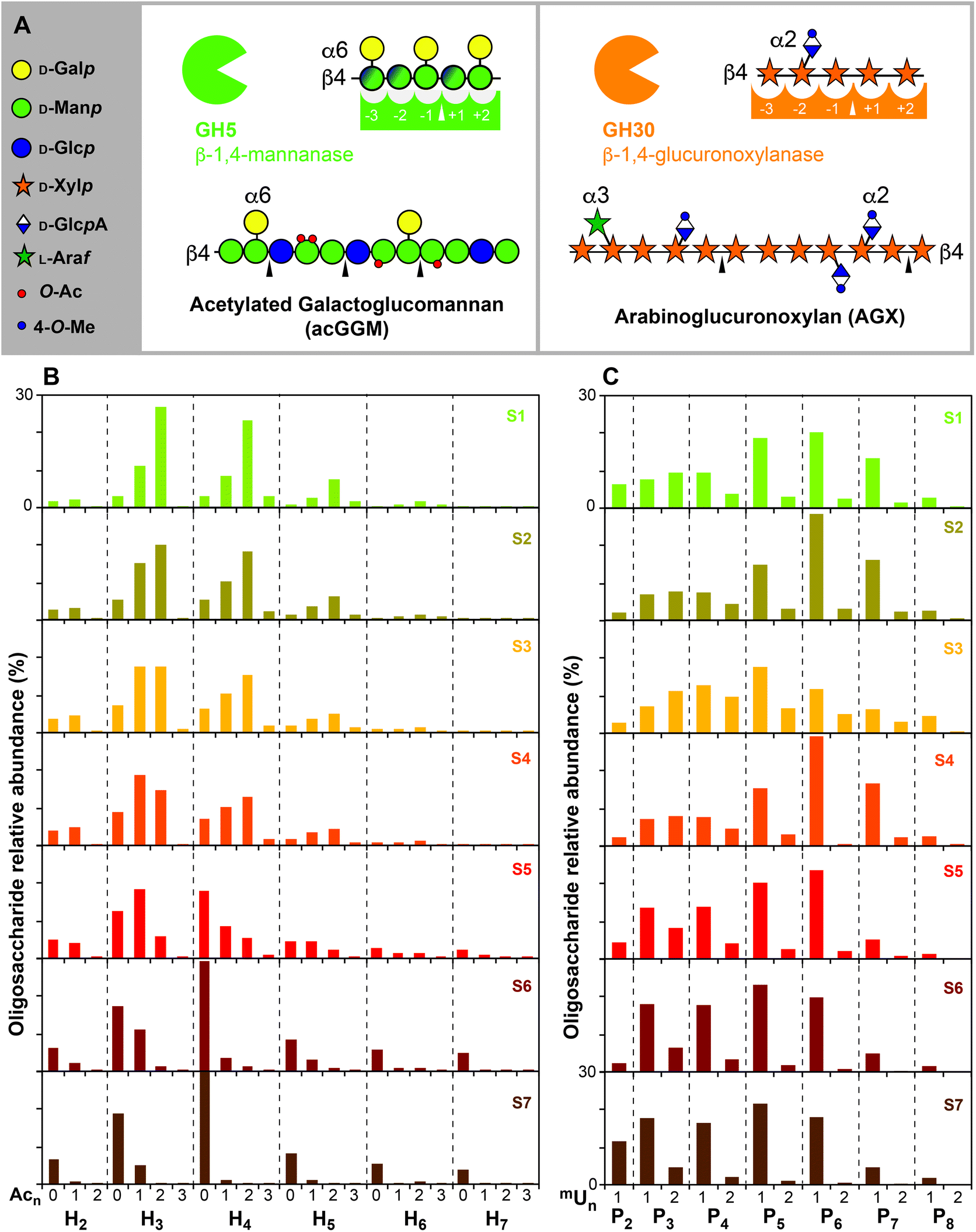 | ||
| Fig. 2 Oligomeric mass profiling (OLIMP) of the spruce hemicelluloses. (A) Substrate recognition by the GH5 β-mannanase and the GH30 β-glucuronoxylanase. (B) Relative abundance (%) of the manno-oligosaccharides calculated from the ESI-MS intensities. (C) Relative abundance (%) of the acidic xylo-oligosaccharides calculated from the ESI-MS intensities. Peak assignment presented in ESI Table S2.† Note: H (hexose: Man, Glc or Gal), Ac (acetyl), X (xylose), mU (mGlcA). | ||
Spruce xylan shows regular motifs with distinct extractability during subcritical water extraction
To further investigate the evolution of the molecular motifs in spruce xylan during subcritical water extraction, the released UXOs after GH30 treatment were chemically-derivatized by reduction and permethylation and analysed by LC-ESI-MS/MS (ESI Fig. S2†). This procedure enables the chromatographic resolution of the complex oligosaccharide mixture, the isobaric selection of the oligosaccharides by their mass-to-charge ratios (m/z) and their individual fragmentation by collision induced dissociation (CID) towards their full sequencing (ESI Fig. S3 and Table S3†). Up to 17 different UXOs were resolved and identified by LC-ESI-MS/MS, which are presented in the single ion monitoring (SIM) chromatograms of the UXOs based on their m/z ratios for selected xylan extracts (Fig. 3A). The identified oligosaccharides agree with the previously reported motifs for AGX in spruce and other conifers,14,15 with the presence of major domains containing regular motifs with an even spacing of glucuronidation (mGlcA) mainly all 6 Xylp units (oligosaccharides 8, 11 and 12) but also all 4 units (oligosaccharides 3 and 6) and Araf substitutions evenly-spaced with respect to the mGlcA substitutions, either at the −2 position from the reducing end (oligosaccharides 6 and 11) or at the −4 position (oligosaccharide 12). The presence of oligosaccharides with an odd spacing of the mGlcA substitutions (oligosaccharides 1, 5 and 10) and a series of oligosaccharides with the presence of glucuronidation at consecutive Xylp units (oligosaccharides 14 and 16) can be observed as well, which corresponds to minor domains in GAX. Interestingly, new oligosaccharides not reported before can be observed, with the occurrence of an mGlcA substitution in the Xylp unit of the reducing end (oligosaccharides 2, 4, 7, 9, 13, 15 and 17). These oligosaccharides cannot be released in principle by the GH30 glucuronoxylanase, due to the required mGlcA in the −2 position. The progressive appearance and enrichment of these oligosaccharide series with an increased extraction time suggest that these oligosaccharides are not released by the enzyme, but they are present in the reducing end of the AGX polymer and are progressively generated by the peeling reactions induced by the long residence times during subcritical water extraction. This indicates that the mGlcA substitutions indeed act as stopping mechanisms for the peeling reactions in AGX during subcritical water treatment.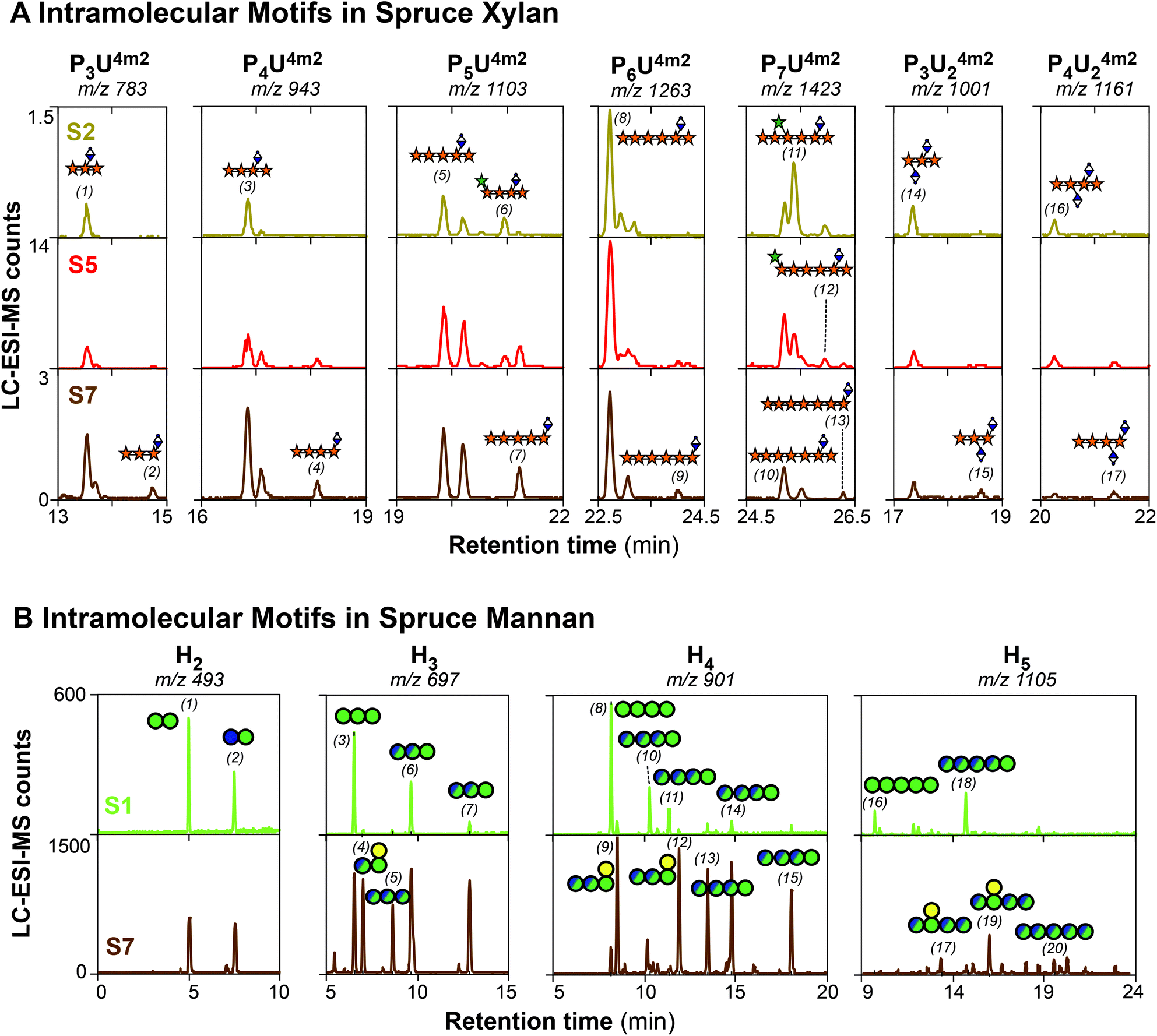 | ||
| Fig. 3 Oligosaccharide profiling of the spruce hemicellulose populations by LC-ESI-MS/MS. (A) Single ion monitoring (SIM) chromatograms for xylan oligosaccharides after β-glucuronoxylanase hydrolysis and reduction/permethylation. Note: P (pentose, could be either a Xylp or Araf), mU (mGlcA). Fragmentation of selected peaks and assignation of the ion fragments is presented in ESI Fig. S3.†59 The proposed assignation of the numbered peaks (1–17) is presented in ESI Table S3.† The sequenced oligosaccharide structures are named following the systematic nomenclature for xylo-oligosaccharides proposed by Fauré et al.60 (B) Single ion monitoring (SIM) chromatograms for the mannan oligosaccharides after β-mannanase hydrolysis and reduction/permethylation. The fragmentation of selected peaks and assignation of the ion fragments is presented in ESI Fig. S5–S7.† The proposed assignation of the numbered peaks (1–20) is presented in ESI Table S4.† Note: H (hexose: either Man, Glc or Gal). | ||
