
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in hydrothermal carbonisation: from tailored carbon materials and biochemicals to applications and bioenergy
Sabina A.
Nicolae
*a,
Heather
Au
 b,
Pierpaolo
Modugno
a,
Hui
Luo
b,
Anthony E.
Szego
c,
Mo
Qiao
b,
Liang
Li
d,
Wang
Yin
e,
Hero J.
Heeres
e,
Nicole
Berge
d and
Maria-Magdalena
Titirici
*ab
b,
Pierpaolo
Modugno
a,
Hui
Luo
b,
Anthony E.
Szego
c,
Mo
Qiao
b,
Liang
Li
d,
Wang
Yin
e,
Hero J.
Heeres
e,
Nicole
Berge
d and
Maria-Magdalena
Titirici
*ab
aSchool of Engineering and Materials Science, Queen Mary University of London, Mile End Road, London E1 4NS, UK
bDepartment of Chemical Engineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK. E-mail: m.titirici@imperial.ac.uk
cMaterials and Environmental Chemistry Department, Stockholm University, SE-106 91, Stockholm, Sweden
dCivil and Environmental Engineering, University of South Carolina, Columbia, SC 29201, USA
eChemical Engineering Department, ENTEG, University of Groningen, Nijenborgh 4, 9747 AG Gronigen, Netherlands
First published on 1st July 2020
Abstract
Introduced in the literature in 1913 by Bergius, who at the time was studying biomass coalification, hydrothermal carbonisation, as many other technologies based on renewables, was forgotten during the “industrial revolution”. It was rediscovered back in 2005, on the one hand, to follow the trend set by Bergius of biomass to coal conversion for decentralised energy generation, and on the other hand as a novel green method to prepare advanced carbon materials and chemicals from biomass in water, at mild temperature, for energy storage and conversion and environmental protection. In this review, we will present an overview on the latest trends in hydrothermal carbonisation including biomass to bioenergy conversion, upgrading of hydrothermal carbons to fuels over heterogeneous catalysts, advanced carbon materials and their applications in batteries, electrocatalysis and heterogeneous catalysis and finally an analysis of the chemicals in the liquid phase as well as a new family of fluorescent nanomaterials formed at the interface between the liquid and solid phases, known as hydrothermal carbon nanodots.
1. Introduction
Our world is facing hard and unprecedented challenges due to the constant increase of population and technology development which implies a growing demand for energy and advanced materials that can no longer rely on fossil fuel exploitation. There is a strong need for a new productive system where clean energy generation and storage go hand in hand with sustainable materials production and studies on Hydrothermal Carbonisation (HTC) of biomass are aimed at developing a clever technology to balance them both. The term hydrothermal carbonisation describes a thermal treatment that takes place in aqueous medium heated at subcritical temperatures (180–250 °C), under self-generated pressure. HTC has been known to scientists over a century ago and it has been used as a way to mimic the natural process of coal formation,1–5 in times when there was only little concern about the temporary shortage of fossil fuel supplies. The last two decades have seen renewed interest in this topic.6–13 The main advantage of HTC is that, as the process takes place in water, biomass does not require a preliminary drying step, thus saving great amounts of energy and time, and furthermore allowing processing of wet mixtures, including aqueous waste and sewage sludge.14Fig. 1 illustrates a schematic of the HTC process and possible products, which will also be covered in this review. In aqueous media, at subcritical temperatures and pressures, lignocellulosic materials undergo a vast series of transformations such as hydrolysis, dehydration, bond cleavage, formation of new bonds, and condensation. The ultimate products of these reactions can be grouped into liquid species (or water-soluble compounds), solid species (primary and secondary chars and carbon dots) and gases (water vapour, CO, CO2, and CH4).15 Water-soluble compounds include furan derivatives (furfural and 5-hydroxymethylfurfural) and carboxylic acids. Many of the soluble chemicals found in the liquid phase after HTC are commercially valuable, and a few of them have been recognized as platform chemicals for a transition to a greener chemistry.16 In some cases, the liquid phase derived from HTC treatment of biowaste feedstocks can also be used in agriculture for irrigation or recycled for a new batch of biomass conversion. Primary chars are the direct product of hydrothermal dehydration and pyrolysis of biomass. They are referred to as biochars or hydrochars to highlight their biological origin, their charred appearance (that corresponds at a physical level to a higher energy density value in terms of the higher heating value compared to that of the original biomass17,18) and the hydrothermal process employed to prepare them. The higher energy density value makes them fit for combustion to generate decentralised energy with lower CO2 emissions when compared with coal power plants, since they are produced from biomass which can, in principle, be regrown to adsorb back the generated CO2. Secondary chars are the result of further dehydration, condensation and polymerization of water dissolved compounds. The resulting particles present a spherical shape and small sizes in the domain of microns. They have long been known to form during HTC of sugars or lignocellulosic biomass. They are also often, confusingly, called humins or hydrochars. The former name has a long documented history of misuse19 and originally referred to the alkali-insoluble fraction of humus. Throughout the times its meaning has shifted to denote the dark-brownish, amorphous products obtained by acid hydrolysis of proteins or sugars. As for the word hydrochars, we already saw that it describes hydrothermally charred biomass. Therefore, in order to avoid any further confusion, we will use the expression hydrothermal carbon (HT carbon) to denote the carbonaceous microspheres obtained via hydrothermal carbonization of cellulosic biomass. Their porosity can be changed using natural templates or activation procedures; their chemical and physical properties can be easily modified by their combination with different components, such as inorganic compounds, and they can be easily functionalized leading to a large number of oxygenated groups present on the surface. Carbon dots are quasi-spherical fluorescent carbon nanoparticles,20,21 with diameters around or below 10 nm,22 an amorphous or nanocrystalline structure with sp2 carbon clusters23 and a high amount of oxygen atoms and carboxylic groups on their surface.24 Although their mechanism of formation is still not very well understood, these nanoparticles have been successfully employed as probes for the detection of metal ions25–28 (due to the quenching effect of these species on the fluorescence of the CDs) and other chemicals,29–31 as catalysts29 or for imaging of cells.32
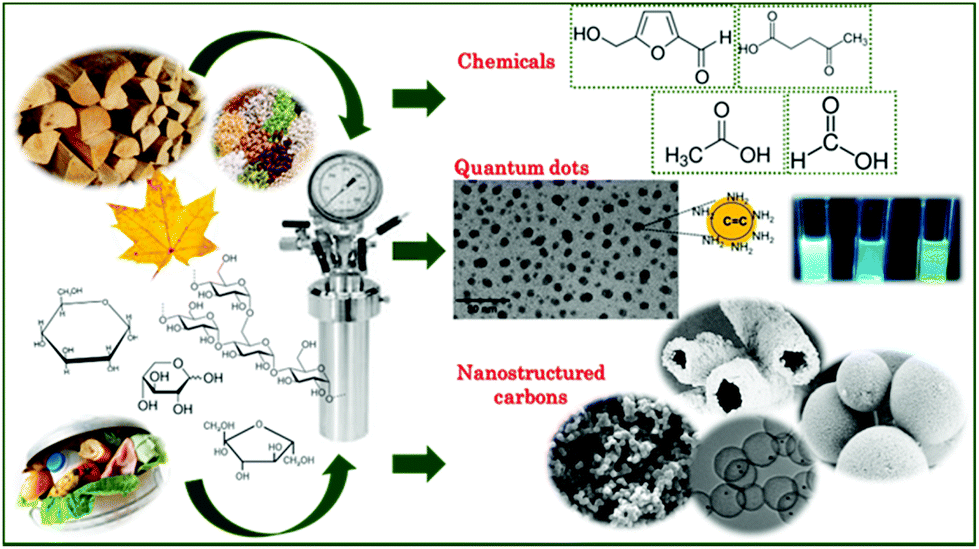 | ||
| Fig. 1 Schematic representation of the HTC process with the main topics represented here. | ||
The first section of this review will be dedicated to the production of advanced carbon materials from biomass or biomass derivatives with focus on applications in energy storage and conversion and environmental production. Following this, we will discuss the composition of the liquid phase depending on the precursors used and the various HTC conditions as well as methods for the separation of these components from the liquid phase and their transformation in other viable chemicals using heterogeneous catalysis. Next, we will address the production of fluorescent nanometre-sized carbons. We will then focus on HTC as a viable route to convert various raw biowaste materials into hydrothermal carbons and provide a thorough characterisation of each product depending on the raw precursors and hydrothermal conditions. Finally, we will present the possibility of upgrading some of the hydrothermal carbons to fuels via reforming over heterogeneous catalysts. For each section we will provide an overview based on recently published papers in the literature and our own perspectives on where scientific knowledge and understanding the fundamentals are still needed for HTC to become a viable technique for both the production of advanced materials and chemicals as well as decentralised bioenergy using biowaste.
2. Advanced hydrothermal carbon materials
2.1. Fundamentals and gaps in hydrothermal carbonisation for advanced carbon production
The three main components of lignocellulosic biomass are cellulose, hemicellulose and lignin. Cellulose is a linear polysaccharide of glucose units bonded together by β-(1,4)-glucosidic bonds. Hemicellulose is a polysaccharide too, but it differs from cellulose in that it is made of a number of different sugar monomers, such as xylose, glucose, mannose and galactose. This variety of monomers makes the overall structure of hemicellulose less stronger than that of cellulose, with shorter branched chains. Lignin is a highly randomly cross-branched polymer of phenylpropane derivatives. Among the three aforementioned biopolymers, lignin is the most stable and least prone33 to hydrolysis under hydrothermal conditions, requiring temperatures as high as 250 °C to start decomposing.34 Therefore, polysaccharides, their monomers and their decomposition products will only be taken into account in the following description of HTC reaction pathways. However, it is important to stress that real biomass always contains lignin to some extent, and its presence is known to hinder the hydrolysis of long saccharide chains under hydrothermal conditions, lowering conversion yields of both soluble and insoluble products.35 On the other hand, some experiments on a mixture of components including saccharides, proteins, lipids and lignin, although performed in a range of temperatures beyond those considered in this review, showed a clear synergistic effect of the different components, with an increased yield of biocrude in the case of the mixture, compared to the average yield obtained by treatment of single precursors.36 Decomposition of cellulosic materials under hydrothermal conditions can be summarized in a few basic steps: hydrolysis of cellulose and hemicellulose chains into their constituting monomers (mainly glucose and other hexose and pentose sugars); dehydration of C6 sugars to 5-hydroxymethylfurfural (5-HMF) and C5 sugars to furfural; decomposition to lower molecular weight compounds or, alternatively, condensation and aromatization reaction, to produce hydrothermal carbon.37 A quite exhaustive scheme of the complex network of reaction pathways of decomposition of cellulose under hydrothermal conditions has recently been proposed by Buendia-Kandia et al.38 (Fig. 2). The scheme was elaborated by studying the decomposition of microcrystalline cellulose under hydrothermal conditions, testing three temperatures (180 °C–220 °C–260 °C), sampling the liquid phase every 20 minutes for an overall reaction time of 120 minutes. Their results confirmed the pathway proposed by Matsumura:39 long chains of cellulose are firstly hydrolysed to smaller oligomers or monomers of glucose. Glucose can undergo isomerization to fructose via the Lobry de Bruyn–Alberda–van Ekenstein transformation or epimerization to mannose.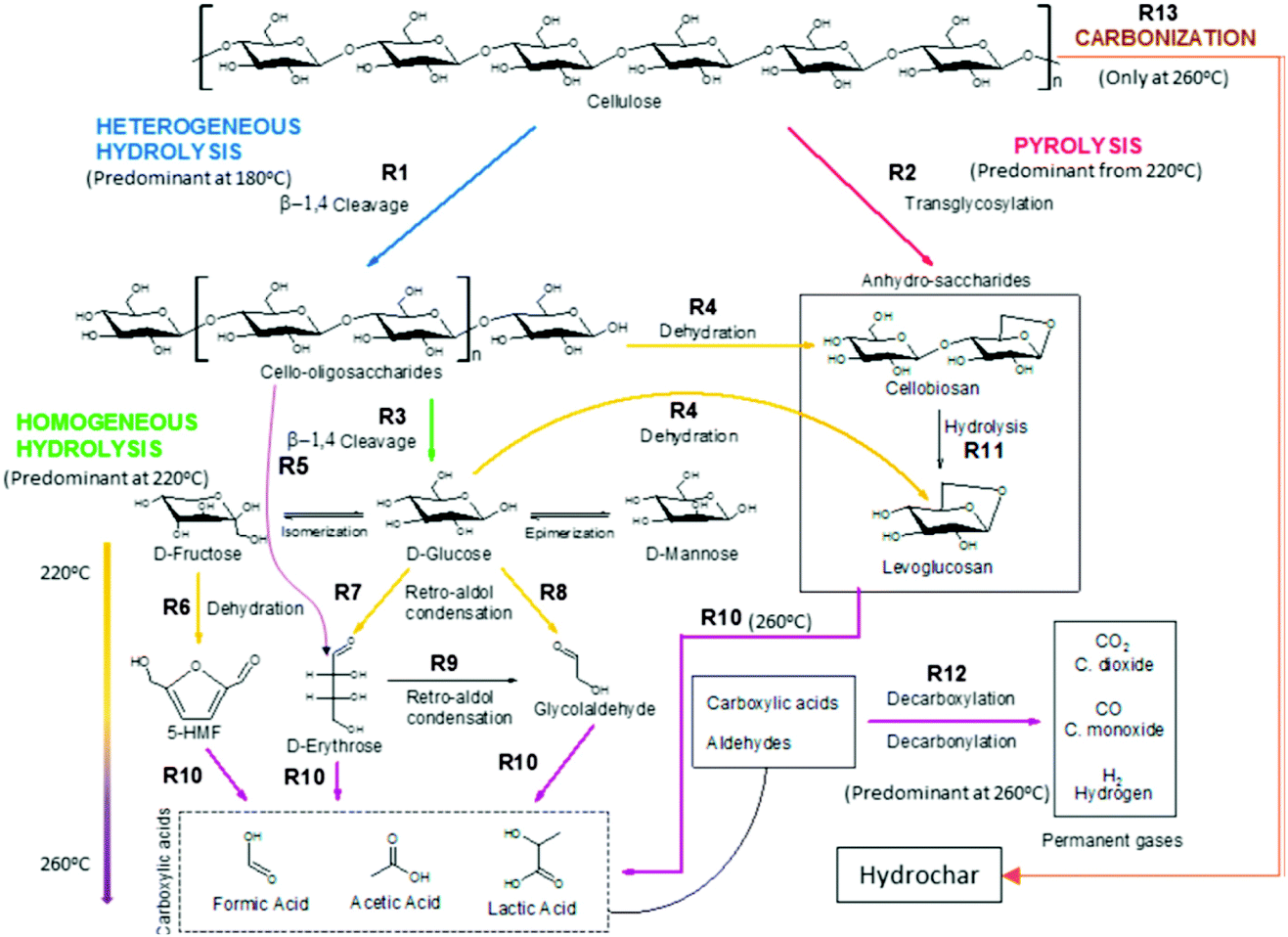 | ||
| Fig. 2 Scheme of the reaction pathways of cellulose decomposition as proposed by Buendia-Kandia et al.; reproduced with permission from ref. 38. | ||
As said before, furfural and 5-HMF derive from dehydration of pentoses (Fig. 3) and hexoses (Fig. 4), respectively. Levulinic acid (LA), also named 4-oxopentanoic acid, is derived from 5-HMF, which, under hydrothermal conditions, is rehydrated to form LA along with formic acid (Fig. 5). Glucose oligomers can undergo dehydration before complete hydrolysis, producing cellobiosan and subsequently levoglucosan. Retro-aldol condensation of glucose produces erythrose and glycolaldehyde from which lactic acid and acetic acid are derived. The presence of lactic acid (LacA) in the hydrothermal conversion of sugars was explained with a mechanism involving an initial retro-aldol condensation of fructose, yielding glyceraldehyde and dihydroxyacetone. Both of these compounds can be dehydrated to pyruvaldehyde, which in turn is converted to LacA with a benzylic acid rearrangement (Fig. 6). The benzylic acid rearrangement requires basic catalysis, and this explains why lactic acid is found in relevant amounts when basic conditions are employed for hydrothermal conversion of cellulosic biomass.40,41 However, it was demonstrated that water under subcritical conditions could provide sufficient hydroxide ions to catalyse the reaction in the absence of any other sources of OH−. In fact, lactic acid can also be found among the HTC products of sugars when an acid is added as a catalyst.42
 | ||
| Fig. 3 Mechanism of dehydration of xylose to furfural via an acyclic pathway; adapted from ref. 43. | ||
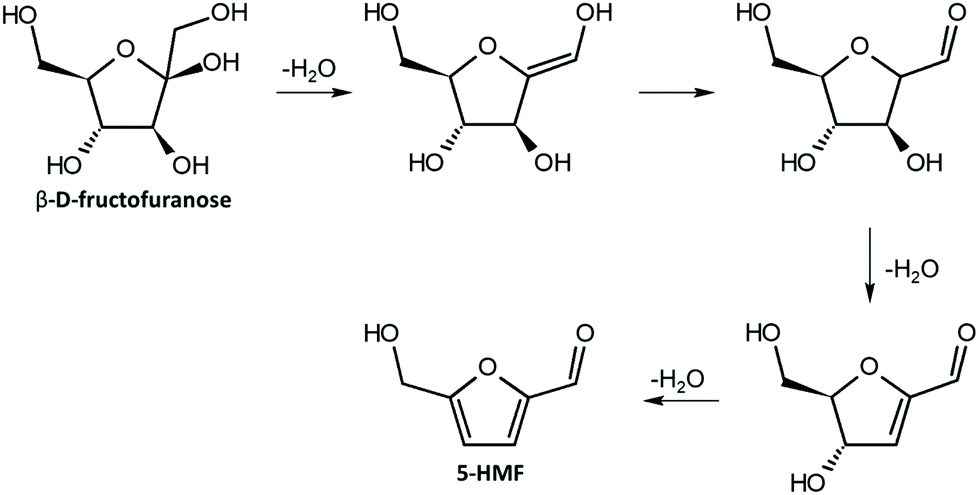 | ||
| Fig. 4 Mechanism of dehydration of fructose to 5-HMF; adapted from ref. 44. | ||
 | ||
| Fig. 5 Mechanism of rehydration of 5-HMF to levulinic acid with loss of a carbon atom in the form of formic acid; adapted from ref. 46. | ||
 | ||
| Fig. 6 Mechanism of formation of lactic acid through retro aldol condensation and a benzylic rearrangement; adapted from ref. 42. | ||
Decarbonylation and decarboxylation reactions account for the production of CO and CO2. Finally, due to limited access of water molecules on the inner cellulose fibres, pyrolysis of cellulose occurs, leading to the direct formation of the primary hydrothermal carbon. As for hydrothermal carbon, this material is structured as spherical microspheres6 whose chemical structure consists of condensed furan rings linked either via the α-carbon or via sp2- or sp3-type carbon.45 This suggests that 5-HMF and furfural are the main building blocks of hydrothermal carbon. 5-HMF is a rather unstable and reactive molecule under hydrothermal conditions. A closer look at its chemical structure is useful in understanding its reactivity and thus to propose hypotheses about its role in the formation of HTC spheres. 5-HMF possesses one aldehyde group and one hydroxymethyl group at positions 2 and 5, respectively, on the furan ring.
The aldehyde group can undergo aldol condensation with α-ketones or aldehydes (Fig. 7) or react with alcohols or diols to give (hemi)acetals. The acetalisation of the aldehyde group activates the furan ring towards electrophilic aromatic substitution. The hydroxymethyl group, on the other hand, can be subject to nucleophilic substitution, as the mildly acidic conditions can protonate the hydroxyl group making it a better leaving group. Finally, the whole furan ring, due to its lower aromaticity when compared to benzene, can behave as a diene, undergoing nucleophilic addition, ring-opening reaction or Diels–Alder cycloaddition.
 | ||
| Fig. 7 Tautomeric forms of HMF and the reaction to a dimer via aldol condensation; adapted from ref. 47. | ||
A recent investigation by van Zandvoort et al.48 has provided very important insights into the chemical structure of hydrothermal carbon. In fact, by means of 1D and 2D solid-state Nuclear Magnetic Resonance (NMR) spectra of 13C-labeled hydrothermal carbon, various different linkages have been identified in the 2D NMR spectra, ranging from the most abundant Cα–Caliphatic and Cα–Cα linkages to minor ones such as Cβ–Cβ and Cβ–Caliphatic cross-links. Furan rings have been found to be linked by short aliphatic chains; levulinic acid is also included in the structure through covalent bonds. A chemical structure (Fig. 8) was proposed.48
 | ||
| Fig. 8 Chemical structure of hydrothermal carbon proposed by van Zandvoort; adapted from ref. 48. | ||
Brown et al.49 have simulated Raman spectra of model constituents of hydrothermal carbon by means of Density Functional Theory (DFT). By fitting the experimental Raman hydrothermal carbon spectrum with a 12-peak fit, they have found that two model structures could reproduce the main features of the hydrothermal carbon: its Raman spectrum and its elemental analysis: (1) a structure consisting of arene domains comprising 6–8 rings connected via aliphatic chains; and (2) a furan/arene structure consisting primarily of single furans and 2 or 3 ring arenes. However, subsequent NMR and infrared (IR) analyses suggested that the latter model is more consistent with the experimental observations. A nucleation pathway was proposed by Sevilla and Fuertes50 which derived observations from the dissolution behaviour of cellulose under hydrothermal conditions, hence known as “the soluble pathway”. This pathway includes: (i) hydrolysis of cellulose chains, (ii) dehydration and fragmentation into soluble products of the monomers that come from the hydrolysis of cellulose, (iii) polymerization or condensation of the soluble products, (iv) aromatization of the polymers thus formed, (v) appearance of a short burst of nucleation and (vi) growth of the nuclei so formed by diffusion and linkage of species from the solution to the surface of the nuclei.50 More recently, Tsilomelekis et al.51 have studied acid catalysed degradation of 5-HMF by means of ATR-FTIR spectroscopy, Scanning Electron Microscopy (SEM) and Dynamic Light Scattering (DLS) to understand the growth mechanism of 5-HMF derived hydrothermal carbon. They have proposed a mechanism based on an initial nucleophilic attack of a 5-HMF carbonyl group to the α- or β-position of the furanic ring, along with aldolic condensation and etherification. Small, soluble oligomers grow heavier until they form small solid nuclei through phase separation. Small particle aggregation and continuous 5-HMF addition lead to the formation of bigger hydrothermal carbon particles. However, Cheng et al.53 have noted that hydrothermal carbon can be partially dissolved by multistage dissolution in an organic solvent to oligomers that have mass numbers ranging from 200 to 600 Da, as detected by liquid chromatography coupled with mass spectrometry (LC-MS). Although this observation has led the authors to speculate that hydrothermal carbon is actually an aggregate of oligomeric species rather than a disordered polymer,53 there might be a different interpretation. In fact, a very recent paper by Higgins et al. reported striking evidence of a core/shell structure in glucose-derived hydrothermal carbon. This evidence, obtained by combined STXM and NEXAFS observations, points to a “harder”, more condensed and hydrophobic core, surrounded by a “softer” shell, rich in aldehydic/carboxylic groups. These reactive functionalities serve as binding sites for the growth of the carbon particles.54 Formic acid and levulinic acid play a key role in this scenario: the former, due to its rather high pKa, significantly increases the rate of conversion of C6 sugar to 5-HMF in an autocatalytic fashion, therefore speeding up the growth of spherical particles. Levulinic acid, on the other hand, has a lower pKa and therefore does not have a strong catalytic effect, but it does affect the growth of the spherical particles taking part in the process as building units and slowing the growth by reducing the surface density.52 Moreover, Jung et al. have stated that significant growth of carbon particles occurs when the 5-HMF concentration has already dramatically declined. This points to an alternative model for growth of particles based on coalescence, which is also supported by the further observation that addition of electrolytes in the reaction medium increases particle sizes by reducing repulsive forces between particles.47 This debate is a proof that a thorough comprehension of the mechanisms underlying HTC has not been achieved yet and there is considerable space for further research.
2.1.1.1. Feedstock. As highlighted in the previous section, furfural and 5-HMF, originating from the dehydration of pentoses and hexoses, respectively, play a key role in the formation of hydrothermal carbon spheres, due to their reactivity. Consequently, it is reasonable to expect chars with a similar morphology and structure, regardless of the carbon precursor, as long as any of the aforementioned compounds are present in the reaction medium. Titirici et al.55 have demonstrated that all hexose (and 5-HMF)-derived carbon spheres have the same morphology. Similarly, carbon spheres obtained from the HTC of xylose and furfural are indistinguishable in shape. Further confirmations come from Falco et al.,8 who have compared the morphology of the HTC secondary char derived from glucose, cellulose and rye straw. While glucose and cellulose-derived carbon spheres appear similar in shape, it is argued that long cellulose fibers are disrupted under hydrothermal conditions, leading to the formation of shorter chains that adopt a spherical shape to minimize contact with water. Rye straw behaviour is similar to that of cellulose, with microspheres appearing on the surfaces of the fibers that suffer only minor disruption.
2.1.1.2. Reaction time and temperature. Reaction time and temperature have been shown to influence the hydrothermal carbon morphology. Sevilla et al.,13 studying hydrothermal carbon yields from the HTC of glucose, sucrose and starch, noted that in any case, a rise of the precursor concentration, reaction time or reaction temperature resulted in an increased yield of hydrothermal carbon and in the diameter of the microspheres. Romero-Anaya56 have substantially confirmed the observation, noting also that carbon sphere growth reaches a maximum (at fixed reaction time and precursor concentration) at 200 °C. Temperature has a big impact on hydrothermal decomposition of cellulose. Depolymerization of microcrystalline cellulose in water-soluble compounds is known to start at 180 °C.57 However, it has been shown that depolymerisation of cellulose to sugar oligomers becomes predominant at 220 °C, while temperatures as high as 260 °C cause a decline of sugar oligomers in favour of decomposition products such as 5-HMF or carboxylic acids.38 Reaction time seems to have an impact on the morphology and size of carbon spheres only up to a certain point. In fact, it has been noted that, over treatment times of 12, 24 and 48 h, sugar derived carbon spheres achieve bigger and more uniform sizes from 12 h to 24 h, but longer reaction times do not produce any change in the size or morphology.56 Moreover, it has been demonstrated that a second subsequent HTC of the carbon spheres results in the uniform growth of the pre-existing particles without any relevant formation of new ones.58 Simsir et al.59 have studied the effects of different reaction times on HTC of different kinds of feedstocks (glucose, cellulose, chitin, chitosan, and wood chips) at a fixed temperature of 200 °C. In this study, glucose-derived carbon spheres are not observed before a 12 hour long treatment, with an average diameter of around 800 nm for a residence time between 12 h and 36 h, and a slightly lower average diameter of 500–600 nm with the increase in residence time to 48 h. This has been explained with the plausible existence of an equilibrium between the growth and decomposition of spherical carbon particles for longer residence times. Chitin is insensitive to HTC, while cellulose and wood chips produce hard carbon spheres, too. Finally, chitosan derived hydrothermal carbon appears in the form of densely aggregated structures.
| f = 50t0.2 × e(−3500/T) |
Fig. 9 presents a comparison of hydrothermal carbon yields associated with literature-collected data. Results from this analysis indicate that the average and median yields obtained when carbonizing the majority of feedstocks range from 40 to 60%. The carbonisation of lignin results in the greatest average and median yields (average: 66% and median: 68%). This observation is not surprising, as lignin has been shown to be only mildly influenced when exposed to HTC.8,35 The carbonisation of municipal solid waste (MSW), digestate, wood, and yard waste also results in average and median yields greater than 60%. Carbonisation of oils (e.g., bio-oil and pyrolysis oil)67,68 results in significantly lower hydrothermal carbon yields. Similarly, the HTC of paper and algae also results in relatively lower yields than those associated with the majority of other feedstocks. In general, as the reaction severity increases, compound volatilisation and solubilisation increase, resulting in decreased solid yields.8,69–71 This general trend is observed for individual feedstocks. However, when collectively investigating trends of all feedstocks, no trend with an increasing severity factor (from left to right on the figure) is observed, suggesting that the feedstock properties significantly influence hydrothermal carbon yields (more than the reaction conditions), consistent with that which has been previously reported.72–75 Lu and Berge72 reported that changes in feedstock properties/complexity influence hydrothermal carbon yield.
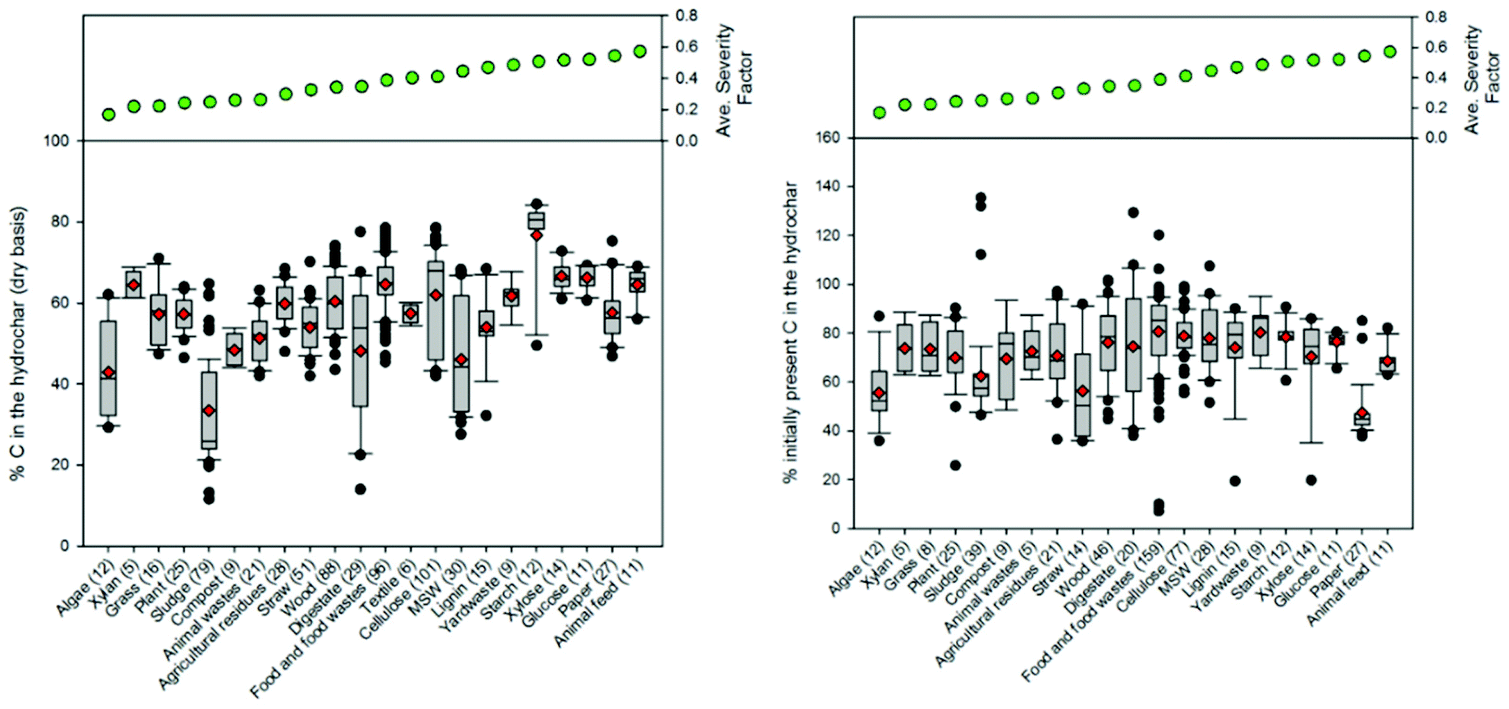 | ||
| Fig. 9 Distributions associated with the hydrothermal carbon yields of different feedstocks based on literature-collected data. The line in each box represents the median value. The ends of each box represent the 25th and 75th percentiles associated with the data. The red diamonds represent the average values. The lines and data points represent the scatter of data beyond the 10th and 90th percentiles. The numbers in parentheses following each feedstock category represent the number of data points represented on the plot. | ||
Fig. 9–12 illustrate how changes in feedstock and process conditions influence the chemical properties of hydrothermal carbon. Based on the compiled data, the majority of the carbon content of the hydrothermal carbon generated from the carbonisation of different organics generally ranges from 40 to 65%, with the hydrothermal carbon generated from starch, xylose, and glucose containing the largest average hydrochar carbon content (Fig. 10 left). Generally, as the reaction severity increases, the carbon content of the hydrothermal carbon increases.8,71,76,77 However, trends in the average hydrothermal carbon content are not observed with increasing severity factor (from left to right on the figure), suggesting that, like that associated with hydrothermal carbon yield, feedstock properties significantly influence the hydrothermal carbon content. Fig. 10 right provides a summary of the fraction of initially present carbon that remains integrated within the hydrothermal carbon following carbonisation. The greatest, on average, loss of carbon from the feedstock to the gas and/or liquid-phases occurs when carbonising paper, straw, and algae. The greatest retention of initially present carbon in the hydrothermal carbon occurs when carbonising food and yard waste (Fig. 10 right), two biomass materials that are ideal candidates for conversion via HTC because of their high initial moisture contents.
 | ||
| Fig. 10 Distributions associated with the (left) percentage of carbon in the hydrothermal carbon and (right) the percentage of initially present carbon that remains in the hydrothermal carbon following carbonisation. The line in each box represents the median value. The ends of each box represent the 25th and 75th percentiles associated with the data. The red diamonds represent the average values. The lines and data points represent the scatter of data beyond the 10th and 90th percentiles. The numbers in parentheses following each feedstock category represent the number of data points represented on the plot. | ||
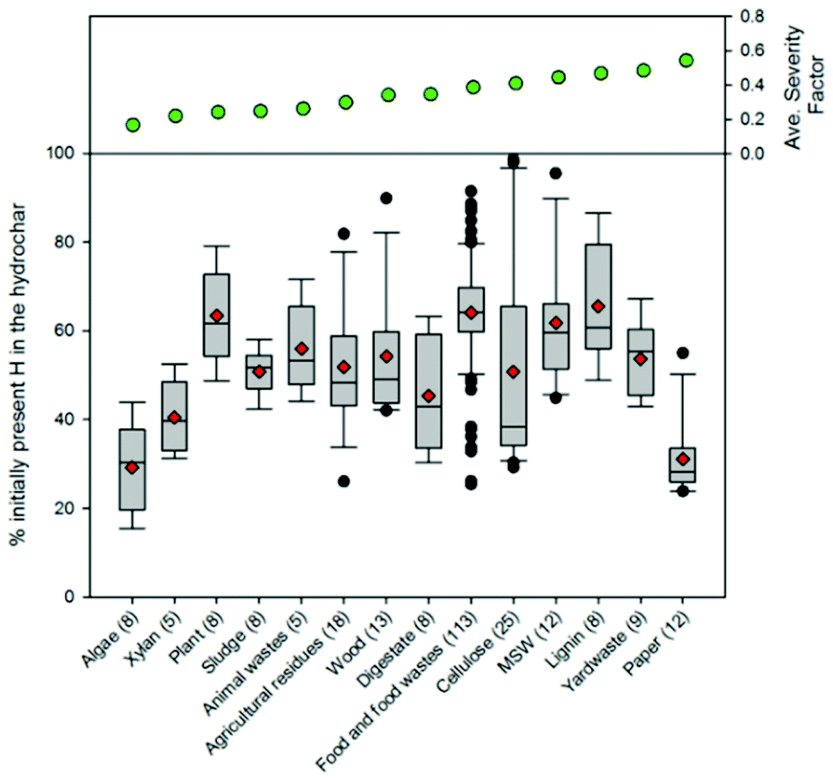 | ||
| Fig. 11 Distributions associated with the percentage of hydrogen in the hydrothermal carbon. The line in each box represents the median value. The ends of each box represent the 25th and 75th percentiles associated with the data. The red diamonds represent the average values. The lines and data points represent the scatter of data beyond the 10th and 90th percentiles. The numbers in parentheses following each feedstock category represent the number of data points represented on the plot. | ||
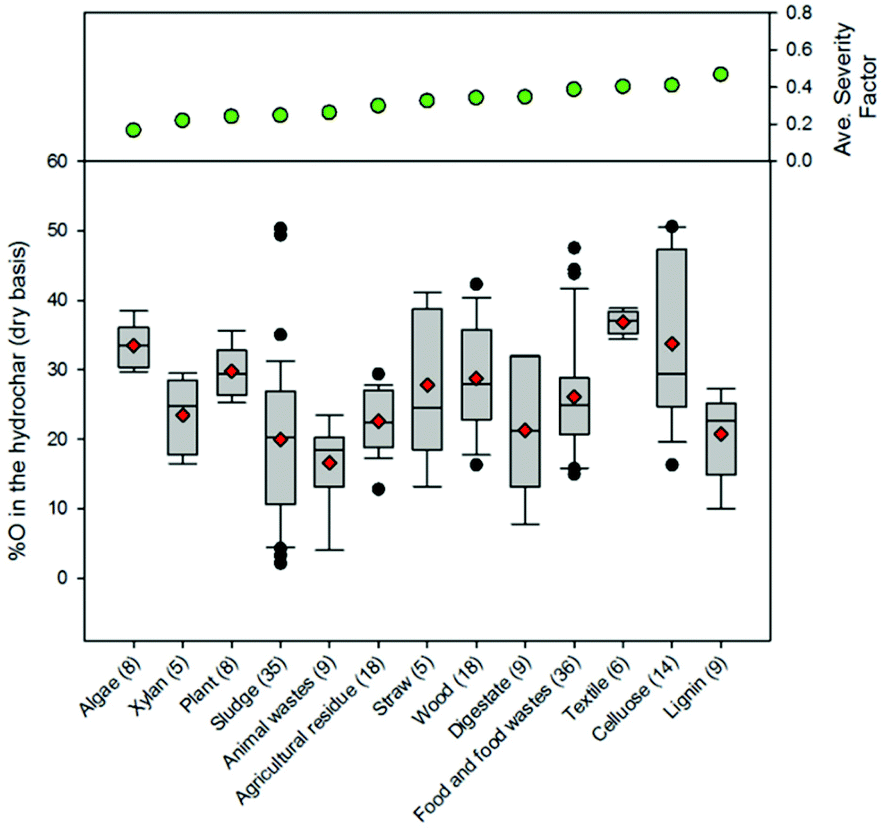 | ||
| Fig. 12 Distributions associated with the percentage of oxygen in the hydrothermal carbon. The line in each box represents the median value. The ends of each box represent the 25th and 75th percentiles associated with the data. The red diamonds represent the average values. The lines and data points represent the scatter of data beyond the 10th and 90th percentiles. The numbers in parentheses following each feedstock category represent the number of data points presented on the plot. | ||
The majority of the generated hydrothermal carbon, regardless of feedstock, contains hydrogen concentrations ranging from approximately 4 to 6% (Fig. 11). An exception to this is the hydrothermal carbon generated when carbonising food waste, which contains an average hydrogen percentage of 7.2%. The feedstocks that result in the greatest average loss of hydrogen to either the gas or liquid-phase are algae and paper, while greater than 60% of the initially present hydrogen remains in the hydrothermal carbon when carbonising food waste, lignin, MSW, and plant materials. At relatively low and high severity factors, slight trends exist, suggesting that at these reaction severities, process conditions are potentially more influential than feedstock properties on the fraction of initially present hydrogen that remains within the hydrothermal carbon. Not surprisingly, a small amount of the oxygen present in the initial feedstocks remains integrated within the hydrothermal carbon following carbonisation (Fig. 12), which is beneficial when considering the use of hydrothermal carbon in different applications, such as an energy source. This observation is consistent with that reported in the literature; the decrease in the solid-phase oxygen content has been reported as being the most influential factor contributing to the decrease in solid recoveries.76 Food waste, lignin, and plant materials, on average, retain the largest fraction of oxygen embedded within the initial feedstocks, when compared to other carbonised feedstocks. Generally, as the reaction severity increases, the hydrothermal carbon oxygen content decreases.76,78,79 Interestingly, trends in the average hydrothermal carbon oxygen content are not observed with increasing severity factors (from left to right on the figure), suggesting that feedstock properties also influence the hydrothermal carbon oxygen content.
2.2. Synthesis of porous carbon materials
Hydrothermal carbon materials have been of great interest, over the years, due to their wide range of applications. Used in many ways, such as for cleaning the dyes present in water and sugar, or for the removal of unpleasant odours,80 present-day applications mention them in industry as adsorbents for gas purification81–83 and water treatment84,85 or a catalyst and catalyst-supports.86,87 They are, also, often used in the fields of energy storage, fuel cells and chromatography technologies.88,89 In order to improve their features, a great number of studies are focused on developing new synthetic approaches for creating porous structures and enhancing the surface area, along with gaining a fundamental understanding of their properties.90,91 As mentioned before, the pristine hydrothermal carbons are nonporous and sometimes not applicable for further usage.92–95 To overcome this, different strategies, including templating methods (soft and hard templating),93,94,96 or chemical activation, using alkaline compounds, KOH,95,97 NaOH,85 and ZnCl298 or acids, such as H3PO4,99 in combination with HTC, have been proposed. | ||
| Fig. 13 Graphical representation of the templating methods: a – soft templating and b – hard templating; reproduced with permission from ref. 104. | ||
2.2.1.1. Soft templating in HTC. Nanostructured HTC–derived materials can be synthesised using polymeric templates of defined size and shape. In soft-templating, an amphiphilic molecule such as a surfactant or block-copolymer self-assembles with a carbon precursor into an organized mesophase, which is stabilised by thermal treatment. The process is controlled by several parameters, such as the concentration, temperature, hydrophilic or hydrophobic reaction, pH, etc. Generally, the templates are polymers such as poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide)triblockcoplymers (PEO-b-PPO-b-PEO) from the Pluronic family,96,105,106 polystyrene-b-poly(ethylene oxide) (PS-b-PEO)107 or polystyrene-b-poly(4-vinlpiridine)(PS-b-P4VP),108 trioctylamine@ferrocene.93 Usually, the carbon precursors consist of small clusters of phenol-formaldehyde, “resol” or phloroglucinol-formaldehyde resins, but many researchers adopted it by using different carbon precursors, including carbohydrates,90,94 nitrogen and carbon-rich compounds,94,96 organic acids,93 and natural polymers (lignin, chitin, and cellulose).109,110 For example, Liang et al.111 synthesised porous carbon spheres via soft templating assisted HTC, using Pluronic F108 as a template and phenol-formaldehyde as a precursor. The mixture was subjected to HTC at 170 °C for 6 h, followed by carbonisation at 600 °C under a N2 atmosphere. Further characterization revealed the formation of spherical carbon particles with a microporous structure. As a result of the high number of micropores, the carbon spheres possessed high specific surface area and high pore volume, with values of about 1481 m2 g−1 and 0.9 cm3 g−1 respectively. Pluronic F127, in combination with xylose, has been reported by Liu et al.112 for the production of carbonaceous long-range ordered mesostructures. The same approach has been used by Xie et al.113 for the synthesis of hierarchically porous materials with tunable properties. In both cases, the obtained materials have been characterized as a mesoporous structure with a surface area of around 450 m2 g−1. Zhou et al.105 reported the synthesis of core-mesoporous shelled carbon spheres via HTC and soft templating. Starting from 2,4-dihydroxybenzoic acid, hexamethylenetetramine and Pluronic P123, they produced uniform carbon spheres with an average diameter of ∼141 nm and shell thicknesses of ∼30 nm (see Fig. 14a and b). Characterisation techniques revealed the porous structure with a surface area of up to 648 m2 g−1 and a large pore volume (1.06 cm3 g−1). In a similar way, Xiao et al.96 reported the synthesis of porous carbon materials starting from glucosamine. During the study, they investigated the influence of pH and the amount of template. By moving from neutral to acidic conditions, the specific surface area significantly increased, from 550 m2 g−1 up to 980 m2 g−1, as a result of P123 micelle formation being favoured at pH 2. Also, an improvement in textural properties was observed on increasing the amount of polymer. SEM micrographs show the formation of agglomerated small particles, confirming the transformation of glucosamine in carbon spheres (Fig. 14c), and TEM reveals the worm-like structure of the sample, related to the formation of micro- or mesopores (Fig. 14d). P123 together with sodium oleate (SO) have been reported by Chen et al.114 in the synthesis of asymmetric flask-like hollow carbon materials. Zhang et al.93 reported the synthesis of hollow carbon spheres starting from ascorbic acid and trioctylamine @ferrocene-in-water. The authors obtained bowl-like hollow carbon spheres (BHCS – see Fig. 14e–i) with a microporous structure, a diameter of approximately 0.53 nm and a specific surface area of about 200 m2 g−1.
 | ||
| Fig. 14 Typical (a) SEM and (b) TEM images of hollow core-mesoporous shelled carbon spheres; reproduced with permission from ref. 105. (c) SEM and (d) TEM images of hydrothermal porous carbons (reproduced with permission from ref. 96). (e) and (f) SEM images with different magnifications of BHCS. (g–i) TEM images of BHCS under different magnifications (reproduced with permission from ref. 93). | ||
The synthesis of ordered porous carbon materials derived from D-fructose has been reported by Kubo et al.90 In this way, the HTC of D-fructose was performed at 130 °C for a long time, in order to ensure the stability of the template. The final composite was characterised by ordered porosity with pore diameters being around 10 nm and wall thicknesses of 6 nm, indicating that the self-assembly of the polymer with fructose was successful (Fig. 15a–c). The material was characterised by a 257 m2 g−1 surface area and a 0.14 cm3 g−1 pore volume. Chen et al.115 synthesised carbon nanospheres starting from a urea-phenol-formaldehyde (UPF) resin and Pluronic F127. The resulting materials had a well-ordered cubic Im3m mesostructure in large domains with spherical shapes with a mean diameter of 240 nm and high surface area, in the range of 446–566 m2 g−1. SEM and TEM results are illustrated in Fig. 15d–g.
 | ||
| Fig. 15 (a) SEM, (b) TEM, and (c) HRTEM micrographs of ordered mesoporous carbons derived from fructose (reproduced with permission from ref. 90). (d and e) SEM images of ordered mesoporous carbons and (f and g) TEM images of the same samples with an Im3m mesoporous structure; reproduced with permission from ref. 115. | ||
2.2.1.2. Hard templating in HTC. The hard-templating method is based on the structure replication approach using sacrificial templates. The advantage of hard templating combined with HTC is that polar functionalities are directly present on the surface of the synthesised material. Different templates can and have been employed, such as silica,116 zeolite,117,118 and metal organic frameworks.119 An advantage of hard templating over soft templating is a better control of the carbon structure, and a higher possibility of obtaining hierarchical structures.120 Xiao et al.94 reported the synthesis of mesoporous carbon materials by combining soft and hard templating strategies. In this way, they used different saccharides (D-glucose_1, D-fructose_2, D-glucosamine hydrochloride_3, D-maltose_4, sucrose_5, and β-cyclodextrin_6), triblock copolymers and tetraethyl orthosilicate (TEOS). During HTC, Pluronic P123 forms micelles that are covered by a silica layer, coming from the hydrolysis of TEOS and the subsequent condensation reaction of orthosilicic acid. The silica interacts with the micelles via hydrogen bonds and acts as a protector for the micelles during the HTC. The carbon precursor reacts on the silica surface, and forms hydrothermal carbon, without harming the micelles. In this way, after the template removal, the carbon presents an ordered structure. The obtained materials were denoted as OHTC-X (where X refers to the carbon source) and were characterised by type IV isotherms, specific for mesoporous structures, with a H2 hysteresis loop, indicating a multilayer adsorption of nitrogen. The surface areas (pore volume) are in between 300 m2 g−1 (0.55 cm3 g−1) and 520 m2 g−1 (0.40 cm3 g−1). TEM images illustrate the ordered structure of the materials (Fig. 16). To assess the ordered character of the pores the authors viewed the samples with and without a silica template (Fig. 16a and b). It was observed that the carbonaceous powders preserve the ordered orientation after template removal. To confirm that the silica formed in situ during the HTC is the structure directing agent, the carbon was burned by calcination, leaving behind ordered silica (Fig. 16c). Hard templated carbons have been prepared by Wang et al.121 using SBA-15 as a sacrificial template for the synthesis of ordered mesoporous doped carbons. Starting from glucose and using HTC, the mixtures are connected through carbon bridges and cover the template uniformly. The final material is highly populated with oxygenated groups, coming from the dehydration compounds of glucose during the HTC. Part of these are removed during further carbonisation, at 900 °C, and the template is removed by NH4HF2 leaching. TEM pictures of the obtained materials show the presence of a well-ordered structure of the composites C/SBA-15 (Fig. 16d) which is stable also after the template removal (Fig. 16e and f). The porosity was confirmed also by N2 sorption isotherms, both doped and undoped carbons showing type IV isotherms characteristic of mesoporous materials with pores ranging from 2 to 8 nm and surface areas of 253 m2 g−1 and 341 m2 g−1, respectively.
 | ||
| Fig. 16 TEM images of (a) OHTC_1, (b) OHTSiC_1 (before etching the silica template) and (c) OHTSi_1 (after burning the carbon at 600 °C), reproduced with permission from ref. 94; TEM pictures of (d) SBA-15/C composites after calcination at 900 °C, (e) pristine carbon materials after template removal and (f) S-doped carbon materials after template removal (reproduced with permission from ref. 121). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 98 and H3PO4.99 Zhu et al.126 synthesised porous carbon materials via the HTC of pineapple waste and chemical activation. In this way, the biomass was washed, pulverized, and sieved, followed by mixing with alkali metal oxalate (Li2C2O4, Na2C2O4 and K2C2O4) in a mass ratio of 1:2 (biomass:alkali metal) and HTC for 10 h at 210 °C. Porosity measurements revealed the formation of nonporous and macroporous structures for the Li2C2O4 and Na2C2O4 activated carbons (ACs) and microporous structures for the ACs derived from the activation with K2C2O4. When comparing the porosity data such as the pore volume (V0) and narrow micropore volume (Vn), Vn is bigger for the K2C2O4 ACs, and the Li2C2O4 and Na2C2O4 ACs present opposite results. The authors explained these textural differences among the AC series with Li2C2O4, Na2C2O4 and K2C2O4 by the different reaction pathways of the oxalates during the activation process. In this way, when K2C2O4 at high temperature is used, a larger amount of CO is released in the third stage, which causes the micropore formation. Sevilla et al.127 reported the synthesis of microporous carbon via the HTC of microalga and glucose mixtures. To ensure the microporosity, the hydrothermal carbon was chemically activated using KOH at elevated temperatures, 650–750 °C, in a mass ratio of 2 KOH/hydrothermal carbon. The obtained materials were characterised by their microporous structure with an apparent surface area of 1800 m2 g−1 to 2200 m2 g−1 at the maximum activation temperature (750 °C). Li et al.128 synthesised a composite structure of a graphene-like nanosheet (GN)/porous carbon framework using waste-peanut skin as a biomass precursor. The procedure was based on a H2SO4 assisted HTC followed by KOH activation, and during the study the concentration of H2SO4 was varied. TEM and SEM results showed that the graphitisation level was very much influenced by the acid concentration. In this way, the sample without H2SO4 presented a typical amorphous carbon structure while the one with the highest H2SO4 concentration possessed three-layer structures with a clear interlayer spacing of 0.38 nm, indicating the graphene-like morphology. The samples were characterized by a micro–mesoporous structure, with a high number of micropores. Shi et al.129 prepared ACs starting from glucose and glucosamine, in parallel, and activation with KOH. During the synthesis, they produced the hydrothermal carbon via HTC followed by chemical activation, as a subsequent step. The resulting materials possessed a porous structure, regardless of the carbon precursor, and the microporosity was observed only after KOH activation. The ACs were characterized by large surface areas, between 778 m2 g−1 and 1513 m2 g−1, depending on the activation parameters. ACs with micro–mesoporous structures have been prepared by Fuertes et al.,95 starting from potato starch and eucalyptus sawdust, via HTC at 250 °C, and activation with KOH and melamine. The powders had a micro–mesoporous bimodal structure. It was observed that melamine has the ability to extend the pore size distribution range of the activated carbons, generating mesopores.95 Again, independent of the biomass source, the surface areas were improved by the melamine incorporation, reaching a maximum of 3420 m2 g−1, and 2.37 cm3 g−1 for pore volume. Boyjoo et al.98 reported the synthesis of ACs starting from Coca-Cola via HTC, followed by chemical activation with ZnCl2, denoted as CMC_1 and CMC_2 (mass ratio of ZnCl2/HTC carbon = 1 and 3, respectively), and with KOH, denoted as CMC_3 (KOH/HTC carbon = 4). Surface area measurements revealed the formation of microporous structures. The nonactivated sample presents a moderate surface area, 405 m2 g−1, and pore sizes in the micropore domain, maybe due to H3PO4 present in the biomass. The samples activated with ZnCl2 achieve surface areas of about 1082 m2 g−1 and 80% of the pore volume consists of micropores. On increasing the amount of activator, the surface area almost doubles, but the pore size distribution shifts towards mesopores, with the percentage of narrow micropores (<0.8 nm) decreasing down to 29%. Plata et al.130 reported the synthesis of ACs prepared through H3PO4-assisted HTC, from different biomass sources (sawdust, almond shells, hemp residues and coconut shells) and H3PO4 for pore development. In a typical synthesis the biomass was dispersed in a certain amount of H3PO4 and deionized water, and HTC was performed at 200 °C for 24 h. Also, in the presence of an activation agent, the carbon yield was higher, probably because H3PO4 acts as a catalyst for the hydrothermal carbon formation.
98 and H3PO4.99 Zhu et al.126 synthesised porous carbon materials via the HTC of pineapple waste and chemical activation. In this way, the biomass was washed, pulverized, and sieved, followed by mixing with alkali metal oxalate (Li2C2O4, Na2C2O4 and K2C2O4) in a mass ratio of 1:2 (biomass:alkali metal) and HTC for 10 h at 210 °C. Porosity measurements revealed the formation of nonporous and macroporous structures for the Li2C2O4 and Na2C2O4 activated carbons (ACs) and microporous structures for the ACs derived from the activation with K2C2O4. When comparing the porosity data such as the pore volume (V0) and narrow micropore volume (Vn), Vn is bigger for the K2C2O4 ACs, and the Li2C2O4 and Na2C2O4 ACs present opposite results. The authors explained these textural differences among the AC series with Li2C2O4, Na2C2O4 and K2C2O4 by the different reaction pathways of the oxalates during the activation process. In this way, when K2C2O4 at high temperature is used, a larger amount of CO is released in the third stage, which causes the micropore formation. Sevilla et al.127 reported the synthesis of microporous carbon via the HTC of microalga and glucose mixtures. To ensure the microporosity, the hydrothermal carbon was chemically activated using KOH at elevated temperatures, 650–750 °C, in a mass ratio of 2 KOH/hydrothermal carbon. The obtained materials were characterised by their microporous structure with an apparent surface area of 1800 m2 g−1 to 2200 m2 g−1 at the maximum activation temperature (750 °C). Li et al.128 synthesised a composite structure of a graphene-like nanosheet (GN)/porous carbon framework using waste-peanut skin as a biomass precursor. The procedure was based on a H2SO4 assisted HTC followed by KOH activation, and during the study the concentration of H2SO4 was varied. TEM and SEM results showed that the graphitisation level was very much influenced by the acid concentration. In this way, the sample without H2SO4 presented a typical amorphous carbon structure while the one with the highest H2SO4 concentration possessed three-layer structures with a clear interlayer spacing of 0.38 nm, indicating the graphene-like morphology. The samples were characterized by a micro–mesoporous structure, with a high number of micropores. Shi et al.129 prepared ACs starting from glucose and glucosamine, in parallel, and activation with KOH. During the synthesis, they produced the hydrothermal carbon via HTC followed by chemical activation, as a subsequent step. The resulting materials possessed a porous structure, regardless of the carbon precursor, and the microporosity was observed only after KOH activation. The ACs were characterized by large surface areas, between 778 m2 g−1 and 1513 m2 g−1, depending on the activation parameters. ACs with micro–mesoporous structures have been prepared by Fuertes et al.,95 starting from potato starch and eucalyptus sawdust, via HTC at 250 °C, and activation with KOH and melamine. The powders had a micro–mesoporous bimodal structure. It was observed that melamine has the ability to extend the pore size distribution range of the activated carbons, generating mesopores.95 Again, independent of the biomass source, the surface areas were improved by the melamine incorporation, reaching a maximum of 3420 m2 g−1, and 2.37 cm3 g−1 for pore volume. Boyjoo et al.98 reported the synthesis of ACs starting from Coca-Cola via HTC, followed by chemical activation with ZnCl2, denoted as CMC_1 and CMC_2 (mass ratio of ZnCl2/HTC carbon = 1 and 3, respectively), and with KOH, denoted as CMC_3 (KOH/HTC carbon = 4). Surface area measurements revealed the formation of microporous structures. The nonactivated sample presents a moderate surface area, 405 m2 g−1, and pore sizes in the micropore domain, maybe due to H3PO4 present in the biomass. The samples activated with ZnCl2 achieve surface areas of about 1082 m2 g−1 and 80% of the pore volume consists of micropores. On increasing the amount of activator, the surface area almost doubles, but the pore size distribution shifts towards mesopores, with the percentage of narrow micropores (<0.8 nm) decreasing down to 29%. Plata et al.130 reported the synthesis of ACs prepared through H3PO4-assisted HTC, from different biomass sources (sawdust, almond shells, hemp residues and coconut shells) and H3PO4 for pore development. In a typical synthesis the biomass was dispersed in a certain amount of H3PO4 and deionized water, and HTC was performed at 200 °C for 24 h. Also, in the presence of an activation agent, the carbon yield was higher, probably because H3PO4 acts as a catalyst for the hydrothermal carbon formation.
| Sample name | Carbon precursor | Synthesis method | Template/activating agent | S BET (m2 g−1) | PV (cm3 g−1) | Ref. |
|---|---|---|---|---|---|---|
| NPCSs | Phenol-formaldehyde | Soft templating-HTC | Pluronic F108 | 1480 | 0.9 | 111 |
| BHCSs | Ascorbic acid | Trioctylamine@ferrocene-in-water emulsion | 199 | NA | 93 | |
| OHTC-1 | Glucose | Soft and hard templating-HTC | Pluronic P123, TEOS | 520 | 0.40 | 94 |
| HPNC-1 | Glucosamine hydrochloride | Soft templating-HTC | Pluronic P123, 0.5 mmol, pH = 7 | 550 | 0.43 | 96 |
| HPNC-2 | Pluronic P123, 0.25 mmol, pH = 2 | 650 | 0.54 | 96 | ||
| HPNC-3 | Pluronic P123, 0.75 mmol, pH = 2 | 710 | 0.54 | 96 | ||
| HPNC-4 | Pluronic P123, 1 mmol, pH = 2 | 980 | 0.78 | 96 | ||
| HPC-3 | Glucose | Pluronic P123, 0.75 mmol, pH = 2 | 490 | 0.29 | 96 | |
| Assai-MW-CO2 | Assai stone | Microwave-assisted HTC and physical activation | CO2 (80 ml min−1) | 1100 | 0.45 | 131 |
| CNSA_6 | Peanut skins | H2SO4 assisted HTC and chemical activation | KOH (1:1 mass ratio) |
1886 | 1.02 | 128 |
| HPCS_1.2 | Starch | HTC-chemical activation | (NH4)2Fe(SO4)2 | 973 | 0.27 | 132 |
| AC hydrochar – dry impregnated KOH | Giant bamboo | KOH (4:1 mass ratio) |
2117 | 1.14 | 133 | |
| Hydrochar – wet impregnated KOH | 2262 | 1.30 | 133 | |||
| BCF | Bamboo sticks | KOH (6:1 mass ratio) |
12 | — | 134 | |
| CMC-1 | Coca Cola waste | ZnCl2 (1:1 mass ratio) |
1082 | 0.43 | 98 | |
| CMC-2 | ZnCl2 (3:1 mass ratio) |
1994 | 0.87 | 98 | ||
| CMC-3 | KOH (4:1 mass ratio) |
1405 | 0.80 | 98 | ||
| COSHTC_3 | Coconut shell | NaOH (3:1 mass ratio) |
876 | 0.44 | 85 | |
| AA-0 | Potato starch | KOH (4:1 mass ratio) |
3000 | 1.41 | 135 | |
| AA-2M | KOH (4:1 mass ratio) and melamine (2:1 mass ratio) |
3280 | 2.37 | 135 | ||
| AA-3M | Potato starch | KOH (4:1 mass ratio) and melamine (3:1 mass ratio) |
3220 | 2.27 | 135 | |
| AC-0 | Cellulose | KOH (4:1 mass ratio) |
3100 | 1.46 | 135 | |
| AC-2M | KOH (4:1 mass ratio) and melamine (2:1 mass ratio) |
3540 | 2.22 | 135 | ||
| AS-0 | Sawdust | KOH (4:1 mass ratio) |
2690 | 1.28 | 135 | |
| AS-2M | KOH (4:1 mass ratio) and melamine (2:1 mass ratio) |
3420 | 2.30 | 135 | ||
| AS-3M | KOH (4:1 mass ratio) and melamine (3:1 mass ratio) |
2990 | 2.35 | 135 |
3. Heteroatom doped carbon materials
As described in the previous section, HTC can be successfully used in the production of porous carbon materials using renewable resources. Furthermore, due to the versatile character of this technique, different strategies can be used in tandem in order to boost the properties of the final materials, as presented in section 2.2. Although the textural properties promote this type of material as a good candidate for application in gas storage fields or water treatment experiments, sometimes its chemical and physical properties are limited, making it a second option candidate for applications such as electrocatalysis or energy storage. A very common method to overcome this consists of functionalization of the carbon structure with various heteroatoms, such as nitrogen,105,136 boron,137,138 sulphur121,139 and phosphorus.140 This brings some advantages including improved catalytic performance and better selectivity for different reactions, such as the oxygen reduction reaction, and higher adsorption capacity when it comes to gas storage and water treatment.3.1. Nitrogen-doped carbon materials
Nitrogen-doped carbon materials (NCs) have received recognition since the earlier studies, due to their remarkable chemical and physical properties. The hydrothermal carbon is highly improved by N incorporation due to the following reasons: the nitrogen atoms are more electronegative than carbon, due to the two lone pair electrons, and this provides a higher electrochemical activity for the NCs, which makes them potential catalysts for the oxygen reduction reaction and CO2 reduction;87 N-doping effectively increases the electrical conductivity and creates defects that can provide enough space for ion, electrolyte or gas diffusion that make them viable for gas storage,141 fuel cells and energy storage.139,142 NCs can have different structures, such as 1-D N-doped carbon nanotubes (NCNTs),143 N-doped nanofibers (NCNFs),144 2-D N-doped graphene (NG)145 or 3-D hierarchical nanostructures with different dimensions.146 Depending on the N-bonding, four main types of N can be distinguished: pyrrolic N, pyridinic N, quaternary N/graphitic N and N oxides of pyridinic N (illustrated in Fig. 17). | ||
| Fig. 17 Schematic structure of N doped carbons; reproduced with permission from ref. 147. | ||
Usually, the carbon is functionalized during HTC followed by high-temperature annealing.148 A wide range of nitrogen precursors can be used, including ammonia,149 urea,148,150 melamine,151 nitric acid152etc. The chemical and physical properties, pore structure, content and types of nitrogen functionalities are all influenced by the nitrogen precursors and heat treatment conditions. Liu et al.153 reported the synthesis of NCs using natural biowaste as a carbon precursor and melamine as a N source, succeeding in incorporating about 1.75 at% N, according to XPS data. Starting from chitosan and gaseous NH3, Wu et al.154 proposed the synthesis of NCs via HTC for energy storage applications and CO2 capture. In this way, they synthesised two sets of materials: NCs derived from chitosan and NCs derived from glucose and gaseous NH3. The nitrogen incorporation was much more successful for the carbons obtained via the HTC of chitosan, about 4.61 wt% N was incorporated, and half of it was attached in the pyrrolic form. When NH3 was used as a N donor, more pyridinic functionalities were reported, with a N content of about 3.58 wt%. Using aqueous NH3, Schipper et al.155 reported the synthesis of NCs via the HTC of glucose, containing about 9 wt% N. Zhang et al.156 synthesised NCs with 5.5 at% N, starting from the HTC of glucose, followed by dry impregnation of the resulting hydrothermal carbon with melamine. Ren et al.157 reported the HTC of NCs derived from microalgae, containing up to 2.89 at% N, coming exclusively from the biomass source. Preuss et al.136 reported the usage of lyophilised ovalbumin from chicken egg white as a N source for the synthesis of NCs, starting from glucose and using HTC. The highest N content obtained in this study was 3.3 at%, which reduces to 2.9 at% after further activation is applied. Qiao et al.158 reported that the NCs improve the electrocatalytic activity of multiwall carbon nanotubes towards oxygen reduction reactions. The N was incorporated mostly as graphitic N (57.16%) and pyridinic N (18.26%), the rest being coordinated with Fe or in an oxidized form. Alatalo et al.159 performed the synthesis of NCs starting from glucose and cellulose in the presence of a soy bean additive as a structure directing agent and a nitrogen precursor. The resulting materials showed good nitrogen incorporation: between 1.0 and 3.7 wt% for pristine materials and 0.2–1.9 wt% for calcined carbons. Table 2 shows a few more examples of NCs synthesised in the literature.
| Sample name | Carbon precursor | Nitrogen source | Nitrogen content (at%) | Ref. |
|---|---|---|---|---|
| AG650 | Glucose and microalgal mixtures | Microalgae | 2.65 | 127 |
| AG700 | 1.43 | |||
| AG750 | 0.68 | |||
| N-GCS-2 | Peach extract | NH4OH | 9.33 | 160 |
| AMBC-600 | Bamboo shoot | Melamine | 12.9 | 161 |
| AMBC-700 | 6.2 | |||
| AMBC-800 | 2.7 | |||
| HPCS-1.2 | Starch | (NH4)2Fe(SO4)2 | 2.3 | 132 |
| H-N400 | Sucrose | (NH4)2SO4 | 6.57 | 162 |
| H-N600 | 7.33 | |||
| H-N800 | 6.54 | |||
| GN-700-4 | Glucose | Urea | 6.20 | 163 |
The examples mentioned above are proof that incorporation of nitrogen atoms within the carbon structure using HTC is possible, but how nitrogen incorporates into the carbon structure is still under debate. So far, the Maillard mechanism164 has been accepted as playing an important role in the process. The mechanism consists of a group of reactions between reducing sugars and amino acids. It is impossible to provide a clear pathway for the formation of NC materials during HTC because throughout these reactions, hundreds of compounds can be formed and react again themselves. The main steps of the Maillard mechanism are described in short in Fig. 18.89
 | ||
| Fig. 18 Some examples of the Maillard reaction; reproduced with permission from ref. 89. | ||
The first products formed during the Maillard reaction are glucosamines via a nucleophilic attack of an amine on the aldehyde of the sugar (Fig. 18, I). The glucosamines are further transformed to a Schiff base, by dehydration (Fig. 18, II) which can, in turn, suffer rearrangements to aminoketones (Fig. 18, III and IV). Compound III can form an α-dicarbonyl species (Fig. 18, V), which after successive dehydration can form 5-HMF, the main intermediate compound formed during HTC.89
3.2. Sulphur doped carbon materials
Together with N, sulphur has gained a lot of attention as a dopant for the carbon structure. Sulphur-doped carbons (SCs) are widely used as cathodes for Li–S batteries, due to the high theoretical capacity of S (around 1675 mA h g−1).165 Other applications of SCs include electrodes for supercapacitors,166 energy storage as hydrogen storage media,166 photoactive materials for light-harvesting and electrocatalysis. SCs can be usually prepared using two strategies: (i) incorporation of S into the structure of synthetic or commercial carbon using a sulphur precursor by sulphurisation,167 and (ii) heating of mixtures of carbon and sulphur precursors.168,169 Generally, the sulphur donor can be any sulphur-containing compound, including elemental sulphur, H2S, SO2, CS2, sulphur-containing organic compounds and polymers.121,139,166,170 Emran et al.171 proposed the synthesis of sulphur-doped carbon microspheres (S-MCMS) via a one-pot HTC for the motorisation of ascorbic acid and lemon juice in food and pharmaceuticals. S-MCMS were prepared from D-glucose and thiourea as the S source, followed by annealing at different temperatures (700 °C, 800 °C, and 900 °C). The obtained materials had an average size of 0.5–5 μM and a highly microporous structure. The annealing temperature had a high influence on the structure, and S-MCMS-900 possessed the highest surface area, with the smallest sphere size and more sp2 carbon chain distortion. Roldán et al.172 synthesised SCs via the HTC of D-glucose activated with ZnCl2 and thiophenecarboxaldehyde as the S source. For comparison, NCs and N, S-doped carbon materials have been prepared using the same approach. The SCs possess C–S–C bonding derived from the thiophenic group and a small amount of S was linked to Zn, forming ZnS sphalerite, inhibiting, in this way, the complete removal of the activating agent. In terms of textural properties, the SCs exhibited surface areas around 550 m2 g−1 with a 0.9 cm3 g−1 pore volume, and according to XPS results 6.7 wt% of the sulphur was incorporated. In an earlier study, the same author prepared SCs for water treatment applications.168 Using the same approach, they prepared mesoporous SCs. For comparison, NCs were also prepared. SCs possessed a mesoporous structure, with 89% mesoporosity and a pore size of approximately 34 nm, containing a high amount of S atoms incorporated onto the carbon structure, 8.3 wt%, which is double that of the N atoms incorporated, 3.9 wt%.3.3. Boron doped carbon materials
Besides nitrogen and sulphur, boron represents a promising dopant for carbon materials. The advantage of boron incorporation lies in the fact that it creates defects, by introducing an uneven charge distribution which enhances the electrochemical performance by improving the charge transfer between neighbouring carbon atoms.173 Also, the incorporation of boron could improve the conductivity of carbon materials by increasing the density of hole-type charge carriers.174 All these improvements make them good candidates for different applications, such as energy storage175 and electrocatalysis.144,173,174 Sun et al.176 reported the synthesis of boron-doped porous graphitic carbon materials (BCs) derived from chitosan (BNGC-2-900). The resulting hydrothermal carbon was chemically activated using ZnCl2 followed by carbonisation at elevated temperatures (900 °C). The produced BCs had high surface areas, up to 1567 m2 g−1, and a pore volume of about 0.48 cm3 g−1. XPS results revealed the presence of carbon, nitrogen (4.99%), boron (3.47%) and oxygen atoms. The successful doping of the materials with boron was confirmed by the presence of peaks from the C 1s spectrum corresponding to C–B (283.4 eV), C–C (284.7 eV), C–N (285.6 eV), C–O (286.1 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (286.4 eV), and C–O–B (288.7 eV). Incorporation of boron atoms in the hydrothermal carbon can affect both the morphology and particle size, according to a study conducted by Kalijadis et al.138 For their synthesis, different concentrations of boric acid (0.1, 0.2, 0.6 and 1 wt%) were mixed with D-glucose and HTC was performed at 180 °C for 24 h. For the undoped sample, the particles present a uniform diameter of about 3 μm and a few particles were about 7 μm. By incorporation of boron atoms, the particle size is increased, up to 20 μm, and the degree of homogeneity is decreased. This enlargement phenomenon is attributed to the catalytic effect that boric acid could have during the HTC, by effective conversion of D-glucose to produce 5-HMF. Based on textural characterisation, BCs consist of a microporous structure, with surface areas between 73 m2 g−1 and 362 m2 g−1 and boron content between 0.05 and 0.19 wt%.
O (286.4 eV), and C–O–B (288.7 eV). Incorporation of boron atoms in the hydrothermal carbon can affect both the morphology and particle size, according to a study conducted by Kalijadis et al.138 For their synthesis, different concentrations of boric acid (0.1, 0.2, 0.6 and 1 wt%) were mixed with D-glucose and HTC was performed at 180 °C for 24 h. For the undoped sample, the particles present a uniform diameter of about 3 μm and a few particles were about 7 μm. By incorporation of boron atoms, the particle size is increased, up to 20 μm, and the degree of homogeneity is decreased. This enlargement phenomenon is attributed to the catalytic effect that boric acid could have during the HTC, by effective conversion of D-glucose to produce 5-HMF. Based on textural characterisation, BCs consist of a microporous structure, with surface areas between 73 m2 g−1 and 362 m2 g−1 and boron content between 0.05 and 0.19 wt%.
3.4. Phosphorus doped carbon materials
Phosphorus (P) is another important heteroatom that gained its place on the list of dopants for the carbon structure. P doping can bring a lot of advantages to the carbon structure, such as improving electrochemical stability by suppressing the formation of electrophilic oxygen species and inhibiting the combustion of the oxygen species, which consequently also contributes to enhancing the cycling stability, capacity retention ratio and operating voltage window for capacitors.140 Regarding their synthesis, P doped carbon materials (PCs) can be prepared either in a two-step synthesis of mesoporous carbon followed by P incorporation,177 or through a one-pot HTC.178 Li et al.177 reported the synthesis of P and N co-doped carbon microspheres following a two-step strategy using D-glucose and (NH4)HPO4. Both doped and undoped materials present a spherical morphology with a smooth surface, as shown by SEM (see Fig. 19a and b). The heteroatom incorporation was confirmed by XPS measurements, with P–O, P–C and C–N bonds being identified. Quantitative XPS analysis revealed that P was incorporated at ∼1.19 at% and N at 1.00 at% in NPCM.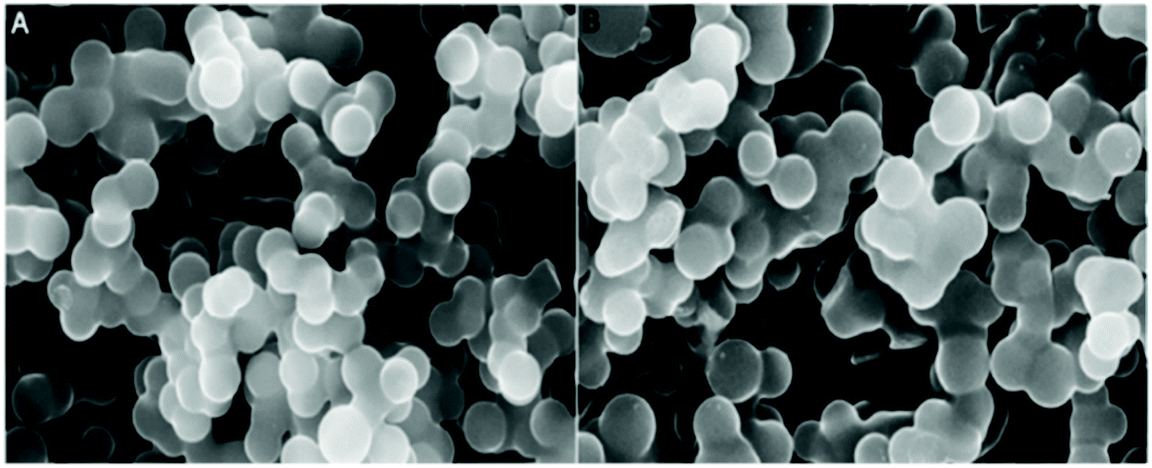 | ||
| Fig. 19 SEM images of (A) CM and (B) NPCM; reproduced with permission from ref. 177. | ||
Wu et al.178 synthesised PCs starting from glucose and tetraphenylphosphonium bromide (C24H20P(Br)) via HTC combined with a soft templating method. The mixture was treated at 180 °C for 4 h during HTC, followed by template removal at 800 °C for 2 h, under an inert atmosphere. During the synthesis, the amount of P donor was varied, and the resulting samples were denoted as P-CHS-1, 2 and 3, with the increasing number referring the increment of the P donor. For comparison, undoped samples were also prepared. P incorporation was examined by FTIR: when comparing CHS and P-CHS-2, similar peaks are identified, at 3060 cm−1 for aromatic C–H stretching vibrations and aromatic C–H out of plane deformation vibrations below 900 cm−1, aliphatic C–H stretching vibrations at 2857 and 2924 cm−1 and C–H scissoring vibrations at 1453 cm−1. This is clear evidence that the HTC of D-glucose is not influenced by P doping. The CC band is present in both samples, with vibrations at 1588 cm−1, and both contain O–H (3440 cm−1), CO (1705 cm−1) and C–O (1278 and 1026 cm−1) bands. Among all these peaks, P-CHS-2 shows a few more, at 1109 cm−1 assigned to P–O combination, at 1085 cm−1 assigned to the ionized linkage P+–O− or P–O–C, and two vibration bands at 725 and 682 cm−1, due to P–C, which confirms the doping of P in the carbon. Guo et al.179 prepared phosphorus-doped carbon nanotubes (PCNTs). For this, the mixture was heated at 170 °C for 12 h. Compared to raw CNTs, PCNTs show a lower surface area and a smaller pore volume of pores, due to the removal of highly disordered amorphous carbon regions. The functional groups on the surface of PCNTs were investigated by XPS. The amount of P was about 1.66 at% and O was about 6.98 at%. Overall, the –P–O groups were dominant, and they formed via the acid reaction with the –OH groups from the edge of the sp2 nanotube layer. The main P containing groups were –PO and –P–C.
3.5. Summary
Heteroatom doped carbon materials showed their potential in a wide range of applications, but they seem a “go to” option in fields such as electrocatalysis for the ORR or CO2 reduction and supercapacitors. As presented above, HTC represents a useful method for the synthesis of NCs, SCs, BCs or PCs either in one pot or two step reactions. The advantage of HTC over other synthesis methods lies in the possibility of being able to use sustainable carbon precursors and any source to dope the heteroatom. Also, HTC implies a small energy consumption and the process itself is easy and eco-friendly.4. Applications
Porous carbon materials have been used in multiple applications since long ago. There is clear evidence of the usage of ACs along the years, for example in ancient times charcoal was used to adsorb unpleasant odours or to clean water. Later, in 1773, AC was mentioned as an adsorptive material in a gas experiment by Scheele. In 1862, Frederick Lipscombe rinsed activated carbon for commercial applications, by using the material to purify potable water, and in 1881 Heinrich Kayser mentioned the ability of charcoal to take up gases. ACs were implemented at the industrial scale at the beginning of the twentieth century, by Chemische Werke, and during the First World War it was used in gas masks by American soldiers to protect them from poison gas.180–183 Due to a series of advantages, such as the low cost of the raw materials, the high degree of microporosity,127 high surface areas (1000 m2 g−1),184 and their good chemical and thermal stability, porous carbon materials are used in a wide range of applications, as described below.4.1. Energy storage
Hydrothermal carbons represent a useful and sustainable class of materials for use in energy storage. They have been extensively explored as graphite replacements in lithium-ion batteries and have enabled the development of sodium-ion technologies which have been previously inaccessible, as well as further the development of supercapacitor electrodes and fuel cells.HTC is an appealing route for obtaining useful carbon materials from cheap and abundant biomass; for example, holly185 and ginkgo leaves,186 sugarcane,187 sweet potato,188 pollen,189 peanut skin128,190 apricot shell,191 and corn stalks192 have all been converted hydrothermally for use in energy storage applications. Several studies have compared hydrothermally treated precursors against directly pyrolysed biomass and found that those materials which have first undergone HTC function significantly better as electrodes in lithium- or sodium-ion batteries. Wang et al.190 attributed the larger initial capacity achieved in their hydrothermally treated peanut skin to the presence of significant micropores and mesopores, and showed that a higher fraction of mesopores was desirable for fast ion diffusion channels. In contrast, two reports by Zheng et al. comparing HTC and direct pyrolysis of holly leaves185 and sweet potato188 found that the greater microporosity introduced by HTC played an important role in the improved electrochemical performances. The discrepancies between these studies likely arise from the intrinsic differences in the nanostructure of the starting biomass. The storage mechanism of sodium in hard carbons is still poorly understood, and hydrothermally treated biomass was used as the precursor for tailored structures in these fundamental studies. Hard carbons derived from reed straw and carbonised at 900–1500 °C exhibited variations in graphitic domains and microporosity, resulting in different contributions to both the sloping and plateau capacity regions.193 Sodium storage in the sloping region was attributed to adsorption in the micropores and defect sites, and Na+ intercalation into the graphite-like layers in the plateau region (Fig. 20 and Table 3).
 | ||
| Fig. 20 LHS: (a) Plateau and sloping curves of RS-x electrodes. (b) Amount and contribution rate of capacity from the plateau and sloping region in RS-x electrodes; capacity contributed at 0.1–4 C by (c) sloping regions and (d) plateau regions. RHS: Schematic illustration of the sodium storage mechanism of RS-x electrodes. Reproduced with permission from ref. 193. | ||
| Precursor | Hydrothermal conditions | Carbonisation temperature | Activation | Template | Reversible capacity (mA h g−1) | FCCE (%) | Application | Ref. |
|---|---|---|---|---|---|---|---|---|
| Glucose | 180 °C, 24 h | 1100 °C (Ar), 800 °C (H2) | — | — | 160 at 50 mA g−1 | — | Na-ion | 194 |
| Glucose | 180 °C, 6 h | 800 °C (N2), 300 °C (air) | — | Na2MoO4 template | 250 at 50 mA g−1 | 50 | Na-ion | 195 |
| Sugar/perylenetetracarboxylic dianhydride (PTCDA) | 195 °C, 5 h | 1100 °C (HTC glucose), 900 °C (HTC glucose + PTCDA) | — | — | 261 at 28 mA g−1 | 67 | K-ion | 196 |
| Sucrose, toluene | 190 °C, 5 h (sucrose) | 1600 °C (HTC sucrose + toluene) | — | — | 312 at 30 mA g−1 | 83 | Na-ion | 197 |
| Sucrose, graphite oxide | 190 °C, 12 h | 700 °C | — | — | 296 at 50 mA g−1 | 44 | Na ion | 198 |
| Sugarcane bagasse, graphene oxide | 150 °C, 4 h | 750 °C (treatment and activation of sugarcane) | KOH | — | 617 at 200 mA g−1 | 56 | Li-ion | 187 |
| Holly leaf | 180 °C, 10 h | 800 °C | — | — | 318 at 20 mA g−1 | 50 | Na-ion | 185 |
| Ginkgo leaf | 250 °C, 12 h | 700 °C | KOH | — | 200 at 200 mA g−1 | 31 | Na-ion | 186 |
| Sweet potato | 160 °C, 10 h | 800 °C | — | — | 320 at 100 mA g−1 | 33 | Li-ion | 188 |
| Rape pollen grains | 180 °C, 24 h | 600 °C | — | — | 145 at 100 mA g−1 | — | Na-ion | 189 |
| Peanut skin | 180 °C, 12 h | 900 °C | KOH | — | 681 at 1000 mA g−1 (Li), 302 at 100 mA g−1 (Na) | 45 (Li), 27 (Na) | Li-ion, Na-ion | 128 |
| Peanut skin | 180 °C, 24 h | 800 °C | KOH | — | 431 at 100 mA g−1 | 34 | Na-ion | 190 |
| Apricot shell | 200 °C, 40 h | 1000 °C | — | — | 184 at 0.1 C | 72 | Na-ion | 191 |
| Reed straw | 200 °C, 24 h | 1300 °C | — | — | 372 at 25 mA g−1 | 77 | Na-ion | 193 |
| Shaddock peel | 180 °C, 24 h | 600 °C | — | — | 287 at 50 mA g−1 | 57 | Na-ion | 199 |
| Phenolic resin | 500 °C, 12 h | 1250 °C | — | — | 311 at 20 mA g−1 | 60 | Na-ion | 200 |
| Nano-Si, sucrose, graphite | 190 °C, 6 h | 1000 °C | — | — | 879 at 30 mA g−1 | 81 | Li-ion | 201 |
| Sn, glucose | 180 °C, 5 h | 500 °C | — | — | 452 at 0.5 C (Li), 487 at 0.5 C (Na) | — | Li-ion, Na-ion | 202 |
| Polyethylene glycol (PEG), SnCl2 | 100 °C, 6 h | 600 °C (carbon template calcination, H2/Ar) | — | Crab-shell template | 298 at 100 mA g−1 | 40 | Li-ion | 203 |
| Graphite oxide, glucose, SnCl4 | 180 °C, 12 h | — | — | — | 1468 at 80 mA g−1 | 64 | Li-ion | 204 |
| Carbon cloth, SnCl4, thioacetamide | 160 °C, 16 h | — | — | — | 1040 at 200 mA g−1 | 33 | Na-ion | 205 |
| SnCl4, apricot shell | 180 °C, 20 h (SnO2 synthesis), 200 °C, 20 h (SnO2-hydrocarbon) | 500 °C | — | — | 288 at 0.1 C | 50 | Na-ion | 191 |
| Na2MoO4, CH3CSNH2, polyvinylpyrrolidone (PVP), SnCl4 | 180 °C, 12 h (MoS2), 120 °C, 10 h (MoS2 + SnO2 + carbon) | 500 °C (MoS2 + SnO2 + carbon) | — | — | 396 at 100 mA g−1 (Na), 312 at 50 mA g−1 (K) | 68 (Na), 73 (K) | Na-ion, K-ion | 206 |
| Corn stalks, Na2MoO4, thiourea | 180 °C, 24 h (MoS2 − carbon) | 600 °C (corn stalks), 650 °C (carbon + MoS2) | — | — | 1231 at 100 mA g−1 | 72 | Li-ion | 192 |
| Loofah sponges, graphene oxide, (NH4)6Mo7O24, thiourea | 160 °C, 6 h (loofah), 200 °C, 24 h (HTC loofah + GO + MoS2) | 400 °C (HTC loofah + GO), 600 °C 2 h (HTC loofah + GO + MoS2) | — | — | 838 at 200 mA g−1 | 55 | Li-ion | 207 |
| Cotton, (NH4)6Mo7O24, thiourea | 220 °C, 24 h | 80 °C (H2/Ar) | — | — | 444 at 100 mA g−1 | 82 | Na-ion | 208 |
| Cotton, (NH4)6Mo7O24, thiourea | 220 °C, 24 h | 80 °C (H2/Ar) | — | — | 444 at 100 mA g−1 | 82 | Na-ion | 208 |
| Citric acid, (NH4)6Mo7O24, ascorbic acid | 180 °C, 5 h (MoO2 + carbon) | 600 °C (carbon template, H2/Ar), 400 °C (MoO2 + carbon) | — | NaCl | 367 at 100 mA g−1 (Na), 213 at 50 mA g−1 (K) | 47 (Na), 45 (K) | Na-ion, K-ion | 209 |
| Avocado, KMnO4, trimethylaluminium | 180 °C, 24 h (avocado) | 600 °C (carbon + MnO) | — | — | 1165 at 150 mA g−1 | 67 | Li-ion | 210 |
| KMnO4, benzoic acid | 160 °C, 10 h | 1200 °C | — | — | 1168 at 50 mA g−1 | 86 | Li-ion | 211 |
Hydrothermally prepared shaddock peel was used to investigate the relationship between capacity and disordered turbostratic nanodomains, with the resulting carbons exhibiting mainly sloping capacity.199 The main mechanism of sodium-ion storage was attributed to the balance of charge conductivity, the sodium-ion diffusion rate, and the number of adsorption sites. Well-defined precursors, such as glucose194 or phenolic resin,200 are also often used for systematic studies. For phenolic resin carbonised at 800–1500 °C, it was found that increasing carbonisation temperatures decreased the d-spacing between graphitic layers, and promoted the shift from microporosity to mesoporosity in the materials, resulting in a decrease in surface area and total pore volume.200 The material carbonised at intermediate temperature (1250 °C) showed the best reversible capacity which was attributed to the balance between optimised d-spacing and the pore structure. Interestingly, the material carbonised at 1500 °C had the highest first cycle coulombic efficiency (62.5%), due to the lower surface area resulting in less significant SEI formation (Fig. 21a), whilst the material carbonised at a lower temperature (1000 °C), despite being less electrically conductive, exhibited a better rate capability at higher C rates, arising from the wider d-spacing (Fig. 21b).
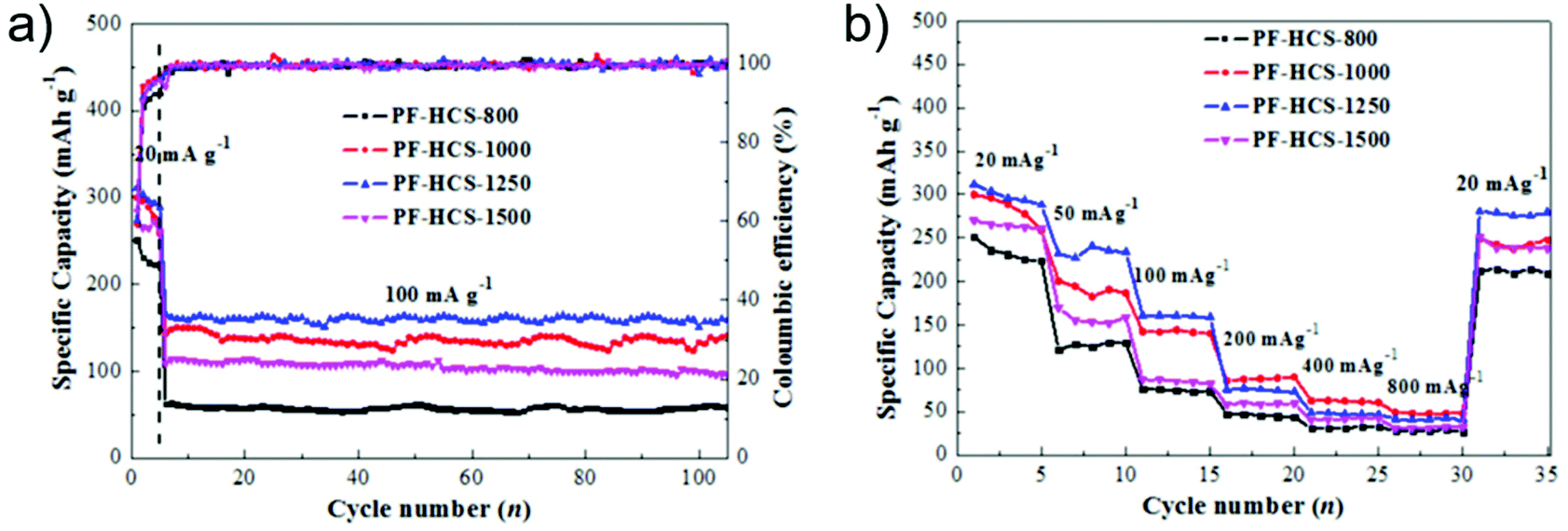 | ||
| Fig. 21 (a) Cycle performances of phenolic resin-based hard carbon sphere electrodes at 100 mA g−1. (b) Rate performances of phenolic resin-based hard carbon sphere electrodes. Reproduced with permission from ref. 200. | ||
Hard carbons exhibit good capacity retention, but often poor rate capability; conversely, pure soft carbons have superior rate capability, but suffer rapid capacity fade. Combining the two can result in a composite anode with improved electrochemical properties. Hard–soft composites prepared for K-ion batteries exhibited a higher rate capability than the pure hard carbon (81 mA h g−1vs. 45 mA h g−1 at 10 C) and superior cycling stability compared to pure soft carbon (93% vs. 55% after 200 cycles at 1 C).196 The hard carbon provided a porous and robust structure to accommodate the intercalation of potassium ions, whilst the conductivity offered by the soft carbon regions facilitated fast ion transport to maintain performance at high current rates. Similarly, hard carbon spheres with a soft carbon coating displayed improved initial coulombic efficiency (54% to 83%) and a capacity retention of 93% after 100 cycles, when tested as anodes in Na-ion batteries.197 In addition, they found that increasing the carbonisation temperature from 1000 °C to 1600 °C could significantly increase the plateau capacity, hypothesising that this effect was due to the larger number of nanopores at higher temperature. The direct addition of graphene derivatives is another common approach for synthesising hard/soft composites; often graphene/graphite oxide is combined with the hard carbon precursor prior to HTC, which serves to reduce the graphene sheets in situ.187,198 In these studies, the enhanced sodium storage capacity and cycling stability of the composite were ascribed to the complementary effect of the different morphological characteristics of the carbons which provided ample sodium storage in the micropores and large interlayer spaces, minimised volume variation during cycling and ensured sufficient contact area with the electrolyte for efficient diffusion and charge transport.198 Aside from pure carbon materials, various carbon/inorganic composites have been explored as electrode materials.195,202,203 For Li-ion batteries, elements such as Si and Sn are promising since lithium can form alloys with either in a ratio of 4.4:1, resulting in a significantly higher theoretical capacity (e.g. Li4.4Si 4200 mA h g−1vs. 372 mA h g−1 for LiC6 in graphite). Unfortunately, formation of these alloys is generally accompanied by severe volume changes, leading to rapid capacity fade with cycling. Furthermore, some of the alloy species are soluble in the electrolyte solution and thus the anode gradually decomposes. In addition, such materials on their own have low intrinsic conductivity which can result in overall poor performance. In several studies, the combination of these inorganic particles with hard or soft carbons serves to stabilise volume changes and improve conductivity, resulting in superior cycling performance. A comparison between hard carbon-coated SnO2 and a mechanically mixed composite demonstrated that the carbon coating could significantly stabilise capacity fading.191 Jeong et al.201 produced silicon coated hard carbon attached to a graphite scaffold; the overall reduced contact area with the electrolyte served to suppress SEI formation, whilst the graphite facilitated electron transport. The report further showed that the electrode thickness and amount of binder strongly affected the lithiation capacity; particularly in the case of alloy materials, the binder content can additionally stabilise volume expansion and contraction on cycling, an important factor which is not often discussed in the literature. Similarly, Zhang et al.204 supported SnO2 particles on rGO and used the growth of hard carbon on the surface to limit the particle size, resulting in a uniform size distribution. Good contact between SnO2 particles and the hard carbon-rGO scaffold was achieved by C–O–Sn covalent bonds which enhanced conductivity, whilst the rGO/hard carbon matrix prevented agglomeration of SnO2 and limited the volume expansion to improve cycling stability. Carbon/transition metal oxide and dichalcogenide electrodes are also being developed for energy storage.207,209 An electrode material derived from avocado and MnO, coated with alumina for stability, was tested in a Li-ion cell, and the high capacity and good rate capability (Table 2) were explained by the structure inherited from the avocado precursor which provided a good buffer for volume expansion, and the Al2O3 coating with greater ionic conductivity.210 In another study with MnO-graphene nanopeapods, structural stability was introduced with a graphene scaffold, which also enhanced electrical conductivity, resulting in high Li storage capacity.211 For Na-ion batteries, analogous carbon-supported MoS2 materials showed promising performance, in which MoS2 nanosheets were vertically grown on cotton-derived carbon fibres which act as a robust conductive support and result in greatly enhanced cycling stability. Even at high current densities, much higher capacities could be maintained, compared to the capacity retention of bare MoS2 nanosheets. In a similar approach, MoS2 nanosheets were grown on the surface of carbonised corn stalks, whereby the intrinsic structure of the biomass precursor served to stabilise the electrode during long cycles and at high current densities.192 Hydrothermally prepared MoS2 was combined with PVP and SnO2 and it also showed excellent cycling stability at high current densities for both Na and K storage206 attributed to the sandwich structure formed by the MoS2 and carbon coating. Well-aligned nanosheets of SnS2 have been grown hydrothermally on a carbon cloth (Fig. 22), and when tested as an anode for sodium-ion batteries they exhibited enhanced electrochemical properties in comparison with pure SnS2.205 The enhanced behaviour was attributed to the introduction of the carbon cloth substrate, which provided the contact area for both the electrolyte and active material and enabled rapid Na-ion transport, improved electrical conductivity, and relieved aggregation and pulverisation of the SnS2 nanosheets.
 | ||
| Fig. 22 (a) Larger-magnification SEM images of SnS2 with a flower-like feature. (b) Typical low-magnification SEM image of SnS2/CC. (c) and (d) Larger-magnification SEM images of SnS2 on CC showing the well-aligned nanosheet architecture. (e) EDS exploration of SnS2/CC. (f) SEM image of the marked region of SnS2/CC and relevant to EDS plots of (g) Sn and (h) S. (i) TEM and (j and k) HRTEM images, with inset showing the FFT pattern, and (l) the corresponding SAED pattern of the SnS2 sample. Reproduced with permission from ref. 205. | ||
Cathode materials have also been developed using similar methodology, for example, the hydrothermally assisted synthesis of Na7V4(P2O7)4(PO4)/C nanorods, which are formed by simultaneous crystallisation of Na7V4(P2O7)4(PO4) and in situ carbonisation of surfactants on the surface.212 The carbon formed as a thin layer around the nanorods, and limited particle growth, resulting in a uniform particle size. With an aqueous electrolyte, a capacity of 51.2 mA h g−1 was achieved at a current density of 80 mA g−1, and 72% of the initial capacity was maintained even at 1000 mA h g−1. The hydrothermal production of a Na3V2O2x(PO4)2F3−2x/carbon composite further demonstrated the importance of the conductive carbon for overall cell performance.213 In a Li–S system, a composite electrode of nickel, sulfur and carbon derived from bamboo exhibited a high initial capacity (1198 mA h g−1 at 0.2 C), good cycling stability (1030 mA h g−1 after 200 cycles) and an improved rate capability.134 These improvements were attributed to the structural stability and high conductivity arising from the Ni particles embedded in the carbon framework. Lastly, peanut shell has been used as both a cathode and anode for a Na-ion hybrid capacitor;214 hydrothermal treatment of the peanut shell for the cathode resulted in a hierarchically porous material with a sheet-like morphology and a surface area comparable to that of graphene materials (2396 m2 g−1) and high levels of oxygen doping (∼13 wt%). The assembled Na-ion capacitor yielded specific energies of 201, 76 and 50 W h kg−1 at specific powers of 285, 8500 and 16500 W kg−1, respectively, and achieved 88% capacity retention after 100000 cycles at 51.2 A g−1.
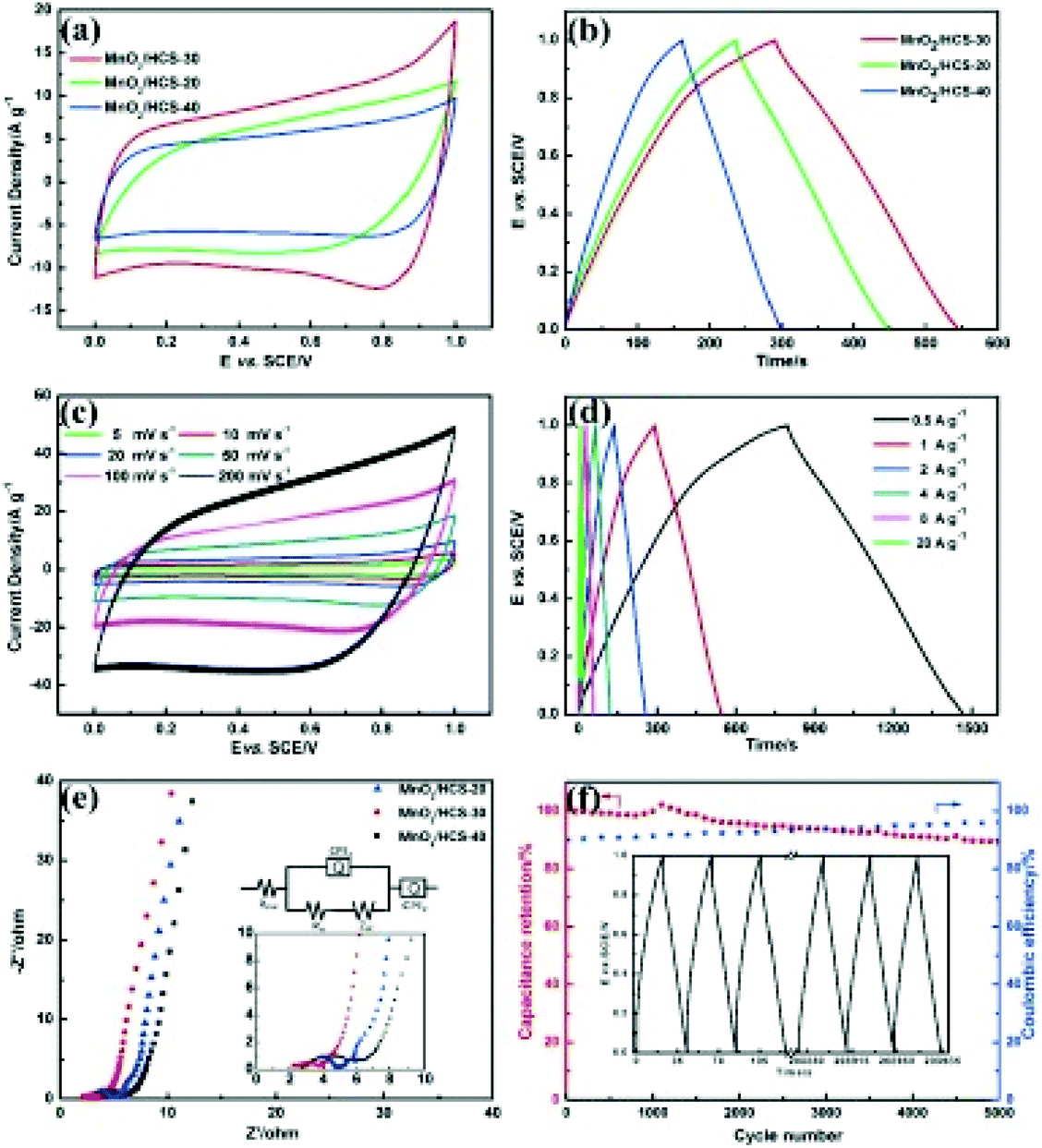 | ||
| Fig. 23 The electrochemical performances of samples in a 1 M Na2SO4 electrolyte. (a) CV curves at a scan rate of 50 mV s−1. (b) GCD curves at a current density of 1 A g−1. (c) CV curves at different scan rates. (d) GCD curves at different current densities of MnO2/HCS-30. (e) Nyquist plots of samples. (f) Cycle life and coulombic efficiency of MnO2/HCS-30 at a current density of 10 A g−1. Reproduced with permission from ref. 224. | ||
| Precursor | Hydrothermal conditions | Carbonisation temperature | Activation/template | Capacitance (F g−1) | Capacitance retention | Ref. |
|---|---|---|---|---|---|---|
| Ginkgo leaf | 250 °C, 12 h | 700 °C (activation) | KOH | 350 at 1 A g−1 | 98% after 30000 cycles at 5 A g−1 |
186 |
| Rye straw | 240 °C, 24 h | 750 °C | KOH | 229 at 1 A g−1 | — | 216 |
| Hemp stems | 160 °C, 12 h | 600 °C (pre-activation), 800 °C (activation) | KOH | 318 at 0.1 A g−1 | 96% after 10000 cycles at 4 A g−1 |
217 |
| Jujun grass | 180 °C, 10 h (ionothermal) | 900 °C (activation) | KOH | 336 at 1 A g−1 | 87% after 2000 cycles at 1 A g−1 | 218 |
| Stem bark | 130 °C, 2 h, KOH | 700 °C | KOH | 320 at 0.5 A g−1 | 94% after 10000 cycles at 2 A g−1 |
219 |
| Rice husk | 180 °C, 10 h, NaOH | 500 °C (pre-activation), 800 °C (activation) | KOH | 302 at 1 A g−1 | 89% after 5000 cycles at 10 A g−1 | 221 |
| Tobacco rods | 200 °C, 12 h | 800 °C | KOH | 287 at 0.5 A g−1 | 96% after 10000 cycles at 5 A g−1 |
227 |
| Starch | 200 °C, 8 h | 800 °C | Ammonium ferrous sulfate (NH4)2Fe(SO4)2 | 248 at 0.5 A g−1 | 93% after 10000 cycles at 2 A g−1 |
132 |
| Lignin | 200 °C, 24 h | 800 °C | KOH | 312 at 1 A g−1 | 98% after 20000 cycles at 10 A g−1 |
220 |
| Glucose | 200 °C, 24 h | 700 °C | KOH | 247 at 0.2 A g−1 | — | 222 |
| Glucosamine | 200 °C, 24 h | 700 °C | KOH | 250 at 0.2 A g−1 | — | 222 |
| Granulated sugar | 200 °C, 24 h | 700 °C (activation), 800 °C (H2 reduction) | ZnCl2 | 136 at 0.1 A g−1 | — | 223 |
| Lignin, graphene oxide | 240 °C, 16 h | — | — | 190 at 0.5 A g−1 | 87% after 10000 cycles at 10 A g−1 |
228 |
| Glucose, graphene oxide | 200 °C, 16 h | 800 °C | KOH | 223 at 0.1 A g−1 | 87% after 10000 cycles at 1 A g−1 |
229 |
| Graphene foam, polyvinyl alcohol (PVA), polyvinylpyrrolidone (PVP) | 190 °C, 12 h | 800 °C | KOH | 188 at 0.5 A g−1 | 100% after 10000 cycles at 2 A g−1 |
230 |
| Distillers dried grains with solubles, graphene oxide | 200 °C, 18 h | 600 °C (activation), then microwave for 30 s at 1200 W | KOH | 102 at 1 A g−1 | 92% after 5000 cycles at 5 A g−1 | 231 |
| Glutaraldehyde, KMnO4 | 180 °C, 5 h (yeast + glutaraldehyde), 160 °C, 3 h (hollow carbon + MnO2) | 850 °C (hollow carbon) | Yeast cells | 255 at 1 A g−1 | 89% after 5000 cycles at 10 A g−1 | 224 |
| Carbon cloth, Co(NO3)2, Na2MoO2, Ni(NO3)2, C6H12N4 | 150 °C, 6 h (CoMoO4 + carbon cloth), 90 °C, 12 h (NiCo-LDH) | 350 °C (CoMoO4 + carbon cloth) | — | 2024 at 1 A g−1 | 91% after 5000 cycles at 1 A g−1 | 225 |
| NiCl2, Se, graphene oxide, hydrazine | 120 °C, 12 h | — | — | 1280 at 1 A g−1 | 98% after 2500 cycles at 5 A g−1 | 226 |
In addition, the introduction of inorganic nanoparticles to carbon225,226 or doping with heteroatoms can introduce complementary pseudocapacitive storage mechanisms, thereby improving the overall performance of the material. Nitrogen,216,218,222,227 oxygen,219,227 sulfur,186 and iron222 have all been incorporated into HTC materials, with the aim of improving capacitance (Table 4). Nitrogen is a common dopant, often arising as a natural component of the precursor.
Interestingly, a comparison of hydrothermally carbonised glucose and glucosamine216 showed that after activation with KOH, the surface area of HTC glucose was higher than that of HTC glucosamine (1766 vs. 1238 m2 g−1), but that there was no significant difference in their capacitances (280 and 287 F g−1, respectively, at 0.2 A g−1). The discrepancy was ascribed to the presence of pyridone and pyridine groups which contribute pseudocapacitance, and of pyrrole groups which improve electron mobility and electron transfer reactions in the carbon matrix. Nitrogen- and oxygen-doped hydrothermally carbonised tobacco rods showed a high capacitance of 286 F g−1 at 0.5 A g−1, and excellent cycling stability.227 The enhanced performance compared to that of commercial activated carbons was attributed to the presence of heteroatoms which provide additional pseudocapacitance, as well as improving electrical conductivity and wettability, enhancing electron and ion transportation. Similarly, N-doping in hydrothermal lignin improved conductivity and surface wettability with electrolyte ions, whilst the hierarchical bowl-like pore structure and large specific surface area (2218 m2 g−1) resulted in a high specific capacitance of 312 F g−1 at 1 A g−1 and excellent cycling stability (98% after 20000 cycles at 10 A g−1, Fig. 24).220 Even at 80 A g−1, 81% capacitance was retained, likely due to the high conductivity and efficient ion transport pathways in the hierarchical pore structure.
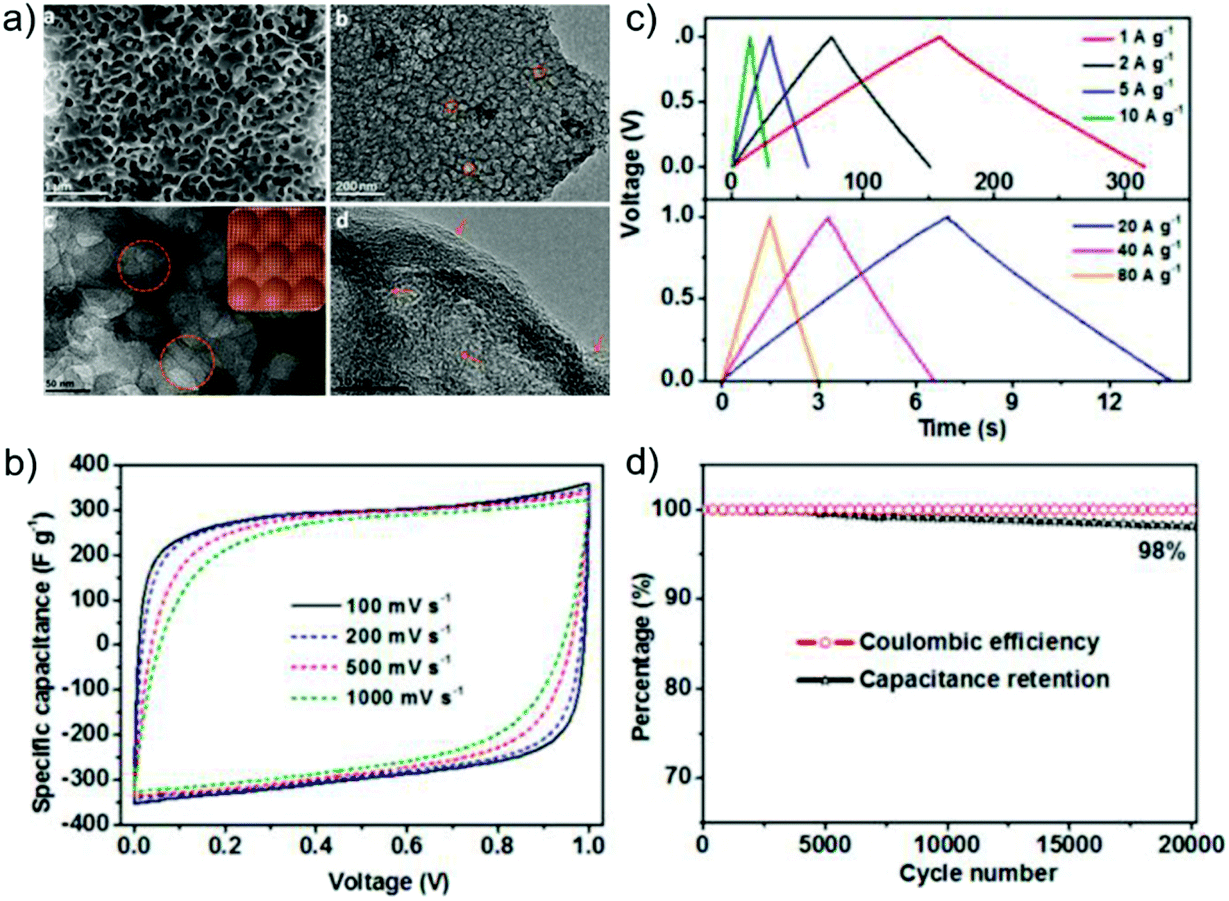 | ||
| Fig. 24 (a) SEM, TEM and HR-TEM images of hierarchical porous nitrogen-doped carbon showing bowl-like pore structures. (b) CV curves, (c) charge/discharge profiles, and (d) cycling stability and coulombic efficiency at 10 A g−1 in a 6 M KOH electrolyte. Reproduced with permission from ref. 220. | ||
Ginkgo leaves have been used as a precursor to sulphur-doped carbons, and highly porous carbons.186 Following sulfuric acid-assisted HTC, and KOH activation, a carbon material with a S-content up to ∼8 wt% was obtained, with a hierarchical porosity, and a specific surface area of 1132 m2 g−1, higher than those of materials produced without activation or treatment with sulfuric acid. Further heating of the sample in air to remove sulphur resulted in an even higher surface area of 1757 m2 g−1. In spite of the lower surface area, the S-doped material showed the best performance, achieving a specific capacitance of 364 F g−1 at 0.5 A g−1, with an aqueous electrolyte. At a high rate of 50 A g−1, the specific capacitance was still 230 F g−1 suggesting that sulphur-doping can increase the pseudocapacitance in the presence of sulfone and sulfoxide moieties and creates a more polarised surface which facilitates charge transfer. The addition of graphene derivatives to biomass precursors results in hybrid xerogel structures with varied morphologies and porosities, depending on the precursor, component ratios, and HTC or activation conditions. In one study, the addition of GO to lignin during HTC was found to modify the pore structure and increase the specific surface area, whilst lignin could effectively reduce the GO sheets under hydrothermal conditions, and prevent aggregation of rGO sheets by interrupting the π–π stacking.228 rGO improved electrical conductivity, and provided structural stability, enabling long cycling. Similarly, xerogels of hydrothermally carbonised glucose-GO followed by KOH activation resulted in cellular structures exhibiting well-connected, continuous and very thin carbon walls (∼5–15 nm) which favoured ionic diffusion and electronic conduction.229 The thickness and connectivity of the walls could be altered by varying the amount of KOH, and it was found that the carbon xerogel with very thin, connected walls and a relatively high graphitic character (from a lower amount of KOH) delivered a much higher specific capacitance than that of a commercial activated carbon (223 vs. 153 F g−1 at 0.1 A g−1) and retained 87% capacity after 10000 cycles at 1 A g−1. The performance was attributed to good conductivity and the hybrid cellular structure. In another work, graphene foam produced by CVD was used as a support for a hybrid hydrogel of polyvinyl alcohol (PVA) and polyvinylpyrrolidone (PVP).230 KOH activation increased the surface area to 2994 m2 g−1, and a gravimetric capacitance of 188 F g−1 at a current density of 0.5 A g−1 was achieved. The hybrid also showed excellent long-term stability at a current density of 2 A g−1, with no capacitance loss after 10000 cycles. The performance can be attributed to the highly porous structure and large accessible surface area, which are stabilised by the graphene foam, and enable rapid and reversible ion transport. The presence of GO in the HTC of distillers dried grains with water-soluble chemicals changed the morphology of the particles from spheres to flakes.231 Additional KOH activation increased the surface area, although it remained low compared to those of other activated carbons (479 m2 g−1). Short microwave irradiation reduced the GO sheets to increase the degree of carbonisation and graphitisation. The symmetric cell showed a specific capacitance of 102 F g−1 at 1 A g−1, whilst 92% capacitance was retained after 5000 cycles at 5 A g−1. The presence of GO is thought to promote biomass carbonisation and graphitisation, as well as increasing electrical conductivity.
4.2. Electrocatalysis
Electrocatalysis plays a crucial role in clean energy conversion to increase the rate, efficiency and selectivity of the chemical transformation.232 This process involves the electrochemical conversion of water, CO2, and N2 into added-value products such as hydrogen, oxygenates and ammonia. Besides, the oxygen reduction reaction (ORR) plays an important role in energy conversion devices such as fuel cells and metal–air batteries. Its sluggish kinetics is a main barrier for the improvement of the overall efficiency of fuel cell devices. Currently commercial catalysts for the ORR are Pt group metals. These metals demonstrated high catalytic activity towards the ORR; nevertheless they are scarce and easy to degrade especially in an acidic electrolyte. Precious metal free nanocarbon with a heteroatom dopant are considered as promising electrocatalysts to replace the current commercial Pt group metals. The heteroatom dopants include but are not limited to non-metal atoms such as N, P, S, B, and transition metals such as Fe, Co, Ni, etc.233,234 These heteroatom dopants within the carbon matrix induce heteroatom–carbon bonds and the polarization of these bonds generates active sites on the adjacent carbon matrix and results in a reduced energy barrier towards electrocatalysis, while simultaneously increasing the electrical conductivity due to the higher concentration of delocalised electrons within the π-system.136,159,235–237Heteroatom doped carbon materials can be obtained from direct hydrothermal carbonisation of carbon and heteroatom precursors. A template can also be involved during the HTC process to help tune the morphology of the as-obtained products. It is worth noting that due to the limited electric conductivity of the HTC carbon, a follow-up carbonisation process is usually applied at high temperature under an inert atmosphere (usually under nitrogen or argon at above 700 °C) to further improve the electric conductivity of the carbon matrix and facilitate the assembly of heteroatom-derived active sites.237,238 For example, nitrogen-doped carbon materials were produced using natural halloysite as a template and urea as the nitrogen source.235 A flaky morphology was obtained with glucose as a precursor, whereas using furfural resulted in rod-like structures (Fig. 25a and b). The metal-free electrocatalysts were tested for the ORR in alkaline aqueous electrolytes, and the rod-like catalyst demonstrated a better performance than the flaky material. Considering similar amounts of N species and graphitization degrees in the as-produced samples, the superior performance of the rod-like catalysts was attributed to the higher surface area and larger pore volume which provided more active sites, a greater complexity in pore size distribution, and a rod-like morphology which facilitated electron transport. Compared to a commercial Pt/C (20 wt%) catalyst, the carbon catalysts demonstrated higher retention in diffusion limiting current density (after 3000) cycles and enhanced methanol tolerances (Fig. 25c). When tested as cathodes in a single cell H2/O2 anion exchange membrane fuel cell, the rod-like catalyst delivered a peak power density as high as 703 mW cm−2 (vs. 1106 mW cm−2 with the commercial Pt/C cathode catalyst) (Fig. 25d). Efforts have also been made to investigate doping with other heteroatoms such as boron, sulfur and phosphorus.121,136,239 Carbogels derived from glucose and ovalbumin were produced to study the synergistic effect of boron and nitrogen and were proved to generate higher electron transfer numbers and lower hydrogen peroxide yields than those observed in purely N-doped systems. The presence of sulfur decreased the surface area and nitrogen content resulting in diminished ORR performance.136 In contrast, a sulfur doping of 5.5 wt% in SiO2-templated mesoporous ordered carbons was found to enhance the electrocatalytic activity in the ORR in alkaline solution, likely due to the fact that the mesoporous structure was retained from the templating method.121 Further study on the N/B/S/P doped carbon matrix from the same research group.
 | ||
| Fig. 25 STEM images of (a) FU and (b) GU in transmission electron mode. (c) The LSV curves of GU, FU and the benchmark Pt/C (20 wt%) catalysts (scan rate = 10 mV s−1, rotation rate = 1600 rpm) obtained in O2 saturated aqueous 0.1 M KOH recorded both before and after 3000 potential cycles (between 0.6 and 1.2 V). (d) Beginning-of-life H2/O2 anion-exchange membrane fuel cell (AEMFC) performance curves recorded at 60 °C for the GU and FU cathode catalysts (along with PtRu/C anodes): inlet gas flows were supplied with flow rates of 1 SLPM (83% relative humidity and without gas back-pressurization); reproduced with permission from ref. 235. | ||
SBA-15 templated carbon was doped with different heteroatom precursors at low and high concentrations (Fig. 26a), as well as a combination of nitrogen/boron, nitrogen/sulfur and nitrogen/phosphorus (Fig. 26b).240 The correlation of material properties (surface area and surface chemistry) with the electrocatalytic activity in an alkaline electrolyte showed that the doping of N via the HTC method is easier than that of B or P (when comparing the yield of the dopant). N doping into the carbon matrix resulted in improved catalytic activity and electron transfer numbers, whilst the other dopants resulted in similar slightly improved performance possibly by creating defects in the carbon matrix. Despite tremendous efforts in this area, most metal-free electrocatalysts demonstrate limited catalytic activity. It is also proved that the influence of trace amounts of metal impurities on the high performance “metal-free” electrocatalysts cannot be ignored.238 Non-precious metal dopants such as Fe, Co, and Ni, in co-existence with non-metal dopants, was demonstrated to boost the catalytic activity of the carbon electrocatalyst. These metal–N–C electrocatalysts have shown comparable catalytic activity, higher stability and tolerance toward MeOH when compared to commercial Pt–C.241 Bifunctional nanocarbon composites can also be obtained via HTC by careful design of the nanocarbon matrix and active sites and tested for the ORR. 2D crystalline carbons were obtained from the HTC of glucose, fructose or cellulose with guanine, which played an important role in producing the 2D-morphology of the resultant carbon materials.242 The porous N-doped carbons were not only found to be highly active towards the ORR but they also showed efficiency for the HER with a very low overpotential of 0.35 V to achieve 10 mA cm−2 in alkaline medium. In the other half of the water splitting reaction, suitable catalysts are also required for the oxygen evolution reaction; in the past, the development of fuel cells was held back due to the slow kinetics of the OER. Non-noble metal alternatives for the OER are often based on transition metal oxides, while carbon-based materials have generally been underexplored due to their relatively poor performance. Nonetheless, hydrothermally prepared transition metal dichalcogenides have also been prepared for the HER, for instance, the in situ hydrothermal synthesis of MoS2/guar gum carbon hybrid nanoflowers possessed an extremely low onset potential of approximately 20 mV, a low overpotential of 125 mV at 10 mA cm−2 and a small Tafel slope of 34 mV dec−1, nearly identical to those of the bulk platinum standard.243 The performance was ascribed to the nanoflower architecture which provided ample active sites; furthermore, strong interactions between the MoS2 nanoflakes and guar gum enabled long-term stability and microstructural integrity, with nearly 100% activity retention after 2000 cycles. In another study, combining MoSe2 with NiSe2 in composite nanowires on a carbon fibre paper skeleton resulted in better performance than either MoSe2 or NiSe2 alone;244 this observation was explained by the abundance of active sites resulting from the suppression of restacking of MoSe2 nanosheets, and the high conductivity of NiSe2 which facilitated the transfer of electrons from the electrode to the active sites. In addition to the heteroatom dopant in the carbon matrix, the HTC approach also activates carbon cloths by creating oxygen-containing functional groups on its surface using peroxovanadium complexes, which results in a higher specific surface area and faster electron transfer rate, when compared to pristine carbon cloths.245 The overpotential (310 mV) at 10 mA cm−2 of the activated carbon cloth is much lower than that of the pristine material and comparable to that of RuO2/C (280 mV), making the carbon cloth a competitive non-metal catalyst for the OER. The HTC process is also a powerful strategy in the assembly of carbon nanostructures of various dimensions (D), with 0D, 1D and 2D nanostructures being able to be assembled into higher 3D structures which possess a well developed hierarchical morphology to both facilitate mass transfer and accommodate the active sites to provide superior electrocatalytic conductivity. Highly porous and granular Ni–Co nanowires were grown hydrothermally on a woven carbon fibre fabric and then coated with a conductive shell by glucose carbonisation.246 The structure of the nanowires greatly increased the catalytic surface area delivering an overpotential of 302 mV at a current density of 10 mA cm−2. The conductive carbon layer not only facilitated electron transport throughout the entire electrode but also prevented fragmentation of the nanowires during the reaction, which translated to an overall increase of the structural integrity and hence a more reliable performance. HTC was also used to prepare 3D hierarchical structures of mesoporous SnO2 nanosheets supported on a flexible carbon cloth, which could efficiently and selectively electrochemically reduce CO2 to formate under aqueous conditions.247 The electrode exhibited a partial current density of 45 mA cm−2 at a moderate overpotential (0.88 V) with high faradaic efficiency (87%), even larger than those of most gas diffusion electrodes. The presence of the SnO2 particles is thought to be the reason why high performances are achieved due to the fact that they show high selectivity in the reduction of CO2, they present a highly porous structure which provides a large surface area increasing the contact surface between the electrode and electrolyte and facilitating mass and charge transfer, and the robustness of their structure allows them to maintain the high stability of the electrocatalyst during long-term operation. Our group reported a series of graphene–HTC carbon hybrids, in which we applied graphene oxide as an electrically conductive substrate, and N-containing biomass derived HTC carbon was used to provide active sites (Fig. 27).
 | ||
| Fig. 26 Combination graph correlating the materials surface area and dopant species with the electrocatalytic activity (alkaline media) for SBA-15 templated carbons (a) doped with different heteroatom precursors in low and high concentrations. (b) Doped with nitrogen/boron, nitrogen/sulfur and nitrogen/phosphorus. Reproduced with permission from ref. 240. | ||
 | ||
| Fig. 27 (a) Schematic illustration of the fabrication procedure of 3D catalytic electrodes. (b) SEM images of CCS Ni-Co Nws on carbon fibre fabric. (c) Polarisation curves of different 3D catalytic electrodes for the OER in 1 m KOH solution; reproduced with permission from ref. 248. | ||
The sandwich structure managed to decouple the influence of electroactivity and active sites by changing the ratio of GO and HTC carbon. It has been reported that a low electrical activity limited the performance of active sites. However, when the electrical activity reached a certain level, active sites play an important role.237 Based on this hybrid structure, we further reported a FeNC electrocatalyst using carbon nanotubes to provide electrical conductivity. The Fe residues in the carbon nanotubes coordinate with N in the hybridized HTC carbon layer to form highly efficient Fe-N based active sites while the HTC carbon layer protects the Fe–N active sites from being poisoned by the by-product and therefore highly improve the long-term stability.158 HTC is an environmentally benign and highly efficient synthesis strategy for electrocatalyst production. However, due to the complicated mechanism during the process, the structural regulation of the well-defined active site configuration remains to be explored. Meanwhile, the HTC synthesis of single-atom catalysts would also be an interesting research direction to further improve the catalytic performance of the catalyst.
4.3. Heterogeneous catalysis
HTC derived carbon materials have been used in catalysis in many ways. On their own, they can be used as catalysts, mostly as solid acid catalysts. This is done by introducing strong Brønsted acidity mainly by the presence of sulfonated groups on the surfaces of the materials. Another widespread use of these materials is as a catalyst support. The tunability of their surface polarity and area facilitates the anchoring of metal nanoparticles, which can then be used in different reactions. Hydrothermal carbon is particularly interesting in this respect because its properties lead to a decrease in leaching of the active species when compared to classic ACs. A last and least studied use of these materials is as templates. Since hydrothermal carbon can be eliminated by combustion in air at temperatures that are not too drastic, they can be used as structure directing agents. | ||
| Fig. 28 Schematic view of sulfonation of porous carbon. | ||
One such case is the one prepared by Roldán et al.249 After the catalyst was tested in the esterification reaction of palmitic acid with methanol, a difference in the deactivation of the catalyst was observed and this depended on the activation temperature employed for each catalyst. When temperatures lower than 500 °C were used, the deactivation of the catalyst was attributed to the formation of sulfonate esters on the surface while for those treated at higher temperatures it is speculated that accumulation of reactants and products in the pores is the main cause of deactivation. Similar catalysts were employed for the esterification of other molecules such as glycerol.252 Glucose was used as a carbon source and the glucose solution was treated for 19 h at 195 °C leading to the preparation of a carbonaceous material which contained 67.9% C and 27.5% of oxygen. It was observed that after sulfonation (150 °C, 15 h) the carbon content decreased to 55.8% and the oxygen content increased to 40.5%. From this change in values, and taking into account the amount of sulphur added, the obvious conclusion is that additional oxygen other than that of the sulfonated groups was added during the treatment with sulphuric acid. This may occur by water addition to double bonds, hydrolysis of furan groups, or addition of ether functionalities. The sulfonated material was tested in the esterification of glycerol with acetic, butyric, and caprylic acids and the catalytic performance was compared to those of commercial sulfonated resins.252 Turnover numbers of glycerol and acetic acid were in the same range for both the commercial and hydrothermal carbon materials. Regeneration of these acid catalysts usually involves the treatment of spent catalysts with acid, cleaving esterified sulphonic groups.253 Pileidis et al.254 also prepared such materials (HTC conditions: 230 °C, 24 h) and used them as catalysts for the esterification of levulinic acid. In this case, not only glucose but also cellulose and rice straw were used as carbon sources. These sources led to the formation of materials with 80, 76, and 70% of carbon (for glucose, cellulose and rice straw, respectively) and were then sulfonated (80 °C, 4 h) introducing 5–6% of sulphur. Esterification was carried out at 60 °C and after 3 h almost full conversion was achieved with the glucose-derived material with a 97% selectivity toward the ester. The second best performance was recorded for the material prepared from rice straw with 92% of both conversion and selectivity. With carbonised and sulfonated cellulose, 89% conversion and selectivity were observed. It is worth noting that it has been reported that sulfonation at high temperature (150 °C) induces changes in the structure of hydrothermal carbon. This is due to a decrease in the abundance of furanic groups and an increase in the presence of benzenic rings.255 Alternative methods of sulfonation have been shown such as the direct HTC in the presence of sulfonic precursors (mainly hydroxyethylsulfonic acid).256 The materials prepared using this method presented very high stability and reusability, enabling future applications.
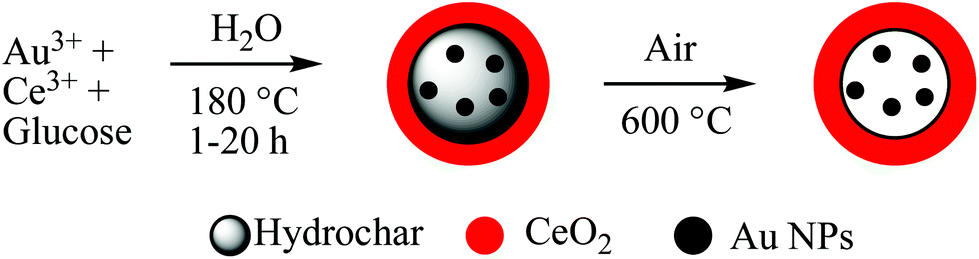 | ||
| Fig. 29 Schematic representation of the synthesis of core–shell distribution of gold particles and cerium oxide. | ||
TEM images (of the calcined samples) showed gold nanoparticles of an average size of 11 nm after 1 h of HTC and the whole diameter of the core–shell structure was about 40 nm. When the HTC was prolonged to 10 h, the gold nanoparticles grew to 16 nm and the shells to 103 nm. The best catalytic performance in carbon monoxide oxidation was shown by the material that was subjected to HTC for 10 h and was calcined subsequently at 600 °C. This catalyst allowed the reduction of the reaction temperature from 300 to 155 °C for full conversion and it was tested on stream for 70 h without any deactivation being evident. In a similar way, cobalt nanoparticles protected within hollow mesoporous silica spheres were synthesized271 starting from hydrothermal carbon spheres with an approximate diameter of 100–150 nm synthesised from glucose carbonised at 180 °C for 4.5 h. After impregnation with cobalt nitrate providing nanoparticles of approx. 4 nm and the synthesis of a mesoporous silica shell (thickness: approx. 20 nm) with CTAB as a soft template, all the organic material was removed by calcination at 430 °C. This synthesis procedure provided a catalytic material with interesting performance in the epoxidation of alkenes. When the cobalt/silica hollow spheres were employed in the epoxidation of styrene with oxygen, a 94% selectivity toward the epoxide was achieved at almost complete conversion (Table 5).
| Sample name | Carbon precursor | Carbonisation conditions | Role of carbon material | Reaction tested | Reaction conditions | Product obtained | Conversion (%) | Selectivity (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| HTC_673_SO3H | Glucose | 673 K, N2 atmosphere, H2SO4, 473 K, Ar atmosphere | Heterogeneous catalyst | Esterification of palmitic acid with methanol | Reflux, 85 °C, 4 h | Methyl palmitate | 90 | — | 249 |
| SHTC | Glucose | HTC 195 °C, 19 h, H2SO4, 150 °C, 15 h | Heterogeneous catalyst | Esterification of glycerol with acetic acid | 115 °C, 10 h, 9:1 AcOH/gly |
Monoacetin | 98 | 5 | 252 |
| Diacetin | 38 | ||||||||
| Triacetin | 57 | ||||||||
| SHTC | Glucose | HTC 195 °C, 19 h, H2SO4, 150 °C, 15 h | Heterogeneous catalyst | Esterification of glycerol with butyric acid | 115 °C, 10 h, 9:1 Butyric OH/gly |
Monoacetin | 99 | 1 | 252 |
| Diacetin | 24 | ||||||||
| Triacetin | 75 | ||||||||
| SHTC | Glucose | HTC 195 °C, 19 h, H2SO4, 150 °C, 15 h | Heterogeneous catalyst | Esterification of glycerol with caprylic acid | 115 °C, 10 h, 9:1 Caprylic OH/gly |
Monoacetin | 98 | 2 | 252 |
| Diacetin | 42 | ||||||||
| Triacetin | 56 | ||||||||
| HTC_Glu_S | Glucose | HTC 240 °C, 24 h, H2SO4, 80 °C, 4 h | Heterogeneous catalyst | Esterification of levulinic acid with ethanol | 60 °C, 3 h | Ethyl levulinate | 97 | 97 | 254 |
| HTC_Cell_S | Cellulose | HTC 240 °C, 24 h, H2SO4, 80 °C, 4 h | Heterogeneous catalyst | Esterification of levulinic acid with ethanol | 60 °C, 3 h | Ethyl levulinate | 89 | 88.9 | 254 |
| HTC_RS_S | Rye straw | HTC 240 °C, 24 h, H2SO4, 80 °C, 4 h | Heterogeneous catalyst | Esterification of levulinic acid with ethanol | 60 °C, 3 h | Ethyl levulinate | 92.2 | 92.2 | 254 |
| Carbon microspheres | Glucose | HTC 180 °C, 10 h | Heterogeneous catalyst | Dehydration of fructose | 100 °C, 90 min, [BMIM][Cl] ionic liquid, | 5-HMF | 89 | 100 | 257 |
| CNS_3OH | Glucose | 160 °C, 12 h, 3 M NaOH solution, RT, 1h | Heterogeneous catalyst | Aldol condensation of benzaldehyde and acetaldehyde | 40 °C, 5h | Cis- and trans-cinnamaldehyde | 33.9 | 94 | 258 |
| Cis- and trans-5-phenyl-2,4-pentadienal | 6 | ||||||||
| Cis- and trans-7-phenyl-2,4, 6-heptatrienal | 0 | ||||||||
| Pd@UCS | Sucrose | 180 °C, 8 h, kayexalate, activated in air, 800 °C, 1h | Catalyst support for Pd NPs | Suzuki coupling reaction | 50 °C, 1 h | Coupling product of bromobenzene and phenylboronic acid | 99 | — | 259 |
| Au-5/C | Glucose | 180 °C, 24 h, modified polymer template, carbonised in 5% H2 with a heat ramp to 700 °C for 5 h | Catalyst support for Au NPs | Hydrogenation of 4-nitrophenol | RT, 180s | 4-Aminophenol | 100 | — | 266 |
| Fe3O4@C-Pd@mSiO2 | Glucose | 180 °C, 4 h | Layer over Fe3O4 and support for Pd NPs | Suzuki–Miyaura crosscoupling reaction | 70 °C, 6 h | Coupling product of phenylboronic acid and p-iodoacetophenone | 99 | — | 267 |
| 20 h-600 °C-Au@CeO2 | Glucose | 180 °C, 20 h, HAuCl4, CeCl3 and CO(NH2)2, calcined at 600 °C, 6 h | Sacrificial component | Low temperature CO oxidation | From RT to 300 °C at 1 °C min−1 | — | 100 | — | 270 |
| Co/HSM | Glucose | 180 °C, 4.5 h, calcined at 400 °C in air for 7 h | Sacrificial component | Epoxidation of styrene with molecular oxygen | 100 °C, 3 h, bubbling with O2 at atmospheric pressure in DMF | Styrene oxide | 98 | 94 | 271 |
| Co/HSM | Glucose | 180 °C, 4.5 h, calcined at 400 °C in air for 7 h | Sacrificial component | Epoxidation of dec-1-en with molecular oxygen | 110 °C, 3 h, bubbling with O2 at atmospheric pressure in DMF | Dec-1-en oxide | 93 | 93 | 271 |
4.4. Gas storage
The porous carbon materials, obtained via HTC combined with templating/activation techniques, have been intensively used for gas capture and separation. Different gases such as CO2,83,98,273–275 H2,135 SO2,82 H2S and CH4,276,277 have a good affinity for the carbon structure, which make the ACs an obvious choice, over the classical gas separation methodologies.278| Sample name | Carbon precursor | Carbonisation conditions | Activation method | S BET (m2 g−1) | V total (cm3 g−1) | V micro (cm3 g−1) | V narrow micro (cm3 g−1) | CO2 uptake (mmol g−1) | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 °C, 1 bar | 25 °C, 1 bar | |||||||||
| a Calculated from the t-plot method. b Pore volume of narrow micropores (<1 nm) obtained from CO2 adsorption in STP. c Micropore volume determined by the DR method. d Micropore volume calculated from the pores <2 nm based on the NLDFT method. e Ultramicropore volume calculated for the pores <0.7 nm based on the NLDFT method. | ||||||||||
| GN-600-1 | Glucose and urea | 600 °C, N2 atmosphere | KOH | 821 | 0.42 | 0.29a | 0.46 | 5.33 | 3.99 | 279 |
| GN-650-1 | Glucose and urea | 650 °C, N2 atmosphere | KOH | 1734 | 0.78 | 0.62a | 0.77 | 6.70 | 4.26 | 279 |
| GN-700-1 | Glucose and urea | 700 °C, N2 atmosphere | KOH | 2394 | 1.13 | 0.93a | 0.87 | 6.46 | 3.92 | 279 |
| C-K-500 | Pineapple waste | 500 °C, N2 atmosphere | K2C2O4 | 422 | 0.04 | — | 0.82 | 2.71 | 2.22 | 126 |
| C-K-600 | Pineapple waste | 600 °C, N2 atmosphere | K2C2O4 | 644 | 0.05 | — | 0.91 | 3.82 | 3.16 | 126 |
| C-K-700 | Pineapple waste | 700 °C, N2 atmosphere | K2C2O4 | 1076 | 0.08 | — | 0.92 | 5.32 | 4.25 | 126 |
| AA-0 | Potato starch | 800 °C, N2 atmosphere | KOH | 3000 | 1.41 | 1.09c | — | — | 2.80 | 135 |
| AA-3M | Potato starch | 800 °C, N2 atmosphere | KOH and melamine | 3220 | 2.37 | 1.07c | — | — | 2.50 | 135 |
| AC-0 | Cellulose | 800 °C, N2 atmosphere | KOH | 3100 | 1.46 | 1.05c | — | — | 2.80 | 135 |
| AC-2M | Cellulose | 800 °C, N2 atmosphere | KOH and melamine | 3540 | 2.22 | 1.28c | — | — | 2.30 | 135 |
| AS-2M | Sawdust | 800 °C, N2 atmosphere | KOH and melamine | 3420 | 2.30 | 1.16c | — | — | 2.20 | 135 |
| CMC-3 | Coca Cola waste | High T°C, inert atmosphere | KOH | 1405 | 0.80 | 0.05d | 0.45e | 6.27 | 5.20 | 98 |
| CMC-2 | Coca Cola waste | High T°C, inert atmosphere | ZnCl2 | 1994 | 0.87 | 0.51d | 0.26e | 4.84 | 3.00 | 98 |
| HPNC-3 | Glucosamine | 600 °C, N2 atmosphere | Pluronic P123 templated | 710 | 0.54 | 0.37d | 0.32e | 4.20 | — | 96 |
| HPNC-4 | Glucosamine | 600 °C, N2 atmosphere | Pluronic P123 templated | 980 | 0.78 | 0.46d | 0.40e | 4.70 | — | 96 |
4.5. Water treatment
Water contamination with organic pollutants or heavy metals attracted significant attention because of their high toxicity and non-degradability.281 Among their applications, porous carbon materials derived from sustainable resources represent a green and effective method to adsorb water pollutants. Several studies reported their ability to take up both organic85,168,282–284 and inorganic molecules.285–2904.6 Bioenergy source–bioenergy potential in hydrothermal carbon
The solid (hydrothermal carbon) and liquid products resulting from the carbonisation of biomass have significant bioenergy potential, which has resulted in numerous studies investigating the use of hydrothermal carbon and/or the process water as fuel sources.72,293,294 Hydrothermal carbons generated from biomass have been reported to have high energy densities (up to 35 MJ kg−1 dry hydrothermal carbon) and structures similar to coal, making them candidates for replacing and/or supplementing fossil fuel-derived energy.37,295–297 Several studies have reported that hydrothermal carbonisation improves the fuel characteristics (e.g., aromaticity) of biowaste, making them more comparable to lignite and bituminous coals.298 Hydrothermal carbon energy content data collected from the literature are summarized in Fig. 30. The results indicate that, on average, hydrothermal carbons generated from the carbonisation of food waste and animal feeds result in the largest energy contents, while the hydrothermal carbon generated from the carbonisation of sludge results in the lowest energy content. Several studies reported that the hydrothermal carbon energy content increases with increasing reaction severity.18,71,76,299–301 It is possible that the high average energy content data associated with hydrothermal carbon generated from animal feed is skewed by process conditions, since the conditions associated with the carbonisation of this feedstock resulted in the highest average severity factor. However, no defined trends associated with the severity factor appear to be present in these data. | ||
| Fig. 30 Distributions associated with the hydrothermal carbon energy content of different feedstocks based on literature-collected data. The line in each box represents the median value. The ends of each box represent the 25th and 75th percentiles associated with the data. The red diamonds represent the average values. The lines and data points represent the scatter of data beyond the 10th and 90th percentiles. The numbers in parentheses following each feedstock category represent the number of data points represented on the plot. | ||
It is important to note that both the hydrothermal carbon energy content and hydrothermal carbon yield (e.g., hydrothermal carbon mass) are important when determining the total energy that can be recovered from hydrothermal carbon. Results from previously conducted studies detailed in the literature indicate that the hydrothermal carbon energy content generally increases with increasing reaction temperature and, in a way, reaction time.17,61,69,71,302–305 Conversely, an increase in temperature and to some degree time generally result in a reduction in hydrothermal carbon generation.8,68,69,71,76,306,307 Therefore, understanding the combined effect of these hydrothermal carbon properties is needed when determining the value of using hydrothermal carbon as a bioenergy source. Combining statistical models for predicting hydrothermal carbon yields and energy contents based on literature-collected data,308 the total energy that may be recovered from different organic waste materials (at a constant reaction temperature, time, and initial solid concentration) can be calculated. Results from this analysis are shown in Fig. 31. In this plot, the feedstocks are listed in order of the lowest carbon content to the greatest carbon content, indicating that there is a relationship between the feedstock carbon content and the total energy that is available for use in the generated hydrothermal carbon. Blending of hydrothermal carbon with coal has been reported to improve the combustion efficiency of low rank coals for energy generation.309–312 Gao et al.313 reported that the presence of a secondary char deposited on the hydrothermal carbon surface is responsible for the high oxidative reactivity of the hydrothermal carbon. The generation of this secondary char (tar-like deposits on the hydrothermal carbon surface) has been reported to increase with the reaction severity (e.g., increase in reaction time and temperature). Gao et al.313 indicate that blending of hydrothermal carbons up to 10% with bituminous coal is possible and does not lead to fuel segregation (e.g., uneven burning) during combustion.
 | ||
| Fig. 31 Calculated total energy recoverable from different organic waste materials. Note that the waste materials are listed in order of their carbon content, ranging from the lowest value (left) to the greatest value (right). Each value represents the average value obtained from different model combinations. The error bars represent the standard deviations associated with these data. | ||
4.7. Catalytic conversions of hydrothermal carbon
Hydrothermal carbons are inevitable byproducts from acid-catalyzed conversions of C5 and C6 sugars to important biobased chemicals such as furfural, 5-hydroxymethylfurfural and levulinic acid. Valorisation of these highly condensed polymeric hydrothermal carbons48,314,315 by catalytic conversions is of high interest but has received limited attention to date. This chapter gives an overview of the research carried out on the catalytic conversion of hydrothermal carbons to liquid products to be used as biofuels or, after fractionation, as a source of valuable biobased chemicals such as alkylphenolics and aromatics. The emphasis will be on four technologies, viz. (i) catalytic hydrotreatments with molecular hydrogen or hydrogen donors such as isoporopanol or formic acid, (ii) catalytic pyrolysis, (iii) aqueous phase reforming and (iv) gasification. | ||
| Fig. 32 Overview of reaction pathways occurring during the catalytic hydrotreatment of hydrothermal carbon in IPA; reproduced with permission from ref. 324. | ||
Further systematic studies to obtain high amounts of alkylphenolics were performed using a Pt/C catalyst in IPA.321 The highest hydrothermal carbon conversion achieved in this study was 72%. The main hydrothermal carbon derived products were aromatics, alkylphenolics and aliphatic hydrocarbons, which was confirmed by performing blank reactions with IPA and Pt/C in the absence of hydrothermal carbon. The highest amount of alkylphenolics was 14% based on GC detectable products in the liquid phase after the reaction. The catalytic hydrotreatment of hydrothermal carbon in the presence of mixtures of FA and IPA using supported Ru catalysts has also been investigated.324 Best results were obtained using Ru/C and hydrothermal carbon conversions up to 69% were achieved using a combination of FA/IPA as the hydrogen donor. Elemental analysis showed that the oils have a considerably reduced oxygen content compared to the hydrothermal carbon feed, with HHVs up to 38 MJ kg−1. The product oils were shown to consist of both monomeric and oligomeric compounds (GPC). Main GC detectable species arising from the hydrochar were substituted alkylphenolics, naphthalenes and cyclic alkanes (GC-MS-FID, GC × GC). Recently Saha and co-workers325 reported the catalytic hydrotreatment of hydrothermal carbons in methanol with additional molecular H2 as the hydrogen source using various noble metals supported on carbon. Experiments were performed in a batch autoclave at 350 °C under 30 bar H2 with a 3 h batch time. The highest yield of the product oil was 43 wt% at 55% conversion of the hydrothermal carbon using Pt/C. All other catalysts were less effective (Ru, Pd and Rh). However, the amounts of GC detectable in the oil (10%) were relatively low. Reaction parameters, including the reaction time, temperature, H2 pressure and catalyst-to-hydrothermal carbon mass ratio were optimized. Higher reaction temperatures were shown to have a positive effect on the hydrothermal carbon conversion. However, the product oil yield does not progressively increase with temperature and shows a maximum at about 400 °C due to excessive gasification at high temperatures. Isotopic-labelling experiments were conducted using 13C-labeled methanol to investigate whether methanol only serves as a hydrogen source or also reacts with the intermediates or products of the liquefaction reaction. The results imply that methanol is incorporated as an –OCH3 group in the products, formed by for instance esterification reactions of hydrothermal carbon-derived fragments and as methyl groups on aromatic units, likely by alkylation reactions.
5. Soluble carbonisation products
The complexity of transformations and reaction pathways occurring during hydrothermal carbonisation of lignocellulosic biomass is reflected in the large number of soluble products found in the aqueous phase. However, all chemicals can be grouped into four classes: aldehydes/ketones (including saccharides), furan derivatives, carboxylic acids and phenols.76,303,330–334 First three classes of compounds can always be found in a relatively high amount or in traces after hydrothermal carbonisation of mono- and polysaccharides, whereas phenols are only found in real biomass HTC, as they arise from hydrolysis of lignin. Xylose, furfural and furfuryl alcohol are derived from the hemicellulosic fraction of biomass.331–334Table 7 shows an extended list of the main carbonisation products grouped in the four above-mentioned classes.| Aldehydes, ketones and monosaccharides | Furan derivatives | Carboxylic acids | Phenols |
|---|---|---|---|
| Glucose | Furfural | Pyruvic acid | Guaiacol |
| Fructose | 5-HMF | Glycolic acid | Catechol |
| Xylose | Furfuryl alcohol | Acetic acid | Cresol |
| Erythrose | 2-Methylbenzofuran | Lactic acid | |
| Dihydroxyacetone | Soluble polymers | Formic acid | |
| Levoglucosan | Levulinic acid | ||
| Pyruvaldehyde | Propionic acid | ||
| Glyceraldehyde | |||
| Formaldehyde | |||
| Acetaldehyde | |||
| 2,5-Dioxo-6-hydroxyhexanal |
5-Hydroxy-methylfurfural (5-HMF) is probably the most renowned and sought-after of all hydrothermal carbonisation products. It is derived from dehydration of C6 monosaccharides. The huge interest around this compound arises from the reactivity of its two functional groups, methoxy- and aldehydic, and the possibilities of derivatisation that they offer. In fact, 5-HMF has been recognized as a valuable bio-based chemical building block16 which can play a key role not only as an intermediate for the production of the biofuel dimethylfuran (DMF), but also for other biomass-derived intermediates, such as 2,5-furan-dicarboxylic acid,339 2,5-dimethylfuran,340,341 adipic acid and levulinic acid (LA).342 As a consequence, a few processes for industrial scale synthesis of 5-HMF from monosaccharides have already been patented. AVA Biochem, a Swiss based company, has been involved in the industrial scale production of 5-HMF derived from biomass since 2013, with an annual production of 20 tonnes. AVA Biochem's approach is based on carbonisation of fructose in water at high pressure. Fructose is in turn obtained from previous treatment of biomass waste. The traditional disadvantage of the production of a remarkable amount of hydrothermal carbon as a byproduct of condensation and polymerization of 5-HMF is avoided by extracting 5-HMF from the system before the condensation and polymerization steps take place. The final product is either a 99.9% pure crystalline solid or is available in solution for further processing. Moreover, as the conversion of fructose in a single step is not complete, the process has been designed to incorporate a recycling step of the unreacted sugar, to enhance overall conversion. These series of improvements have allowed the process to be scaled to the industrial level.343
Levulinic acid (LA) is considered as one of the most promising platform chemicals produced from lignocellulosic biomass for fuels and chemicals.344,345 It is regarded as a speciality chemical that finds applications for several purposes, such as a source of polymer resins, animal feed and food as well as components of the flavouring and fragrance industry, textile dyes, additives, extenders for fuels, antimicrobial agents, herbicides and plasticizers.346,347 The biorefinery process employs cellulosic biomass for the synthesis of levulinic acid and formic acid. The conversion takes place in two steps: the ground biomass is injected into the first reactor, where it is then mixed with dilute sulfuric acid at a temperature of 210–220 °C and a pressure of 25 bar. A residence time of 12 seconds is sufficient for the hydrolysis and dehydration reaction to occur, leading to the synthesis of a 5-HMF intermediate. Subsequently, the mixture is moved to a second, larger reactor. Here the acidic catalyst concentration is the same, while temperature and pressure are milder. A longer residence time of 20 minutes is necessary to complete the reaction of rehydration of 5-HMF to levulinic acid and formic acid. Furfural and other by-products are removed at this stage. Water is subsequently boiled off, along with the remaining volatiles, and levulinic acid is extracted from the resulting mixture by distillation. In this process, approximately 50% of the initial mass of 6-carbon sugars is converted to LA, 20% to formic acid and 30% to tar.348
Furfural is largely produced by dehydration of pentose sugars rich in hemicellulose. In fact, currently, commercial production of furfural exclusively relies on dehydration of lignocellulosic biomass. The typical process involves the breaking down of the hemicellulose fraction of a lignocellulosic biomass in a 3% sulfuric acid solution for 3 hours, in a range of temperatures between 170 and 185 °C. Under these conditions, 40–50% of the potential furfural is obtained. The undissolved residual solid, composed of lignin and cellulose, is recovered and employed for energy recovery, while the acid catalyst is recycled.349 Several mechanisms have been speculated to explain pentose dehydration to furfural including both acyclic and cyclic intermediates. However, it is reasonable to think that the most plausible mechanism under acidic conditions involves acyclic xylose isomerization to xylulose followed by further dehydration and cyclization to furfural.43 The interest in bio-derived furfural arises from the many possibilities that this molecule offers in terms of catalytic upgrading to fuel additives.350
The abovementioned compounds have received a great deal of attention because of their primary role in the transition to sustainable, bio-based fuels and materials. However, among the numerous other chemical compounds, some organic acids such as lactic acid, formic acid and acetic acid are worth mentioning. Despite their limited strategic relevance, they still have commercial value due to their abundance as by-products in the hydrothermal conversion of biomass and levulinic acid synthesis. Lactic acid (LacA) is a promising platform chemical for the synthesis of solvents (ethyl lactate), biodegradable plastics (polylactic acid, PLA) and other polymer precursors (acrylic acid).16,351 Formic acid can serve as a starting compound for the synthesis of green solvents,348 whereas acetic acid could replace inorganic acids for the synthesis of roadway deicer salts.352
5.1. Element distribution in the liquid and gas-phases
Significantly less composition data have been reported for the gas and liquid carbonisation products. Fig. 33 provides a summary of the fraction of initially present carbon transferred to the gas and liquid-phases following carbonisation. As shown, a small fraction of initially present carbon is transferred to the gas-phase, which is consistent with that reported in the literature. Generally, as the reaction severity increases, the fraction of carbon in the gas-phase increases.71,76,303,353 The carbonisation of paper results in the greatest transfer of carbon to the gas, which is not surprising because the conditions associated with the carbonisation of paper also represent the largest average severity factor. Conversely, a significant fraction of carbon is transferred to the liquid-phase as a result of carbonisation, which may have significant implications associated with using the liquid-phase as an energy source via anaerobic digestion.354,355 The distribution of data describing this fraction of carbon is quite large, likely because of the significant influence process conditions have on liquid-phase carbon contents.68,71,72,76,353,356 Unlike that associated with other carbonisation products, there is a trend associated with the severity factor, which is consistent with that reported in the literature. At low average severity factors, large fractions of carbon are transferred to the liquid-phase. As average severity factors increase, the transfer of carbon to the liquid-phase decreases. These trends are consistent with those reported in the literature.71,76 | ||
| Fig. 33 Distributions associated with the percentage of initially present carbon in the (a) gas and (b) liquid-phases following the carbonisation of different feedstocks. The line in each box represents the median value. The ends of each box represent the 25th and 75th percentiles associated with the data. The red diamonds represent the average values. The lines and data points represent the scatter of data beyond the 10th and 90th percentiles. The numbers in parentheses following each feedstock category represent the number of data points represented on the plot. | ||
5.2. Influence of feedwater and catalysts on soluble carbonisation products
HTC of biomass is usually performed under acidic conditions, because hydrolysis and dehydration reactions are catalyzed by hydronium ions. As mentioned before, 5-HMf is the pivotal compound in the whole process of transformation, as it is the starting point for both conversions to dissolved products (levulinic acid) and solid products (hydrothermal carbon). Therefore, any consideration of the influence of reaction parameters on the final products of hydrothermal carbonisation must take into account the reactivity of 5-HMF. The rate-limiting step in the dehydration of glucose to 5-HMF in acidic medium is the isomerization of glucose to fructose.44,357 This explains the reluctance of glucose, compared to fructose, in dehydrating to 5-HMF: in fact, isomerization of glucose to fructose is base catalyzed and therefore it is slower under typical acidic conditions for the synthesis of 5-HMF.44The nature of the acid used to catalyse the dehydration of saccharides plays a major role in the final yield and distribution of products. Lu et al. noted faster kinetics in HCl catalysed hydrolysis of cellulose compared to that of H2SO4 in the same concentration.356 Reiche et al. described a different behaviour in HTC of 20 wt% glucose solutions treated with HCl or HNO3 with the same synthesis pH. Nitric acid, in fact, due to the oxidizing properties of NO3−, drives the conversion towards higher yields of hydrothermal carbon in spite of the formation of 5-HMF, levulinic acid and formic acid. Hydrochloric acid, conversely, catalyses more efficient conversion of glucose to HMF and levulinic acid subsequently, with much lower HTC carbon yields. It is also worth pointing out a strong imbalance between levulinic acid and formic acid, in favour of the former, for both HCl and HNO3 acidified samples, despite a theoretical ratio of 1:1. This effect is particularly strong at a synthesis pH of 0. Under these conditions, the strong acidity makes formic acid and other carboxylic acids break down into gaseous species (CO and CO2), also causing a slight rise in the final pH.358
Hydrothermal treatment of poly- and monosaccharides under basic conditions leads to a quite significant change in the scenario. In fact, although bases such as NaOH, KOH and Ca(OH)2 are all able to accelerate the hydrolysis of cellulose to glucose, they somehow change the pathway of carbonisation, slowing down its dehydration to 5-HMF, with a consequent buildup of the glucose concentration and lower yields of levulinic acid.338,356 On the other hand, a rise in formic acid and lactic acid concentrations is observed with higher synthesis pH,338 pointing out the preferred pathway of degradation of the hexoses, involving retro aldol condensation, as shown in Fig. 6.
Salt anions too can affect the conversion of sugars under hydrothermal conditions. In fact, anions with good leaving group qualities can strongly accelerate the rate of dehydration of fructose to 5-HMF, provided that they are also small and good nucleophiles. This is due to their intervention in the first step of dehydration of fructose, which involves substitution and elimination reactions on the C2 carbon.359 Lewis acids are also of interest in the homogeneous catalysis of dehydration reaction as substitutes of traditional Brønsted acids, due to their lower corrosivity. Various Lewis acids have been studied for carbohydrate conversion. Dehydration of glucose has been achieved using CrCl2 in an ionic liquid with 70% yield of HMF;360 CrCl3 and HCl with 59% yield of HMF in water (NaCl)/THF biphasic medium;361 and SnCl4 in an ionic liquid to afford a 64% yield of HMF.362 More recently, Jiang et al.363 have achieved a yield of levulinic acid of 88% in biphasic medium (water:MeTHF) at 200 °C in 60 min (pH 1) with FeCl3 and a yield of 56% for 5-HMF in a biphasic solution at higher pH (pH = 2) in 180 min with FeSO4. Weiqi et al.364 have studied the synergistic effect of coupling CrCl3 and H3PO4 on glucose conversion to LA compared to a single use of CrCl3 or H3PO4. The highest LA yield of 54.24% was obtained from 100% glucose conversion at 170 °C for 240 min. CO2 in hydrothermal applications can be considered as both a Lewis acid in gaseous form or Brønsted acid in its hydrated form. In any case, it has been proved to be effective at catalysing carbohydrates to 5-HMF. Lin et al.365 have obtained a 5-HMF yield of 60.33% from fructose conversion at 190 °C for 20 min with a CO2 pressure of 2 MPa. There is proof that compressed CO2 is also effective at hindering the formation of undesired 5-HMF oligomers, thus improving the conversion yield of sugar to 5-HMF.366 Using heterogeneous catalysis has the significant advantage of avoiding tedious recovery steps that are necessary for homogeneous catalysts. The most recent heterogeneous catalysts proposed include transition metal oxides,367 phosphates,368,369 zeolites,370 organic polymers,371 and carbon sulfates.372–374
5.3. Influence of reaction time and temperature on soluble carbonisation products
Several studies have investigated the effect of reaction time and temperature on product yields in the liquid phase after hydrothermal carbonisation of carbohydrates and lignocellulosic feedstock. Table 8 provides a summary of the reaction conditions used in the papers taken into account. Seen from the perspective of reaction time, in the early stages of polysaccharide conversion, the HTC liquid phase is characterised by a relatively high concentration of sugars (glucose) and aromatics (5-HMF). As a general trend, it appears that hydrolysis of polysaccharides to simple sugars consistently increases with temperature, with the maximum yield shifting to shorter reaction time as temperature increases.303,330 The highest sugar yields are generally found between 180 °C and 230 °C and between 1 hour and 2 hours.331,333,334 Fructose yields are generally lower than glucose yields330,331 and this may serve as proof that the reaction of fructose dehydration to 5-HMF is kinetically faster than the isomerization of glucose to fructose. Therefore, in the early stages of hydrolysis of cellulose, the concentration of glucose builds up, whereas fructose is readily consumed. 5-HMF yield is very dependent on both the reaction time and temperature and its production is strictly connected to the sugar content in the liquid phase. Although the time scale of the 5-HMF concentration peak may vary from a few minutes to hours, depending on the type and size of the reactor,330,331,333,334 it always follows the maximum in monosaccharide concentration. 5-HMF prefers moderate temperature for optimal yields (180–220 °C),331,333,334 while higher temperatures cause faster conversion but lower yields, due to the increase of secondary reactions. Experiments on the conversion of simple sugar monomers (glucose and fructose) and dimers (sucrose), although being conducted in the presence of an acid catalyst and therefore being kinetically faster, lead to a similar conclusion.357 Xylose and furfural are only minor products in the hydrothermal carbonisation of hexoses.357 In contrast, furfural becomes much more relevant in hemicellulose-rich biomass and its decline throughout the process is slower compared to that of 5-HMF, which indicates a slightly higher stability.331 Finally, organic acids tend to prevail as the reaction time proceeds and higher temperatures are used, with acetic acid being particularly high in concentration in HTC of lignin-rich biomass.303,331–334| Carbon precursor | Biomass/water ratio (wt%) | Reaction temperature (°C) | Reaction time | Ref. |
|---|---|---|---|---|
| Starch | 10% | 180, 200, 220, 240 | 2 to 40 min | 330 |
| Biomass (Jeffrey pine and white fir) | 12.5% | 215, 235, 255 275, 295 | 30 min | 303 |
| 255 | 5 to 60 min | |||
| Cellulose | 20% | 225, 250, 275 | 0 to 96 h | 76 |
| Cellulose, wheat straw, poplar | 12.5% | 200, 230, 260 (fixed) | 0 to 8 h | 331 |
| 160–260 (dynamic) | — | |||
| Coconut husk, rice husk | 10% | 140, 160, 180, 200 | 4 h | 332 |
| 200 | 1, 2, 3 h | |||
| Cassava rhizome | 20%, 10%, 6.6% | 160, 180, 200 | 1, 2, 3 h | 333 |
| Corncomb | 20%, 10%, 6.6% | 160, 180, 200 | 1, 2, 3 h | 334 |
| Sucrose, glucose, fructose | 2% (sucrose) 1.05% (glu., fru.) | 180 | 2 to 24 min | 357 |
| 200, 220 | 2 to 18 min |
5.4. Chemical isolation
HTC typically occurs in water or aqueous medium, where the chemical products of interest are found at the end of the reaction. Typical compounds found in the water phase are carboxylic acids, aldehydes, ketones and furfural derivatives. Although the yields of conversion may vary, the concentrations of these products are quite diluted, making separation by direct distillation unfavourable. Therefore, every process designed to convert cellulosic biomass into platform chemicals must deal with a liquid–liquid extraction step. A good solvent for liquid–liquid extraction must fulfil some basic requirements: it must extract effectively and selectively the compound of interest; it must be recoverable by distillation, in order to be recycled, thus lowering the production costs; it must be poorly soluble in water, in order to minimize losses during extraction; it must have a fairly different density from water, to allow quick separation. In addition to these fundamental features, from the perspective of a greener process, it should also be inexpensive and non-toxic.375 Several organic solvents have been evaluated for the liquid–liquid extraction of 5-HMF, levulinic acid and furfural. Methyl isobutyl ketone (MIBK),376,377 2-butanol,340,376 THF,378 and 2-MTHF378 are most frequently used for the extraction of HMF, although some other alternative solvents such as dimethylcarbonate,369 o-propylphenol379 or hexafluoroisopropanol380 have been proposed due to their remarkably high partitioning coefficients. MIBK in particular has been proven to be the best solvent for the extraction of 5-HMF in a counter current, due to the combined effect of its high partition coefficient and its lower solubility in water, compared to other solvents. Addition of sodium chloride further improves the partition coefficient.381 Exploiting the salting-out effect of some common salts is an effective method to maximize the liquid–liquid extraction. Salting-out efficiency has been demonstrated to be roughly independent of the nature of the cations; in fact, it only depends on the anion used. Moreover, the partitioning behaviour of the salt is roughly independent of the extraction solvent used.382 Pentasodium phytate, a green and cheap salting-out agent derived from cereals and currently considered waste, has also been considered as an alternative to inorganic salts. In a comparative study with other inorganic salts in a water/1-butanol/HMF system, the most pronounced separation of HMF than the other tested sulfates has been shown.383 Salts can also increase the separation efficiency when a system of two mixed solvents is used instead of one single pure solvent. However, subsequent recovery of the solvents can be reduced upon addition of salts due to the formation of complexes.384 When ionic liquids are used as reaction media for dehydration of sugars to HMF and levulinic acid, extraction with the aforementioned solvent leads to very poor yields. This has been explained with the strong H-bond interaction between the hydroxyl group of HMF and the anion of the IL.385 In fact, it has been observed that the addition of small chain alcohols, by interfering with the aforementioned H-bond, results in a significant improvement of the extraction efficiency of HMF by MIBK.386 Compressed CO2 as the extraction solvent of HMF from ILs has also been considered, proving to be a promising approach due to the combination of both the extraction efficiency and enhanced solubility of glucose in the IL medium.387 Brouwer et al.388 have recently proposed a method to separate furfural, levulinic and formic acid in different stages from the same acidic aqueous stream. Furfural is extracted in the first stage with toluene. Subsequently, levulinic acid and formic acid are extracted with a mixture of 30% TOPO (trioctylphosphine oxide) in MIBK, with a high partition coefficient and selectivity over sulfuric acid. A double temperature swing back extraction has been performed to concentrate LA and FA, therefore lowering the energy required for the final distillation step to obtain levulinic acid.6. Carbon dots
Apart from the formation of new chemical compounds in the liquid phase, there is an interface grown between the solid carbon microsphere and liquid chemicals, which is considered as a third product. This is where the nucleation process takes place as a new phase is formed, leading to thermodynamic instability. The small nuclei formed at this interface represent the fluorescent carbon dots (CDs) with a size generally below 10 nm, which were previously reported via the HTC process.389 Because of some unique properties, such as low toxicity, high photoluminescence (PL) quantum yield, facile modification and excellent electron donating/accepting abilities, CDs have found applications in many fields, ranging from bioimaging to sensors, LEDs, photocatalysis, solar cells and more.24,390,391The electronic states and optical properties that make CDs suitable for the above-mentioned applications are strictly connected to their structural features, which can be controlled during synthesis. On that note, a detailed mechanism for the formation of CDs from molecular precursors is of vital importance to better understand the unique properties of these materials and also facilitate control of their key characteristics.
6.1. Formation mechanism
So far, two different mechanisms have been proposed in the literature. One suggests a growth according to the classical La Mer model,392 where the resulting nuclei grow uniformly and isotropically by diffusion of solutes towards the particle surface until the final size is attained.392 It is usually divided into several stages, which depending on the classification can be summarized as decomposition, condensation, polymerisation, nucleation (aromatization) and growth. Wang et al. reported a simple model of the different stages in 2012.393 Some people also infer the final step as surface passivation by the residue precursor.394,395 Liu and co-workers prepared nitrogen and sulfur co-doped CDs via hydrothermal treatment of L-cys and NH3·H2O.394 They have claimed that L-cys molecules went through a few steps from decomposition to passivation, as shown in Fig. 34A. The HRTEM images of CDs being impeccable gradually support the possible formation mechanism. In one of the earlier reports, Sahu et al. have proposed a simplified 3-step explanation for the formation mechanism of hydrothermally synthesized CDs.20 The authors have concluded that the dehydration and decomposition of fructose/glucose gave rise to different soluble chemical compounds, as mentioned before in the liquid phase. Hydronium ions formed from these acids acted as a catalyst in subsequent decomposition reaction stages. The polymerisation and condensation of these soluble chemicals gave rise to soluble polymers where aromatization took place via aldol condensation, cycloaddition and hydroxymethyl-mediated furan resin condensation.50 When the concentration of aromatic clusters reached a critical supersaturation point, burst nucleation took place, and carbon dots were grown. Similarly, the particle formation of CDs reported from banana juice396 and sweet potato397 also follows the three steps of carbonisation of carbohydrates. Later Qu et al. synthesized N-doped graphene quantum dots (GQDs) with a hydrothermal method with citric acid and urea.398 They concluded that the formation of N-doped GQDs involved two steps. In the first step, citric acid molecules self-assembled into sheet structures and dehydrolyzed to form a graphene framework, and the reaction between citric acid and amines occurred. Through the second step of intramolecular dehydrolysis, amides reacted with carboxylic groups and formed pyrrolic N. Similar methods have also been reported by Bai et al. for N-doped CDs produced from citric acid and ethylenediamine,407 and Atchudan et al. with Chionanthus retusus fruit extract.400 Our group has been pioneering in the field of HTC for the last decade and based on the structural investigation of CDs, we propose an evolutional mechanism, which involves the influence of the gas phase during the reaction.401,402 We believe that some of the polymeric nuclei formed initially during the hydrothermal process act as hot spots (due to temperature gradients existing in the autoclave during the HTC process). At the same time, CO/CO2 gas along with H2 are formed in situ in the autoclave from partial gasification of glucose under subcritical conditions. These gases deposit onto the hot spot nuclei in a CVD-like process via CO2 reduction under autogenic pressures (30 bars) leading to the formation of crystalline carbons. With the observation from the HRTEM images in Fig. 34B, we hypothesize that the first crystalline form of CDs is sp3-bonded carbon obtained from an amorphous/carbon black type which then can further convert into carbon anions which subsequently form graphitic structures.403 During the incipient stage of glucose transformation via the “hot spot CVD” mechanism mentioned above, a mixture of sp2/sp3 carbon nanostructures are initially formed which are later converted into predominately sp2 carbon nanostructures.403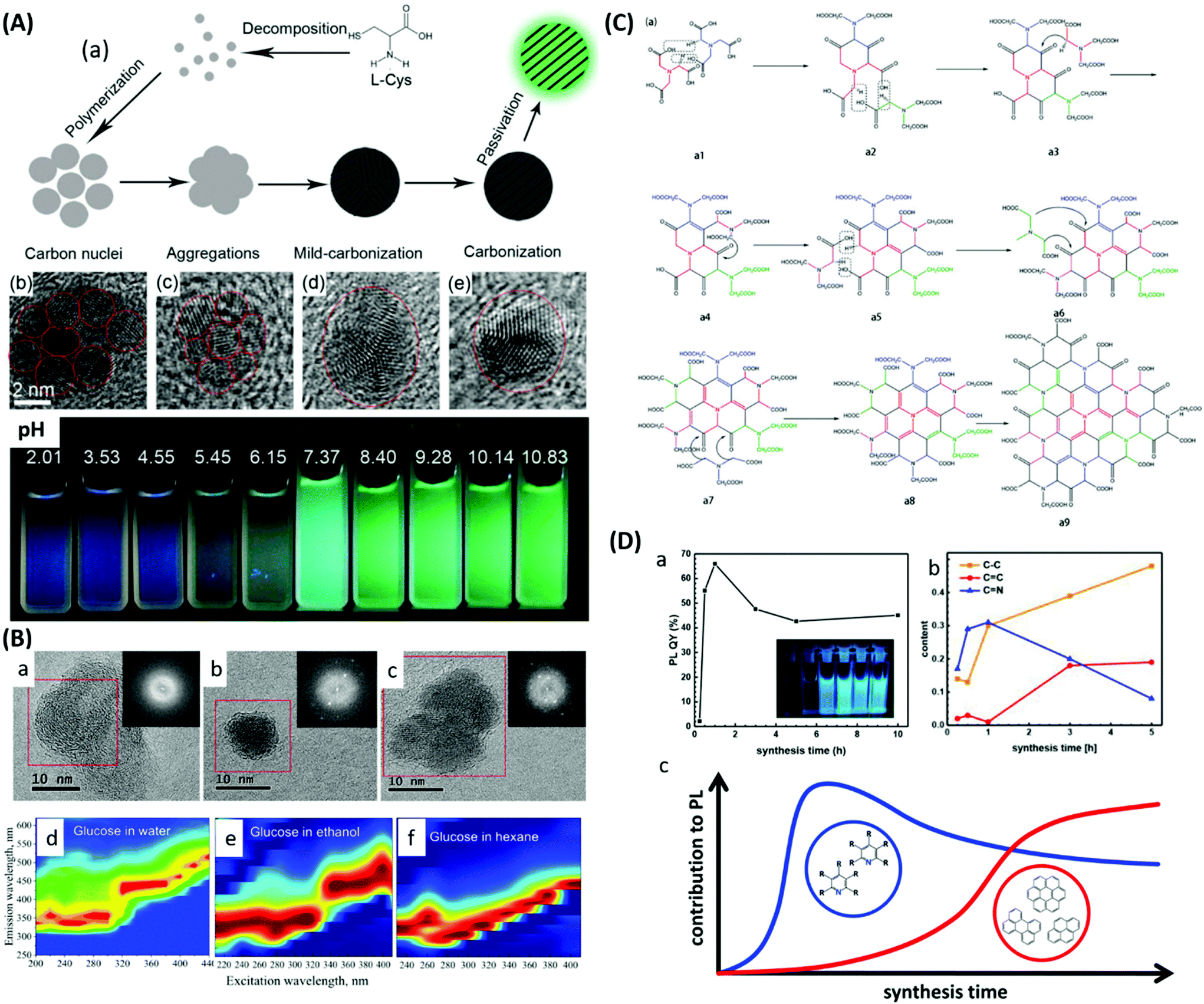 | ||
| Fig. 34 (A) Schematic illustration of the possible mechanism for the formation of N,S-CDs using L-cys as the carbon source, HR-TEM of CDs at different formation stages, and the PL behaviour under different pH values; reproduced with permission from ref. 394. (B) HR-TEM images showing the evolution of glucose derived CDs and the influence of solvent polarity on the excitation-dependent PL properties; reproduced with permission from ref. 401. (C) Mechanism of the reaction from NTA to N-doped GQDs; reproduced with permission from ref. 404. (D) (a) PL QY of CDs as a function of reaction time, (b) evolution of the relative amount of C–C, CC, and CN bonded C atoms inside the CDs during the synthesis, and (c) the scheme of different structures contributing to PL with respect to time; reproduced with permission from ref. 405. | ||
Jiang and co-workers modelled the polymerization process of nitrilotriacetic acid (NTA) to form N-doped GQDs under hydrothermal conditions (250 °C, 4 h), and concluded that the mechanism involved dehydration and nucleophilic addition between different NTA molecules to form a conjugated system, as shown in Fig. 34C, which grows into larger CQDs as the reaction continues.404 The other formation mechanism involves local nucleation on some high energy areas. Dai et al. synthesized hydrothermal CDs and proposed a formation mechanism based on the temperature increase.406 At first, formation of large-sized polymeric nanoparticles through intermolecular dehydration occurred upon high temperature and pressure exposure of the precursor. As the reaction proceeded, nucleation started as the aforementioned polymeric nanoparticles partitioned, resulting in the coexistence of polymers and CDs. Upon further increase in heating time, CDs constantly surpassed the polymer nanoparticles until the latter disappeared. Similar processes have also been reported by Yang et al. with amino acid with respect to the time increase.407 Unfortunately, in all the above cases, the formation mechanism appears as a hypothetical suggestion and there is no clear evidence via in situ characterisation to support it. Therefore, an in-depth insight involving in situ techniques is crucial for the survival of these systems initially and later to accomplish their commercialization.
6.2. Reaction parameters
At this stage, it is crucial to explore the parameters affecting the properties of the resulting CDs during their synthesis. It is well-known that surface groups in particular are highly dependent on the conditions of the synthesis, such as the solvent, temperature and pH. The solvents can affect the excitation dependent PL properties, as shown in the maps in Fig. 34B, and this is generally attributed to the presence or absence of oxygen-containing functional groups. Such groups are formed easily in solvents with stronger hydrophilicity and higher polarity (such as water and ethanol), enhancing blue emission. In contrast, low polarity solvents such as cyclohexane and toluene show a negligible amount of oxygen-related groups, decreasing blue emission, while increasing the red one.408,409 Temperature is another key factor in the preparation of CDs. Several physical, chemical and optical properties of the resulting products can be affected by the reaction temperature such as the particle size, crystallization and surface functionality and as a consequence the PL intensity and peak position.410,411 The different pH environment during HTC can largely affect the PL properties of CDs by affecting the reversible protonation and deprotonation of the abundant functional groups.412 Each functional group contributes to the overall emission in a different way in different pH ranges, therefore leading to intensity variation or even shifting the peak position. Compared to the other parameters, the reaction time can influence the CD formation stage, which changes the structural and optical properties accordingly. Bai and co-workers changed the reaction time of metronidazole derived CDs and found that the formation of CDs starts after 3 h. They monitored the surface functional group changes using FTIR techniques and learned that the surface groups of CDs changed gradually, with –NO2 reduced to N–H, while C–OH and C–H oxidized to C–O–C and COOH. The abundant surface groups enabled further carbonisation of the CDs, as well as granted good solubility in water. After 8 h reaction, the PL intensity became much stronger than that at 3 h, while the chemical groups changed a little.399 The effect of reaction time on the QY is also investigated, which showed a volcano-shaped trend with the highest QY obtained at a 6 h reaction time.413 Jiang and co-workers combined experimental and theoretical calculations on nitrogen-doped GQDs to study the formation mechanism. As the reaction proceeded, the size of the planar structure increased and a size change occurred from non-uniform to uniform. This phenomenon was explained by the stability of the different products: the less stable intermediate small fractions would decompose into the original precursor while the relatively stable N-doped graphene layers would grow at high temperature and pressure.404 A different formation behaviour of CDs has been reported by Feldmann and co-workers using citric acid and ethylenediamine as the precursors. They have found that the CDs grew rapidly during the first 30 min and the size remained constant for the rest of the synthesis. Instead of growing in size, they undergo a substantial transformation of the internal structure, with larger aromatic domains and a higher level of graphitization, as seen in Fig. 34D. The PL QY decreases accordingly, with a shorter PL lifetime, which implies an increase in the nonradiative recombination process.4056.3. Photo-related applications
By understanding the formation mechanism and controlling the reaction parameters to manipulate the optical properties of CDs, researchers have been pushing forward to utilize this material for practical applications. Due to their easy and cheap synthesis, low toxicity, high (aqueous) solubility, good optical properties with high fluorescence quantum yield, facile modification and stability against photobleaching, CDs made from HTC are superior to the traditional semiconductor quantum dots. Interesting facts about this material have been discovered from research over the past few years, which makes it a good candidate for various applications, such as bio-imaging, optronics, photocatalysis and photovoltaics. Our group has been working on applying CDs as sustainable alternatives for some hazardous materials in solar cells and photocatalysis. In an early report, biomass derived CDs have been used to sensitize ZnO nanorods to visible light, allowing their use in solid-state nanostructured solar cells.389 The performance of the devices depended on the functional groups on the CD surface, and it is likely that the functionalisation determines the binding of CDs to the ZnO surface and the nature of the charge transportation and recombination. Later we further investigated the influence of functionalisation on solar cell performance.415 Six types of CDs were produced from different precursors and used as sensitizers for TiO2-based solar cells. By investigating the structural and optical properties of different CDs we have shown that the combination of amine and carboxylic acid groups is more beneficial for enhancing the solar cell performance. In a more recent work, chitosan derived CDs with nitrogen doping were studied as potential photocatalysts.414 The CDs were covalently bonded on a conductive substrate and tested as a photoanode. As shown in Fig. 35, the CD electrode is capable of producing a photocurrent under visible light, owing to the visible light absorption, easily accessed energy states and good charge transfer ability, which can be used to sensitize other semiconductor photocatalysts or it can act as a photocatalyst itself to drive chemical reactions. | ||
| Fig. 35 (a) TEM images of as-prepared CDs; (b) fabrication of the CD electrode; (c) chopped photocurrent generation under visible light illumination and (d) the proposed reaction mechanism of CDs as the photocatalyst; reproduced with permission from ref. 414. | ||
7. Summary and perspective
HTC represents a useful strategy for a variety of present-day applications. The high flexibility in reaction parameters, including the reaction time and temperature, plus the easiness in combining the feedstock with different catalysts, on the one hand, in order to enhance the valuable chemicals that can be found in the liquid phase and/or the hydrothermal carbon yield, or various templates, activators and/or heteroatom doping, on the other hand, in order to produce porous carbon structures decorated with functionalities, make the HTC one of the most used approaches, around the research world, when it comes to transforming waste into valuable products via a sustainable route.As is summarised here, HTC can be used in degradation of biomass and biomass derivatives in three different products, such as liquid phase containing chemicals (5-HMF, levulinic acid, formic acid, and acetic acid), solid carbon materials and fluorescent carbons. Regarding the biomass decomposition and hydrothermal carbon formation, several mechanisms have been proposed and the main steps are hydrolysis, dehydration, polymerization, aromatization and nucleation. Many studies have been focused on the influence of the feedstock, reaction time and temperature on the materials properties and carbon yield, summarizing that as the reaction severity increases, compound solubilisation and volatilisation increase, decreasing the solid yields. Also, when the trends of many feedstocks were collectively investigated, it was observed that feedstock properties have a significant influence on the hydrothermal carbon yield. When it comes to the solid composition, it was observed that regardless of the biomass precursor, the resulting hydrothermal carbons contain between 4 and 6% hydrogen; meanwhile the oxygen content seems to be highly dependent on the precursor. Regarding their structural properties, the resulting powders consist mainly of nonporous spherical particles, with variation in sizes and shape depending on the starting material. The advantage of combining HTC with side strategies, such as templating methods (soft or hard templating) or chemical activation, results in the production of highly porous carbon structures characterised by their large specific surface area and large pore volume, as detailed in section 2. This is a great achievement for further involvement of these materials in environmental applications such as air depollution and water treatment or in energy storage applications, including Li-ion batteries and supercapacitors, as electrode materials. Carbon-based materials have superior cycling stability and rate capability, and so can be operated at higher charge and discharge rates and have much longer lifetimes. Moreover, the hierarchical pore structures enable ion mass transport, and therefore porous carbon is an important material for supercapacitor electrodes. Other applications of the HTC carbon include their usage as electrocatalysts in the ORR. Due to the easiness of combining the carbon precursor (biomass) with different chemicals, a wide range of heteroatom doped carbons can be synthesised via HTC, as discussed in section 3. The heteroatoms doped within the carbon matrix induce heteroatom–carbon bonds, and the polarisation of these bonds generates active sites on the adjacent carbon matrix resulting in a reduced energy barrier towards electrocatalysis. Simultaneously, the electrical conductivity is increased due to the higher concentration of delocalised electrons within the π-system. In addition, the HTC carbon powders can be successfully employed as a catalyst support as the tunability of their surface polarity and area facilitates the anchoring of metal nanoparticles, which can then be used in different reactions.
Chemicals in the liquid phase such as 5-HMF, levulinic acid, formic acid, lactic acid and acetic acid, represent a huge opportunity as a source of bio-based materials and fuel additives. Their synthesis is strictly connected to that of hydrothermal carbons and therefore their joint production represents an added value. Of course, this opportunity comes with several challenges. Yields of conversion are still relatively poor, separation from reaction medium is difficult and corrosion risk due to highly acidic process water is high. All of these issues must be addressed to allow the successful transit of these processes to industrial scale production.
The newly discovered third product, carbon dots, holds great potential in many applications, especially in photocatalysis and photovoltaics. In section 6, the formation mechanism and the reaction parameters were discussed, giving guidance to future investigations on this intriguing material. Given the fact that HTC is an easy and low-cost process, and the CDs formed from this method show excellent bio-compatible and optical properties, producing high quality CDs with HTC to replace the traditional hazardous semiconductor quantum dots could be one of the future directions towards sustainability.
Overall, publications from the past few years show that HTC has been intensively researched and upgraded to be used in novel and up-to-date applications. Despite the fact that the process has been known since 100 years ago, this review reveals that the research community has high interest in HTC, and there are still gaps to be fulfilled and debates to be solved, in terms of the mechanism of carbon sphere formation and carbon quantum dot formation. Moreover, considerable attention should be devoted to upscaling the HTC process, especially related to the advanced materials production requiring advanced reactor design and process engineering. This includes also the energetic and techno-economic analysis along with their environmental impact at each life stage.
A great deal of fundamentals in HTC have already been understood but there is still a lack of details. For example, the mechanism of nucleation, the rate-limiting step, the kinetics of the reaction, and the mechanism of the reaction still remain ambiguous. Something that is not well understood so far is the role and influence of pressure during the HTC process and how the reaction mechanism/nucleation change in the presence of external gases, i.e. when the HTC process is run under an inert gas or CO2. In addition, it is still not clear what dictates the formation of the amorphous HTC spheres and what dictates the formation of the carbon dots in solution. We assume that a gas phase reaction is involved in the formation of the carbon dots which is yet to be proved. To understand the missing fundamentals, in situ/operando characterisation using synchrotron facilities (Small Angle X-Ray/Neutron Diffraction and Pair Distribution Function) is crucial. In addition, on-line monitoring using NMR or HPLC-MS of the reaction products will be able to shed light on the formation mechanism of these challenging materials. Understanding the fundamentals will allow better control over the synthesis and upscaling of these materials. This is valid not only for the carbon dots, but also for the HTC materials, where still certain fundamental aspects such as nucleation, growth, aggregation, and the role of water remain to be elucidated and where in operando characterisation techniques during the HTC materials formation will play an important role in overcoming these major challenges.
Of crucial importance in the future, an area where not many scientific breakthroughs have been seen, is processing HTC materials. This could involve techniques such as electrospinning, electrowriting or 3D printing to create 3D architectures with tunable properties for a wide range of applications from energy storage to wearable electronics. Finally, new applications of these materials must be explored along with already emerging applications, with focus on energy and the environment.
New applications will emerge especially in the case of carbon dots, in particular in photocatalysis, biosensors and optoelectronic applications. Emerging applications of HTC in batteries and electrocatalysis will continue to rise. In particular, the development of multi-ion batteries based on dual carbon electrodes designed with the right configuration for both alkali metals and anion storage as well as other hybrid supercapacitor-battery systems will be crucial in the near future. These batteries could become compostable, whereby after the end of life, a battery containing no current collector but only carbon electrodes could be mixed with soil using the concept of Terra Pretta. In terms of electrocatalysis using HTC supports to anchor single atom sites or dimer sites to control the selectivity to various products, especially for oxygen, CO2 and nitrogen, reaction will be crucial. Particularly promising will be the use of the resulting modified HTC materials as electrocatalysts to further convert the liquid chemicals from the aqueous phase into high end products and/or fuels. If we could create a circular process where the HTC products could be tailor made with the right electrochemical properties to be able to further catalyze under the application of an external bias the conversion of liquid phase chemicals such as HMF and LA into other valuable components, the economic value of the HTC process would increase.
Other applications of increasing importance will be based on using HTC adsorbents for critical metal recovery to recover metals of crucial economic importance from batteries, fuel cells, electrolysers, solar panels or wind turbines and facilitate the movement towards a circular economy.
Finally, clearer correlations, especially when using real biomass, must be made between the (bio)chemistry of the biomass precursor and the resulting carbon materials given that biomass is variable. For the production of advanced materials, it is desired that raw biomass is separated into pure components before being transformed by means of hydrothermal processes. For bioenergy, the biomass-final product characteristics must be understood and optimised to ensure reproducibility.
Life cycle assessment and life cycle costing along with a careful estimation of energy balances must become an integral part of future studies reporting either advanced materials production or biomass to bioenergy processes. Only then, we can ensure truly sustainable, environmentally benign and economically viable processes. So far almost none of the reports on HTC and products are accompanied by LCA; however this must change and become the new norm.
Conflicts of interest
There are no conflicts to declare.References
- F. Bergius and H. Specht, Verlag Wilhelm Knapp, Halle der Saale, Ger, 1913, p. 58 Search PubMed.
- E. Berl and A. Schmidt, Justus Liebigs Ann. Chem., 1932, 493, 97–123 CrossRef CAS.
- E. Berl and A. Schmidt, Justus Liebigs Ann. Chem., 1932, 493, 124–135 CrossRef CAS.
- E. Berl and A. Schmidt, Justus Liebigs Ann. Chem., 1932, 493, 135–152 CrossRef CAS.
- E. Berl and A. Schmidt, Justus Liebigs Ann. Chem., 1932, 496, 283–303 CrossRef CAS.
- Q. Wang, H. Li, L. Chen and X. Huang, Carbon, 2001, 39, 2211–2214 CrossRef CAS.
- C. Falco, F. Perez Caballero, F. Babonneau, C. Gervais, G. Laurent, M.-M. Titirici and N. Baccile, Langmuir, 2011, 27, 14460–14471 CrossRef CAS PubMed.
- C. Falco, N. Baccile and M.-M. Titirici, Green Chem., 2011, 13, 3273–3281 RSC.
- C. Falco, J. P. Marco-Lozar, D. Salinas-Torres, E. Morallon, D. Cazorla-Amorós, M.-M. Titirici and D. Lozano-Castelló, Carbon, 2013, 62, 346–355 CrossRef CAS.
- C. Falco, J. M. Sieben, N. Brun, M. Sevilla, T. der Mauelen, E. Morallón, D. Cazorla-Amorós and M.-M. Titirici, ChemSusChem, 2013, 6, 374–382 CrossRef CAS PubMed.
- M.-M. Titirici, A. Thomas and M. Antonietti, New J. Chem., 2007, 31, 787–789 RSC.
- M.-M. Titirici, A. Thomas and M. Antonietti, J. Mater. Chem., 2007, 17, 3412–3418 RSC.
- M. Sevilla and A. B. Fuertes, Chem. – Eur. J., 2009, 15, 4195–4203 CrossRef CAS PubMed.
- L. Wang, Y. Chang and A. Li, Renewable Sustainable Energy Rev., 2019, 108, 423–440 CrossRef CAS.
- M.-M. M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103–116 RSC.
- T. Werpy and G. Petersen, Other Inf. PBD 1 Aug 2004, 2004, Medium: ED; Size: 76 pp.
- S. Román, J. M. V. Nabais, C. Laginhas, B. Ledesma and J. F. González, Fuel Process. Technol., 2012, 103, 78–83 CrossRef.
- A. Alvarez-Murillo, S. Roman, B. Ledesma and E. Sabio, J. Anal. Appl. Pyrolysis, 2015, 113, 307–314 CrossRef CAS.
- J. A. Rice and P. MacCarthy, Geoderma, 1988, 43, 65–73 CrossRef CAS.
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatra, Chem. Commun., 2012, 48, 8835 RSC.
- Z. C. Yang, M. Wang, A. M. Yong, S. Y. Wong, X. H. Zhang, H. Tan, A. Y. Chang, X. Li and J. Wang, Chem. Commun., 2011, 47, 11615–11617 RSC.
- X. T. Zheng, A. Ananthanarayanan, K. Q. Luo and P. Chen, Small, 2015, 11, 1620–1636 CrossRef CAS PubMed.
- S. C. Ray, A. Saha, N. R. Jana and R. Sarkar, J. Phys. Chem. C, 2009, 113, 18546–18551 CrossRef CAS.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- Y. Chen, Y. Wu, B. Weng, B. Wang and C. Li, Sens. Actuators, B, 2016, 223, 689–696 CrossRef CAS.
- N. Wang, Y. Wang, T. Guo, T. Yang, M. Chen and J. Wang, Biosens. Bioelectron., 2016, 85, 68–75 CrossRef CAS PubMed.
- T. N. J. I. Edison, R. Atchudan, J. J. Shim, S. Kalimuthu, B. C. Ahn and Y. R. Lee, J. Photochem. Photobiol., B, 2016, 158, 235–242 CrossRef CAS PubMed.
- J. Liao, Z. Cheng and L. Zhou, ACS Sustainable Chem. Eng., 2016, 4, 3053–3061 CrossRef CAS.
- M. Jahanbakhshi and B. Habibi, Biosens. Bioelectron., 2016, 81, 143–150 CrossRef CAS PubMed.
- J. Li, L. Zhang, P. Li, Y. Zhang and C. Dong, Sens. Actuators, B, 2018, 258, 580–588 CrossRef CAS.
- Y. Guo, L. Yang, W. Li, X. Wang, Y. Shang and B. Li, Microchim. Acta, 2016, 183, 1409–1416 CrossRef CAS.
- R. Atchudan, T. N. J. I. Edison, M. G. Sethuraman and Y. R. Lee, Appl. Surf. Sci., 2016, 384, 432–441 CrossRef CAS.
- H. Wikberg, T. Ohra-aho, F. Pileidis and M.-M. Titirici, ACS Sustainable Chem. Eng., 2015, 3, 2737–2745 CrossRef CAS.
- D. Kim, K. Lee and K. Y. Park, J. Ind. Eng. Chem., 2016, 42, 95–100 CrossRef CAS.
- E. Dinjus, A. Kruse and N. Tröger, Chem. Eng. Technol., 2011, 34, 2037–2043 CrossRef CAS.
- J. Lu, Z. Liu, Y. Zhang and P. E. Savage, ACS Sustainable Chem. Eng., 2018, 6, 14501–14509 CrossRef CAS.
- A. Funke and F. Ziegler, Biofuels, Bioprod. Biorefin., 2010, 4, 160–177 CrossRef CAS.
- F. Buendia-Kandia, G. Mauviel, E. Guedon, E. Rondags, D. Petitjean and A. Dufour, Energy Fuels, 2018, 32(4), 4127–4138 CrossRef CAS.
- Y. Matsumura, M. Sasaki, K. Okuda, S. Takami, S. Ohara, M. Umetsu and T. Adschiri, Combust. Sci. Technol., 2006, 178, 509–536 CrossRef CAS.
- X. Yan, F. Jin, K. Tohji, T. Moriya and H. Enomoto, J. Mater. Sci., 2007, 42, 9995–9999 CrossRef CAS.
- X. Yan, F. Jin and K. Tohji, AIChE J., 2010, 56, 2727–2733 CrossRef CAS.
- F. Jin, Z. Zhou, H. Enomoto, T. Moriya and H. Higashijima, Chem. Lett., 2004, 33, 126–127 CrossRef CAS.
- B. Danon, G. Marcotullio and W. De Jong, Green Chem., 2014, 16, 39–54 RSC.
- R. J. Van Putten, J. N. M. Soetedjo, E. A. Pidko, J. C. Van Der Waal, E. J. M. Hensen, E. De Jong and H. J. Heeres, ChemSusChem, 2013, 6, 1681–1687 CrossRef CAS PubMed.
- N. Baccile, G. Laurent, F. Babonneau, F. Fayon, M.-M. Titirici and M. Antonietti, J. Phys. Chem. C, 2009, 113, 9644–9654 CrossRef CAS.
- G. Yang, E. A. Pidko and E. J. M. Hensen, J. Catal., 2012, 295, 122–132 CrossRef CAS.
- D. Jung, M. Zimmermann and A. Kruse, ACS Sustainable Chem. Eng., 2018, 6, 13877–13887 CrossRef CAS.
- I. Van Zandvoort, E. J. Koers, M. Weingarth, P. C. A. Bruijnincx, M. Baldus and B. M. Weckhuysen, Green Chem., 2015, 17, 4383–4392 RSC.
- A. B. Brown, B. J. McKeogh, G. A. Tompsett, R. Lewis, N. A. Deskins and M. T. Timko, Carbon, 2017, 125, 614–629 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Carbon, 2009, 47, 2281–2289 CrossRef CAS.
- G. Tsilomelekis, M. J. Orella, Z. Lin, Z. Cheng, W. Zheng, V. Nikolakis and D. G. Vlachos, Green Chem., 2016, 18, 1983–1993 RSC.
- Y. Qi, B. Song and Y. Qi, RSC Adv., 2016, 6, 102428–102435 RSC.
- Z. Cheng, J. Everhart, G. Tsilomelekis, V. Nikolakis, B. Saha and D. Vlachos, Green Chem., 2018, 20, 997–1006 RSC.
- L. J. R. Higgins, A. P. Brown, J. P. Harrington, A. B. Ross, B. Kaulich and B. Mishra, Carbon, 2020, 161, 423–431 CrossRef CAS.
- M. Titirici, M. Antonietti and N. Baccile, Green Chem., 2008, 10, 1204–1212 RSC.
- A. J. Romero-Anaya, M. Ouzzine, M. A. Lillo-Ródenas and A. Linares-Solano, Carbon, 2014, 68, 296–307 CrossRef CAS.
- Y. Yu and H. Wu, Ind. Eng. Chem. Res., 2010, 49, 3902–3909 CrossRef CAS.
- M. Li, W. Li and S. Liu, Carbohydr. Res., 2011, 346, 999–1004 CrossRef CAS PubMed.
- H. Simsir, N. Eltugral and S. Karagoz, Bioresour. Technol., 2017, 246, 82–87 CrossRef CAS PubMed.
- S.-M. Alatalo, E. Repo, E. Mäkilä, J. Salonen, E. Vakkilainen and M. Sillanpää, Bioresour. Technol., 2013, 147, 71–76 CrossRef CAS PubMed.
- L. Li, J. R. V. Flora, J. M. Caicedo and N. D. Berge, Bioresour. Technol., 2015, 187, 263–274 CrossRef CAS PubMed.
- S. Nizamuddin, H. A. Baloch, G. J. Griffin, N. M. Mubarak, A. W. Bhutto, R. Abro, S. A. Mazari and B. S. Ali, Renewable Sustainable Energy Rev., 2017, 73, 1289–1299 CrossRef CAS.
- I. Oliveira, D. Blöhse and H.-G. Ramke, Bioresour. Technol., 2013, 142, 138–146 CrossRef CAS PubMed.
- K. U. Suwelack, D. Wüst, P. Fleischmann and A. Kruse, Biomass Convers. Biorefin., 2016, 6, 151–160 CrossRef CAS.
- K. Suwelack, D. Wust, M. Zeller, A. Kruse and J. Krumpel, Biomass Convers. Biorefin., 2016, 6, 347–354 CrossRef CAS.
- H. P. Ruyter, Fuel, 1982, 61, 1182–1187 CrossRef CAS.
- Y. Fu, J. Ye, J. Chang, H. Lou and X. Zheng, Fuel, 2016, 180, 591–596 CrossRef CAS.
- D. Knežević, W. van Swaaij and S. Kersten, Ind. Eng. Chem. Res., 2010, 49, 104–112 CrossRef.
- M. K. Akalın, K. Tekin and S. Karagoz, Bioresour. Technol., 2012, 110, 682–687 CrossRef PubMed.
- D. Knežević, W. P. M. van Swaaij and S. R. A. Kersten, Ind. Eng. Chem. Res., 2009, 48, 4731–4743 CrossRef.
- L. Li, R. Diederick, J. R. V. Flora and N. D. Berge, Waste Manage., 2013, 33, 2478–2492 CrossRef CAS PubMed.
- X. Lu and N. D. Berge, Bioresour. Technol., 2014, 166, 120–131 CrossRef CAS PubMed.
- K. Aydıncak, T. Yumak, A. Sınag and B. Esen, Ind. Eng. Chem. Res., 2012, 51, 9145–9152 CrossRef.
- N. D. Berge, K. S. Ro, J. Mao and J. R. V. Flora, Environ. Sci. Technol., 2011, 45, 5696–5703 CrossRef CAS PubMed.
- S. Kang, X. Li, J. Fan and J. Chang, Ind. Eng. Chem. Res., 2012, 51, 9023–9031 CrossRef CAS.
- X. Lu, P. Pellechia, J. R. V. Flora and N. Berge, Bioresour. Technol., 2013, 138, 180–190 CrossRef CAS PubMed.
- S. S. Jamari and J. R. Howse, Biomass Bioenergy, 2012, 47, 82–90 CrossRef CAS.
- X. Cao, K. S. Ro, M. Chappell, Y. Li and J. Mao, Energy Fuels, 2011, 25, 388–397 CrossRef CAS.
- C. Areeprasert, P. Zhao, D. Ma, Y. Shen and K. Yoshikawa, Energy Fuels, 2014, 28, 1198–1206 CrossRef CAS.
- M. Sevilla, G. A. Ferrero and A. B. Fuertes, Carbon, 2017, 114, 50–58 CrossRef CAS.
- R. Azargohar, Production of Activated Carbon and its Catalytic Application for Oxidation of Hydrogen Sulphide, 2009 Search PubMed.
- E. Atanes, A. Nieto-Márquez, A. Cambra, M. C. Ruiz-Pérez and F. Fernández-Martínez, Chem. Eng. J., 2012, 211–212, 60–67 CrossRef CAS.
- K. V. Kumar, K. Preuss, L. Lu, Z. X. Guo and M. M. Titirici, J. Phys. Chem. C, 2015, 119, 22310–22321 CrossRef CAS.
- M. A. Islam, A. Benhouria, M. Asif and B. H. Hameed, J. Taiwan Inst. Chem. Eng., 2015, 52, 57–64 CrossRef CAS.
- M. A. Islam, M. J. Ahmed, W. A. Khanday, M. Asif and B. H. Hameed, J. Environ. Manage., 2017, 203, 237–244 CrossRef CAS PubMed.
- P. P. Sharma, J. Wu, R. M. Yadav, M. Liu, C. J. Wright, C. S. Tiwary, B. I. Yakobson, J. Lou, P. M. Ajayan and X.-D. D. Zhou, Angew. Chem., Int. Ed., 2015, 54, 13701–13705 CrossRef CAS PubMed.
- H. Wang, J. Jia, P. Song, Q. Wang, D. Li, S. Min, C. Qian, L. Wang, Y. F. Li, C. Ma, T. Wu, J. Yuan, M. Antonietti and G. A. Ozin, Angew. Chem., Int. Ed., 2017, 56, 7847–7852 CrossRef CAS PubMed.
- G. W. Liu, T. Y. Chen, C. H. Chung, H. P. Lin and C. H. Hsu, ACS Omega, 2017, 2, 2106–2113 CrossRef CAS PubMed.
- M.-M. M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- S. Kubo, R. J. White, N. Yoshizawa, M. Antonietti and M. M. Titirici, Chem. Mater., 2011, 23, 4882–4885 CrossRef CAS.
- K. V. Kumar, S. Gadipelli, K. Preuss, H. Porwal, T. Zhao, Z. X. Guo and M. M. Titirici, ChemSusChem, 2017, 10, 199–209 CrossRef CAS PubMed.
- M.-M. M. Titirici, R. J. White, C. Falco and M. Sevilla, Energy Environ. Sci., 2012, 5, 6796–6822 RSC.
- Z. Zhang, M. Qin, B. Jia, H. Zhang, H. Wu and X. Qu, Chem. Commun., 2017, 53, 2922–2925 RSC.
- P. W. Xiao, L. Zhao, Z. Y. Sui, M. Y. Xu and B. H. Han, Microporous Mesoporous Mater., 2017, 253, 215–222 CrossRef CAS.
- A. B. Fuertes and M. Sevilla, Carbon, 2015, 94, 41–52 CrossRef CAS.
- P. W. Xiao, D. Guo, L. Zhao and B. H. Han, Microporous Mesoporous Mater., 2016, 220, 129–135 CrossRef CAS.
- L. Zhu, F. Shen, R. L. Smith and X. Qi, Energy Technol., 2017, 5, 452–460 CrossRef CAS.
- Y. Boyjoo, Y. Cheng, H. Zhong, H. Tian, J. Pan, V. K. Pareek, S. P. Jiang, J. F. Lamonier, M. Jaroniec and J. Liu, Carbon, 2017, 116, 490–499 CrossRef CAS.
- Q.-P. Fabian, R.-R. Ramiro, M. Emilia and C.-A. Diego, ChemPlusChem, 2016, 81, 1349–1359 CrossRef PubMed.
- M. T. Gilbert, J. H. Knox and B. Kaur, Chromatographia, 1982, 16, 138–146 CrossRef CAS.
- S. Kubo, R. Demir-Cakan, L. Zhao, R. J. White and M. M. Titirici, ChemSusChem, 2010, 3, 188–194 CrossRef CAS PubMed.
- S. Z. Niu, S. Da Wu, W. Lu, Q. H. Yang and F. Y. Kang, Xinxing Tan Cailiao/New Carbon Mater., 2017, 32, 289–296 CrossRef.
- C. Liang, K. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785–5789 CrossRef CAS PubMed.
- Y. Zhang, L. Liu, B. Van Der Bruggen and F. Yang, J. Mater. Chem. A, 2017, 5, 12673–12698 RSC.
- T. Zhou, R. Ma, Y. Zhou, R. Xing, Q. Liu, Y. Zhu and J. Wang, Microporous Mesoporous Mater., 2018, 261, 88–97 CrossRef CAS.
- S. Wang, C. Han, J. Wang, J. Deng, M. Zhu, J. Yao, H. Li and Y. Wang, Chem. Mater., 2014, 26, 6872–6877 CrossRef CAS.
- S. A. Bagshaw, E. Prouzet, T. J. Pinnavaia, S. A. Bagshaw, E. Prouzet and T. J. Pinnavaia, Science, 1995, 269, 1242–1244 CrossRef PubMed.
- J. Texter, L. Zhao, P. W. Xiao, F. P. Caballero, B. H. Han and M. M. Titirici, Carbon, 2017, 112, 117–129 CrossRef CAS.
- Suhas, V. K. Gupta, P. J. M. Carrott, R. Singh, M. Chaudhary and S. Kushwaha, Bioresour. Technol., 2016, 216, 1066–1076 CrossRef CAS PubMed.
- J. Deng, M. Li and Y. Wang, Green Chem., 2016, 18, 4824–4854 RSC.
- Z. Liang, L. Zhang, H. Liu, J. Zeng, J. Zhou, H. Li and H. Xia, Results Phys., 2019, 12, 1984–1990 CrossRef.
- J. Liu, L. Xie, Z. Wang, S. Mao, Y. Gong and Y. Wang, Chem. Commun., 2019, 56, 229–232 RSC.
- L. Xie, Z. Wang, J. Liu, Y. Gong, S. Mao, G. Lü, X. Ma, L. Yu and Y. Wang, Carbon, 2019, 155, 611–617 CrossRef CAS.
- C. Chen, H. Wang, C. Han, J. Deng, J. Wang, M. Li, M. Tang, H. Jin and Y. Wang, J. Am. Chem. Soc., 2017, 139, 2657–2663 CrossRef CAS PubMed.
- A. Chen, Y. Yu, Y. Zhang, W. Zang, Y. Yu, Y. Zhang, S. Shen and J. Zhang, Carbon, 2014, 80, 19–27 CrossRef CAS.
- Q. Kong, L. Zhang, M. Wang, M. Li, H. Yao and J. Shi, Sci. Bull., 2016, 61, 1195–1201 CrossRef CAS.
- N. P. Stadie, S. Wang, K. V. Kravchyk and M. V. Kovalenko, ACS Nano, 2017, 11, 1911–1919 CrossRef CAS PubMed.
- H. Nishihara and T. Kyotani, Chem. Commun., 2018, 54, 5648–5673 RSC.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed.
- S. Dutta, A. Bhaumik and K. C.-W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 RSC.
- L. P. Wang, W. S. Jia, X. F. Liu, J. Z. Li and M. M. Titirici, J. Energy Chem., 2016, 25, 566–570 CrossRef.
- F. Rodríguez-Reinoso and M. Molina-Sabio, Carbon, 1992, 30, 1111–1118 CrossRef.
- J. Guo, Y. Luo, A. C. Lua, R.-A. Chi, Y.-L. Chen, X.-T. Bao and S.-X. Xiang, Carbon, 2007, 45, 330–336 CrossRef CAS.
- J. Jin, S. Tanaka, Y. Egashira and N. Nishiyama, Carbon, 2010, 48, 1985–1989 CrossRef CAS.
- X. Zhou, Z. Jia, A. Feng, X. Wang, J. Liu, M. Zhang, H. Cao and G. Wu, Carbon, 2019, 152, 827–836 CrossRef CAS.
- M. Zhu, W. Cai, F. Verpoort and J. Zhou, Chem. Eng. Res. Des., 2019, 146, 130–140 CrossRef CAS.
- M. Sevilla, W. Gu, C. Falco, M. M. Titirici, A. B. Fuertes and G. Yushin, J. Power Sources, 2014, 267, 26–32 CrossRef CAS.
- J. Li, H. Qi, Q. Wang, Z. Xu, Y. Liu, Q. Li, X. Kong and J. Huang, Electrochim. Acta, 2018, 271, 92–102 CrossRef CAS.
- J. Shi, N. Yan, H. Cui, Y. Liu, Y. Weng, D. Li and X. Ji, J. CO2 Util., 2017, 21, 444–449 CrossRef CAS.
- F. Quesada-Plata, R. Ruiz-Rosas, E. Morallón and D. Cazorla-Amorós, ChemPlusChem, 2016, 81, 1349–1359 CrossRef CAS PubMed.
- O. F. Cruz, J. Silvestre-albero, M. E. Casco, D. Hotza and C. R. Rambo, Mater. Chem. Phys., 2018, 216, 42–46 CrossRef CAS.
- X. Lu, C. Jiang, Y. Hu, H. Zhong, Y. Zhao, X. Xu and H. Liu, J. Appl. Electrochem., 2018, 48, 233–241 CrossRef CAS.
- C. Rodríguez Correa, C. Ngamying, D. Klank and A. Kruse, Biomass Convers. Biorefin., 2018, 8, 317–328 CrossRef.
- X. Zhang, Y. Zhong, X. Xia, Y. Xia, D. Wang, C. Zhou, W. Tang, X. Wang, J. B. Wu and J. Tu, ACS Appl. Mater. Interfaces, 2018, 10, 13598–13605 CrossRef CAS PubMed.
- M. Sevilla, W. Sangchoom, N. Balahmar, A. B. Fuertes and R. Mokaya, ACS Sustainable Chem. Eng., 2016, 4, 4710–4716 CrossRef CAS.
- K. Preuss, L. C. Tǎnase, C. M. Teodorescu, I. Abrahams and M. M. Titirici, J. Mater. Chem. A, 2017, 5, 16336–16343 RSC.
- L. S. Panchakarla, A. Govindaraj and C. N. R. Rao, Inorg. Chim. Acta, 2010, 363, 4163–4174 CrossRef CAS.
- A. Kalijadis, J. DorCevidć, T. Trtić-Petrović, M. Vukčević, M. Popović, V. Maksimović, Z. Rakočević and Z. Laušević, Carbon, 2015, 95, 42–50 CrossRef CAS.
- J. Wu, J. Hu, K. Song, J. Xu and H. Gao, J. Alloys Compd., 2017, 704, 1–6 CrossRef CAS.
- W. Ma, L. Xie, L. Dai, G. Sun, J. Chen, F. Su, Y. Cao, H. Lei, Q. Kong and C. M. Chen, Electrochim. Acta, 2018, 266, 420–430 CrossRef CAS.
- B. Ashourirad, A. K. Sekizkardes, S. Altarawneh and H. M. El-Kaderi, Chem. Mater., 2015, 27, 1349–1358 CrossRef CAS.
- E. Antolini, Renewable Sustainable Energy Rev., 2016, 58, 34–51 CrossRef CAS.
- O. S. G. P. Soares, R. P. Rocha, A. G. Gonçalves, J. L. Figueiredo, J. J. M. Órfão and M. F. R. Pereira, Appl. Catal., B, 2016, 192, 296–303 CrossRef CAS.
- L. Wang, Y. Wang, M. Wu, Z. Wei, C. Cui, M. Mao, J. Zhang, X. Han, Q. Liu and J. Ma, Small, 2018, 1800737, 1800737 CrossRef PubMed.
- K. Sharma, D. Hui, N. H. Kim and J. H. Lee, Nanoscale, 2019, 11, 1205–1216 RSC.
- A. Zhang, S. Cao, Y. Zhao, C. Zhang and A. Chen, New J. Chem., 2018, 42, 6903–6909 RSC.
- A. Sarapuu, E. Kibena-Põldsepp, M. Borghei and K. Tammeveski, J. Mater. Chem. A, 2018, 6, 776–804 RSC.
- M. Borghei, N. Laocharoen, E. Kibena-Põldsepp, L. S. Johansson, J. Campbell, E. Kauppinen, K. Tammeveski and O. J. Rojas, Appl. Catal., B, 2017, 204, 394–402 CrossRef CAS.
- Y. Song, G. Liu and Z.-Y. Y. Yuan, RSC Adv., 2016, 6, 94636–94642 RSC.
- T. Berthold, C. R. Castro, M. Winter, G. Hoerpel, M. Kurttepeli, S. Bals, M. Antonietti and N. Fechler, ChemNanoMat, 2017, 3, 311–318 CrossRef CAS.
- A. L. Yaumi, M. Z. A. Bakar and B. H. Hameed, Energy, 2018, 155, 46–55 CrossRef CAS.
- W. He, P. Xue, H. Du, L. Xu, M. Pang, X. Gao, J. Yu, Z. Zhang and T. Huang, Int. J. Hydrogen Energy, 2017, 42, 4123–4132 CrossRef CAS.
- F. Liu, Y. Gao, C. Zhang, H. Huang, C. Yan, X. Chu, Z. Xu, Z. Wang, H. Zhang, X. Xiao and W. Yang, J. Colloid Interface Sci., 2019, 548, 322–332 CrossRef CAS PubMed.
- Q. Wu, G. Zhang, M. Gao, L. Huang, L. Li, S. Liu, C. Xie, Y. Zhang and S. Yu, J. Alloys Compd., 2019, 786, 826–838 CrossRef CAS.
- F. Schipper, S. Kubo and T. P. Fellinger, J. Sol-Gel Sci. Technol., 2019, 89, 101–110 CrossRef CAS.
- W. Zhang, Z. Ren, Z. Ying, X. Liu and H. Wan, J. Alloys Compd., 2018, 743, 44–51 CrossRef CAS.
- M. Ren, Z. Jia, Z. Tian, D. Lopez, J. Cai, M. M. Titirici and A. B. Jorge, ChemElectroChem, 2018, 5, 2686–2693 CrossRef CAS.
- M. Qiao, S. S. Meysami, G. A. Ferrero, F. Xie, H. Meng, N. Grobert and M. M. Titirici, Adv. Funct. Mater., 2017, 1707284, 1–7 Search PubMed.
- S. M. Alatalo, K. Qiu, K. Preuss, A. Marinovic, M. Sevilla, M. Sillanpää, X. Guo and M. M. Titirici, Carbon, 2016, 96, 622–630 CrossRef CAS.
- R. Atchudan, T. N. J. I. Edison, S. Perumal and Y. R. Lee, Appl. Surf. Sci., 2017, 393, 276–286 CrossRef CAS.
- G. Huang, Y. Wang, T. Zhang, X. Wu and J. Cai, J. Taiwan Inst. Chem. Eng., 2019, 96, 672–680 CrossRef CAS.
- K. G. Latham, W. M. Dose, J. A. Allen and S. W. Donne, Carbon, 2018, 128, 179–190 CrossRef CAS.
- L. Rao, R. Ma, S. Liu, L. Wang, Z. Wu, J. Yang and X. Hu, Chem. Eng. J., 2019, 362, 794–801 CrossRef CAS.
- F. C. de Oliveira, J. S. dos R. Coimbra, E. B. de Oliveira, A. D. G. Zuñiga and E. E. G. Rojas, Crit. Rev. Food Sci. Nutr., 2016, 56, 1108–1125 CrossRef PubMed.
- X. Ma, G. Ning, Y. Wang, X. Song, Z. Xiao, L. Hou, W. Yang, J. Gao and Y. Li, Electrochim. Acta, 2018, 269, 83–92 CrossRef CAS.
- A. Elmouwahidi, J. Castelo-Quibén, J. F. Vivo-Vilches, A. F. Pérez-Cadenas, F. J. Maldonado-Hódar and F. Carrasco-Marín, Chem. Eng. J., 2018, 334, 1835–1841 CrossRef CAS.
- K. Anoop Krishnan, K. G. Sreejalekshmi, V. Vimexen and V. V. Dev, Ecotoxicol. Environ. Saf., 2016, 124, 418–425 CrossRef CAS PubMed.
- L. Roldán, Y. Marco and E. García-Bordejé, Microporous Mesoporous Mater., 2016, 222, 55–62 CrossRef.
- S. M. Alatalo, E. Mäkilä, E. Repo, M. Heinonen, J. Salonen, E. Kukk, M. Sillanpää and M. M. Titirici, Green Chem., 2016, 18, 1137–1146 RSC.
- S. S. Zhang, Inorg. Chem. Front., 2015, 2, 1059–1069 RSC.
- M. Y. Emran, M. A. Shenashen, A. A. Abdelwahab, M. Abdelmottaleb and S. A. El-Safty, New J. Chem., 2018, 42, 5037–5044 RSC.
- L. Roldán, L. Truong-Phuoc, A. Ansón-Casaos, C. Pham-Huu and E. García-Bordejé, Catal. Today, 2018, 301, 2–10 CrossRef.
- M. H. Yeh, Y. S. Li, G. L. Chen, L. Y. Lin, T. J. Li, H. M. Chuang, C. Y. Hsieh, S. C. Lo, W. H. Chiang and K. C. Ho, Electrochim. Acta, 2015, 172, 52–60 CrossRef CAS.
- M. H. Yeh, Y. A. Leu, W. H. Chiang, Y. S. Li, G. L. Chen, T. J. Li, L. Y. Chang, L. Y. Lin, J. J. Lin and K. C. Ho, J. Power Sources, 2018, 375, 29–36 CrossRef CAS.
- J. Jiang, P. Nie, S. Fang, Y. Zhang, Y. An, R. Fu, H. Dou and X. Zhang, Chin. Chem. Lett., 2018, 29, 624–628 CrossRef CAS.
- L. Sun, Y. Fu, C. Tian, Y. Yang, L. Wang, J. Yin, J. Ma, R. Wang and H. Fu, ChemSusChem, 2014, 7, 1637–1646 CrossRef CAS PubMed.
- Y. Li, Z. Wang, L. Li, S. Peng, L. Zhang, M. Srinivasan and S. Ramakrishna, Carbon, 2016, 99, 556–563 CrossRef CAS.
- J. Wu, C. Jin, Z. Yang, J. Tian and R. Yang, Carbon, 2015, 82, 562–571 CrossRef CAS.
- M. Q. Guo, J. Q. Huang, X. Y. Kong, H. J. Peng, H. Shui, F. Y. Qian, L. Zhu, W. C. Zhu and Q. Zhang, Xinxing Tan Cailiao/New Carbon Mater., 2016, 31, 352–362 CrossRef CAS.
- R. C. Bansai and M. Goyal, Activated Carbon Adsorption, CRC Press, Boca Raton, 2005 Search PubMed.
- F. Y. Chang, K. J. Chao, H. H. Cheng and C. S. Tan, Sep. Purif. Technol., 2009, 70, 87–95 CrossRef CAS.
- H. Marsh and F. Rodríguez-Reinoso, Characterization of Activated Carbon, 2006 Search PubMed.
- T. Gupta, Carbon Black, Gray Transparent, 2017, pp. 1–319 Search PubMed.
- C. Laginhas, J. M. V. Nabais and M. M. Titirici, Microporous Mesoporous Mater., 2016, 226, 125–132 CrossRef CAS.
- P. Zheng, T. Liu, X. Yuan, L. Zhang, Y. Liu, J. Huang and S. Guo, Sci. Rep., 2016, 6, 1–9 CrossRef PubMed.
- E. Hao, W. Liu, S. Liu, Y. Zhang, H. Wang, S. Chen, F. Cheng, S. Zhao and H. Yang, J. Mater. Chem. A, 2017, 5, 2204–2214 RSC.
- M. Imtiaz, C. Zhu, Y. Li, M. S. Pak, I. Zada, S. W. Bokhari, Z. Chen, D. Zhang and S. Zhu, J. Alloys Compd., 2017, 724, 296–305 CrossRef CAS.
- P. Zheng, T. Liu, J. Zhang, L. Zhang, Y. Liu, J. Huang and S. Guo, RSC Adv., 2015, 5, 40737–40741 RSC.
- W. Li, J. Huang, L. Feng, L. Cao, Y. Ren, R. Li, Z. Xu, J. Li and C. Yao, J. Alloys Compd., 2017, 716, 210–219 CrossRef CAS.
- H. Wang, W. Yu, J. Shi, N. Mao, S. Chen and W. Liu, Electrochim. Acta, 2016, 188, 103–110 CrossRef CAS.
- E. Demir, M. Aydin, A. A. Arie and R. Demir-Cakan, J. Alloys Compd., 2019, 788, 1093–1102 CrossRef CAS.
- L. Ma, B. Zhao, X. Wang, J. Yang, X. Zhang, Y. Zhou and J. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 22067–22073 CrossRef CAS PubMed.
- J. Wang, L. Yan, Q. Ren, L. Fan, F. Zhang and Z. Shi, Electrochim. Acta, 2018, 291, 188–196 CrossRef CAS.
- R. Väli, A. Jänes, T. Thomberg and E. Lust, J. Electrochem. Soc., 2016, 163, A1619–A1626 CrossRef.
- H. Tang, M. Wang, T. Lu and L. Pan, Ceram. Int., 2017, 43, 4475–4482 CrossRef CAS.
- Z. Jian, S. Hwang, Z. Li, A. S. Hernandez, X. Wang, Z. Xing, D. Su and X. Ji, Adv. Funct. Mater., 2017, 27, 1–6 Search PubMed.
- Y. Li, S. Xu, X. Wu, J. Yu, Y. Wang, Y. S. Hu, H. Li, L. Chen and X. Huang, J. Mater. Chem. A, 2015, 3, 71–77 RSC.
- L. Yin, Y. Wang, C. Han, Y. M. Kang, X. Ma, H. Xie and M. Wu, J. Power Sources, 2016, 305, 156–160 CrossRef CAS.
- R. Li, J. Huang, Z. Xu, H. Qi, L. Cao, Y. Liu, W. Li and J. Li, Energy Technol., 2018, 6, 1080–1087 CrossRef CAS.
- H.-L. Wang, Z.-Q. Shi, J. Jin, C.-B. Chong and C.-Y. Wang, J. Electroanal. Chem., 2015, 755, 87–91 CrossRef CAS.
- S. Jeong, X. Li, J. Zheng, P. Yan, R. Cao, H. J. Jung, C. Wang, J. Liu and J. G. Zhang, J. Power Sources, 2016, 329, 323–329 CrossRef CAS.
- J.-S. Choi, H.-J. Lee, J.-K. Ha and K.-K. Cho, J. Nanosci. Nanotechnol., 2018, 18, 6459–6462 CrossRef CAS PubMed.
- S. Y. Son, S.-A. Hong, S. Y. Oh, Y.-C. Lee, G.-W. Lee, J. W. Kang, Y. S. Huh and I. T. Kim, J. Nanosci. Nanotechnol., 2018, 18, 6463–6468 CrossRef CAS PubMed.
- Q. Zhang, Q. Gao, W. Qian, H. Zhang, Y. Tan, W. Tian, Z. Li and H. Xiao, J. Mater. Chem. A, 2017, 5, 19136–19142 RSC.
- L. Wang, J. Yuan, Q. Zhao, Z. Wang, Y. Zhu, X. Ma and C. Cao, Electrochim. Acta, 2019, 308, 174–184 CrossRef CAS.
- Z. Chen, D. Yin and M. Zhang, Small, 2018, 14, 1–9 Search PubMed.
- Y. Guo, Y. Zhang, Y. Wang, D. Zhang, Y. Lu, R. Luo, Y. Wang, X. Liu, J. K. Kim and Y. Luo, Electrochim. Acta, 2019, 296, 989–998 CrossRef CAS.
- X. Li, Y. Yang, J. Liu, L. Ouyang, J. Liu, R. Hu, L. Yang and M. Zhu, Appl. Surf. Sci., 2017, 413, 169–174 CrossRef CAS.
- S. Bao, S.-H. Luo, S.-X. Yan, Z.-Y. Wang, Q. Wang, J. Feng, Y.-L. Wang and T.-F. Yi, Electrochim. Acta, 2019, 307, 293–301 CrossRef CAS.
- W. ur Rehman, Y. Xu, X. Du, X. Sun, I. Ullah, Y. Zhang, Y. Jin, B. Zhang and X. Li, Appl. Surf. Sci., 2018, 445, 359–367 CrossRef CAS.
- Z. Xiao, G. Ning, Z. Yu, C. Qi, L. Zhao, Y. Li, X. Ma and Y. Li, Nanoscale, 2019, 11, 8270–8280 RSC.
- C. Deng, S. Zhang and Y. Wu, Nanoscale, 2015, 7, 487–491 RSC.
- V. Palomares, M. Blas, P. Serras, A. Iturrondobeitia, A. Peña, A. Lopez-Urionabarrenechea, L. Lezama and T. Rojo, ACS Sustainable Chem. Eng., 2018, 6, 16386–16398 CrossRef CAS.
- J. Ding, H. Wang, Z. Li, K. Cui, D. Karpuzov, X. Tan, A. Kohandehghan and D. Mitlin, Energy Environ. Sci., 2015, 8, 941–955 RSC.
- H. Lu and X. S. Zhao, Sustainable Energy Fuels, 2017, 1, 1265–1281 RSC.
- D. Salinas-Torres, D. Lozano-Castelló, M. M. Titirici, L. Zhao, L. Yu, E. Morallón and D. Cazorla-Amoros, J. Mater. Chem. A, 2015, 3, 15558–15567 RSC.
- Y. Wang, R. Yang, M. Li and Z. Zhao, Ind. Crops Prod., 2015, 65, 216–226 CrossRef CAS.
- Y. Liu, B. Huang, X. Lin and Z. Xie, J. Mater. Chem. A, 2017, 5, 13009–13018 RSC.
- T. Wei, X. Wei, Y. Gao and H. Li, Electrochim. Acta, 2015, 169, 186–194 CrossRef CAS.
- L. Zhang, T. You, T. Zhou, X. Zhou and F. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 13918–13925 CrossRef CAS PubMed.
- C. Li, D. He, Z.-H. Huang and M.-X. Wang, J. Electrochem. Soc., 2018, 165, A3334–A3341 CrossRef CAS.
- K. K. Lee, W. Hao, M. Gustafsson, C. W. Tai, D. Morin, E. Björkman, M. Lilliestråle, F. Björefors, A. M. Andersson and N. Hedin, RSC Adv., 2016, 6, 110629–110641 RSC.
- M. Härmas, T. Thomberg, T. Romann, A. Jänes and E. Lust, J. Electrochem. Soc., 2017, 164, A1866–A1872 CrossRef.
- W. Du, X. Wang, J. Zhan, X. Sun, L. Kang, F. Jiang, X. Zhang, Q. Shao, M. Dong, H. Liu, V. Murugadoss and Z. Guo, Electrochim. Acta, 2019, 296, 907–915 CrossRef CAS.
- Y. Zhao, X. He, R. Chen, Q. Liu, J. Liu, J. Yu, J. Li, H. Zhang, H. Dong, M. Zhang and J. Wang, Chem. Eng. J., 2018, 352, 29–38 CrossRef CAS.
- B. Kirubasankar, V. Murugadoss, J. Lin, T. Ding, M. Dong, H. Liu, J. Zhang, T. Li, N. Wang, Z. Guo and S. Angaiah, Nanoscale, 2018, 10, 20414–20425 RSC.
- Y. Q. Zhao, M. Lu, P. Y. Tao, Y. J. Zhang, X. T. Gong, Z. Yang, G. Q. Zhang and H. L. Li, J. Power Sources, 2016, 307, 391–400 CrossRef CAS.
- W. Ye, X. Li, J. Luo, X. Wang and R. Sun, Ind. Crops Prod., 2017, 109, 410–419 CrossRef CAS.
- M. Enterría, F. J. Martín-Jimeno, F. Suárez-García, J. I. Paredes, M. F. R. Pereira, J. I. Martins, A. Martínez-Alonso, J. M. D. Tascón and J. L. Figueiredo, Carbon, 2016, 105, 474–483 CrossRef.
- F. Barzegar, A. Bello, O. O. Fashedemi, J. K. Dangbegnon, D. Y. Momodu, F. Taghizadeh and N. Manyala, Electrochim. Acta, 2015, 180, 442–450 CrossRef CAS.
- Y. Wang and L. Jiang, ACS Sustainable Chem. Eng., 2017, 5, 5588–5597 CrossRef CAS.
- Z. W. She, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355(6321), eaad4998 CrossRef PubMed.
- C. Tang, M. M. Titirici and Q. Zhang, J. Energy Chem., 2017, 26, 1077–1093 CrossRef.
- F. Cao, M. Zhao, Y. Yu, B. Chen, Y. Huang, J. Yang, X. Cao, Q. Lu, X. Zhang, Z. Zhang, C. Tan and H. Zhang, J. Am. Chem. Soc., 2016, 138, 6924–6927 CrossRef CAS PubMed.
- Y. Lu, L. Wang, K. Preuß, M. Qiao, M. M. Titirici, J. Varcoe and Q. Cai, J. Power Sources, 2017, 372, 82–90 CrossRef CAS.
- H. Yu, L. Shang, T. Bian, R. Shi, G. I. N. Waterhouse, Y. Zhao, C. Zhou, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2016, 28, 5140–5140 CrossRef CAS PubMed.
- M. Qiao, C. Tang, G. He, K. Qiu, R. Binions, I. P. Parkin, Q. Zhang, Z. Guo and M. M. Titirici, J. Mater. Chem. A, 2016, 4, 12658–12666 RSC.
- K. Preuss, M. Qiao and M. Titrici, Hydrothermal Carbon Materials for the Oxygen Reduction Reaction, in Carbon-Based Metal-Free Catalysts, ed. L. Dai, Wiley, 2018, vol. 2, pp. 369–401 Search PubMed.
- W. Wan, Q. Wang, L. Zhang, H. W. Liang, P. Chen and S. H. Yu, J. Mater. Chem. A, 2016, 4, 8602–8609 RSC.
- K. Preuss, A. M. Siwoniku, C. I. Bucur and M.-M. Titirici, ChemPlusChem, 2019, 84, 457–464 CrossRef CAS PubMed.
- G. He, M. Qiao, W. Li, Y. Lu, T. Zhao, R. Zou, B. Li, J. A. Darr, J. Hu, M. M. Titirici and I. P. Parkin, Adv. Sci., 2017, 4, 1600214 CrossRef PubMed.
- B. Huang, Y. Liu and Z. Xie, J. Mater. Chem. A, 2017, 5, 23481–23488 RSC.
- Y. Cheng, K. Pang, X. Wu, Z. Zhang, X. Xu, J. Ren, W. Huang and R. Song, ACS Sustainable Chem. Eng., 2018, 6, 8688–8696 CrossRef CAS.
- L. Zhang, T. Wang, L. Sun, Y. Sun, T. Hu, K. Xu and F. Ma, J. Mater. Chem. A, 2017, 5, 19752–19759 RSC.
- D. Huang, S. Li, X. Zhang, Y. Luo, J. Xiao and H. Chen, Carbon, 2018, 129, 468–475 CrossRef CAS.
- S. H. Bae, J. E. Kim, H. Randriamahazaka, S. Y. Moon, J. Y. Park and I. K. Oh, Adv. Energy Mater., 2017, 7, 1–11 CAS.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505–509 CrossRef CAS PubMed.
- S.-H. H. Bae, J.-E. E. Kim, H. Randriamahazaka, S.-Y. Y. Moon, J.-Y. Y. Park and I.-K. K. Oh, Adv. Energy Mater., 2017, 7, 1601492 CrossRef.
- L. Roldán, E. Pires, J. M. Fraile and E. García-Bordejé, Catal. Today, 2015, 249, 153–160 CrossRef.
- D. R. Lathiya, D. V. Bhatt and K. C. Maheria, Bioresour. Technol. Rep., 2018, 2, 69–76 CrossRef.
- Z.-E. Tang, S. Lim, Y.-L. Pang, H.-C. Ong and K.-T. Lee, Renewable Sustainable Energy Rev., 2018, 92, 235–253 CrossRef CAS.
- C. De La Calle, J. M. Fraile, E. García-Bordejé, E. Pires and L. Roldán, Catal. Sci. Technol., 2015, 1–7 Search PubMed.
- S. Kang and J. Yu, Sugar Technol., 2018, 20, 182–193 CrossRef CAS.
- F. D. Pileidis, M. Tabassum, S. Coutts, M. M. Ttitirici and M.-M. Titirici, Cuihua Xuebao/Chin. J. Catal., 2014, 35, 929–936 CrossRef CAS.
- J. M. Fraile, E. García-Bordejé, E. Pires and L. Roldán, Carbon, 2014, 77, 1157–1167 CrossRef CAS.
- I. F. Nata, M. D. Putra, C. Irawan and C.-K. Lee, J. Environ. Chem. Eng., 2017, 5, 2171–2175 CrossRef CAS.
- X. Qi, N. Liu and Y. Lian, RSC Adv., 2015, 5, 17526–17531 RSC.
- Y. Yan, Y. Dai, S. Wang, X. Jia, H. Yu and Y. Yang, Appl. Catal., B, 2016, 184, 104–118 CrossRef CAS.
- J. Chen, J. Zhang, W. Sun, K. Song, D. Zhu and T. Li, Appl. Organomet. Chem., 2017, 32, 3 Search PubMed.
- A. Eiad-ua, B. Jomhataikool, W. Gunpum, N. Viriya-empikul and K. Faungnawakij, Mater. Today: Proc., 2017, 4, 6153–6158 Search PubMed.
- J. Liu, N. Wickramaratne and S. Qiao, et al. , Nat. Mater., 2015, 14, 763–774 CrossRef CAS PubMed.
- C. Gai, F. Zhang, T. Yang, Z. Liu, W. Jiao, N. Peng, T. Liu, Q. Lang and Y. Xia, Green Chem., 2018, 20, 2788–2800 RSC.
- P. Wataniyakul, P. Boonnoun, A. T. Quitain, M. Sasaki, T. Kida, N. Laosiripojana and A. Shotipruk, Catal. Commun., 2018, 104, 41–47 CrossRef CAS.
- Y. M. Lu, H. Z. Zhu, W. G. Li, B. Hu and S. H. Yu, J. Mater. Chem. A, 2013, 1, 3783–3788 RSC.
- S. P. Dubey, A. D. Dwivedi, I.-C. Kim, M. Sillanpaa, Y.-N. Kwon and C. Lee, Chem. Eng. J., 2014, 244, 160–167 CrossRef CAS.
- J. Cheng, Y. Wang, C. Teng, Y. Shang, L. Ren and B. Jiang, Chem. Eng. J., 2014, 242, 285–293 CrossRef CAS.
- Z. Sun, J. Yang, J. Wang, W. Li, S. Kaliaguine, X. Hou, Y. Deng and D. Zhao, J. Mater. Chem. A, 2014, 2, 6071–6074 RSC.
- J. Yu, L. Yan, G. Tu, C. Xu, X. Ye, Y. Zhong, W. Zhu and Q. Xiao, Catal. Lett., 2014, 144, 2065–2070 CrossRef CAS.
- F. Zhang, H. Hu, H. Zhong, N. Yan and Q. Chen, Dalton Trans., 2014, 43, 6041 RSC.
- B. He, Q. Zhao, Z. Zeng, X. Wang and S. Han, J. Mater. Sci., 2015, 50, 6339–6348 CrossRef CAS.
- Z. Q. Shi, L. X. Jiao, J. Sun, Z. B. Chen, Y. Z. Chen, X. H. Zhu, J. H. Zhou, X. C. Zhou, X. Z. Li and R. Li, RSC Adv., 2014, 4, 47–53 RSC.
- K. Huang, J. Liu, L. Wang, G. Chang, R. Wang, M. Lei, Y. Wang and Y. He, Appl. Surf. Sci., 2019, 487, 1145–1151 CrossRef CAS.
- G. Singh, K. S. Lakhi, I. Y. Kim, S. Kim, P. Srivastava, R. Naidu and A. Vinu, ACS Appl. Mater. Interfaces, 2017, 9, 29782–29793 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. De Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- N. Querejeta, M. V. Gil, C. Pevida and T. A. Centeno, J. CO2 Util., 2018, 26, 1–7 CrossRef CAS.
- S. Sethupathi, M. Zhang, A. U. Rajapaksha, S. S. R. Lee, N. M. Nor, A. R. Mohamed, M. Al-Wabel, S. S. R. Lee and Y. S. Ok, Sustainability, 2017, 9, 1–10 CrossRef.
- M. C. Castrillon, K. O. Moura, C. A. Alves, M. Bastos-Neto, D. C. S. Azevedo, J. Hofmann, J. Möllmer, W. D. Einicke and R. Gläser, Energy Fuels, 2016, 30, 9596–9604 CrossRef CAS.
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, Renewable Sustainable Energy Rev., 2014, 39, 426–443 CrossRef CAS.
- L. Rao, R. Ma, S. Liu, L. Wang, Z. Wu, J. Yang and X. Hu, Nitrogen enriched porous carbons from D-glucose with excellent CO 2 capture performance, Elsevier B.V., 2019 Search PubMed.
- G. Shang, Q. Li, L. Liu, P. Chen and X. Huang, J. Air Waste Manage. Assoc., 2016, 66, 8–16 CrossRef CAS PubMed.
- M. I. Inyang, B. Gao, Y. Yao, Y. Xue, A. Zimmerman, A. Mosa, P. Pullammanappallil, Y. S. Ok and X. Cao, Crit. Rev. Environ. Sci. Technol., 2016, 46, 406–433 CrossRef CAS.
- X. He, K. B. Male, P. N. Nesterenko, D. Brabazon, B. Paull and J. H. T. Luong, ACS Appl. Mater. Interfaces, 2013, 5, 8796–8804 CrossRef CAS PubMed.
- M. A. Islam, I. A. W. Tan, A. Benhouria, M. Asif and B. H. Hameed, Chem. Eng. J., 2015, 270, 187–195 CrossRef CAS.
- T. Xiong, X. Yuan, X. Chen, Z. Wu, H. Wang, L. Leng, H. Wang, L. Jiang and G. Zeng, Appl. Surf. Sci., 2018, 427, 1107–1117 CrossRef CAS.
- X. F. Yu, Y. H. Liu, Z. W. Zhou, G. X. Xiong, X. H. Cao, M. Li and Z.-B. Zhang, J. Radioanal. Nucl. Chem., 2014, 300, 1235–1244 CrossRef CAS.
- K. Sun, J. Tang, Y. Gong and H. Zhang, Environ. Sci. Pollut. Res., 2015, 22, 16640–16651 CrossRef CAS PubMed.
- H. Cai, X. Lin, L. Tian and X. Luo, Ind. Eng. Chem. Res., 2016, 55, 9648–9656 CrossRef CAS.
- L. Han, H. Sun, K. S. Ro, K. Sun, J. A. Libra and B. Xing, Bioresour. Technol., 2017, 234, 77–85 CrossRef CAS PubMed.
- J. Lu, F. Fu, Z. Ding, N. Li and B. Tang, J. Hazard. Mater., 2017, 330, 93–104 CrossRef CAS PubMed.
- B. Han, E. Zhang, G. Cheng, L. Zhang, D. Wang and X. Wang, Chem. Eng. J., 2018, 338, 734–744 CrossRef CAS.
- Z. Zheng, Y. Wang, W. Zhao, G. Xiong, X. Cao, Y. Dai, Z. Le, S. Yu, Z. Zhang and Y. Liu, J. Radioanal. Nucl. Chem., 2017, 312, 187–198 CrossRef CAS.
- G. Deng, Y. Zhang, X. Luo and J. Yang, Appl. Organomet. Chem., 2018, e4473 CrossRef.
- P. Biller and A. B. Ross, Biofuels, 2012, 3, 603–623 CrossRef CAS.
- S. M. Heilmann, L. R. Jader, L. A. Harned, M. J. Sadowsky, F. J. Schendel, P. A. Lefebvre, M. G. von Keitz and K. J. Valentas, Appl. Energy, 2011, 88, 3286–3290 CrossRef CAS.
- M. Escala, T. Zumbühl, C. Koller, R. Junge and R. Krebs, Energy Fuels, 2013, 27, 454–460 CrossRef CAS.
- D. Kim, K. Yoshikawa and K. Park, Energies, 2015, 8, 12412 Search PubMed.
- E. Steurer and G. Ardissone, Energy Procedia, 2015, 79, 47–54 CrossRef CAS.
- X. Zhuang, H. Zhan, Y. Huang, Y. Song, X. Yin and C. Wu, Bioresour. Technol., 2018, 267, 17–29 CrossRef CAS PubMed.
- D. Basso, F. Patuzzi, D. Castello, M. Baratieri, E. C. Rada, E. Weiss-Hortala and L. Fiori, Waste Manage., 2016, 47, 114–121 CrossRef CAS PubMed.
- V. Benavente, E. Calabuig and A. Fullana, J. Anal. Appl. Pyrolysis, 2015, 113, 89–98 CrossRef CAS.
- M. T. Reza, W. Yan, M. H. Uddin, J. G. Lynam, S. K. Hoekman, C. J. Coronella and V. R. Vásquez, Bioresour. Technol., 2013, 139, 161–169 CrossRef CAS PubMed.
- J. Chen, Z. Chen, C. Wang and X. Li, Mater. Lett., 2012, 67, 365–368 CrossRef CAS.
- S. K. Hoekman, A. Broch and C. Robbins, Energy Fuels, 2011, 25, 1802–1810 CrossRef CAS.
- I. H. Hwang, H. Aoyama, T. Matsuto, T. Nakagishi and T. Matsuo, Waste Manage., 2012, 32, 410–416 CrossRef CAS PubMed.
- S. Kieseler, Y. Neubauer and N. Zobel, Energy Fuels, 2013, 27, 908–918 CrossRef CAS.
- L. Garcia Alba, C. Torri, C. Samorì, J. Van Der Spek, D. Fabbri, S. R. A. Kersten and D. W. F. Brilman, Energy Fuels, 2012, 26, 642–657 CrossRef CAS.
- S. M. Heilmann, H. T. Davis, L. R. Jader, P. A. Lefebvre, M. J. Sadowsky, F. J. Schendel, M. G. von Keitz and K. J. Valentas, Biomass Bioenergy, 2010, 34, 875–882 CrossRef CAS.
- L. Li, Y. Wang, J. Xu, J. R. V. Flora, S. Hoque and N. D. Berge, Bioresour. Technol., 2018, 262, 284–293 CrossRef CAS PubMed.
- C. He, K. Wang, Y. Yang and J.-Y. Wang, Energy Fuels, 2014, 28, 6140–6150 CrossRef CAS.
- C. Liu, J. Xu, J. Hu, H. Zhang and R. Xiao, Chem. Eng. Technol., 2017, 40, 1092–1100 CrossRef CAS.
- G. K. Parshetti, S. K. Hoekman and R. Balasubramanian, Bioresour. Technol., 2013, 135, 683–689 CrossRef CAS PubMed.
- A. M. Smith, S. Singh and A. B. Ross, Fuel, 2016, 169, 135–145 CrossRef CAS.
- L. Gao, M. Volpe, M. Lucian, L. Fiori and J. L. Goldfarb, Energy Convers. Manage., 2019, 181, 93–104 CrossRef CAS.
- I. Van Zandvoort, E. R. H. Van Eck, P. De Peinder, H. J. Heeres, P. C. A. Bruijnincx and B. M. Weckhuysen, ACS Sustainable Chem. Eng., 2015, 3, 533–543 CrossRef CAS.
- I. Van Zandvoort, Y. Wang, C. B. Rasrendra, E. R. H. Van Eck, P. C. A. Bruijnincx, H. J. Heeres and B. M. Weckhuysen, ChemSusChem, 2013, 6, 1745–1758 CrossRef CAS PubMed.
- M. Kleinert and T. Barth, Energy Fuels, 2008, 22, 1371–1379 CrossRef CAS.
- A. Kloekhorst and H. J. Heeres, ACS Sustainable Chem. Eng., 2015, 3, 1905–1914 CrossRef CAS.
- A. L. Jongerius, R. Jastrzebski, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2012, 285, 315–323 CrossRef CAS.
- M. Trautmann, A. Löwe and Y. Traa, Green Chem., 2014, 16, 3710–3714 RSC.
- M. Trautmann, S. Lang and Y. Traa, Fuel, 2015, 151, 102–109 CrossRef CAS.
- Y. Wang, S. Agarwal, Z. Tang and H. J. Heeres, RSC Adv., 2017, 7, 5136–5147 RSC.
- A. Effendi, H. Gerhauser and A. V. Bridgwater, Renewable Sustainable Energy Rev., 2008, 12, 2092–2116 CrossRef CAS.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M. C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 RSC.
- Y. Wang, S. Agarwal, A. Kloekhorst and H. J. Heeres, ChemSusChem, 2016, 9, 951–961 CrossRef CAS PubMed.
- Z. Cheng, B. Saha and D. G. Vlachos, ChemSusChem, 2018, 11, 3545 CrossRef CAS.
- C. B. Rasrendra, M. Windt, Y. Wang, S. Adisasmito, I. G. B. N. Makertihartha, E. R. H. Van Eck, D. Meier and H. J. Heeres, J. Anal. Appl. Pyrolysis, 2013, 104, 299–307 CrossRef CAS.
- S. Agarwal, D. van Es and H. J. Heeres, J. Anal. Appl. Pyrolysis, 2017, 123, 134–143 CrossRef CAS.
- T. M. C. Hoang, L. Lefferts and K. Seshan, ChemSusChem, 2013, 6, 1651–1658 CrossRef CAS PubMed.
- T. M. C. Hoang, E. R. H. Van Eck, W. P. Bula, J. G. E. Gardeniers, L. Lefferts and K. Seshan, Green Chem., 2015, 17, 959–972 RSC.
- M. Nagamori and T. Funazukuri, J. Chem. Technol.Biotechnol., 2004, 79, 229–233 CrossRef CAS.
- M. T. Reza, B. Wirth, U. Lüder and M. Werner, Bioresour. Technol., 2014, 169, 352–361 CrossRef CAS PubMed.
- K. Nakason, B. Panyapinyopol, V. Kanokkantapong, N. Viriya-empikul, W. Kraithong and P. Pavasant, J. Energy Inst., 2017, 1–11 Search PubMed.
- K. Nakason, B. Panyapinyopol, V. Kanokkantapong, N. Viriya-empikul, W. Kraithong and P. Pavasant, J. Energy Inst., 2018, 91, 184–193 CrossRef CAS.
- K. Nakason, B. Panyapinyopol, V. Kanokkantapong, N. Viriya-empikul, W. Kraithong and P. Pavasant, Biomass Convers. Biorefin., 2018, 8, 199–210 CrossRef CAS.
- F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2006, 45, 2163–2173 CrossRef CAS.
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS.
- R. Becker, U. Dorgerloh, E. Paulke, J. Mumme and I. Nehls, Chem. Eng. Technol., 2014, 37, 511–518 CrossRef CAS.
- M. T. Reza, E. Rottler, L. Herklotz and B. Wirth, Bioresour. Technol., 2015, 182, 336–344 CrossRef CAS PubMed.
- J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363–8367 CrossRef CAS PubMed.
- Y. Román-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef PubMed.
- J. Jae, W. Zheng, R. F. Lobo and D. G. Vlachos, ChemSusChem, 2013, 6, 1158–1162 CrossRef CAS PubMed.
- G. M. G. Maldonado, R. S. Assary, J. Dumesic and L. A. Curtiss, Energy Environ. Sci., 2012, 5, 6981–6989 RSC.
- S. Krawielitzki and T. M. Kläusli, Ind. Biotechnol., 2015, 11, 6–8 CrossRef.
- T. Werpy, G. Petersen, A. Aden, J. Bozell, J. Holladay, J. White, A. Manheim, D. Eliot, L. Lasure and S. Jones, Top value added chemicals from biomass. Volume 1-Results of screening for potential candidates from sugars and synthesis gas, 2004 Search PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- Q. Fang and M. A. Hanna, Bioresour. Technol., 2002, 81, 187–192 CrossRef CAS PubMed.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato and S. Hayashi, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef CAS PubMed.
- D. J. Hayes, S. Fitzpatrick, M. H. B. Hayes and J. R. H. Ross, in Biorefineries-Industrial Processes and Products: Status Quo and Future Directions, ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley, 2006, pp. 139–164 Search PubMed.
- A. S. Mamman, J. Lee, Y. Kim, I. T. Hwang, N. Park, Y. K. Hwang and J. Chang, Biofuels, Bioprod. Biorefin., 2008, 438–454 CrossRef CAS.
- J. P. Lange, E. Van Der Heide, J. Van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS PubMed.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
- F. Jin, Z. Zhou, A. Kishita and H. Enomoto, J. Mater. Sci., 2006, 41, 1495–1500 CrossRef CAS.
- S. K. Hoekman, A. Broch, C. Robbins, B. Zielinska and L. Felix, Biomass Convers. Biorefin., 2013, 3, 113–126 CrossRef CAS.
- B. Wirth and J. Mumme, Appl. Bioenergy, 2013, 1–10 Search PubMed.
- E. Erdogan, B. Atila, J. Mumme, M. T. Reza, A. Toptas, M. Elibol and J. Yanik, Bioresour. Technol., 2015, 196, 35–42 CrossRef CAS PubMed.
- X. Lu, J. R. V. Flora and N. D. Berge, Bioresour. Technol., 2014, 154, 229–239 CrossRef CAS PubMed.
- D. Steinbach, A. Kruse, J. Sauer and P. Vetter, Energies, 2018, 11, 645 CrossRef.
- S. Reiche, N. Kowalew and R. Schlögl, ChemPhysChem, 2014, 16, 579–587 CrossRef PubMed.
- P. Körner, S. Beil and A. Kruse, React. Chem. Eng., 2019, 4, 747–762 RSC.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597 LP – 1600 CrossRef PubMed.
- V. Choudhary, S. H. Mushrif, C. Ho, A. Anderko, V. Nikolakis, N. S. Marinkovic, A. I. Frenkel, S. I. Sandler and D. G. Vlachos, J. Am. Chem. Soc., 2013, 135, 3997–4006 CrossRef CAS PubMed.
- S. Hu, Z. Zhang, J. Song, Y. Zhou and B. Han, Green Chem., 2009, 11, 1746–1749 RSC.
- Y. Jiang, L. Yang, C. M. Bohn, G. Li, D. Han, N. S. Mosier, J. T. Miller, H. I. Kenttämaa and M. M. Abu-Omar, Org. Chem. Front., 2015, 2, 1388–1396 RSC.
- W. Weiqi and W. Shubin, Chem. Eng. J., 2017, 307, 389–398 CrossRef.
- H. Lin, Q. Xiong, Y. Zhao, J. Chen and S. Wang, AIChE J., 2016, 63, 257–265 CrossRef.
- X. Fu, J. Dai, X. Guo, J. Tang, L. Zhu and C. Hu, Green Chem., 2017, 19, 3334–3343 RSC.
- L. Yang, J. Su, S. Carl, J. G. Lynam, X. Yang and H. Lin, Appl. Catal., B, 2015, 162, 149–157 CrossRef CAS.
- Q. Hou, M. Zhen, L. Liu, Y. Chen, F. Huang, S. Zhang, W. Li and M. Ju, Appl. Catal., B, 2018, 224, 183–193 CrossRef CAS.
- A. Dibenedetto, M. Aresta, L. Di Bitonto and C. Pastore, ChemSusChem, 2016, 9, 118–125 CrossRef CAS PubMed.
- H. Li, S. Saravanamurugan, S. Yang and A. Riisager, Green Chem., 2016, 18, 726–734 RSC.
- K. Dong, J. Zhang, W. Luo, L. Su and Z. Huang, Chem. Eng. J., 2018, 334, 1055–1064 CrossRef CAS.
- X. Qi, H. Guo, L. Li and R. L. Smith, ChemSusChem, 2012, 5, 2215–2220 CrossRef CAS PubMed.
- J. Wang, L. Zhu, Y. Wang, H. Cui, Y. Y. Zhang and Y. Y. Zhang, J. Chem. Technol. Biotechnol., 2017, 92, 1454–1463 CrossRef CAS.
- J. Howard, D. W. Rackemann, J. P. Bartley, C. Samori and W. O. S. Doherty, ACS Sustainable Chem. Eng., 2018, 6, 4531–4538 CrossRef CAS.
- A. G. Demesa, A. Laari, E. Tirronen and I. Turunen, Chem. Eng. Res. Des., 2015, 93, 531–540 CrossRef CAS.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1938 CrossRef PubMed.
- T. Shimanouchi, Y. Kataoka, M. Yasukawa, T. Ono and Y. Kimura, Solvent Extr. Res. Dev., 2013, 20, 205–212 CrossRef CAS.
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2013, 15, 85–90 RSC.
- L. C. Blumenthal, C. M. Jens, J. Ulbrich, F. Schwering, V. Langrehr, T. Turek, U. Kunz, K. Leonhard and R. Palkovits, ACS Sustainable Chem. Eng., 2016, 4, 228–235 CrossRef CAS.
- E. Weingart, L. Teevs, R. Krieg and U. Prüße, Energy Technol., 2018, 6, 432–440 CrossRef CAS.
- E. C. Sindermann, A. Holbach, A. de Haan and N. Kockmann, Chem. Eng. J., 2016, 283, 251–259 CrossRef CAS.
- S. Mohammad, G. Grundl, R. Müller, W. Kunz, G. Sadowski and C. Held, Fluid Phase Equilib., 2016, 428, 102–111 CrossRef CAS.
- G. Grundl, M. Müller, D. Touraud and W. Kunz, J. Mol. Liq., 2017, 236, 368–375 CrossRef CAS.
- F. Liu, S. Sivoththaman and Z. Tan, Sustainable Environ. Res., 2014, 24, 149–157 Search PubMed.
- H. Wang, S. Liu, Y. Zhao, H. Zhang and J. Wang, ACS Sustainable Chem. Eng., 2016, 4, 6712–6721 CrossRef CAS.
- J. Zhou, T. Huang, Y. Zhao, Z. Xia, Z. Xu, S. Jia, J. Wang and Z. C. Zhang, Ind. Eng. Chem. Res., 2015, 54, 7977–7983 CrossRef CAS.
- X. Sun, Z. Liu, Z. Xue, Y. Zhang and T. Mu, Green Chem., 2015, 17, 2719–2722 RSC.
- T. Brouwer, M. Blahusiak, K. Babic and B. Schuur, Sep. Purif. Technol., 2017, 185, 186–195 CrossRef CAS.
- J. Briscoe, A. Marinovic, M. Sevilla, S. Dunn and M. Titirici, Angew. Chem., Int. Ed., 2015, 54, 4463–4468 CrossRef CAS PubMed.
- R. Wang, K. Q. Lu, Z. R. Tang and Y. J. Xu, J. Mater. Chem. A, 2017, 5, 3717–3734 RSC.
- G. A. M. Hutton, B. C. M. Martindale and E. Reisner, Chem. Soc. Rev., 2017, 46, 6111–6123 RSC.
- X. Sun and Y. Li, Angew. Chem., Int. Ed., 2004, 43, 597–601 CrossRef PubMed.
- X. Jia, J. Li and E. Wang, Nanoscale, 2012, 4, 5572–5575 RSC.
- Z. Song, F. Quan, Y. Xu, M. Liu, L. Cui and J. Liu, Carbon, 2016, 104, 169–178 CrossRef CAS.
- M. L. Liu, B. Bin Chen, C. M. Li and C. Z. Huang, Green Chem., 2019, 21, 449–471 RSC.
- B. De and N. Karak, RSC Adv., 2013, 3, 8286–8290 RSC.
- J. Shen, S. Shang, X. Chen, D. Wang and Y. Cai, Mater. Sci. Eng., C, 2017, 76, 856–864 CrossRef CAS PubMed.
- D. Qu, M. Zheng, L. Zhang, H. Zhao, Z. Xie, X. Jing, R. E. Haddad, H. Fan and Z. Sun, Sci. Rep., 2015, 4, 5294 CrossRef PubMed.
- J. Liu, S. Lu, Q. Tang, K. Zhang, W. Yu, H. Sun and B. Yang, Nanoscale, 2017, 9, 7135–7142 RSC.
- R. Atchudan, T. N. J. I. Edison, D. Chakradhar, S. Perumal, J. J. Shim and Y. R. Lee, Sens. Actuators, B, 2017, 246, 497–509 CrossRef CAS.
- N. Papaioannou, A. Marinovic, N. Yoshizawa, A. E. Goode, M. Fay, A. Khlobystov, M. Titirici and A. Sapelkin, Sci. Rep., 2018, 8, 6559 CrossRef PubMed.
- H. Luo, N. Papaioannou, E. Salvadori, M. M. Roessler, G. Ploenes, E. R. H. van Eck, L. C. Tanase, J. Feng, Y. Sun, Y. Yang, M. Danaie, A. Belen Jorge, A. Sapelkin, J. Durrant, S. D. Dimitrov and M. M. Titirici, ChemSusChem, 2019, 12, 4432–4441 CrossRef CAS PubMed.
- H. Shengliang, T. Fei, B. Peikang, C. Shirui, S. Jing and Y. Jing, Mater. Sci. Eng., B, 2009, 157(1–3), 11–14 Search PubMed.
- R. Tian, S. Zhong, J. Wu, Y. Geng, B. Zhou, Q. Wang and W. Jiang, J. Mater. Chem. C, 2017, 5, 9174–9180 RSC.
- F. Ehrat, S. Bhattacharyya, J. Schneider, A. Löf, R. Wyrwich, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2017, 17, 7710–7716 CrossRef CAS PubMed.
- B. Dai, C. Wu, Y. Lu, D. Deng and S. Xu, J. Lumin., 2017, 190, 108–114 CrossRef CAS.
- J. Yang, W. Chen, X. Liu, Y. Zhang and Y. Bai, Mater. Res. Bull., 2017, 89, 26–32 CrossRef CAS.
- Z. Tian, X. Zhang, D. Li, D. Zhou, P. Jing, D. Shen, S. Qu, R. Zboril and A. L. Rogach, Adv. Opt. Mater., 2017, 5, 1700416 CrossRef.
- T. Zhang, J. Zhu, Y. Zhai, H. Wang, X. Bai, B. Dong, H. Wang and H. Song, Nanoscale, 2017, 9, 13042–13051 RSC.
- S. K. Bhunia, A. Saha, A. R. Maity, S. C. Ray and N. R. Jana, Sci. Rep., 2013, 3, 1473 CrossRef PubMed.
- R. Purbia and S. Paria, Biosens. Bioelectron., 2016, 79, 467–475 CrossRef CAS PubMed.
- P. Yang, J. Zhao, L. Zhang, L. Li and Z. Zhu, Chem. – Eur. J., 2015, 21, 8561–8568 CrossRef CAS PubMed.
- Q. Xu, Y. Liu, C. Gao, J. Wei, H. Zhou, Y. Chen, C. Dong, T. S. Sreeprasad, N. Li and Z. Xia, J. Mater. Chem. C, 2015, 3, 9885–9893 RSC.
- D.-W. Zhang, N. Papaioannou, N. M. David, H. Luo, H. Gao, L. C. Tanase, T. Degousée, P. Samorì, A. Sapelkin, O. Fenwick, M.-M. Titirici and S. Krause, Mater. Horiz., 2018, 5, 423–428 RSC.
- A. Marinovic, L. S. Kiat, S. Dunn, M. M. Titirici and J. Briscoe, ChemSusChem, 2017, 10, 1004–1013 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |