
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTandem catalytic aromatization of volatile fatty acids†
Egor V.
Fufachev
 a,
Bert M.
Weckhuysen
a and
Pieter C. A.
Bruijnincx
*ab
a,
Bert M.
Weckhuysen
a and
Pieter C. A.
Bruijnincx
*ab
aUtrecht University, Inorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, The Netherlands. E-mail: p.c.a.bruijnincx@uu.nl
bUtrecht University, Organic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, The Netherlands
First published on 27th April 2020
Abstract
The transition towards a circular economy requires closing the carbon loop, e.g. by the development of new synthesis routes to valuable intermediates and products from organic-rich waste streams. Volatile fatty acids (VFA) can be fermentatively produced from wastewater and serve as circular platform chemicals. We show that these VFA can be catalytically upgraded to light aromatics (i.e., benzene, toluene, ethylbenzene and xylenes, BTEX) via a tandem catalytic reaction involving TiO2-catalyzed ketonization and zeolite ZSM-5 catalyzed aromatization. Including this intermediate ketonization step is demonstrated to be much more efficient than direct VFA aromatization, as direct acid conversion mainly gave rise to short-chain olefins by decarboxylation and low BTEX yields of 1%. A one-reactor, tandem catalytic conversion instead significantly improved the yield to 45% when zeolite Ga/ZSM-5 was used. Furthermore, the effect of VFA-derived ketone composition, a process parameter set by the fermentation process, on aromatics production efficiency and product distribution was found to be very pronounced for zeolite Ga/ZSM-5, but not for non-promoted zeolite HZSM-5. This suggests a different reaction mechanism to dominate on zeolite Ga/ZSM-5, involving dehydration on the Brønsted acid sites and cyclization/aromatization on the Ga sites. Finally, water, expected to be present in the feed during VFA upgrading, caused the activity of zeolite Ga/ZSM-5 to drop reversibly, but also led to lower coke buildup. Analysis of the spent catalyst with solid-state 27Al nuclear magnetic resonance spectroscopy and temperature-programmed reduction with H2 showed that the catalyst structure remained intact, also with water present in the feed. Together, the results demonstrate that catalytic ketonization/aromatization is an attractive circular approach for converting waste-derived carboxylic acids into renewable aromatics.
1. Introduction
The production of bulk chemicals, such as benzene, toluene, ethylbenzene and xylenes (BTEX), from renewable feedstock has been the focus of much current research in both academia and industry. As an alternative to the well-studied routes for aromatics production from biomass, e.g., via pyrolysis,1 lignin depolymerization2 or sugar-derived furan aromatization,3 BTEX production from fermented waste-water streams provides an attractive circular option. This way, valuable chemicals production is combined with simultaneous minimization of the amount of waste.Fermentation is a well-established route for the production of various chemicals at low cost and high efficiency.4 The production of short-chain carboxylic acids, so-called volatile fatty acids (VFA), by fermentation has been reported for various feedstocks ranging from industrial wastewater5 to urban6 and food7 waste. The main carboxylic acids produced by such a fermentation process are acetic, propionic and butyric acids, versatile chemicals with myriad applications. The complexity of a typical fermentation broth,8 containing considerable amounts of salts and the VFA at low concentration make direct catalytic upgrading of VFA in the broth unfeasible. Instead, isolation of the VFA by a robust extraction technique prior to further conversion is required. VFA can be selectively extracted from dilute aqueous streams either by liquid–liquid extraction or by adsorption on a resin.5,8–11 Upon thermal treatment, the extracted VFA can be liberated into the gas phase and isolated or can undergo an in-line, gas-phase catalytic conversion. An example of the latter approach is VFA upgrading by ketonization over a metal oxide catalyst9,12 to a mixture of ketones, a versatile platform for the synthesis of various value-added chemicals or fuels.13 As an alternative to the typical feedstock used for oxygenates-to-aromatics processes, these VFA-derived ketones can, for example, be converted to aromatic hydrocarbons. Alternatively, the recovered VFA could in principle also be converted directly to aromatics (Scheme 1).
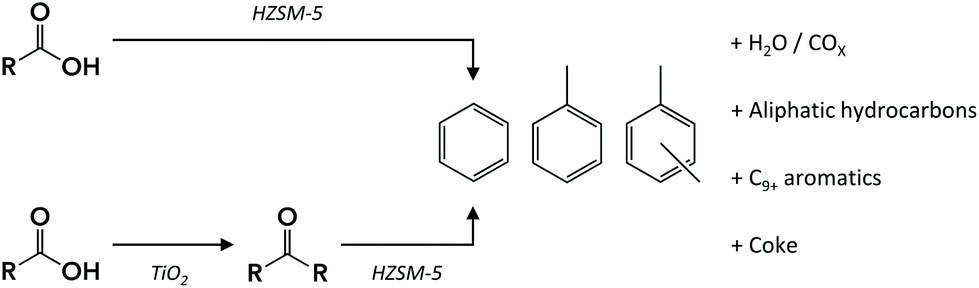 | ||
| Scheme 1 Synthesis routes for the production of BTEX from waste-derived VFA. | ||
Indeed, the zeolite-catalyzed conversion of short-chain oxygenates (including alcohols,14–16 esters,16 aldehydes,15–19 furans20 and acids15,17,21) into (aromatic) hydrocarbons has been extensively investigated, particularly in model compound studies of the chemistry shown by individual components during bio-oil upon upgrading.16–18,21,22 Zeolite HZSM-5 is then commonly used for the deoxygenation of such biomass-derived oxygenates. Its high surface area together with a high density of strong acid sites, appropriate pore size and (shape) selectivity can allow for efficient conversion of the oxygenates to light aromatics. Several authors15,16,23 have compared the reactivities of various oxygenates and found the selectivity to BTEX to follow the sequence: acids < alcohols/ethers/esters < ketones/aldehydes. However, while the literature on conversion of alcohols, furans and aldehydes over zeolite HZSM-5 is vast, the number of studies focused on the conversion of ketones is very limited.
The direct transformation of acids over zeolite catalysts (Fig. 1) is proposed to proceed via two major pathways: decarboxylation to CO2 and hydrocarbon gases16,17,23 or ketonization of two acid molecules to form a ketone, carbon dioxide and water.21,23 This ketonization reaction has been much more extensively studied on metal oxides such as TiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12,24 and ZrO2,25 with far fewer examples existing for zeolite-mediated ketonization.26 Gumidyala et al.27 reported on almost fully selective ketone production over HZSM-5 at temperatures of 270–330 °C. Upon further temperature increase, the selectivity shifted to sequential aldol condensation reactions of the ketone. Cyclization and aromatization reactions of those aldol products produce C10+ aromatics that upon dealkylation can yield BTEX.21 Direct cracking of aldols to C4–C7 hydrocarbons and subsequent oligomerization to aromatics is a concurrent route.15,16,21 Thus, the conversion of acids to aromatics is thought to proceed via sequential acid to ketone and ketone to BTEX reactions. Alternatively, ketones also can undergo hydrogenation/dehydration or decarboxylation/decarbonylation reactions, depending on the reaction temperature,17 forming aliphatic hydrocarbons that contribute to aromatics formation by cracking, oligomerization and aromatization reactions. All these reactions result in a complex product mixture of paraffins (C1–C4), olefins (C2–C4), aliphatic compounds (C5+) and alkylated aromatics (C6–C10+). Side reactions contribute to coke formation, which is often considerable for oxygenates such as furans,20 aldehydes18,19 and alcohols.28 Indeed, Adjaye et al.16 studied the conversion of various oxygenates over HZSM-5 and reported a fivefold higher amount of coke with propionic acid than with 2-methylcyclopentanone. More recently, Wang et al.21 reported that HZSM-5 deactivated faster with propionic acid than with 3-pentanone, attributing the difference to stronger adsorption of the acid on the zeolite acid sites. Taken together, ketones in fact seem to be more desirable reactants for aromatics production than their acid precursors.
12,24 and ZrO2,25 with far fewer examples existing for zeolite-mediated ketonization.26 Gumidyala et al.27 reported on almost fully selective ketone production over HZSM-5 at temperatures of 270–330 °C. Upon further temperature increase, the selectivity shifted to sequential aldol condensation reactions of the ketone. Cyclization and aromatization reactions of those aldol products produce C10+ aromatics that upon dealkylation can yield BTEX.21 Direct cracking of aldols to C4–C7 hydrocarbons and subsequent oligomerization to aromatics is a concurrent route.15,16,21 Thus, the conversion of acids to aromatics is thought to proceed via sequential acid to ketone and ketone to BTEX reactions. Alternatively, ketones also can undergo hydrogenation/dehydration or decarboxylation/decarbonylation reactions, depending on the reaction temperature,17 forming aliphatic hydrocarbons that contribute to aromatics formation by cracking, oligomerization and aromatization reactions. All these reactions result in a complex product mixture of paraffins (C1–C4), olefins (C2–C4), aliphatic compounds (C5+) and alkylated aromatics (C6–C10+). Side reactions contribute to coke formation, which is often considerable for oxygenates such as furans,20 aldehydes18,19 and alcohols.28 Indeed, Adjaye et al.16 studied the conversion of various oxygenates over HZSM-5 and reported a fivefold higher amount of coke with propionic acid than with 2-methylcyclopentanone. More recently, Wang et al.21 reported that HZSM-5 deactivated faster with propionic acid than with 3-pentanone, attributing the difference to stronger adsorption of the acid on the zeolite acid sites. Taken together, ketones in fact seem to be more desirable reactants for aromatics production than their acid precursors.
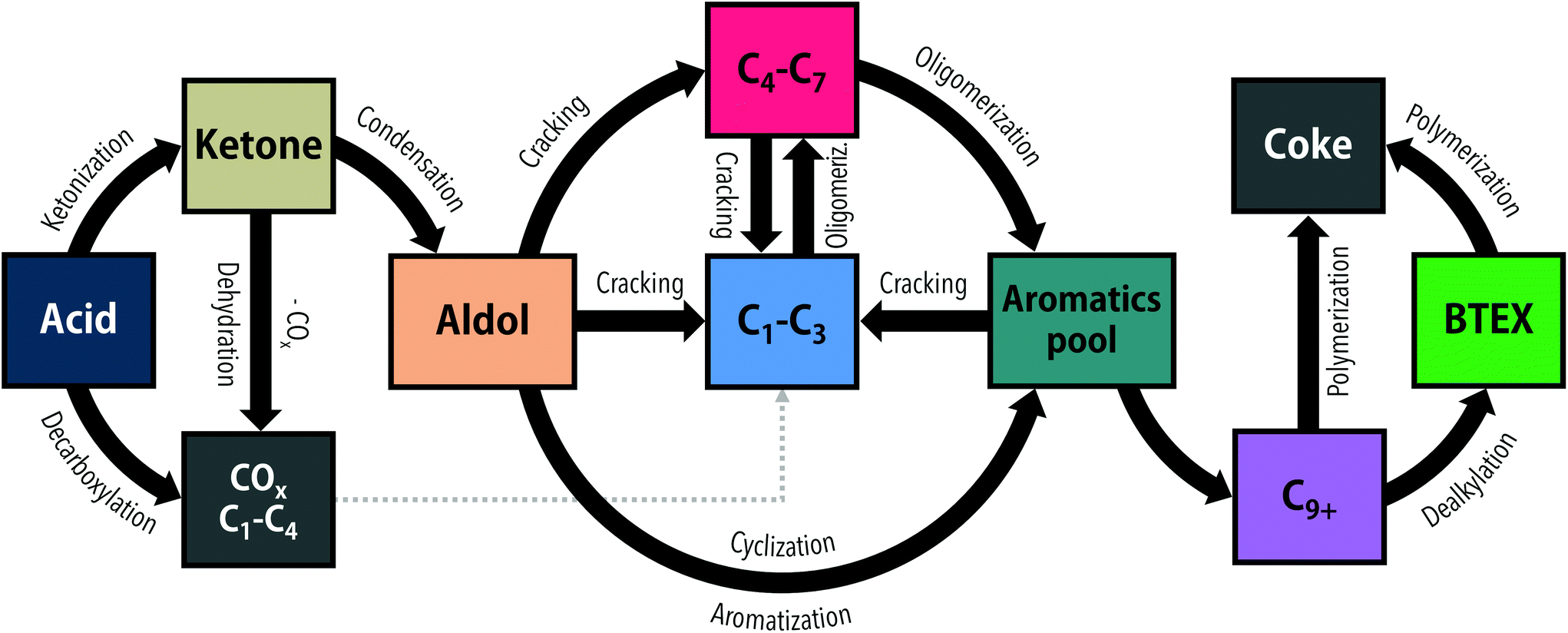 | ||
| Fig. 1 General reaction pathways for the conversion of acids and ketones over a HZSM-5 catalyst. | ||
Incorporation of transition metals, such as Ga and Zn, into zeolites, is a well-known method to enhance selectivity towards aromatics and is widely applied for the conversion of paraffins and olefins.29–33 The number of studies on the conversion of oxygenates over metal-promoted zeolites, however, is again rather limited. Yu et al.14 reported on a significant enhancement of the BTEX yield from isobutyl alcohol upon addition of Zn. A beneficial effect of Ga-promotion on BTEX selectivity in the co-aromatization reaction of 2,5-dimethylfuran with ethylene was in turn observed by Uslamin et al.20 These metal promoters decrease Brønsted acidity, thus suppressing cracking reactions, and introduce strong Lewis acid sites with strong dehydrogenation activity.
When integrated recovery and catalytic conversion of extracted VFA is targeted, one has to deal with a mixture of liberated acids or generated ketones as well as with co-liberated water.12 Fuhse et al.23 observed an effect of acid carbon chain length on ketonization catalyst deactivation, but little is known on how substrate variation influences subsequent ketone aromatization efficiency. For example, acetone was found to be selectively converted to i-butene, while the higher ketones produced a significant amount of aromatics.34 The effect of co-extracted water is also anticipated to be significant,8,12 as water is known to improve the long-term stability of zeolites17,35 by suppressing coke formation. Hensen et al.,36 for example, observed that co-feeding of water led to a substantial increase in the propane dehydrogenation rate over zeolite Ga/HZSM-5 and improved catalyst stability by the formation of reactive binuclear hydroxyl-bridged Ga species. On the other hand, the presence of water can also lead to dealumination and structure collapse of a zeolite material.37
Here, we demonstrate efficient aromatics production from circular VFA building blocks by tandem catalytic ketonization–aromatization. We also show that VFA ketonization prior to aromatization of VFA is much more efficient in terms of BTEX yield than direct VFA aromatization. Furthermore, a significant effect of feed complexity, i.e. the use of acid and ketone mixtures and adding water to the feed, on activity and aromatic product distribution is presented. Catalyst characterization provided insight into the stability of the zeolites under these conditions. Taken together, this first example of tandem catalytic VFA valorization is shown to be an attractive strategy for the conversion of waste-derived carboxylic acids into light aromatics of direct use in the chemical industry.
2. Experimental
2.1. Catalyst preparation
Protonated HZSM-5 catalyst (Si/Al = 25) was prepared by deammonization of the corresponding NH4ZSM-5 (Zeolyst, CBV 5524G) at 550 °C for 6 h in stagnant air. This catalyst is denoted as HZ. Ga-Doped HZSM-5 was prepared by wet impregnation: 3 g of HZ was crushed by pestle and mortar and dried at 120 °C for 2 h. The dried zeolite was dispersed in 150 mL of MilliQ water (18,2 MΩ cm at 25 °C) and mechanically stirred at 1000 rpm for 30 min. An aqueous solution of Ga(NO3)3·xH2O (Acros, 99.9998% trace metal basis) was added dropwise to the suspended HZSM-5, to obtain a Ga-loading of 4.5 wt% and mixed for 3 h. Water was removed using a rotary evaporator. The resulting solid was dried overnight at 120 °C followed by calcination at 550 °C in air for 5.5 h with a temperature ramp of 5 °C min−1. The catalysts were subjected to two reduction/oxidation cycles. The reduction was performed under 150 mL min−1 flow of H2 at 510 °C (5 °C min−1) for 4 h and reoxidation under 150 mL min−1 flow of air at 510 °C (5 °C min−1) for 4 h. The resulting catalyst material is denoted as Ga/HZ.2.2. Catalyst characterization
Fourier-transform infrared spectra of adsorbed pyridine were recorded on a Thermo IS5 spectrometer with a resolution of 4 cm−1 over 32 scans. The sample was pressed into a self-supported pellet and placed into an FT-IR cell with CaF2 windows. Prior to pyridine adsorption, the sample was dried at 450 °C for 1 h under ultra-high vacuum (10−7–10−9 mbar). Pyridine (18–20 mbar) was introduced to the cell at 150 °C, and after 30 min the spectrum was recorded. The sample was heated to 550 °C with a temperature ramp of 5 °C min−1, with a spectrum measured every 150 °C. The number of acid sites was calculated based on the equation (eqn (1)) derived from Beer's law: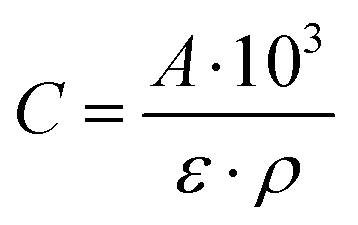 | (1) |
240 scans were accumulated using an inter-scan delay of 1 s. The 27Al chemical shift was externally referenced to an aqueous aluminium(III) nitrate solution. Temperature-programmed reduction (TPR-H2) analysis was performed on a Micrometics Autochem II analyser. About 50 mg of a sample was dried at 450 °C for 1 h under N2 flow. Subsequently, the sample was heated to 1000 °C with a rate of 5 °C min−1 under a flow of 5% H2 in He. H2 consumption was measured with a TCD-detector.
2.3. Catalyst testing
Gas-phase aromatization reactions were studied in a conventional fixed bed reactor (Fig. S1†). The liquid feed was pumped by an HPLC pump (LC-20AT, Shimadzu) with a weight hourly space velocity (WHSV) of 1 h−1. For acid conversion reactions, butyric acid (Acros Organics, 99+%) was used as the feed. For ketone conversion reactions, 4-heptanone (Alfa Aesar, 98%), 3-hexanone (Acros Organics, 98+%) or a ketones mixture with an 1:6.4:19.9:36.7 molar ration of 2-butanone (Sigma-Aldrich, ≥99.5%), 3-pentanone (Alfa Aesar, ≥99%), 3-hexanone (Acros Organics, 98+%) and 4-heptanone (Alfa Aesar, 98%) were fed to evaporator #1 (Fig. S1†). For the reactions with water, 60 vol% of MilliQ water (18.2 MΩ cm at 25 °C) was co-fed via evaporator #2 (Fig. S1†). Cyclooctane (Sigma-Aldrich, ≥99%) was used as an internal standard. A mass flow controller (F-201CV, Bronkhorst) was used to control the flow of N2 carrier gas (100 mL min−1). A quartz reactor (i.d. 8 mm) was loaded with a zeolite catalyst (particle size of 75–38 μm) mixed with two volumes of silicon carbide. For the ketonization and the tandem ketonization/aromatization reactions, TiO2 P25 (Aeroxide, Evonik) with a particle size of 212–150 μm mixed with two volumes of SiC was used. Prior to the reaction, a catalyst bed was dried at 450 °C for 1 h under N2 flow (100 mL min−1). The catalytic studies were performed at atmospheric pressure. The reaction products were analysed with an on-line gas chromatograph (GC, Bruker, 430-GC) equipped with flame ionization detector (FID) and a PoraPLOT Q-HT 25 × 0.32 analytical column. Ketone conversion and products yield were calculated using eqn (S1)–(S3) as reported in the ESI.†
3. Results and discussion
3.1. Direct and tandem-catalytic conversion of butyric acid
Zeolite-catalyzed aromatics production from waste-derived VFA can either be attempted directly or in stepwise fashion, e.g. by inclusion of a catalytic ketonization step prior to the aromatization reaction (Scheme 1). The direct conversion of butyric acid, the major component of a typical VFA mixture,12 over HZ zeolite (Fig. 2a) mainly produced a mixture of short-chain olefins, ∼90% of which is propylene, with only traces of aromatic products, corresponding to a carbon yield of 1%. The unidentified products denoted as “others” in Fig. 2 include the COx gases and any coke formed. That decarboxylation is the main route under these conditions is in line with previous reports.17 For example, Adjaye et al.16 reported that propionic acid conversion over HZSM-5 (Si/Al = 28) at 410 °C gave significant amounts of coke (∼26%) and gaseous products (∼38%, of which 50–80% CO2) and an aromatics yield of only ∼8%. Similarly, Chang et al.15 and Gayubo et al.17 observed acetone and i-butene as the main products during the conversion of acetic acid over HZSM-5, formed via ketonization and subsequent aldol condensation/cracking reactions, respectively. More recently, Wang et al.21 initially observed a high aromatics yield (C6–C10+) in the reaction of propionic acid over HZSM-5 (Si/Al = 40) at 350 °C, but the catalyst was found to rapidly deactivate due to the strong adsorption of the acid on the zeolite.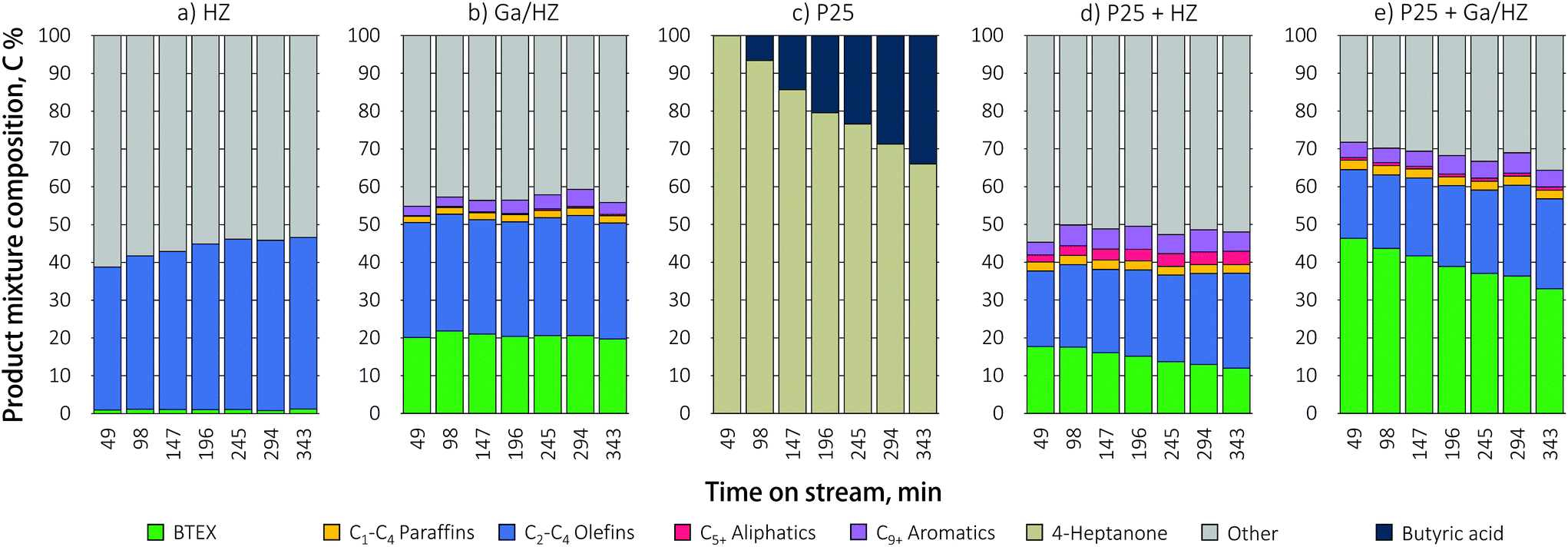 | ||
| Fig. 2 Product mixture composition during butyric acid conversion over TiO2, (promoted) zeolite and a mixed TiO2/(promoted) zeolite catalytic bed (450 °C; WHSV = 1 h−1). | ||
The use of a Ga-promoted zeolite (Fig. 2b) improved the efficiency of direct butyric acid conversion, giving a BTEX yield of 20%. This considerable increase is attributed to the introduction of dehydrogenating (GaO)+ Lewis acid sites at the expense of strong Brønsted sites, thus suppressing cracking reactions.20,31
Tandem conversion by ketonization/aromatization over a dual catalytic bed packed in one reactor was studied as an alternative to the direct, low yielding conversion of butyric acid. Including such an intermediate ketonization step indeed proved highly beneficial. First, the conversion of butyric acid over TiO2 P25, a prototypical ketonization catalyst,26 is shown to proceed efficiently, giving the 4-heptanone product at high selectivity (Fig. 2c). However, catalyst deactivation was observed with time on stream at 450 °C for this single catalyst, probably due to coking. The reason for this deactivation is that the residence time, set such to allow comparison with the direct acid conversion route, is rather high for this ketonization catalyst at this temperature. Next, the TiO2 catalyst was combined with a zeolite catalyst in a stacked beds configuration with the zeolite bed located downstream of the titania catalyst. A TiO2 to HZSM-5 weight ratio of 1:6 was used to obtain an WHSV of 1 h−1 of the formed ketone(s) with respect to the zeolite. A stacked bed rather than a mixed bed configuration was chosen as this allows for the selective primary conversion of the acid to the ketone. The amount of TiO2 P25 catalyst was then adapted such that the conditions allowed for full conversion of the acid, but not for any sequential ketone reduction/dehydration reactions that would give olefinic byproducts.39 The results in Fig. 2d and e demonstrate that the conversion over dual metal oxide/zeolite catalytic bed gave considerably higher aromatic yields over both the zeolites, with the BTEX yield increasing from 1% to 18% over P25/HZ and from 20% to 46% upon combining the Ga-doped zeolite with titania. Tandem catalytic conversion of the acid over the dual bed reactor showed a shift in selectivity towards toluene formation compared to Ga/HZ alone, which produced more or less equal amounts of benzene and toluene (Fig. 3). The propylene selectivity in the olefin fraction dropped (Fig. 3), implying a more significant contribution of C9+ aromatic cracking reactions upon combining the zeolite with TiO2. Fig. 2e shows the aromatics yield to drop gradually, but at a much slower rate than observed for the TiO2 only example (Fig. 2c). The shift in product distribution from aromatics to olefins is thought to be due decarboxylation or strong adsorption of unreacted acid reaching the zeolite bed, this as a result of gradual deactivation of the ketonization catalyst. Overall, the results clearly show the advantage of converting the acid to aromatics via an intermediate ketonization step. Further optimization of the process by, e.g. conversion over separate ketonization and aromatization reactors with different temperature regimes would allow the stability of the primary ketonization catalyst to be improved, allowing the BTEX yields to be further increased.
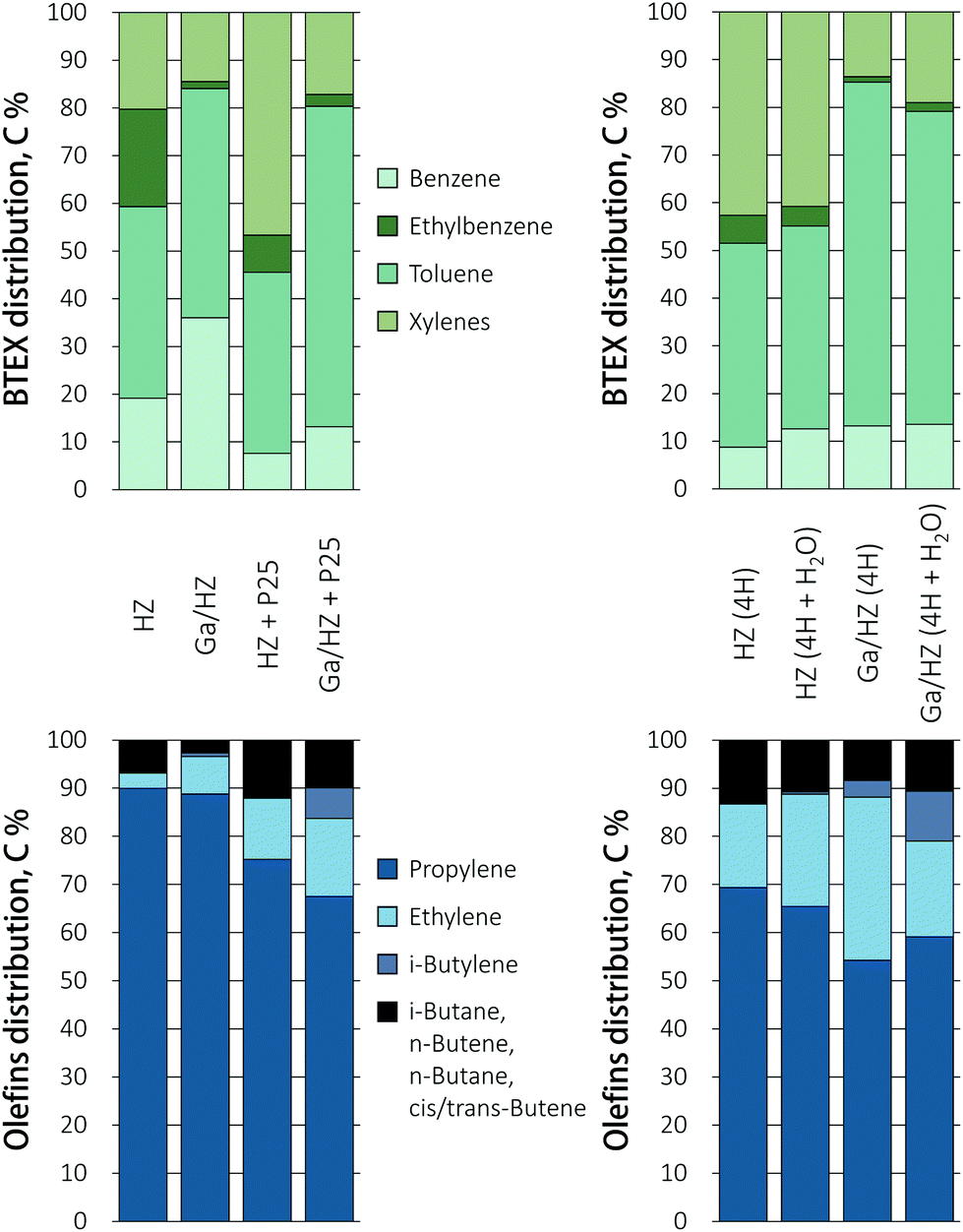 | ||
| Fig. 3 Distribution of aromatic and olefin products in the conversion of butyric acid and 4-heptanone (+H2O). TOS = 49 min. | ||
3.2. Zeolite-catalyzed ketone conversion
The effect of using ketones with odd and even numbered carbon chain lengths on the distribution of BTEX, was investigated over the zeolite catalysts. Previously, conversion of various ketones over HZSM-5 (Si/Al = 65) was shown to result in some small, non-linear changes in BTEX distribution upon increasing the carbon chain.23 We studied feeds consisting of 4-heptanone, 3-hexanone or a ketone mixture of 4-heptanone, 3-hexanone, 3-pentanone and 2-butanone (in a molar ratio of 23:14:5:1, reflecting the product of VFA ketonization). The product mixture composition as function of feed composition over HZ is illustrated in Fig. 4. Notably, no deactivation of the catalyst was observed with time on stream for any of the ketone reactants, supporting the notion that the deactivation seen in Fig. 2 is acid induced.
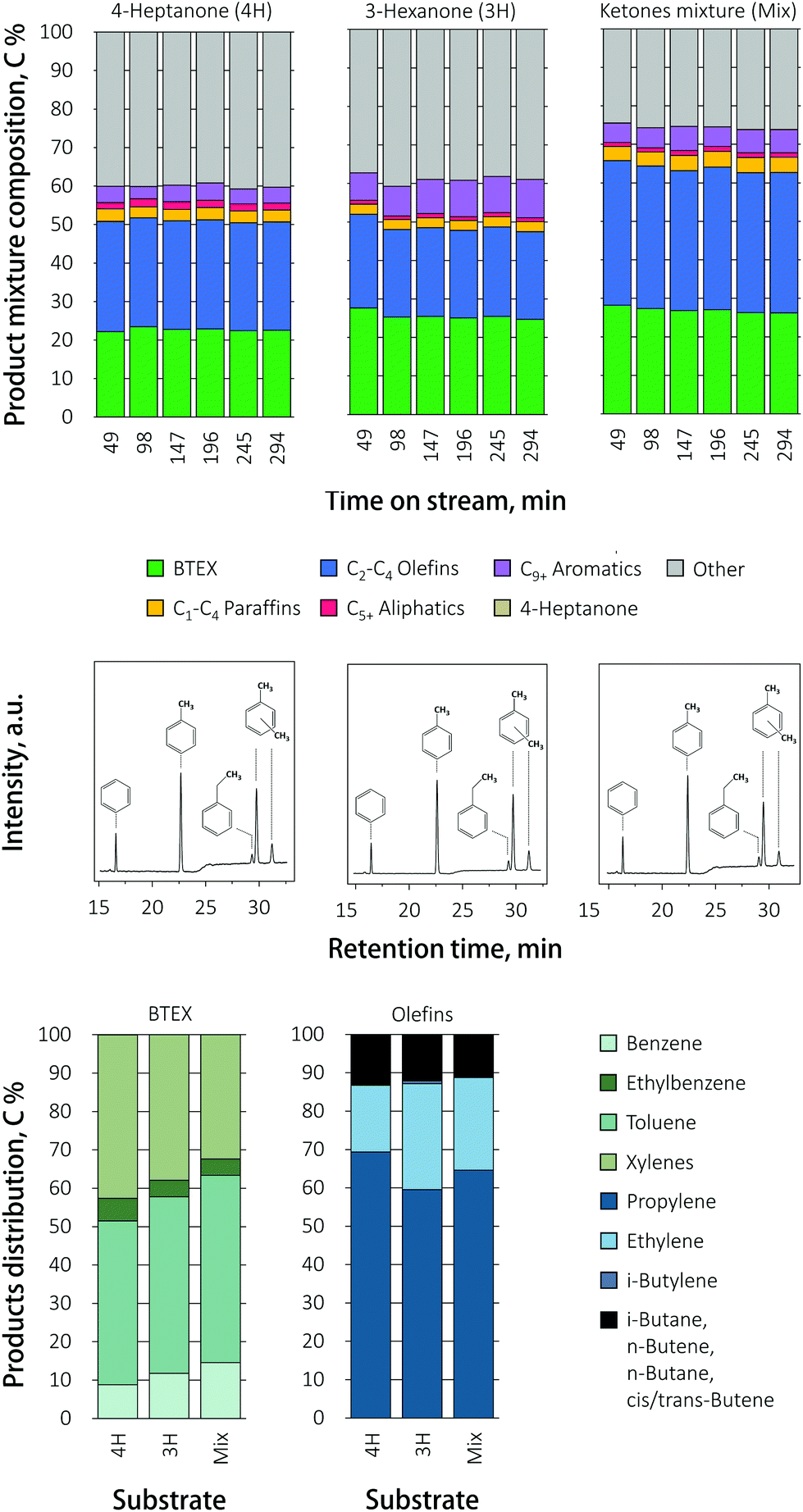 | ||
| Fig. 4 Product mixture composition, BTEX distribution and corresponding gas chromatograms (time-on-stream, TOS = 49 min) recorded during conversion of various ketones over HZ catalyst (450 °C; WHSV = 1 h−1). | ||
The total mass balance did increase when the ketones mixture was fed to the reactor, as slightly did the BTEX yield, in comparison to pure 3-hexanone or 4-heptanone. Selectivity shifted from benzene and toluene to ethylbenzene for 3-hexanone and the ketone mixture, while the production of xylenes stayed almost the same. The olefin distribution shifted from C3 propylene to C2 ethylene for 3-hexanone and the ketone mixture, compared to 4-heptanone.
The influence of the ketone feed composition on product distribution over the promoted Ga/HZ zeolite is presented in Fig. 5. As with undoped HZ, the conversion of 3-hexanone and the mixture proceeded with higher selectivity to ethylene compared to 4-heptanone. The total BTEX yields were the same for the various ketones. Interestingly, the BTEX yield obtained during the direct conversion of 4-heptanone over Ga/HZ (55%) is higher than the highest BTEX yield (45%) seen with butyric acid over the dual bed configuration. This difference is thought to be caused by the water that is produced in stoichiometric amounts in the first ketonization reaction. The distribution of aromatic products was now found to be more strongly governed by the feed composition: while toluene C7 is the primary product in the reaction of 4-heptanone (C7), the selectivity shifted to benzene C6 when 3-hexanone (C6) was fed. The conversion of the ketone mixture also gave a higher benzene yield; this shift in BTEX product distribution is also evident from the clean gas chromatograms (Fig. 5). Since the influence of feed composition is different for pristine and promoted zeolites, it is reasonable to assume that the reaction proceeds via different (dominant) mechanisms over these two catalysts. Possible pathways for ketone conversion over unpromoted HZSM-5 were reported before and are discussed above. FT-IR spectra after pyridine adsorption showed that the addition of Ga results in the conversion of up to 60% of the Brønsted acid sites on pristine HZ zeolite (111.5 μmol g−1 (BAS) and 17.7 μmol g−1 (LAS)) to Lewis acid sites on Ga/HZ zeolite (40 μmol g−1 (BAS) and 75 μmol g−1 (LAS)) in the form of (GaO)+ cations, as also observed by TPR (see below). The Brønsted acid sites are considered the active sites for the primary aldol condensation reaction.21 It is then reasonable to assume that the substitution of most of the BAS with (GaO)+ leads to a shift in dominant deoxygenation mechanism, changing the final product mixture. Notably, Rane et al.33 previously observed toluene C7 as the only aromatic product during the aromatization of n-heptane over a Ga/HZSM-5 catalyst prepared by chemical vapour deposition. All Brønsted acid sites of the zeolites were found to be replaced by monovalent Ga+ species by this method, suggesting that Ga+ cations are thus able to catalyse the required dehydrogenation reactions, including the necessary ring-closure step in the absence on Brønsted acid sites for this alkane. Therefore, it is assumed that the conversion of the ketone over Ga/HZSM-5 could proceed (partially) via the formation of an alkene that undergoes subsequent dehydrogenation/cyclization/aromatization on the Ga sites. These olefins can be produced via a secondary reduction of ketones to alcohols, and subsequent dehydration to the olefin, with the hydrogen source for this reduction assumed to come from cyclization of the ketone condensation product.39 An alternative mechanism for olefin formation involves secondary (cross) condensation to a diketone alcohol that decomposes to the corresponding iso-olefin and acid.40 The distribution of aromatics would then be determined by the structure of ketone on the Ga/HZ catalyst, while the effect of ketone structure is negligible over HZ due to the formation of bulky C9+ aromatics first that dealkylate to light BTEX molecules. While this fits the data presented, clearly, further investigations are required to unravel the complex mechanisms at play.
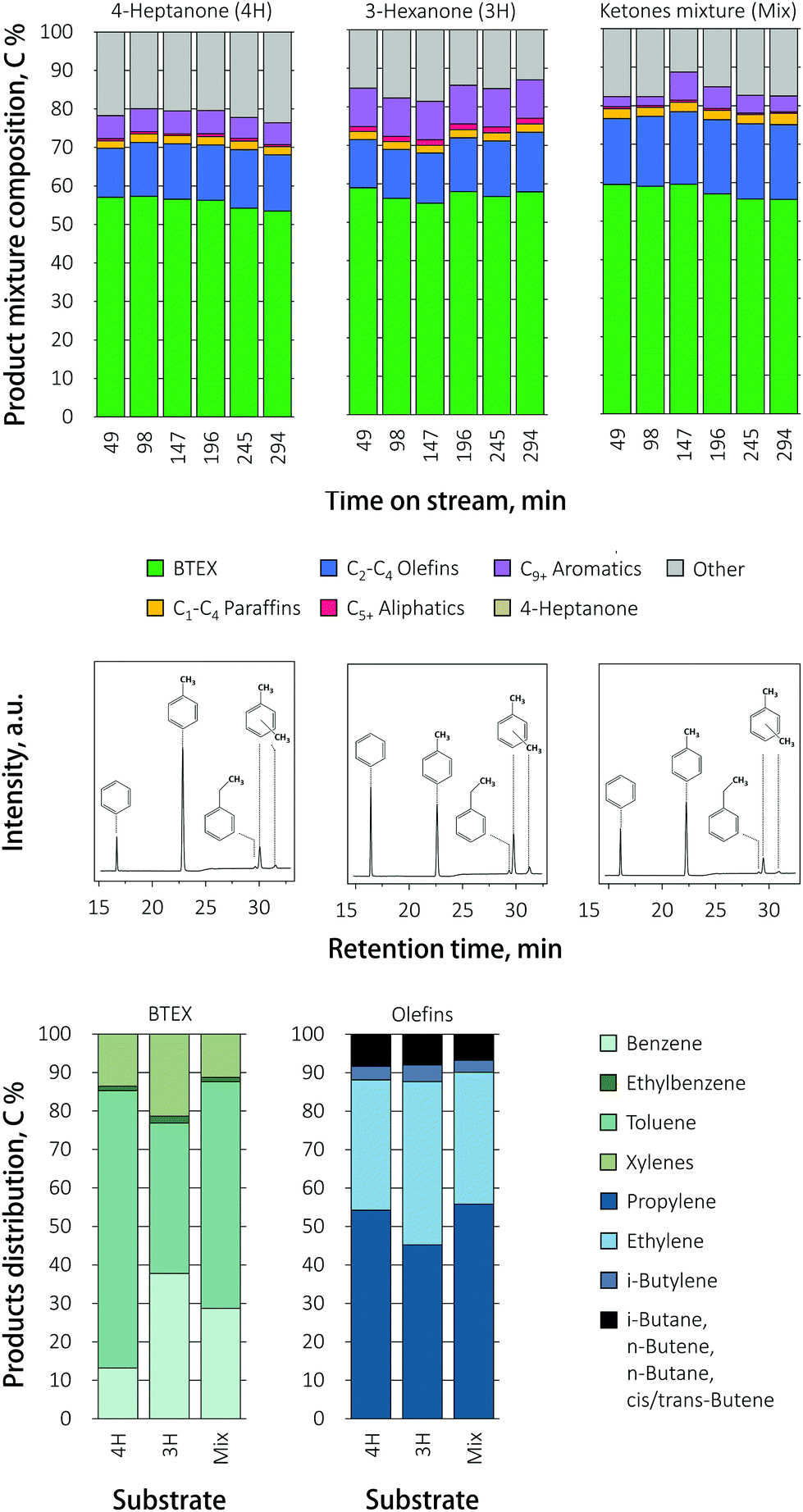 | ||
| Fig. 5 Product mixture composition, BTEX distribution and corresponding gas chromatograms (time-on-stream, TOS = 49 min) recorded during conversion of various ketones over Ga/HZ catalyst (450 °C; WHSV = 1 h−1). | ||
3.3. Effect of water on catalytic performance
The extraction of VFA from a highly dilute fermentation broth is accompanied by a significant amount of co-extracted water (up to 90%),8 which may affect the performance of a catalyst positioned downstream. Up to 80% of this water can be removed by drying of a VFA-saturated adsorbent with residual process heat prior to the desorption of acid, but this still leaves a considerable amount of water to be dealt with.12 The effect of water on zeolite stability, activity and product mixture distribution was studied with time on stream in the reaction of 4-heptanone with co-fed 60 vol% of water, the amount of H2O expected to co-desorb with VFA from a dried adsorbent.12 The results in Fig. 6 illustrate that while water does not affect the catalytic performance of the HZ catalyst, it does result in a drop of aromatics yield the Ga-promoted zeolite. Such an inhibiting effect of water is not unexpected as water would competitively adsorb on the Ga Lewis acid sites, leading to active site blockage. To check whether water inhibition is reversible, the liquid feed was switched off and the catalytic bed purged with nitrogen for 1 h at 450 °C. After this treatment, a water-free 4-heptanone solution was fed to the reactor. The original BTEX yield was restored, indicating that water inhibition is indeed reversible.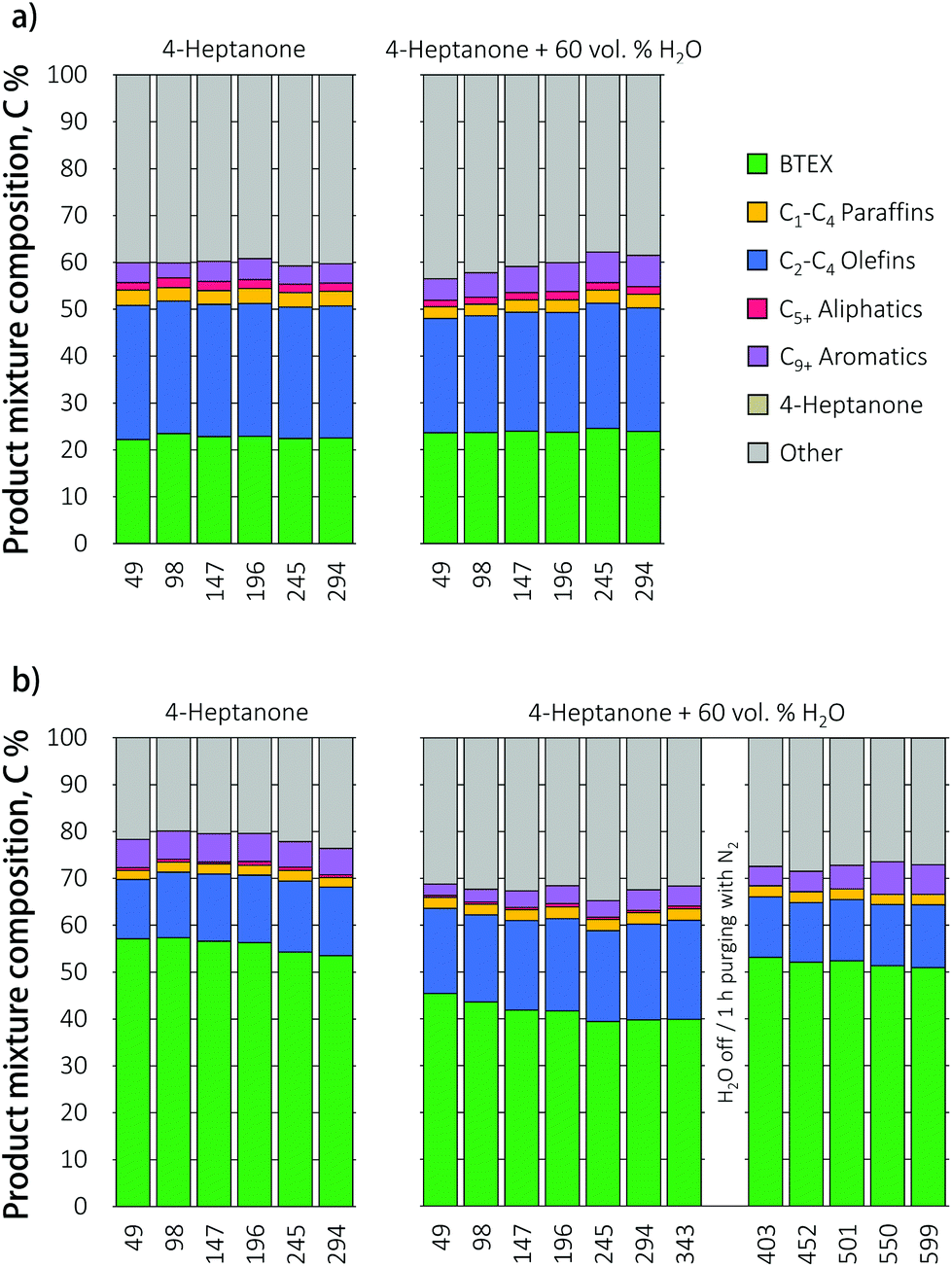 | ||
| Fig. 6 Product mixture composition during the conversion of 4-heptanone and 4-heptanone/water mixture over (a) HZ and (b) Ga/HZ catalysts (450 °C; WHSV = 1 h−1). | ||
The hydrothermal stability of the zeolites was further studied by 27Al NMR of the fresh and spent catalysts. The spectrum of the fresh HZ catalyst (black, Fig. 7) consists of two peaks assigned to framework tetrahedral AlIV (56 ppm) and extra-framework octahedral AlVI species (0 ppm).41,42 Partial dealumination of the framework is expected upon oxidative deammonization of NH4ZSM-5.43 The 27Al NMR analysis of spent HZ catalyst showed no significant changes in zeolite structure after 14 h reaction with either the 4-heptanone reagent or the 4-heptanone/water mixture, demonstrating the high hydrothermal stability of the catalyst under the tested conditions. The NMR spectrum of the fresh Ga/HZ catalyst (Fig. 7) shows that modification by Ga led to a higher degree of dealumination, expected due to the thermal treatments involved in catalyst synthesis. The observed small shift to lower chemical shifts (from 56 ppm to 54 ppm) and peak broadening of the tetrahedral Al peak is attributed to chemical shielding of the 27Al nucleus by the ion-exchanged Ga cations.32 Moreover, a new peak around 32 ppm corresponding to extra-framework penta-coordinate AlV was observed for the Ga-promoted zeolite.42 The spectra of the spent catalysts are very similar to the fresh catalyst, indicating the course of reaction did not lead to a significant redistribution of tetrahedral aluminium species.
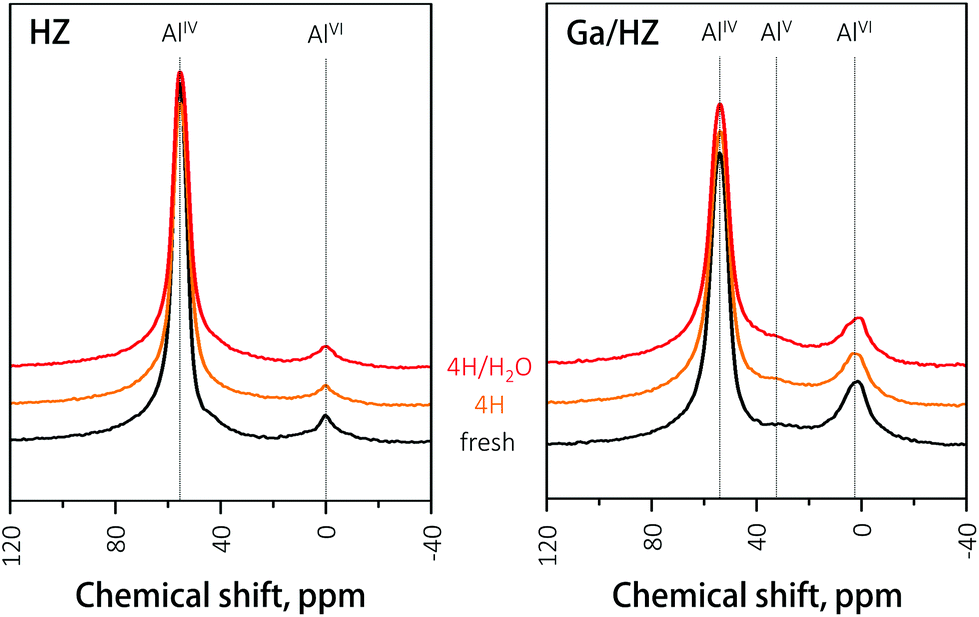 | ||
| Fig. 7 27Al-NMR spectra of zeolite catalysts. | ||
The nature of gallium on the fresh and spent Ga/HZ catalysts was studied by TPR-H2. The deconvoluted reduction profiles (Fig. 8) show four distinct peaks.44 The TPR peaks around 450 °C and 800 °C are assigned to highly dispersed, small Ga2O3 particles and segregated bulk Ga2O3, respectively. The reduction peak at 700 °C corresponds to highly dispersed (GaO)+ species strongly interacting with the zeolite.30–32 The high dehydrogenation activity of these (GaO)+ species originates from the ability of these Lewis acid–base pairs to catalyse C–H bond cleavage (eqn (S4)–(S7)†).36
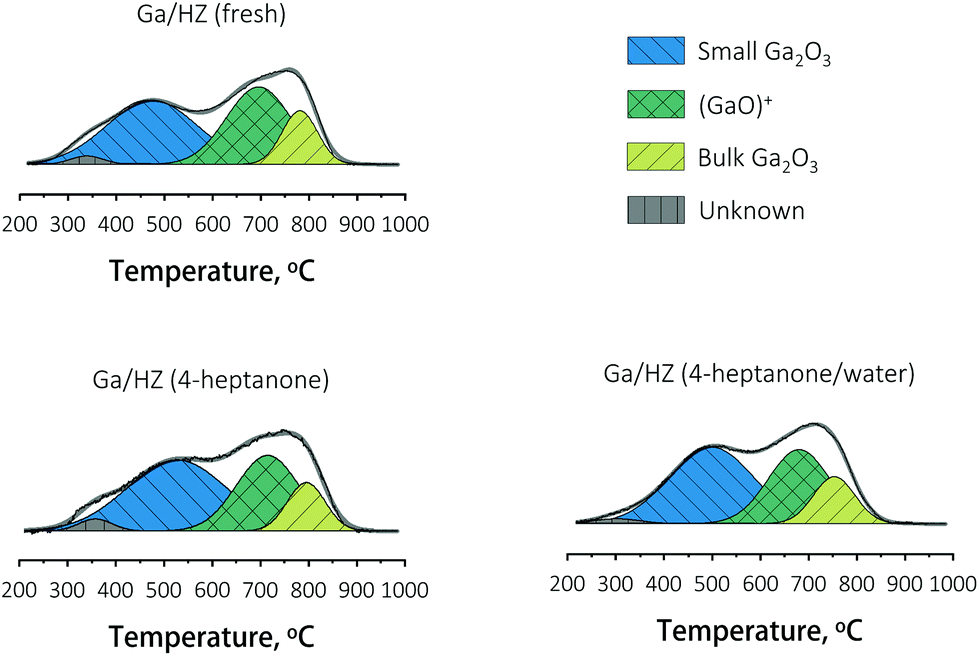 | ||
| Fig. 8 TPR profiles of fresh and spent Ga/HZ catalysts. | ||
Hensen et al.36 reported an increase in propane dehydrogenation activity and change in product distribution upon addition of steam to a change in Ga speciation, with added water preventing deactivation by reduction and dehydration (eqn (S7)†). In our case the distribution of aromatic products (Fig. 4) did not change significantly upon addition of water, even though the composition of the olefin products was affected. A comparison of the reduction profiles of the fresh and spent catalysts, illustrated in Fig. 8, reveals that the nature of the Ga species did not change significantly over the course of both the reaction with or without water: neither via reduction to Ga+ species,36 nor via Brønsted acid proton regeneration upon hydrolysis of gallium species.
The coke content on the spent HZ and Ga/HZ catalysts was determined by TGA-MS, as illustrated in Fig. 9. Little coke was found on both catalysts after 14 h of reaction with 4-heptanone, with the coke content being lower on Ga-promoted HZSM-5 (∼2 wt%) than on the undoped zeolite (∼3 wt%). On both spent catalysts the little coke formed was of a “hard” nature, desorbing at >350 °C. This type of coke consists of highly hydrogen-deficient polyaromatic molecules.45 The amount of formed coke is noticeably lower than reported in the literature. For example, 16 wt% of coke on HZSM-5 (Si/Al = 20) was reported in the conversion of acetone/n-butanol mixture (1:2 w/w) at 400 °C after 210 min (ref. 35) and 4 wt% on HZSM-5 after just 4 h reaction of acetaldehyde at 400 °C.17 Importantly, the presence of water in the feed further decreased the amount of coke by almost a factor of two, contributing to a higher long-term stability of the catalyst. This decrease in coke formation as a result of water in the feed is proposed to be due to competitive adsorption between water and the hydrocarbon intermediates involved in coke formation.17
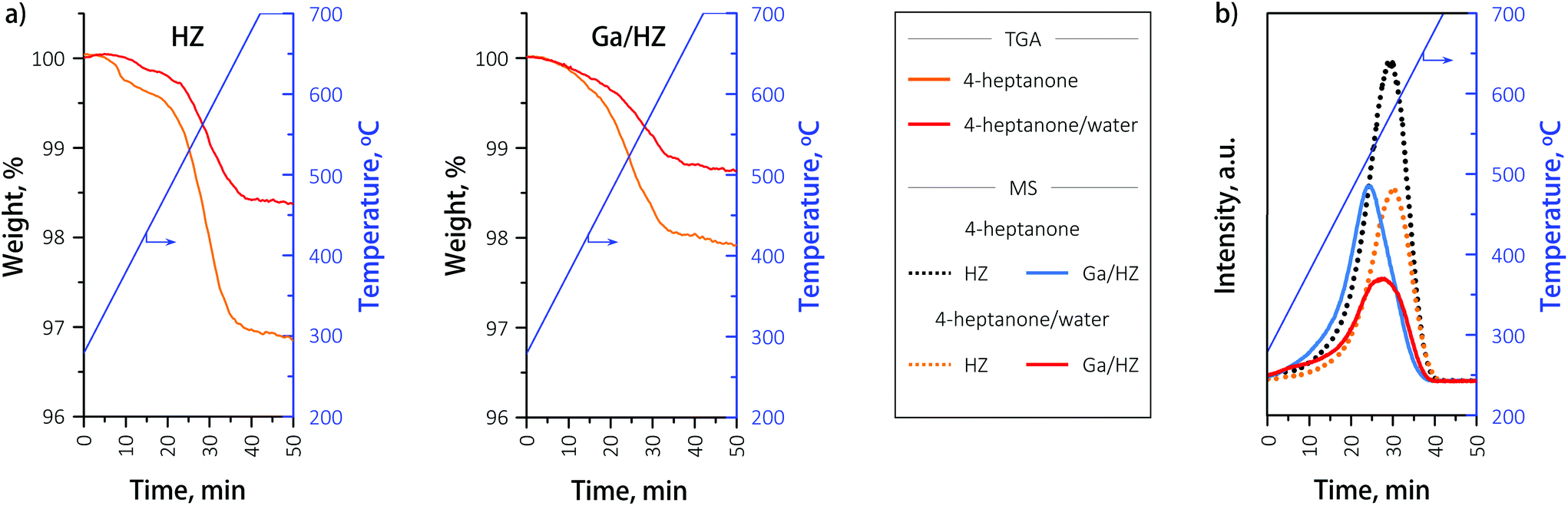 | ||
| Fig. 9 (a) TGA-MS analyses of spent catalysts in the range 300–700 °C. (b) MS signals of CO2 as a function of temperature for spent catalysts. | ||
4. Conclusions
The conversion of waste-derived carboxylic acids towards benzene, toluene, ethylbenzene and xylene (BTEX) by successive ketonization and aromatization reactions is shown to be a feasible route for VFA upgrading. The direct reaction of butyric acid over a zeolite catalyst predominately proceeded via decarboxylation yielding propylene as the main product, with only traces of aromatics being formed. In contrast, acid conversion over a combination of a TiO2 P25 ketonization catalyst and downstream (promoted) zeolite catalyst does allow for the one-step formation of BTEX in good yield. The effect of ketone feed on the product mixture distribution was found to be negligible for the HZSM-5 zeolite, but strong for the Ga-promoted HZSM-5 one. This suggests that different reaction mechanisms govern the reaction, as a result of substitution of up to 60% of Brønsted acid sites with (GaO)+ species. An inhibiting effect of water was observed for the reactions over zeolite Ga/HZSM5. The inhibition, however, was found to be reversible and thought to originate from competitive adsorption and active sites blockage. Importantly, characterization of spent catalysts by 27Al-NMR, TGA-MS and TPR-H2 show that the presence of water has no effect on the catalyst's structure. Water does reduce the extent of coke deposition, coke that is of a highly hydrogen-deficient polyaromatic nature. Overall, the results show the integrated catalytic approach reported here to be an attractive strategy for the efficient upgrading of waste-derived VFA to valuable circular aromatics. The reactions sequence involving the integrated intermediate ketonization step increases the carbon efficiency of the whole process without introducing intermediate separation steps.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support provided by NWO-TTW and Paques through the STW-Paques Program. J. J. B. van Duin (Utrecht University, UU) is acknowledged for his valuable research contributions during his MSc thesis studies. J. L. Weber (UU) is acknowledged for his contribution to the development of the reaction setup. A. Lucini Paioni (UU) and Prof. Dr M. Baldus (UU) are thanked for performing the 27Al NMR measurements. D. Wezendonk (UU) is acknowledged for performing the TGA-MS measurements.References
- S. Kelkar, C. M. Saffron, Z. Li, S. S. Kim, T. J. Pinnavaia, D. J. Miller and R. Kriegel, Green Chem., 2014, 16, 803–812 RSC.
- Z. Sun, B. Fridrich, A. De Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- S. Thiyagarajan, H. C. Genuino, J. C. Van Der Waal, E. De Jong, B. M. Weckhuysen, J. Van Haveren, P. C. A. Bruijnincx and D. S. Van Es, Angew. Chem., Int. Ed., 2016, 55, 1368–1371 CrossRef CAS PubMed.
- M. Atasoy, I. Owusu-agyeman, E. Plaza and Z. Cetecioglu, Bioresour. Technol., 2018, 268, 773–786 CrossRef CAS PubMed.
- S. Bengtsson, J. Hallquist, A. Werker and T. Welander, Biochem. Eng. J., 2008, 40, 492–499 CrossRef CAS.
- C. Sans, J. Mata-Alvarez, F. Cecchi, P. Pavan and A. Bassetti, Bioresour. Technol., 1995, 51, 89–96 CrossRef CAS.
- S. Dahiya, O. Sarkar, Y. V. Swamy and S. V. Mohan, Bioresour. Technol., 2015, 182, 103–113 CrossRef CAS PubMed.
- E. Reyhanitash, S. R. A. Kersten and B. Schuur, ACS Sustainable Chem. Eng., 2017, 5, 9176–9184 CrossRef CAS PubMed.
- E. Reyhanitash, E. Fufachev, K. D. van Munster, M. B. M. van Beek, L. M. J. Sprakel, C. C. N. Edelijn, B. M. Weckhuysen, S. R. A. Kersten, P. C. A. Bruijnincx and B. Schuur, Green Chem., 2019, 21, 2023–2034 RSC.
- W. S. Lee, A. S. M. Chua, H. K. Yeoh and G. C. Ngoh, Chem. Eng. J., 2014, 235, 83–99 CrossRef CAS.
- C. S. López-Garzón and A. J. J. Straathof, Biotechnol. Adv., 2014, 32, 873–904 CrossRef PubMed.
- E. Fufachev, Chemical Building Blocks from Fermented Wastewater by Chemocatalytic Conversion, PhD thesis, Utrecht University, 2019 Search PubMed.
- Y. Wang, M. Peng, J. Zhang, Z. Zhang, J. An, S. Du, H. An, F. Fan, X. Liu, P. Zhai, D. Ma and F. Wang, Nat. Commun., 2018, 9, 5183 CrossRef PubMed.
- L. Yu, S. Huang, S. Zhang, Z. Liu, W. Xin, S. Xie and L. Xu, ACS Catal., 2012, 2, 1203–1210 CrossRef CAS.
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249–259 CrossRef CAS.
- J. D. Adjaye and N. N. Bakhshi, Biomass Bioenergy, 1995, 8, 131–149 CrossRef CAS.
- A. G. Gayubo, A. T. Aguayo, A. Atutxa, R. Aguado, M. Olazar and J. Bilbao, Ind. Eng. Chem. Res., 2004, 43, 2619–2626 CrossRef CAS.
- X. Zhu, L. L. Lobban, R. G. Mallinson and D. E. Resasco, J. Catal., 2010, 271, 88–98 CrossRef CAS.
- J. Deischter, K. Schute, D. S. Neves, B. E. Ebert, L. M. Blank and R. Palkovits, Green Chem., 2019, 21, 1710–1717 RSC.
- E. A. Uslamin, B. Luna-Murillo, N. Kosinov, P. C. A. Bruijnincx, E. A. Pidko, B. M. Weckhuysen and E. J. M. Hensen, Chem. Eng. Sci., 2018, 198, 305–316 CrossRef.
- X. Wang, S. Ding, H. Wang, X. Liu, J. Han, Q. Ge and X. Zhu, Appl. Catal., A, 2017, 545, 79–89 CrossRef CAS.
- S. Vichaphund, D. Aht-Ong, V. Sricharoenchaikul and D. Atong, Renewable Energy, 2015, 79, 28–37 CrossRef CAS.
- J. Fuhse and F. Bandermann, Chem. Eng. Technol., 1987, 10, 323–329 CrossRef.
- T. N. Pham, D. Shi, T. Sooknoi and D. E. Resasco, J. Catal., 2012, 295, 169–178 CrossRef CAS.
- A. Pulido, B. Oliver-Tomas, M. Renz, M. Boronat and A. Corma, ChemSusChem, 2013, 6, 141–151 CrossRef CAS PubMed.
- T. N. Pham, T. Sooknoi, S. P. Crossley and D. E. Resasco, ACS Catal., 2013, 3, 2456–2473 CrossRef CAS.
- A. Gumidyala, T. Sooknoi and S. Crossley, J. Catal., 2016, 340, 76–84 CrossRef CAS.
- E. C. Nordvang, E. Borodina, J. Ruiz-Martínez, R. Fehrmann and B. M. Weckhuysen, Chem. – Eur. J., 2015, 21, 17324–17335 CrossRef CAS PubMed.
- V. O. Rodrigues, J. G. Eon and A. C. Faro, J. Phys. Chem. C, 2010, 114, 4557–4567 CrossRef CAS.
- R. Fricke, H. Kosslick, G. Lischke and M. Richter, Chem. Rev., 2000, 100, 2303–2405 CrossRef CAS PubMed.
- H. Xiao, J. Zhang, X. Wang, Q. Zhang, H. Xie, Y. Hana and Y. Tan, Catal. Sci. Technol., 2015, 5, 4081–4090 RSC.
- I. Nowak, J. Quartararo, E. G. Derouane and J. C. Védrine, Appl. Catal., A, 2003, 251, 107–120 CrossRef CAS.
- N. Rane, M. Kersbulck, R. A. van Santen and E. J. M. Hensen, Microporous Mesoporous Mater., 2008, 110, 279–291 CrossRef CAS.
- K. Nedomová, S. Beran and P. Jíru, React. Kinet. Catal. Lett., 1986, 32, 353–357 CrossRef.
- A. de Lucas, P. Cañizares and A. Durán, Appl. Catal., A, 2001, 206, 87–93 CrossRef CAS.
- E. J. M. Hensen, E. A. Pidko, N. Rane and R. A. Van Santen, Angew. Chem., Int. Ed., 2007, 46, 7273–7276 CrossRef CAS PubMed.
- S. M. Campbell, D. M. Bibby, J. M. Coddington, R. F. Howe and R. H. Meinhold, J. Catal., 2002, 161, 350–358 CrossRef.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- B. Oliver-Tomas, M. Renz and A. Corma, J. Mol. Catal. A: Chem., 2016, 415, 1–8 CrossRef CAS.
- R. A. L. Baylon, J. Sun, K. J. Martin, P. Venkitasubramanian and Y. Wang, Chem. Commun., 2016, 52, 4975–4978 RSC.
- L. Rodríguez-González, F. Hermes, M. Bertmer, E. Rodríguez-Castellón, A. Jiménez-López and U. Simon, Appl. Catal., A, 2007, 328, 174–182 CrossRef.
- A. W. Chester and E. G. Derouane, Zeolite characterization and Catalysis: A tutorial, Springer, Berlin, 2010 Search PubMed.
- E. Bourgeat-Lami, P. Massiani, F. Di Renzo, F. Fajula and T. Des Courieres, Catal. Lett., 1990, 5, 265–271 CrossRef CAS.
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43(5), 1126–1128 CrossRef CAS.
- M. Guisnet and P. Magnoux, Appl. Catal., A, 2001, 212, 83–96 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc00964d |
| This journal is © The Royal Society of Chemistry 2020 |