The evolution of the reported intramolecular motifs of AGX with the extraction time shows interesting trends. The oligosaccharides with an even spacing of mGlcA mainly all 6 Xylp units (oligosaccharides 8 and 11) are the most abundant in the xylan populations at shorter extraction times (extracts S1–S4); this indicates that these motifs are present in major domains in AGX.14,15 Interestingly, the relative abundance of XXA3XmU2X (oligosaccharide 11) decreases with the extraction time together with the relative increase of the intensity of the XXXXU4m2X counterpart (oligosaccharide 8). This trend is also observed for the remaining arabinose-containing oligosaccharides (oligosaccharides 6 and 12), since their intensity drastically decreases in the latter extracts (S5–S7). This indicates that Araf moieties are more sensitive to the hydrolytic conditions during subcritical water extraction and they are progressively cleaved at longer residence times, as we have previously proposed for arabinoxylans from cereal tissues.29 The relative abundance of the series of oligosaccharide motifs with consecutive mGlcA substitution in the xylan backbone (e.g. mU2mU2X, XmU2mU2X) remains fairly constant for the sequential extracts. However, the presence of smaller UXOs (e.g. XmU2X, XXmU2X) becomes progressively enriched in the extracts at longer residence times, revealing the presence of recalcitrant AGX populations with a larger relative abundance of the minor domains with odd and tighter glucuronidation. This suggests that the glucuronidation content and the presence of specific motifs with tighter and consecutive spacing play a large role in controlling the recalcitrance of xylan to extraction and their connectivity to cellulose and lignin, in agreement with previous studies.10,11,20,58
Glycomic profiling reveals the presence of two mannan populations with distinct patterns of glucosylation and galactosylation in spruce softwood
Unlike for xylans in the secondary cell walls of flowering plants and conifers, no regular motifs have been reported so far for the intramolecular distribution of glycosyl units (e.g. glucose and mannose in the backbone, and galactose decorations) and acetyl substitutions in secondary cell wall glucomannans.16,23 The identification of such patterns is hampered by the lack of specific enzymes identifying specific motifs in mannans, and due to the isobaric nature of the Glc, Man, and Gal units that makes it impossible to distinguish them by mass spectrometric approaches. In this study, we tackled this issue by using a two-pronged strategy combining complementary derivatization procedures of the enzymatically-digested manno-oligosaccharides (MOs) prior to LC-ESI-MS/MS separation and fragmentation analysis. First, we studied the glycosyl patterns by reduction and permethylation, which enables the identification of the substitution points in the mannan backbone by their specific fragmentation scars, but at the expense of cleaving the acetyl groups during the derivatization procedure thus simplifying the spectra. Then, we studied the pattern of acetylation of the native oligosaccharides by reductive amination in order to selectively label the reducing end, thus breaking the molecular symmetry of the MOs during tandem MS fragmentation.The successful isomeric resolution of the reduced and permethylated manno-oligosaccharides during chromatographic separation was demonstrated by single ion monitoring (SIM) (Fig. 3B and ESI Fig. S4†), exhibiting baseline separation from the smallest disaccharides (H2) to the largest detected penta-saccharides (M5). The complexity of the released isobaric (non-acetylated) reduced and permethylated MOs is majestic, as up to 20 separate peaks could be identified by the SIM procedure and subsequently fragmented by collision induced dissociation (CID). The fragmentation spectra by LC-ESI-MS/MS of the series of detected MOs (ESI Fig. S5 to S7†) enable their sequencing in selected cases, and the assignation of the 20 oligosaccharides is presented in ESI Table S4.† The linear manno-oligosaccharide standards (e.g. oligosaccharides 1, 3, 8, and 16) always eluted earlier than the oligosaccharides containing Gal decorations and Glc units in the backbone. The presence of Gal substitutions in the Manp reducing end was confirmed for oligosaccharides (4), (9), (12), (17) and (19) by the fragmentation scars of the reduced and permethylated oligosaccharides (ESI Fig. S5 and S6†), in agreement with the cleavage mechanism reported for the selected β-mannanase. Interestingly, the existence of internal Gal substitutions was proposed for the longer oligosaccharides (15) and (17), always placed in the −3 position from the reducing end (ESI Fig. S5†). These specific patterns were only observed for the mannan populations in the extracts at longer residence times, which had a higher galactose content as reported by the monosaccharide compositional analysis (Table 1). Unfortunately, the specific sequence of Glc and Man units in the oligosaccharide backbone could not be univocally determined from the tandem MS/MS fragmentation spectra, as the linear oligosaccharides exhibited very similar fragmentation patterns. Specific labelling techniques for the Man and/or Glc units in the oligosaccharides would be therefore required for the univocal assignation of the backbone sequence, which is not a trivial analytical task that we are currently undertaking. However, based on the potential number of combinations of Glc and Man in the linear H2–H4 isomers, most combinations are present in the oligosaccharides from the recalcitrant GGM population.
Despite the lack of obvious motifs in the sequence of Man and Glc units in the mannan backbone in the length scale of the enzymatically-released oligosaccharides, the oligomeric profiles show significant differences for the mannan populations extracted sequentially using subcritical water. The initial mannan population (S1) shows a much simpler oligomeric profile, with mainly linear oligosaccharides with a larger Man content, and just few decorated oligosaccharides containing Gal. Linear mannobiose (M2), mannotriose (M3) and mannotetraose (M4) constitute the main oligosaccharides detected, with minor abundance of linear glucose-containing manno-gluco-oligosaccharides (MGOs). On the other hand, the more recalcitrant mannan populations extracted at longer residence times (fractions S6 and S7) exhibit a much richer oligosaccharide profile, with the presence of Gal substitutions and a higher abundance of the linear glucose-containing MGOs. Indeed, the presence of the linear manno-oligosaccharides (M1–M5) is limited, and the presence of complex galactosylated and glucosylated MGO becomes significant. The higher content of Glc in the backbone drastically affects the hydrolytic action of the β-mannanase, as the enzyme does not tolerate the presence of a Glc unit at the +2 or −1 positions. The presence of Gal substitutions was evidenced both at the reducing end and at the −3 position of the MGO oligosaccharides. Interestingly, in the H3 series (m/z 697), only one of the two possible isomers with a Gal in the reducing end (ML or GL) was detected. As the presence of Gal decorations has been ascribed to the Man units in the backbone, this again hints at the potential occurrence of an even pattern of decorations in the mannan backbone although in this case in minor domains compared to those existing in spruce AGX. The occurrence of this even pattern of galactosylation has been previously reported for a seed mucilage galactoglucomannan from Arabidopsis thaliana.61
Softwood mannans do not display a regular acetylation pattern
The initial mannan population extracted at short residence times exhibited a relatively high degree of acetylation (DSac = 0.4) and lower content of Gal substitutions and Glc in the backbone, as previously described. In order to obtain more detailed information about the position of the acetyl groups in spruce acGGM, the enzymatically-hydrolysed extracts by β-mannanase were subjected to a mild derivatization process by reductive amination using anthranilic acid (AA), which on one side labelled the reducing end thus breaking the symmetry of the manno-oligosaccharides preserving at the same time the native acetylation (ESI Fig. S8a†). Comprehensive separation with baseline resolution was achieved for the acetylated MOs released by the β-mannanase treatment. Indeed, the SIM of the series of isomers corresponding to H4Acn reveals a rich and complex isomeric profile with over 20 isomeric peaks for the H4Ac1 and H4Ac2 oligosaccharides (Fig. S8b†). This rich and complex isomeric profile suggests that the position of the acetyl groups can occur in multiple positions of the oligosaccharides, in contrast to what was observed for acetylated glucuronoxylans in flowering plants (e.g. in Arabidopsis thaliana19,62 and Betula pendula11) where the isomeric acetylation patterns were much simpler, indicating the presence of preferred positions of the acetyl groups. The evolution of the H4Ac2 series for the different extracts reveals the reduction of acetylation with the extraction time (Fig. S8c†), which could be due to the fact that the acetylation is progressively lost during the harsh extraction conditions or that the recalcitrant mannan population has natively less acetylation.In order to ascertain the position of the acetyl groups, fragmentation of the isomerically-resolved MOs was performed by tandem MS/MS. Similar fragmentation patterns resulting from glycosidic linkage cleavages were observed for the H4Ac2 isomers, which reveal the putative position of the acetyl groups in the −2, −3, and −4 positions (see selected fragmentations for different isomers in Fig. S9†). The lack of acetylated fragments in the −1 position suggests that the β-mannanase cannot perform its catalytic activity when an acetyl group is present in the structure. This comprehensive fragmentation study indicates that the acetyl groups can be placed in either of the sugar units in the backbone, therefore suggesting the absence of an even pattern of acetylation as it has been recently shown for hardwood xylans. Unfortunately, the regioselectivity of acetylation in the O-2 and/or O-3 positions could not be assigned by LC-ESI-MS/MS, due to the absence of diagnostic cross-ring fragments in the MS/MS spectra. Previous studies by nuclear magnetic resonance indicated that acetylation in the O-2 position occurs in a two-fold ratio compared to the O-3 position.34
Surprisingly, we had difficulties in assigning the internal position of the acetyl groups in the oligosaccharides in some cases, since conflicting fragments were observed from glycosidic bond cleavage that could arise from multiple internal positions in the oligosaccharide (Fig. S7b and c†). This indicates the occurrence of acetyl degradation and/or migration during the ionization and fragmentation procedure by LC-ESI-MS/MS,63 even if the ionization and dissociation were performed with as mild conditions as possible. In any case, despite these analytical challenges, the complex isomeric pattern and the sequences from the fragmentation indicate that the acetylation can occur both in alternating and consecutive sugar units, thus suggesting the absence of a controlled acetylation pattern in the length scale of the oligosaccharides analysed here.
Conformation of mannan motifs and their interactions with cellulose surfaces
As the knowledge about both composition and internal monosaccharide sequence in hemicelluloses has increased dramatically over the last few years, there has been an active discussion about the possible influence of details in the chemical structure on the strength of the interaction with cellulose fibrils in the cell wall. One idea that has been advocated primarily by Dupree and co-workers14,18,61 is that polysaccharides whose backbone can adopt a cellulose-like structure may adsorb onto the fibrils in a highly ordered fashion that resembles co-crystallization. This picture is applicable to both xylans and mannans as long as they can adopt a 2-fold screw symmetry, and as long as the molecular fitting is not obstructed by side-group substitutions and acetylation. However, due to the 2-fold screw, this can be avoided when such substitutions are present in evenly-spaced patterns, as in the case of acetylated glucuronoxylan in flowering plants11,19,62 and arabinoglucuronoxylan in conifers.14,15 Indeed, in our previous publication15 we performed comprehensive molecular dynamics simulations of the regular oligosaccharide motifs sequenced in spruce AGX both in solution and onto cellulose surfaces, demonstrating that the presence of regularly-spaced substitutions do not hinder interactions with cellulose and the occurrence of the 2-fold screw conformation. In the case of mannans, the situation is complicated by the fact that the hydroxyl group in the C2 position in mannose has an axial configuration, as opposed to equatorial in glucose. This obstructs the possibility of a perfect fit to the cellulose surface, which suggests that the mannose content in GGM would have a negative influence on the affinity for cellulose. In this section, we use molecular dynamics simulations to investigate the binding strength of glucomannan motifs to the “hydrophilic” 110 face of a cellulose crystal.Our previous studies on hemicellulose motifs64 show that both mannans as well as glucomannans (GM) show similar conformational properties in solution, with respect to the conformation of glycosidic linkages. Specifically, they all can adopt a 2-fold screw conformation, just like the glucans, which hints at the fact that they could potentially bind in a crystal-like fashion to cellulose. In this study, three model GM motifs were placed on top of the cellulose 110 surface in a cellulose Iβ crystal-like conformation. Specifically, the GM hydroxyls were placed such as to extend the Iβ hydrogen bond network and the structures were allowed to relax for 10 ns. The final structures and the free energy charts show large differences between the all-glucose (GGGG) and all-mannose (MMMM) structures (Fig. 4B). While GGGG retains its cellulose-like structure, MMMM adopts a more twisted one. The conformational space of the GGGG glycosidic linkages is highly localized and positioned at the diagonal, which corresponds to a perfect 2-fold screw.65 The MMMM glycosidic linkages, on the other hand, display a more distributed conformational space, with the minimum placed beside the diagonal. This means that this motif did not retain the 2-fold symmetry, likely due to the mismatch caused by the C2 hydroxyls. The third, mixed motif, MGMG, was placed such that the C2 hydroxyls of the mannose units were pointing away from the cellulose surface, and consequently did not disrupt the hydrogen bond network. Indeed, this motif retained its 2-fold screw conformation, just as GGGG.
 | ||
| Fig. 4 Molecular dynamics simulation of the interaction of mannan oligosaccharide motifs with cellulose surfaces: (A) Definition of dihedral angles of the glycosidic linkage: ϕ = O5′–C1′–O4–C4 and ψ = C1′O4–C4–C3. (B). Conformation and free energy maps of the three selected manno-gluco-tetrasaccharides interacting with a cellulose surface (1–10): cellotetraose (GGGG), mannotetraose (MMMM) and an alternating MGO (GMGM). (C) Pulling-out experiments of selected MGOs on cellulose surfaces: snapshot and desorption energies per backbone sugar residue. | ||
To find out what possible effect this could have on the binding strength, simulations were performed in which the three motifs were pulled off the cellulose surface, and the free energy of binding was calculated as a function of distance from the surface. As can be seen (Fig. 4C), there is a clear correlation between the mannan backbone structure and its binding strength, where GGGG and MGMG display similar desorption energies, whereas for MMMM it is decreased by almost one half. Simulations were also performed for the additional motifs MMGM and MLMM (ESI Fig. S10†) where L stands for a mannose with an α-(1→6)-linked galactose unit. Both these motifs have axial C2 hydroxyls that hinder a perfect fit with cellulose, and consequently the desorption energies are lower than those for GGGG and MGMG (Table 2). For comparison purposes, the desorption energies we previously calculated for xylan are presented here.15 The xylan backbone has all its hydroxyls in the equatorial position, just as in glucose, but lacks the C6 hydroxymethyl group. Interestingly, the desorption energy for xylan ends up in between the GGGG and MMMM motifs (Table 2). This indicates that the composition and backbone sequence in hemicelluloses (xylans and mannans) modulates the intensity of the interactions with cellulose surfaces.
Insights into the influence of the mannan and xylan molecular structure on the supramolecular architecture and recalcitrance of softwoods
The sequential subcritical water extraction of spruce wood, together with the advanced characterization of the fractions by mass spectrometry (MS) based carbohydrate sequencing and molecular dynamics simulations, offer new insights into the influence of the hemicellulose molecular structure on the recalcitrance and organization of lignocelluloses from softwoods. The occurrence of mannan and xylan populations with distinct molecular structures based on their extractability and recalcitrance is revealed here (Fig. 5A). An accessible acetylated mannan population is extracted under milder conditions; this first population exhibits a relatively high degree of acetylation (DSac = 0.4) and a lower relative content of Glc in the backbone and of Gal substitutions. Acetylation plays a role in the biological function and properties of plant cell wall polysaccharides, although the individual contribution of the acetylation patterns for each polysaccharide type is not clear yet.66–68 Acetylation may influence the solubility of mannan polymers and increase their mobility in the cell wall; indeed, the deacetylation of mannans promotes their aggregation and reduces their solubility in water69,70 and favours the interaction with bacterial cellulose microfibrils in vitro.71 MS-based glycomics reveals that the acetylation pattern in spruce mannan is not regularly patterned, unlike what has been previously observed for acetylated glucuronoxylans from flowering plants.11,19,62 This patterned acetylation in glucuronoxylan is essential for their interaction with cellulose surfaces as a two-fold screw,17 contributing to the mechanical integrity of secondary cell walls. This suggests that this accessible acetylated mannan population may not have a structural function by interacting with cellulose surfaces. | ||
| Fig. 5 (a) Correlation between the molecular features of spruce hemicelluloses and their recalcitrance. (b) Proposed supramolecular organization of the mannan and xylan populations in softwoods. Spruce wood contains cellulose microfibrils of 4 nm composed of 18 chains.25,73 The accessible acetylated mannan population is integrated in the macrofibrillar matrix without contact with the cellulose surfaces. Both the xylan and the recalcitrant mannan populations display direct interactions with cellulose and lignin, mediated by their distinct molecular structures. | ||
After the extraction of this accessible mannan, the extraction of xylan becomes predominant. The molecular structure of spruce AGX shows major domains with even spacing of the mGlcA (predominantly all 6 Xylp units) and Araf substitutions, together with minor domains with uneven and consecutive mGlcA substitutions.15 The sequential extraction of spruce softwoods reveals that the major domains with an even spacing structure of AGX are more abundant at the initial extraction times, whereas the more recalcitrant xylan populations exhibit a higher relative abundance of the minor domains with more clustered mGlcA substitutions. This again suggests the role of glucuronidation in regulating the interconnectivity of xylan and lignin units through lignin–carbohydrate complexes,11,72 influencing the recalcitrance of lignocellulose to deconstruction in agreement with previous studies.10,58 Finally, a recalcitrant mannan population with a possibly lower acetylation (DSac = 0.1) and higher relative content of Glc and Gal is extracted at higher residence times. This mannan population shows more complex patterns of glucosylation in the backbone and galactosylation as substitutions, as revealed by enzymatic profiling and carbohydrate sequencing by LC-ESI-MS/MS. This distinct intramolecular structure of the recalcitrant mannan might have an important function in modulating the interaction with cellulose surfaces and therefore influencing the architecture of softwood lignocellulose.
Molecular modelling provides insightful details about the influence of the distinct intramolecular motifs of spruce mannans and xylans on the adsorption onto cellulose surfaces. The in silico simulations show that the content of mannose and its distribution in the backbone influence the binding strength of GGM to cellulose. The explanation offered by the simulations is that the axial conformation of the C2 hydroxyl groups in mannose hinders it from adsorbing in a cellulose-like conformation onto the fibrils, which of course glucose can. However, the simulations also show that for an even spacing of the mannose residues alternated by Glc units, the spruce mannan can be positioned such as the mannose C2 hydroxyl groups point away from the cellulose, in which case it adsorbs just as strongly as a pure β-glucan chain does. These results are in perfect analogy with the earlier results from molecular modelling of mannans61 and xylans,14,15,19 where an even placement of decorations favoured interactions with both the hydrophilic and the hydrophobic surfaces of celluloses. In our previous study on the molecular dynamics of spruce AGX motifs onto cellulose surfaces,15 we demonstrated that the presence of an even pattern of Araf and mGlcA substitutions on the backbone is not only sterically tolerated on both hydrophilic and hydrophobic cellulose surfaces but it also favours adsorption onto cellulose surfaces. When we normalize the free energy of adsorption calculated from our pull-out studies (Table 2) for GGGG (13–15 kJ per mol per residue), MMMM (7–8 kJ per mol per residue), and GMGM (14–15 kJ per mol per residue), and we compare it from the normalized pull-out results from our previous study for xylohexaose (12 kJ per mol per residue), we can conclude that the presence of consecutive mannosyl residues affects drastically the adsorption energy on cellulose surfaces due to the presence of the OH-group in the axial position. On the other hand, the normalized energy of adsorption of the xylan backbone falls between the fully mannosylated backbone and rather close to the theoretical value for a β-glucan backbone, which indicates that the lack of a C6 carbon in Xylp has a lower effect than the epimeric C2 hydroxyl in mannose on the adsorption onto cellulose. Finally, as previously reported, the occurrence of alternating GMGM residues shows similar free adsorption energies to the β-glucan backbone, which indicates the importance of the Glc content and patterning in the glucomannan backbone to modulate the interactions with cellulose. These values are in agreement with the relative extractability/recalcitrance of the distinct mannan and xylan populations in spruce softwoods, which again reflects the importance of the molecular motifs of hemicelluloses in tuning their interactions with cellulose surfaces, and the overall lignocellulose connectivity and recalcitrance.
This study provides molecular insights about the supramolecular architecture and organization of the polymeric components in spruce secondary cell walls. In softwoods from conifers, the individual cellulose microfibrils of 3–4 nm in diameter25,74 can be organized in larger aggregates known as macrofibrils, with average diameters of 10–20 nm,25,73 but that can reach up to 60 nm as revealed by cryo-SEM.24 In secondary cell walls from Arabidopsis thaliana, it has been reported that xylan and lignin contribute to the macrofibril size, but the role of mannans in the overall organization of plant secondary cell wall macrofibrils is not clear yet.24 Here we propose a model for the architecture of spruce softwoods where the different hemicellulose populations have distinct organization and arrangement (Fig. 5B). The accessible acGGM populations with high mobility might be arranged in the macrofibre wall matrix with no direct interaction with the cellulose microfibre surfaces, since the interactions are prevented by the high acetylation and high mannose content. On the other hand, spruce AGX shows major domains with an even pattern of glucuronidation and arabinosylation, which favours their interaction with cellulose surfaces. The minor xylan domains with clustered and consecutive glucuronidation show higher recalcitrance, which could be caused by their closer interaction with lignin residues. Indeed, there is indirect evidence that (m)GlcA can act as a crosslinking point with lignin units in secondary cell walls, since a reduced mGlcA content decreases recalcitrance10,58 and glucuronyl esterases assist in releasing carboxylic acids in model compounds and enriched birch75 and spruce76 extracts. Finally, the recalcitrant GGM population might be directly interacting with the cellulose surfaces, mediated by the higher content of Glc in the backbone, and the presence of even motifs of alternating Man units with higher content of Gal substitutions. This agrees well with a recent study by the solid-state NMR of never-dried spruce softwoods,23 which reveals the existence of rigid mannan and xylan populations both closely interacting with cellulose microfibrils, and also matrix mannan and xylan populations not directly bound with cellulose.
This study reveals the distinct molecular structures of these mannan populations in spruce softwoods, and provides technical evidence that sequential subcritical water extraction can provide targeted hemicellulose fractions with controlled purity and molecular structures. This is of large technical importance for the preparation of hemicellulose-based materials with controlled properties, since wood hemicelluloses are a widely available but largely unexploited source of biopolymers due to their heterogeneous molecular composition.
Conclusions
The implementation of sequential subcritical water extraction without previous delignification shows great potential for the targeted isolation of hemicellulose fractions from spruce softwoods with high purities and high molar masses. The control of the extraction times enables the targeted isolation of mannan and xylan populations with distinct molecular structures. An accessible acetylated mannan population with high mannose content could be isolated at short residence times (0–60 min), followed by an enrichment of the xylan populations at moderate extraction times (60–180 min), and finally a recalcitrant mannan population with higher glucose and galactose content was isolated at much longer residence times (180–300 min).The combination of enzymatic deconstruction and mass-spectrometric based carbohydrate sequencing reveals the presence of distinct intramolecular motifs in spruce mannans and xylans and the effect of extraction. The accessible mannan population shows the occurrence of major domains with linear mannosyl and gluco-mannosyl oligosaccharides, with minor substituted motifs. This mannan population does not show a regular acetylation pattern, contrary to what has been previously described for hardwood glucuronoxylan. The major and minor domains in spruce AGX show different recalcitrance to sequential extraction, where the even motifs present in the major domains are more abundant at shorter extraction times and the minor domains with clustered glucuronidation are enriched at longer times. Finally, the recalcitrant mannan domains display a complex pattern of glucosylation and galactosylation, where the presence of minor domains with an even placement of the mannose residues can be detected.
Molecular dynamics simulations reveal the importance of the backbone sequence in spruce mannan to modulate the interaction with cellulose surfaces. The presence of consecutive mannosyl units hinders direct interaction with cellulose, due to the presence of the C2 hydroxyl group in axial configuration. On the other hand, the occurrence of an even glucosylation pattern in the GGM backbone allows the close interaction with cellulose surfaces, with similar free energies to that of the pure β-glucan backbone.
The integration of sequential subcritical water extraction, MS-based carbohydrate sequencing and molecular dynamics simulations provides novel molecular insights into the heterogeneity of the hemicellulose populations in softwoods, their contribution to lignocellulose recalcitrance and their putative role in the supramolecular architecture and the organization in conifer secondary cell walls. This has large implications for the sustainable exploitation of softwoods for material applications, and for overcoming the innate recalcitrance of lignocellulosic biomass through the design of integral biorefineries.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Prof. James F. Preston (University of Florida) for providing the GH30 glucuronoxylanase. MD simulations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at the PDC Center for High Performance Computing, KTH Royal Institute of Technology, partially funded by the Swedish Research Council through grant agreement 2016-07213. FV and AMA thank the Swedish Research Council (Project 621-2014-5295) for the financial support. The Knut and Alice Wallenberg (KAW) Foundation is acknowledged for the resources and financial support to JW within the Wallenberg Wood Science Centre.References
- Y. M. Bar-On, R. Phillips and R. Milo, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6506–6511 CrossRef CAS PubMed.
- J. Philp, New Biotechnol., 2018, 40, 11–19 CrossRef CAS PubMed.
- D. Keegan, B. Kretschmer, B. Elbersen and C. Panoutsou, Biofuels, Bioprod. Biorefin., 2013, 7, 193–206 CrossRef CAS.
- D. Cosgrove and M. Jarvis, Front. Plant Sci., 2012, 3, 204 Search PubMed.
- L. Petridis and J. C. Smith, Nat. Rev. Chem., 2018, 2, 382–389 CrossRef CAS.
- R. A. Burton, M. J. Gidley and G. B. Fincher, Nat. Chem. Biol., 2010, 6, 724–732 CrossRef CAS PubMed.
- M. C. McCann and N. C. Carpita, J. Exp. Bot., 2015, 66, 4109–4118 CrossRef CAS PubMed.
- M. E. Himmel, S.-Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, science, 2007, 315, 804–807 CrossRef CAS PubMed.
- S.-Y. Ding, Y.-S. Liu, Y. Zeng, M. E. Himmel, J. O. Baker and E. A. Bayer, Science, 2012, 338, 1055–1060 CrossRef CAS PubMed.
- J. C. Mortimer, G. P. Miles, D. M. Brown, Z. Zhang, M. P. Segura, T. Weimar, X. Yu, K. A. Seffen, E. Stephens, S. R. Turner and P. Dupree, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17409–17414 CrossRef CAS PubMed.
- A. Martínez-Abad, N. Giummarella, M. Lawoko and F. Vilaplana, Green Chem., 2018, 20, 2534–2546 RSC.
- R. L. Silveira, S. R. Stoyanov, S. Gusarov, M. S. Skaf and A. Kovalenko, J. Am. Chem. Soc., 2013, 135, 19048–19051 CrossRef CAS PubMed.
- H. V. Scheller and P. Ulvskov, Annu. Rev. Plant Biol., 2010, 61, 263–289 CrossRef CAS PubMed.
- M. Busse-Wicher, A. Li, R. L. Silveira, C. S. Pereira, T. Tryfona, T. C. F. Gomes, M. S. Skaf and P. Dupree, Plant Physiol., 2016, 171, 2418–2431 CAS.
- A. Martínez-Abad, J. Berglund, G. Toriz, P. Gatenholm, G. Henriksson, M. Lindström, J. Wohlert and F. Vilaplana, Plant Physiol., 2017, 175, 1579–1592 CrossRef.
- J. Liu, A. S. Leppanen, V. Kisonen, S. Willfor, C. Xu and F. Vilaplana, Int. J. Biol. Macromol., 2018, 112, 616–625 CrossRef CAS PubMed.
- N. J. Grantham, J. Wurman-Rodrich, O. M. Terrett, J. J. Lyczakowski, K. Stott, D. Iuga, T. J. Simmons, M. Durand-Tardif, S. P. Brown, R. Dupree, M. Busse-Wicher and P. Dupree, Nat. Plants, 2017, 3, 859–865 CrossRef CAS PubMed.
- T. J. Simmons, J. C. Mortimer, O. D. Bernardinelli, A.-C. Pöppler, S. P. Brown, E. R. de Azevedo, R. Dupree and P. Dupree, Nat. Commun., 2016, 7, 13902 CrossRef CAS PubMed.
- M. Busse-Wicher, T. C. F. Gomes, T. Tryfona, N. Nikolovski, K. Stott, N. J. Grantham, D. N. Bolam, M. S. Skaf and P. Dupree, Plant J., 2014, 79, 492–506 CrossRef CAS PubMed.
- N. Giummarella and M. Lawoko, ACS Sustainable Chem. Eng., 2017, 5, 5156–5165 CrossRef CAS.
- M. Lawoko, G. Henriksson and G. r. Gellerstedt, Biomacromolecules, 2005, 6, 3467–3473 CrossRef CAS.
- X. Kang, A. Kirui, M. C. Dickwella Windanage, F. Mentink-Vigier, D. J. Cosgrove and T. Wang, Nat. Commun., 2019, 10, 347 CrossRef CAS PubMed.
- O. M. Terrett, J. J. Lyczakowski, L. Yu, D. Iuga, W. T. Franks, S. P. Brown, R. Dupree and P. Dupree, Nat. Commun., 2019, 10, 4978 CrossRef PubMed.
- J. J. Lyczakowski, M. Bourdon, O. M. Terrett, Y. Helariutta, R. Wightman and P. Dupree, Front. Plant Sci., 2019, 10, 1398 CrossRef PubMed.
- A. N. Fernandes, L. H. Thomas, C. M. Altaner, P. Callow, V. T. Forsyth, D. C. Apperley, C. J. Kennedy and M. C. Jarvis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, E1195–E1203 CrossRef PubMed.
- A. Martínez-Abad, A. C. Ruthes and F. Vilaplana, J. Appl. Polym. Sci., 2016, 133, 42523 Search PubMed.
- J. V. Rissanen, H. Grénaman, C. Xu, S. Willför, D. Y. Murzin and T. Salmi, ChemSusChem, 2014, 7, 2947–2953 CrossRef CAS.
- S. S. Toor, L. Rosendahl and A. Rudolf, Energy, 2011, 36, 2328–2342 CrossRef CAS.
- A. C. Ruthes, A. Martinez-Abad, H.-T. Tan, V. Bulone and F. Vilaplana, Green Chem., 2017, 19, 1919–1931 RSC.
- K. S. Mikkonen, K. Parikka, A. Ghafar and M. Tenkanen, Trends Food Sci. Technol., 2013, 34, 124–136 CrossRef CAS.
- W. Xu, X. Zhang, P. Yang, O. Långvik, X. Wang, Y. Zhang, F. Cheng, M. Österberg, S. Willför and C. Xu, ACS Appl. Mater. Interfaces, 2019, 11, 12389–12400 CrossRef CAS PubMed.
- V. Kisonen, C. Xu, R. Bollström, J. Hartman, H. Rautkoski, M. Nurmi, J. Hemming, P. Eklund and S. Willför, Cellulose, 2014, 21, 4497–4509 CrossRef CAS.
- T. Hannuksela and C. Hervé du Penhoat, Carbohydr. Res., 2004, 339, 301–312 CrossRef CAS PubMed.
- J. Lundqvist, A. Teleman, L. Junel, G. Zacchi, O. Dahlman, F. Tjerneld and H. Stålbrand, Carbohydr. Polym., 2002, 48, 29–39 CrossRef CAS.
- A. Escalante, A. Goncalves, A. Bodin, A. Stepan, C. Sandström, G. Toriz and P. Gatenholm, Carbohydr. Polym., 2012, 87, 2381–2387 CrossRef CAS.
- L. S. McKee, H. Sunner, G. E. Anasontzis, G. Toriz, P. Gatenholm, V. Bulone, F. Vilaplana and L. Olsson, Biotechnol. Biofuels, 2016, 9, 1–13 CrossRef PubMed.
- F. Bertaud, A. Sundberg and B. Holmbom, Carbohydr. Polym., 2002, 48, 319–324 CrossRef CAS.
- J. F. Saeman, W. E. Moore, R. L. Mitchell and M. A. Millett, Tappi J., 1954, 37, 336–343 CAS.
- S. Willför, A. Pranovich, T. Tamminen, J. Puls, C. Laine, A. Suurnäkki, B. Saake, K. Uotila, H. Simolin, J. Hemming and B. Holmbom, Ind. Crops Prod., 2009, 29, 571–580 CrossRef.
- A. G. J. Voragen, H. A. Schols and W. Pilnik, Food Hydrocolloids, 1986, 1, 65–70 CrossRef CAS.
- P. Mischnick, in Mass Spectrometry of Polymers, ed. M. Hakkarainen, Springer, Berlin Heidelberg, 2012, vol. 248, ch. 134, pp. 105–174 Search PubMed.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- K. N. Kirschner, A. B. Yongye, S. M. Tschampel, J. González-Outeiriño, C. R. Daniels, B. L. Foley and R. J. Woods, J. Comput. Chem., 2008, 29, 622–655 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- T. Song, A. Pranovich and B. Holmbom, Bioresour. Technol., 2011, 102, 10518–10523 CrossRef CAS PubMed.
- P. O. Kilpeläinen, S. S. Hautala, O. O. Byman, L. J. Tanner, R. I. Korpinen, M. K. J. Lillandt, A. V. Pranovich, V. H. Kitunen, S. M. Willför and H. S. Ilvesniemi, Green Chem., 2014, 16, 3186–3194 RSC.
- F. Bertaud and B. Holmbom, Wood Sci. Technol., 2004, 38, 245–256 CAS.
- S. Willför, R. Sjöholm, C. Laine, M. Roslund, J. Hemming and B. Holmbom, Carbohydr. Polym., 2003, 52, 175–187 CrossRef.
- F. J. St John, J. C. Hurlbert, J. D. Rice, J. F. Preston and E. Pozharski, J. Mol. Biol., 2011, 407, 92–109 CrossRef CAS PubMed.
- J. R. Bromley, M. Busse-Wicher, T. Tryfona, J. C. Mortimer, Z. Zhang, D. M. Brown and P. Dupree, Plant J., 2013, 74, 423–434 CrossRef CAS PubMed.
- J. J. Lyczakowski, K. B. Wicher, O. M. Terrett, N. Faria-Blanc, X. Yu, D. Brown, K. B. R. M. Krogh, P. Dupree and M. Busse-Wicher, Biotechnol. Biofuels, 2017, 10, 224 CrossRef PubMed.
- B. Domon and C. E. Costello, Glycoconjugate J., 1988, 5, 397–409 CrossRef CAS.
- R. Fauré, C. M. Courtin, J. A. Delcour, C. Dumon, C. B. Faulds, G. B. Fincher, S. Fort, S. C. Fry, S. Halila, M. A. Kabel, L. Pouvreau, B. Quemener, A. Rivet, L. Saulnier, H. A. Schols, H. Driguez and M. J. O'Donohue, Aust. J. Chem., 2009, 62, 533–537 CrossRef.
- L. Yu, J. J. Lyczakowski, C. S. Pereira, T. Kotake, X. Yu, A. Li, S. Mogelsvang, M. S. Skaf and P. Dupree, Plant Physiol., 2018, 178, 1011–1026 CrossRef CAS PubMed.
- S.-L. Chong, L. Virkki, H. Maaheimo, M. Juvonen, M. Derba-Maceluch, S. Koutaniemi, M. Roach, B. Sundberg, P. Tuomainen, E. J. Mellerowicz and M. Tenkanen, Glycobiology, 2014, 24, 494–506 CrossRef CAS PubMed.
- M. U. Roslund, O. Aitio, J. Wärnå, H. Maaheimo, D. Y. Murzin and R. Leino, J. Am. Chem. Soc., 2008, 130, 8769–8772 CrossRef CAS PubMed.
- J. Berglund, T. Angles d'Ortoli, F. Vilaplana, G. Widmalm, M. Bergenstråhle-Wohlert, M. Lawoko, G. Henriksson, M. Lindström and J. Wohlert, Plant J., 2016, 88, 56–70 CrossRef CAS PubMed.
- A. D. French and G. P. Johnson, Cellulose, 2009, 16, 959–973 CrossRef CAS.
- P. Pawar, S. Koutaniemi, M. Tenkanen and E. Mellerowicz, Front. Plant Sci., 2013, 4, 118 Search PubMed.
- S. Gille and M. Pauly, Front. Plant Sci., 2012, 3, 12 CAS.
- M. Pauly and V. Ramírez, Front. Plant Sci., 2018, 9, 1210 CrossRef PubMed.
- S. Willför, K. Sundberg, M. Tenkanen and B. Holmbom, Carbohydr. Polym., 2008, 72, 197–210 CrossRef.
- S. Kishani, F. Vilaplana, W. Xu, C. Xu and L. Wågberg, Biomacromolecules, 2018, 19, 1245–1255 CrossRef CAS PubMed.
- S. E. C. Whitney, J. E. Brigham, A. H. Darke, J. S. G. Reid and M. J. Gidley, Carbohydr. Res., 1998, 307, 299–309 CrossRef CAS.
- N. Takahashi and T. Koshijima, Wood Sci. Technol., 1988, 22, 231–241 CrossRef CAS.
- M. C. Jarvis, Philos. Trans. R. Soc., A, 2018, 376, 20170045 CrossRef PubMed.
- L. Donaldson, Wood Sci. Technol., 2007, 41, 443 CrossRef CAS.
- C. Mosbech, J. Holck, A. S. Meyer and J. W. Agger, Biotechnol. Biofuels, 2018, 11, 71 CrossRef PubMed.
- J. Arnling Bååth, N. Giummarella, S. Klaubauf, M. Lawoko and L. Olsson, FEBS Lett., 2016, 590, 2611–2618 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc01207f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |