
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpruce milled wood lignin: linear, branched or cross-linked?
Mikhail
Balakshin
 *a,
Ewellyn Augsten
Capanema
b,
Xuhai
Zhu†
a,
Irina
Sulaeva
c,
Antje
Potthast
c,
Thomas
Rosenau
c and
Orlando J.
Rojas
ad
*a,
Ewellyn Augsten
Capanema
b,
Xuhai
Zhu†
a,
Irina
Sulaeva
c,
Antje
Potthast
c,
Thomas
Rosenau
c and
Orlando J.
Rojas
ad
aDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, Vuorimiehentie 1, Espoo, 02150, Finland. E-mail: mikhail.balakshin@aalto.fi; Tel: +358-(0)50-308-6570
bRISE Bioeconomy, Box 5604, SE-114 86 Stockholm, Sweden
cDepartment of Chemistry, Institute for Chemistry of Renewable Resources, University of Natural Resources and Life Sciences (BOKU), Muthgasse 18, 1190, Vienna, Austria
dDepartments of Chemical and Biological Engineering, Chemistry and Wood Science, University of British Columbia, 2360 East Mall, Vancouver, BC V6T 1Z4, Canada
First published on 17th June 2020
Abstract
The subject of lignin structure, critical for fundamental and practical reasons, is addressed in this study that includes a review of the methods applied to elucidate macromolecular branching. The recently available approaches for determination of the absolute molecular mass of spruce milled wood lignin (MWL) along with the quantification of terminal groups clearly indicate that MWL is significantly branched and cross-linked (with ∼36% lignin units partaking in these linkages). Results from independent methods imply that about half of the branching and crosslinking linkages involve aromatic rings, predominantly 5–5′ etherified units; meanwhile, a significant number of linkages are located in the side chains. Quantitative 13C NMR analyses suggest that the branches involve different aliphatic ether (alkyl-O-alkyl) types at the α- and γ-positions of the side chain, with intact β-O-4 linkages. While the exact structures of these moieties require further investigation, our results point to the fact that conventional lignification theory disagrees with the presence of such key moieties in softwood MWL and the observed high degree of branching/crosslinking. Potential reasons for the noted discrepancies are discussed.
Introduction
Understanding the molecular structure of lignins is of primary importance not only for fundamental reasons, but also to access lignin reactivity and valorization. A more cross-linked and branched lignin is associated with higher steric hindrance, inaccessibility and lower molecular flexibility than a strictly linear one, all of which should be considered in any reaction involving lignin's reactive centers. Moreover, a greater difficulty is expected, for example, in breaking the cross-linked macromolecule into low molecular mass products and in extracting them from the cell wall matrix. Certain potential lignin-derived materials, such as carbon fibers, benefit from a precursor that is more linear. In contrast, branched and cross-linked lignins would be beneficial in sorbents, hydrogels or filters. Adhesive formulations might require an optimal branching/cross-linking degree. In general, branching affects polymer flow in solution and as a melt. Higher branching results in smaller hydrodynamic radii and therefore lower viscosity compared to the non-branched polymers with the same molar mass.Despite the lack of direct evidence, lignin is usually considered as a 3-dimensional, cross-linked natural macromolecule.1 Goring provided three indirect pieces of evidence for 3-dimensional native lignin (protolignin) networks,1 namely: (1) insolubility of native lignin in neutral solvents (however, this may also be the result of chemical linkages between lignin and polysaccharides); (2) wide dispersity of isolated lignins and, (3) the shape of lignin molecules in solution.
Recently, Milled Wood Lignin (MWL), a preparation used as a model for native lignin, was proposed to be linear;2 such assertion should be subjected to scrutiny, as we attempt in the present study. In fact, linearity has been concluded based on the observed negligible amounts (<1/100 C9-units) of 5–5′ and 4-O-5′ etherified units, as quantified by DFRC (derivatization followed by reductive cleavage) method coupled with 31P NMR. Related propositions of rather linear lignins have been forwarded in a recent review by Ralph et al.3 who assumed models of native lignins, including a softwood, based on the best knowledge available on lignification theory along with 2D NMR quantification.
It can be reasonably stated that the recent claims of strictly linear lignin are in conflict with classical theories,1,4–7 a subject that has not been addressed so far. Our goal is to critically discuss the different methods used for the quantification of lignin branching and crosslinking points, such as etherified 5–5′, 4-O-5′, and other lignin units (Fig. 1), and to contest the amounts reported for spruce MWL, which leads to a revision of the accepted view of lignin structure.
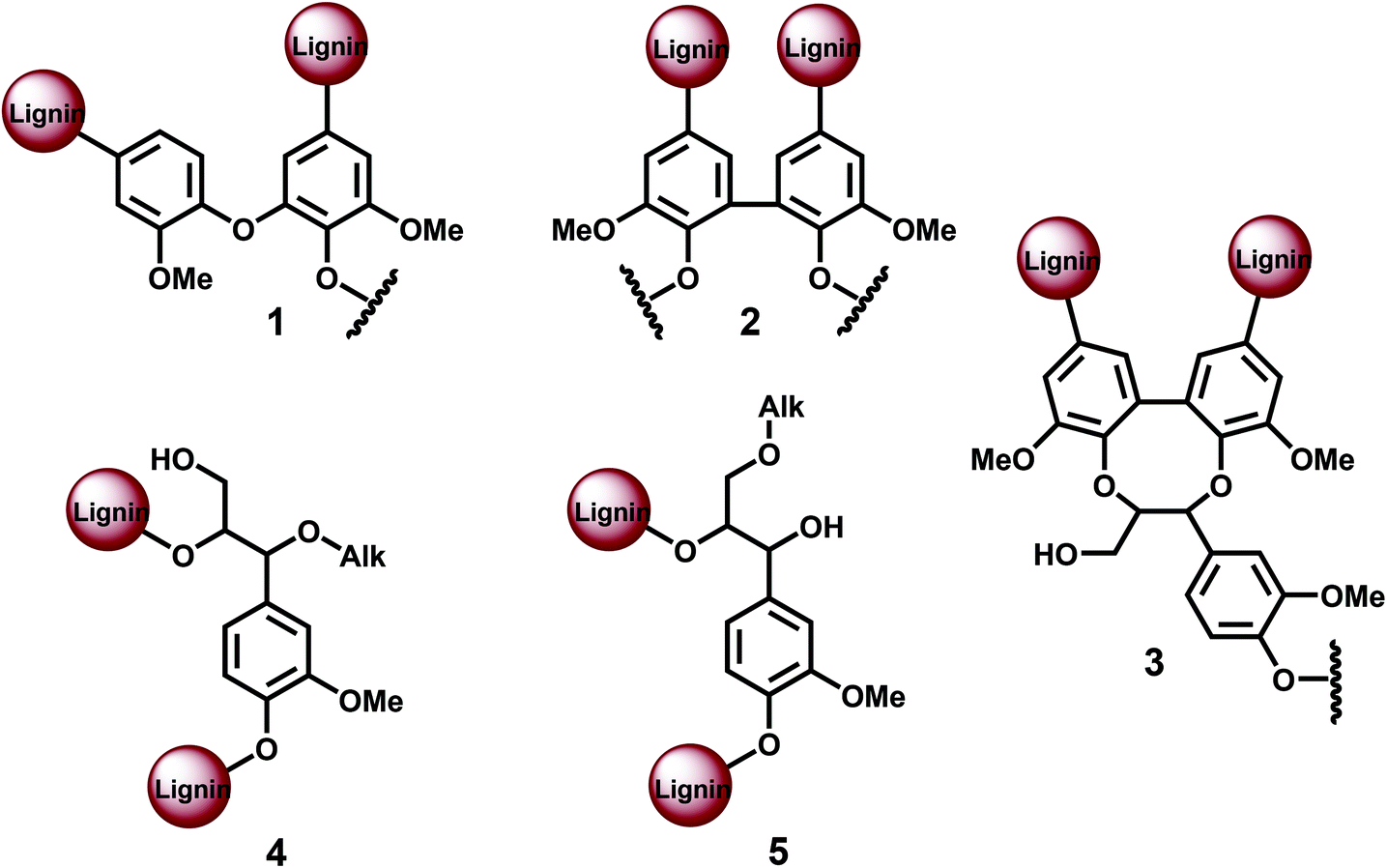 | ||
| Fig. 1 Different lignin branching/cross-linking units: 1. etherified 4-O-5′, 2. etherified 5–5′, 3. dibenzodioxocin (DBDO), 4. α-O-Alk/β-O-4 5. γ-O-Alk/β−O−4. | ||
Discussion
In the current study, we focused on a spruce MWL, given that it has been investigated widely as a model of native softwood (SW) lignin. In addition to our measurements, literature data on other softwood lignins is included for discussion. It should be noted that the structural variations of different softwood lignins are rather subtle8,9 suggesting the convenience of discussing the main structural features independently of the softwood species. First, we evaluate the degree of branching by sum parameters, such as lignin degree of polymerization (DP) and the proportion of polymer terminal units. We use the term DP as a first approximation and as a synonym of the number of C9-units in softwood lignin.Lignin as a classical biopolymer with a broad molar mass distribution is to be considered as a mixture of diverse macromolecular chains, with different molar and structural features, i.e. differing functional groups and branching types as well as side chain composition per main chain. Hence, a disperse mixture of chain lengths and structural features should be considered. Importantly, multi-angle light scattering in the infrared-range (MALS(IR)) has been introduced recently to rather precisely quantify the molar mass distribution of lignin, allowing quite precise absolute figures of molecular weight.10 While such information is now accessible, any structural understanding of lignin is still challenged by the rather broad distribution of molecular features. The aim of this work is to combine complementary data of a more precise molar mass and of structural elements that have been inaccessible so far, allowing a tentative lignin structural model. Such information is used to address the question whether the structure of MWL lignin is linear or rather cross-linked. Light scattering is a method of choice for such inquiries since it is sensitive to cross-linking of macromolecules and is able to measure the extent of such a feature. Given that MALS(IR) has become more widely available recently, it seems straightforward to apply this approach to lignin. Unfortunately, lignin is an isotropic scatterer at the wavelengths suitable for analysis. Isotropic scatterers do not show angular dependency of the scattered light below λ/20, i.e., at a diameter of a molecule in solution of 785 nm/20 ≈ 40 nm. This angular dependency, however, is required to calculate the radius of gyration <rg2>, which can be used to judge compactness and also molecular cross-linking. The lignin molecules in solution are too small to evoke an angular dependency of the scattered light, thus rendering a direct approximation of the crosslinking – simply by light scattering – impossible.
The analysis of lignins by SEC-MALS(IR) yields the most reliable molar mass data, because neither fluorescence nor absorption phenomena are negatively influencing the results. Based on these methodologies10 the Mn of the spruce MWL is found to be about 4500 (Fig. 2a); therefore a DPn = 4500/180 = 25 can be assumed. Thus, as a first approximation, we will relate MWL to a model consisting of 25 monomeric units, being well aware that we deal with an average molar mass and an averaged structure.
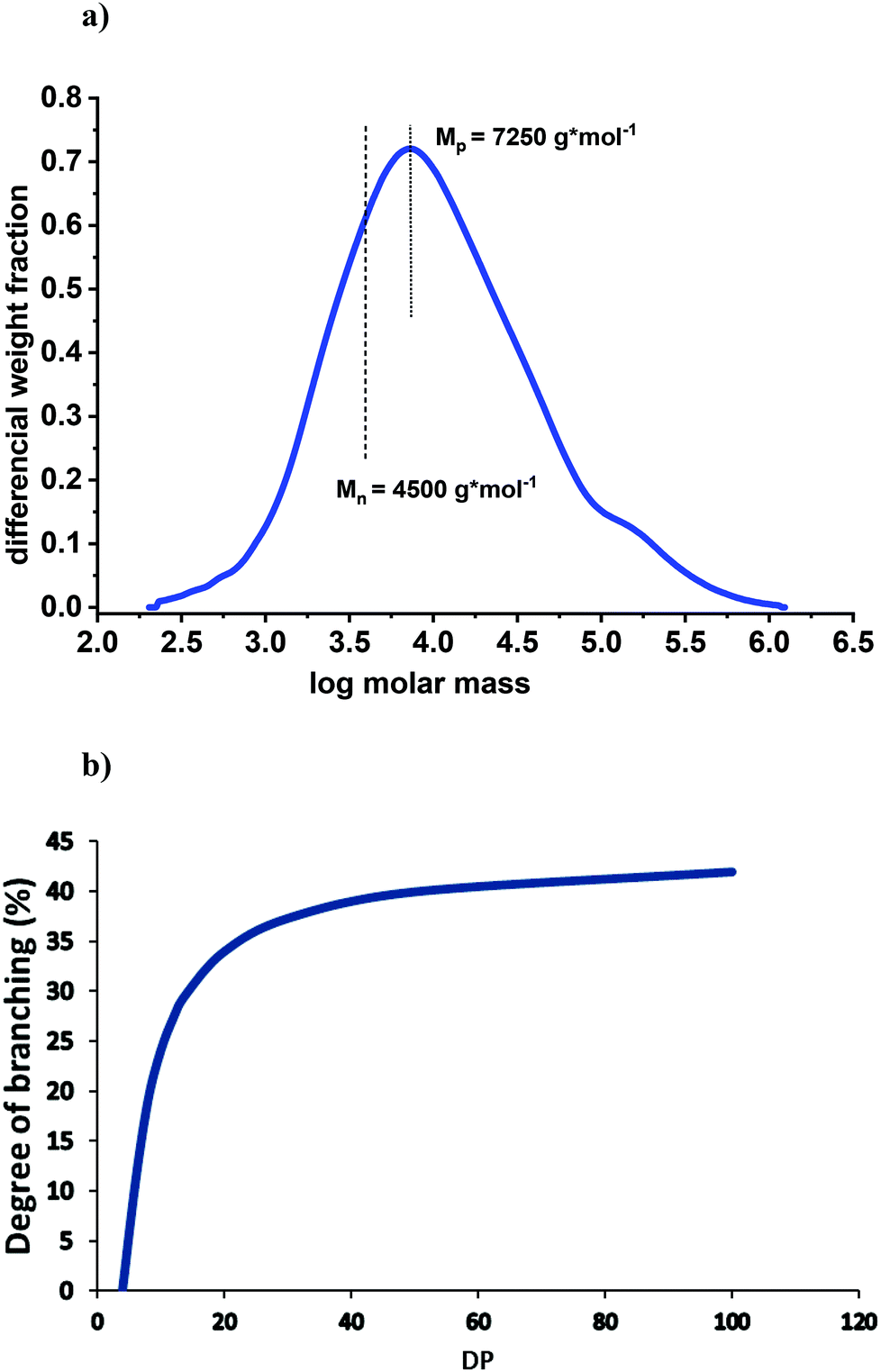 | ||
| Fig. 2 (a) Molar mass distribution of spruce MWL measured by SEC-MALS(IR); the analysis was performed according to the conditions described in detail by Zinovyev et al.10 (b) Theoretical correlation between DP and degree of branching at the proportion of terminal units of 44%. | ||
The analysis of the amounts of terminal lignin units (T) along with DP evaluation gives an idea on lignin branching. There is a variety of terminal units in the side chain11,12 (Table 1), in addition to small amounts of traditionally considered coniferyl alcohol and aldehyde. The presence of small amounts carboxyl moieties in MWL is well known;6,12–14 although their exact structures are not established; in any case, these units are terminal by definition. In total, the amount of terminal units in the side chain is about 0.18/Ar (Table 1). In the aromatic ring, about 0.05/Ar of 0.31/Ar total PhOH are in condensed lignin structures (mostly 5–5′ types),12i.e. about 0.26/Ar phenolic units are non-condensed and therefore terminal. Thus, the amounts of terminal units in spruce MWL are well established by a large number of independent studies.4–7,11–14 The proportion of overall terminal units is about 0.18 + 0.26 = 0.44, corresponding to 44% or 11 units in our lignin model (per 25 Ar).
| Units | Freudenberg4 | Adler5 | Sakakibara7 | Ralph model3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
Zhang11 | This paper | This paper, per 25 Ar |
|---|---|---|---|---|---|---|---|
| a Values in parenthesis are per description to the model, the other are directly from the model.3 b Amount of C9-units involved; in a case of symmetric moieties, the amount of these structures is 1/2 of the value shown in the Table. | |||||||
| Structures: | |||||||
| Internal | |||||||
| β-O-4/α-OH | 34 | 55 (60) | 36 | 36 | 9 | ||
| β-O-4/α-CO | – | 1 | 2 | 0.5 | |||
| DBDO | 5 | 5 | 4 | 1 | |||
| Phenylcoumaran | 9 | 9–12 | 10 | 10 | 12 | 10 | 2.5 |
| Pinoresinol | 3.5 | 5 | 2.5 | 2 | 0.5 | ||
| β-1/α-OH | 7 | 9 | – | 2 | 1 | 0.25 | |
| Spirodienone | 5(1–2) | 2 | 2 | 0.5 | |||
| Secoisolariciresinol (SILR) | – | 1 | 1 | 0.25 | |||
| Terminal | 10 | 18 | 4.5 | ||||
| Coniferyl alcohol | 4 | 10 | 1 | 2 | 0.5 | ||
| Coniferaldehyde | 4 | 4 | – | 5 | 4 | 1 | |
| Dihydroconiferyl alcohol | 4 | – | 1 | 2 | 0.5 | ||
| Ar–CO–CH2–CH2OH | – | 3 | 2 | 0.5 | |||
| Vanillin | – | 3 | 4 | 1 | |||
| Ar–COOH | – | 1 | 0.25 | ||||
| Ar–CH2–COOH | – | 3 | 0.75 | ||||
| Functional groups: | |||||||
| Aliphatic OH | 100 | 100 | 145 | 107 | |||
| Primary | 75 | 90 | 68 | 17 | |||
| Secondary | 25 | 55 | 39 | 10 | |||
| Phenolic OH | 32 | 20–25 | 31 | 10 (6) | 31 | 8 | |
| Carbonyl | 20 | 20 | 21 | – | 21 | ||
| COOR | 4 | 2 | – | 4 | 1 | ||
| Interunit linkages: | |||||||
| β-O-4 total | 44 | 50 | 45 | 65 | 50 | 12.5 | |
| α-O-4/β-O-4 (non-cyclic) | 8 | 6–8 | 11 | – | <1 | na | |
| Alk-O-Alk totalb | 18 | – | 32 | 8 | |||
| α-O-Alk/β-O-4/γ-OH | – | 8 | 2 | ||||
| α-OH/β-O-4/γ-O-Alk | – | 8 | 2 | ||||
| α-O-Alk/β-CH(?)/γ-O-Alk | – | 8 × 2 | 2 × 2 | ||||
| 5–5′ totalb | 19–22 | 21 | 10 | 27 | 7 | ||
| Etherifiedb | 14–16 | 10 | 23 | 6 | |||
| Phenolicb | 5–6 | — | 4 | 1 | |||
| 4-O-5′ | 6 | 4 | 7 | 5 (1) | 2 | 0.5 | |
| Degree of condensation | 45 | 25 (21) | 40 | ||||
The main polymer chain contains two terminal units. Additional 9 terminal units in the 25-units model result from branching or/and crosslinking. Each new branched chain (such as 4-O-5′ etherified or 5–5′ semi-etherified units) introduces one additional terminal unit (Fig. 1). Thus, the proposed 25-units lignin model containing 11 terminal units has, therefore, one main chain (2T) and nine side chains (remaining 9T). This would give an average chain length of 25:10 = 2.5 units. In the case of exclusive cross-linking (such as by 5–5′ etherified units), each cross-linking introduces two additional terminal units (Fig. 1), resulting in an average of 11:2 = 5.5 individual chains per 25 units, with 4.5 resulting cross-linking points. In this case, the average chain length would be 25:5.5 = 4.5 units. In general, the numbers of branching (B) and cross-linking (X) points in the lignin (average) model are:
| B + 2X = T − 2 = 9. |
More general, for different percentage of terminal units (T%), is:
| B = DP × T%/100 − 2 = MW/180 × T%/100 − 2. |
The degree of branching (B%) is, therefore:
| B% = B/DP × 100 = (DP × T%/100 − 2)/DP × 100. |
When DP (i.e. molecular weight) is large, follows
| DP × T%/100 ≫ 2 |
| B% ∼ T%. |
The presented approach is rather practical since it allows judging the degree of lignin branching directly from the determination of terminal units.
Fig. 2b illustrates the correlation between DP and degree of branching at a fixed proportion of terminal units, namely 44%. At this ratio, lignin would be linear (no branching) at a DP of about 4. The degree of branching significantly increases with an increased DP until a value of ∼20; then, the effect levels off. For example, the degree of branching at DP = 40 (Mp = 7250) is 39% vs. 36% at DP = 25.
Applying GPC analyses of other softwood MWLs (Table 2) we were able to show some molar mass deviation that likely depends on the conditions of MWL isolation and purification. Even the lowest value of Mn of 3700 g mol−1 corresponds to a DP of about 21. Assuming a similar chemical structure, this value would result in B% of 35%, i.e. practically the same as B% of our core MWL.
Branching/cross-linking at the aromatic ring
The main potential branching points at the aromatic ring are semi-etherified 5–5′, dibenzodioxocin (DBDO) and etherified 4-O-5′ moieties; 5–5′ linkages in fully etherified moieties are cross-linking points (Fig. 1). Most of the methods for their quantification are based on the analysis of different types of phenolic units, either directly by 31P NMR spectroscopy or by the permanganate oxidation method (PO) followed by gas chromatographic (GC) analysis of the generated monomeric and dimeric products. A limitation of these methods is that only free phenolic C9-units are covered. As native lignins consist of predominantly etherified C9-units, a comprehensive and selective cleavage of Alk-O-Ar ether linkages should be performed beforehand to access the originally non-phenolic lignin units through their analysis by 31P NMR spectroscopy or PO.A few methods for this cleavage have been suggested, namely, CuO oxidation,15 DFRC16 and thioacidolysis (TA).17 Thus, the following combinations have been used: CuO-PO,5,18 TA-31P NMR,19 and DFRC-31P NMR2,20 (Fig. 3).
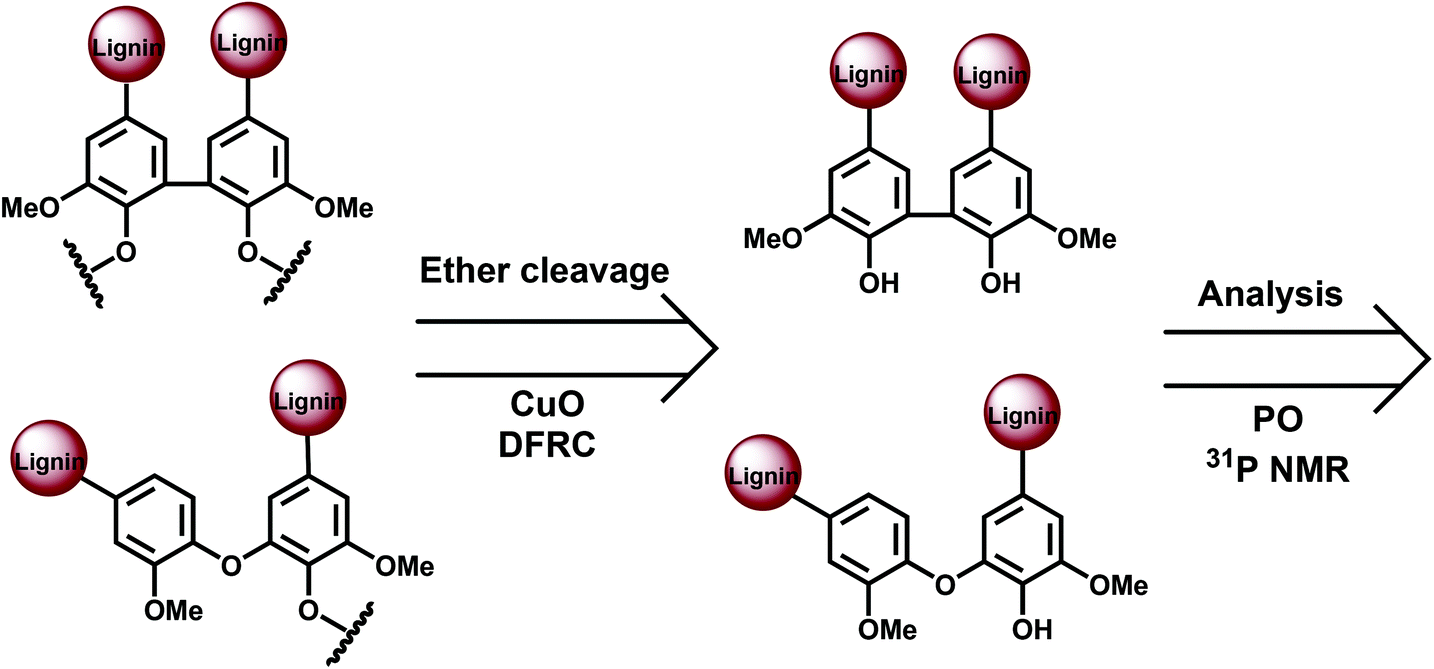 | ||
| Fig. 3 General approach for the analysis of phenolic and etherified lignin moieties. | ||
13C NMR spectroscopic studies of lignins were also applied, after selective modification by mild and selective chemical pretreatments. This aimed at shifting the resonance of undesirable moieties away from those of the units of interest in order to reduce signal overlap.12,21 The results from these methods are summarized in Table 3.
| Moieties | CuO-PO12,17 | TA-31P NMR13 | DFRC-31P NMR2 | 13C NMR (A)15 | 13C NMR (B)16 | HSQC |
|---|---|---|---|---|---|---|
| a Expressed as number of C9-units involved in 5–5′ structures. b After correction (see text). c Claimed to be DBDO only. “ne” and “et” correspond to non-etherified and etherified units. | ||||||
| 5–5′ ne | 5–6 | 5–8 | 5 | |||
| 4-O-5′ ne | 2 | |||||
| 5–5′ et | 14–16 | 6–8c | 19 | |||
| Including DBDO | 6–8 | 8 | ||||
| 4-O-5′ et | 2 | Not reported | Below LoD | |||
| 5–5′ + 4-O-5′ et. (branching/cross-linking units) | 16–18 | Not reported | 6–8c | 23 | ||
| Total 5–5′ | 19–22 | 16 (21b) | 24–27 | 27 | ||
| Total 4-O-5′ | 4 | 5 (7b) | ∼2 | |||
 | ||
| Fig. 4 CuO-permanganate oxidation approach for the analysis of lignin branching points. | ||
CuO degradation15 of Alk-O-Ar bonds has been used to access originally non-phenolic lignin units.5,18,22 Also a pretreatment under kraft cooking conditions can be used for this purpose, although it is less selective and therefore not further considered here. PO without any pretreatment (CuO or kraft) yields data on the structure of phenolic moieties, whereas the CuO-PO approach covers all lignin moieties, i.e. originally phenolic and etherified ones. The content of the etherified moieties is simply derived by calculating the difference between the PO and CuO-PO values.
Norway spruce (Picea abies) softwood MWL analyzed by the CuO-PO method contained about five 5–5′ units per 100 aromatic rings (100 Ar) in originally phenolic and twenty 5–5′ units per 100 aromatic rings in non-phenolic units,18 the latter also includes DBDO moieties. The amounts of 4-O-5′ units were rather small: about 2/100 Ar for phenolic and 2/100 Ar for non-phenolic units, i.e. 4/100 Ar in total.5,6,18
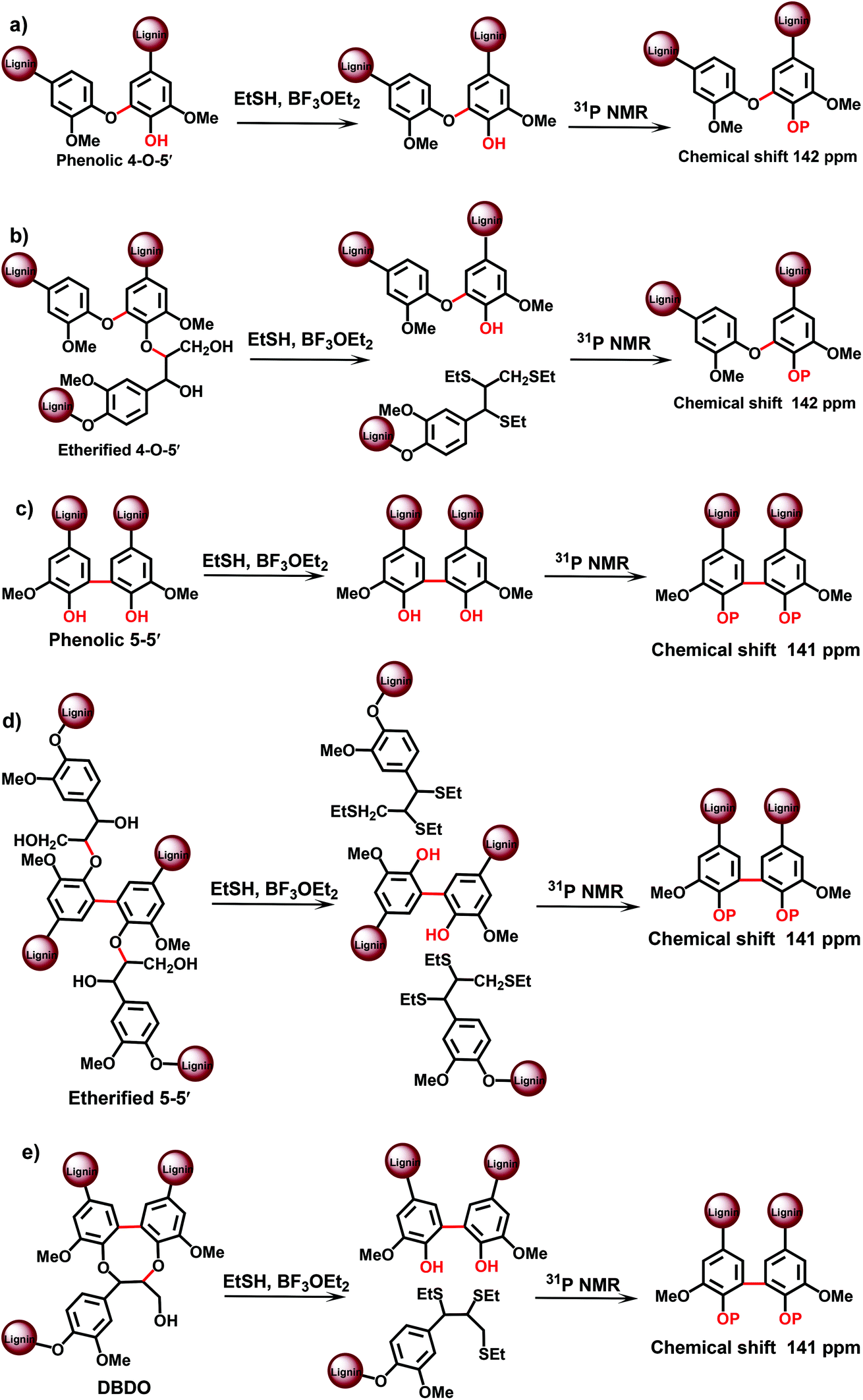 | ||
| Fig. 5 TA-31P NMR approach for the analysis of lignin branching/crosslinking points. | ||
An advantage of the method is the much better resolution of the resonances of various condensed lignin moieties in the 31P NMR spectra after TA degradation. It becomes therefore possible to separate different types of condensed structures, assigned to β-5, 5–5′ and 4-O-5′ moieties.19 This cannot be done directly with the original MWL, because the resolution of 31P NMR spectra for the different condensed moieties is insufficient:13,19 the condensed moieties can be analyzed as a sum, but an accurate differentiation between (originally) phenolic and etherified condensed moieties is not possible. Recently, a modified TA method has been suggested,24 which involves pre-methylation of the originally phenolic OH groups in lignin with diazomethane prior to the actual TA step. This allows for the analysis of originally etherified lignin moieties. The information on the phenolic lignin moieties is thus obtained by the difference between the total and etherified substructures. Unfortunately, only the sum of all condensed structures has been reported24 by this modified method – without the distinction of different types (β-5, 5–5′, 4-O-5′) of the condensed structures.
The total amount of 5–5′ and 4-O-5′ was similar, although slightly lower, to the values obtained by the CuO-PO approach: about 16/100 Ar and 5/100 Ar, respectively (Table 3). However, as the 31P NMR method underestimated the amount of phenolic OH units by about 25%,19 it was reasonable to introduce a correction coefficient that gives the values of 21/100 Ar for 5–5′ and 7/100 Ar for 4-O-5′ units, correspondingly (Table 3).
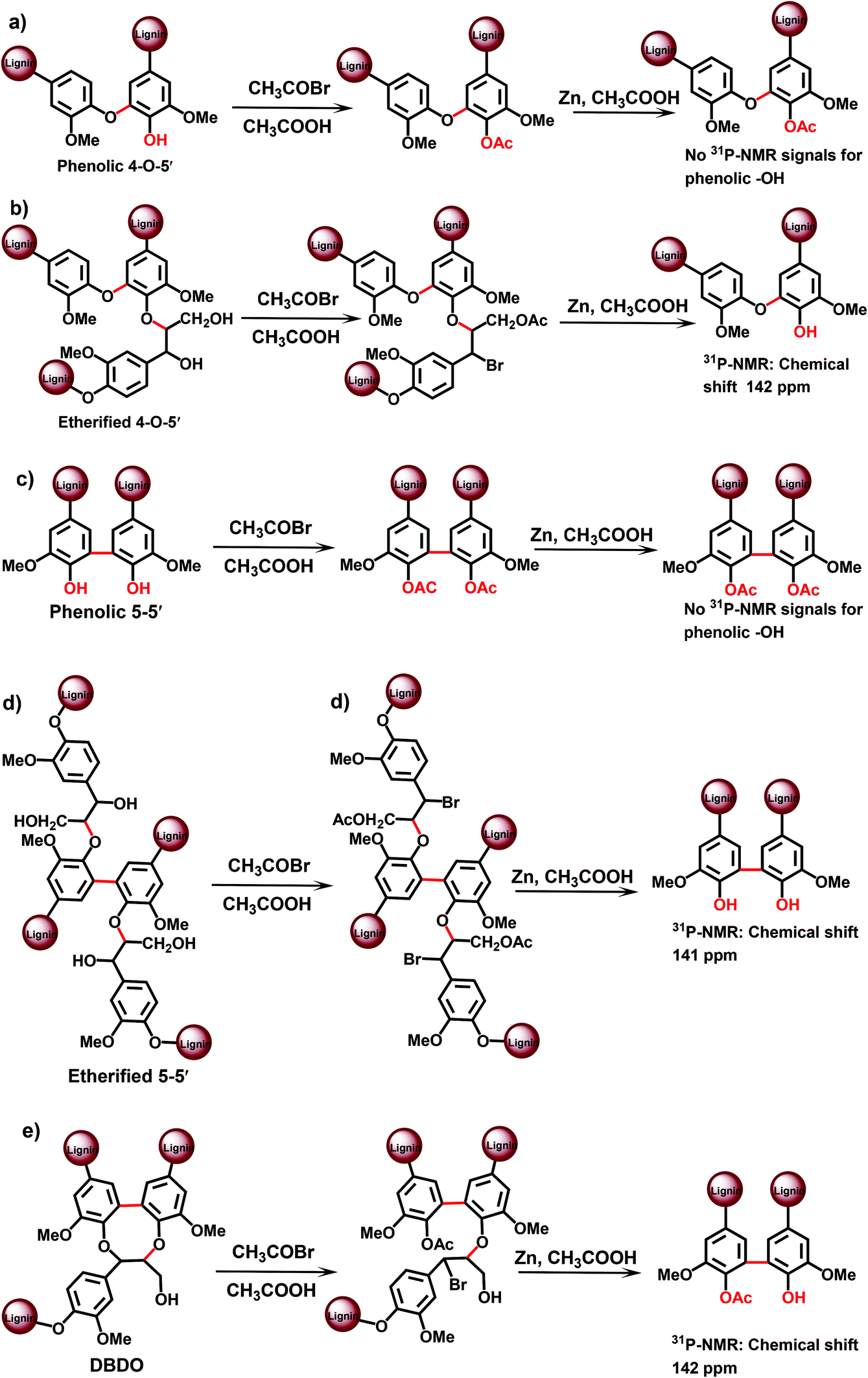 | ||
| Fig. 6 DFRC-31P NMR approach for the analysis of lignin branching/crosslinking points. | ||
| 5–5′et = 1 − (I162–148)ac + (I157–151)na − β-5 − SD, | (1) |
According to this approach (13C NMR with pre-acetylation, method A) the amount of 5–5′ etherified units in the spruce MWL was determined as 19/100 Ar.12
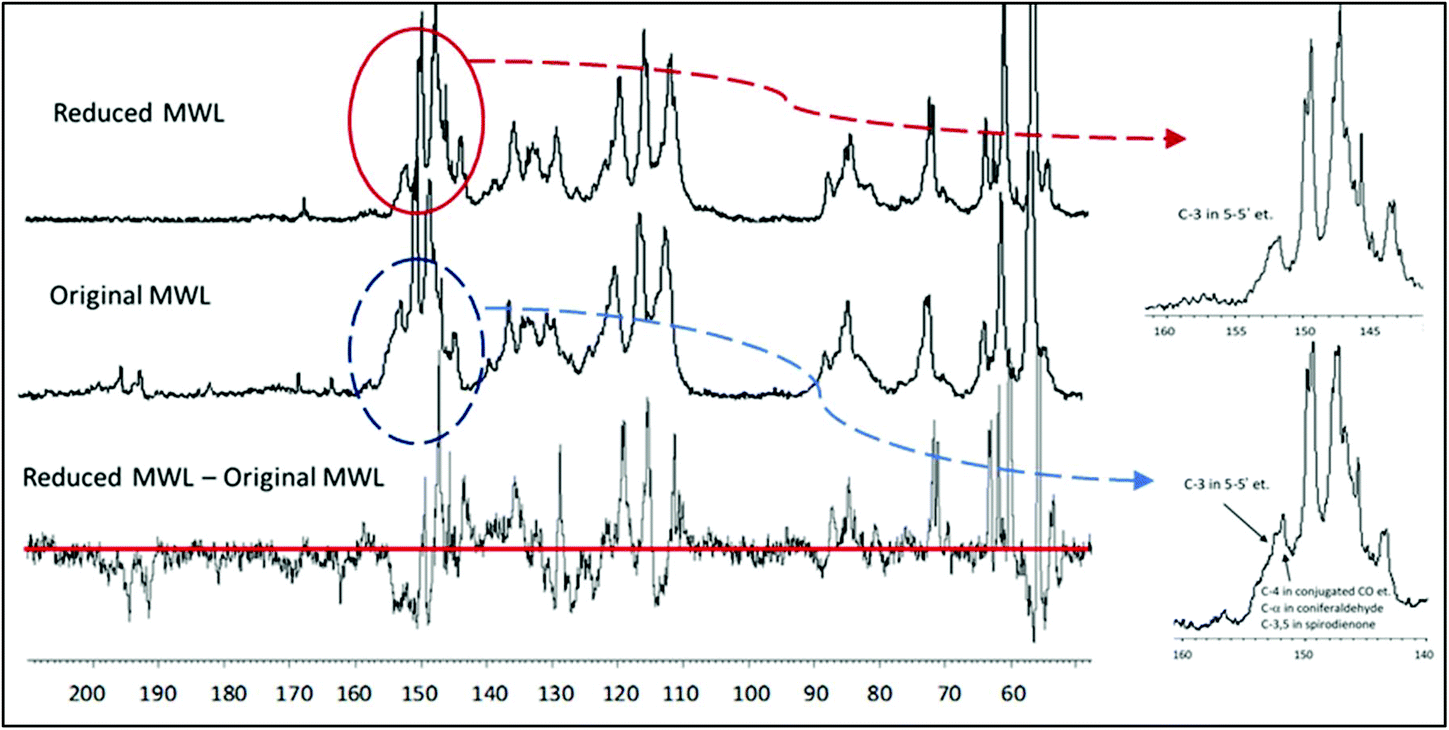 | ||
| Fig. 7 Quantitative 13C NMR spectra of the original MWL (middle trace), the MWL reduced with NaBH4 (upper trace) and the difference spectrum (lower trace). Quantification per 100 Ar (i.e. per 100 monomeric units or mol%) is done normalizing the resonance of aromatic and vinylic lignin moieties in the 13C NMR spectra (ca. 163–100 ppm) as 612.6,12 | ||
The reduction of MWL with NaBH4 is very useful for the estimation of 5–5′ etherified moieties. The reduction of aromatic α-carbonyl structures shifts their C4 resonance upfield, out of the critical region, so that the integral at 155–151 ppm now gives only the value for 5–5′ and 4-O-5′ etherified moieties. The amount of conjugated carboxylic acid/ester moieties (which are not reduced by NaBH4) is below 1% (Table 1) and therefore does not significantly impair the quantification. The value for 5–5′ and 4-O-5′ etherified moieties, obtained by the 13C NMR/reduction approach, was 23/100 Ar,21 and thus rather close to the number calculated according to the rather complex semi-quantitative Method A (19/100 Ar of 5–5′ etherified units).12
A hypothesis expressed in earlier work12 suggested that the majority of non-etherified 5–5′ moieties had a conjugated carbonyl group, their amount having been semi-quantitatively estimated as 5/100 Ar. This suggestion was confirmed by lignin reduction, which shifted the resonance of C4 of these moieties from 149–148 ppm (negative peak in the difference spectrum) to 144–142 ppm (positive peak) (Fig. 7). The difference in the resonance was about 0.05/Ar. Additional confirmation was obtained after acetylation of the reduced lignin which resulted in the decrease of the resonance at 144–142 ppm for these 5/100 Ar structures, due to upfield shift of the C4 in the acetylated 5–5′ (originally phenolic) moieties. Thus, reduction of lignin with NaBH4 allows a quite precise quantification of 5–5′ and 4-O-5′ etherified (23/100 Ar) and 5–5′ non-etherified (5/100 Ar) moieties.21
In terms of the analysis of phenolic lignin moieties (original or/and formed during the pretreatment), the PO method offers the most detailed information on various types of substitutions at the aromatic ring. However, some correction coefficients are used when calculating the amount of lignin substructures based on the yields of monomeric and dimeric degradation products analysed by GC.23 In contrast, 31P NMR analysis is able to quantify various lignin moieties directly. Still, the limited resolution of 31P NMR spectra of lignins, especially of the original, not pretreated ones, does not allow for such detailed information as, for instance, the PO method. In addition, the amount of 4-O-5′ units reported by TA-31P NMR method is apparently overestimated (Table 3) due to insufficient spectral separation of these moieties from other condensed lignin units.
An advantage of wet-chemical methods, i.e., acetylation and NaBH4 reduction, used prior to 13C NMR lignin analysis is the completeness of the conversion and the absence of side reactions under the very mild reactions conditions used.12,14,21 However, Method A12 is semiquantitative as it requires certain calculations (see eqn (1)) increasing the experimental error.
From the methods reviewed, 13C NMR analysis of the NaBH4-reduced lignin (Method B)21 is apparently the most accurate approach for the analysis of etherified 5–5′ and 4-O-5′ units (as a sum) in softwood lignins. The amount of these structures can be directly retrieved from the spectra of the reduced lignins without any additional calculations. The reproducibility of the measurements of this type of signals was estimated to be about 2.5% (relative),31i.e., about ± 1/100 Ar. Moreover, 13C NMR allows for expression of the values obtained directly per monomeric lignin unit (Ar) without deduction of C9-formulae used in the other methods.
In summary, four independent methods (CuO-PO; TA-31P NMR, 13C NMR A and B) showed significant amount of lignin branching/cross-linking points to be located at the aromatic ring, i.e., in etherified 5–5′ (major) and 4-O-5′ (minor) structures, being in the range of about 20/100 Ar (Table 3). Only the DFRC-31P NMR method afforded much lower numbers, obviously due to incomplete cleavage of Alk–O–Ar linkages. Importantly, the value of ∼20/100 Ar correlates well with the total amount of condensed structures (∼40% of all lignin units) quantified by various other methods5,6,9,12,26,32 (Table 1), whereas a lower amount of 5–5′ units as reported by Crestini et al.2 would result in a significant gap.
Thus, the presence of higher amounts of 5–5′ etherified units in the complex lignin structure was deduced from different independent methods, which clearly substantiates the evidence by their combination and the common, consistent message.
Branching and cross-linking points in the side chain
Etherified 5–5′ and 4-O-5′ units contribute one branching point (DBDO structure) and two cross-linking points (5–5′ etherified) in the 25-unit model (Table 1). However, according to the above calculation, the total number of branching/cross-linking points in MWL lignin is almost twice as high. Based on the total degree of condensation (∼40%),12 there is almost no room for additional cross-linking points at the aromatic ring (there are some β-6 structures, but at rather low amounts5–7,9). Therefore, the additional branching/cross-linking points must come from the side chains. To gain better insights into this issue, we review the general lignin structure in more detail.We are able to identify about 80% of the specific structures in the side chains (Table 1); none of them present in branching. The remaining structures, apparently, have high structural diversity, and are difficult to identify with 2D NMR methods because each of them is present only in very small amounts. This also results in the broad 13C NMR signals observed.12 The amount of these structures can be deducted from the material balance of different lignin functionalities.
The amount of Alk–O-moieties is:
| Alk–O- = oxygenated aliphatic − free aliphatic OH = 2.15–1.07 = 1.08/Ar; |
| Position | Moieties | Amounts |
|---|---|---|
| γ | OH-total | 68 |
| Pinoresinol | 4 | |
| Coniferaldehyde | 4 | |
| Short chain (vanillin + COOR) | 8 | |
| Total identified | 84 | |
| γ-O-Alk (tentative) | 16 | |
| β | β-O-4/α-OH | 36 |
| β-O-4/α-CO | 2 | |
| DBDO | 4 | |
| Phenylcoumaran | 10 | |
| β-1/α-OH | 1 | |
| Spirodienone | 2 | |
| Secoisolariciresinol | 2 | |
| Coniferyl alcohol | 2 | |
| Dihydroconiferyl alcohol | 2 | |
| Ar–CO–CH2–CH2OH | 2 | |
| Pinoresinol | 4 | |
| Coniferaldehyde | 4 | |
| Short chain (vanillin + COOR) | 8 | |
| Total identified | 79 | |
| Carbonyl (non-conjugated) | 4 | |
| Other β-O-4 (tentative) | 8 | |
| β-Aliphatic (tentative) | 9 | |
| α | OH | 38 |
| β-O-4/α-CO | 2 | |
| DBDO | 4 | |
| Phenylcoumaran | 10 | |
| Spirodienone | 4 | |
| Secoisolariciresinol | 2 | |
| Coniferyl alcohol | 2 | |
| Dihydroconiferyl alcohol | 2 | |
| Ar–CO–CH2–CH2OH | 2 | |
| Pinoresinol | 4 | |
| Coniferaldehyde | 4 | |
| Short chain (vanillin + COOR) | 8 | |
| Total identified | 82 | |
| α-O-Alk (tentative) | 18 | |
| Ar-4 | OH | 31 |
| Spirodienone | 2 | |
| Etherified in: | 67 | |
| β-O-4 total in: | 50 | |
| β-O-4/α-OH | 36 | |
| β-O-4/α-CO | 2 | |
| DBDO-β | 4 | |
| Other β-O-4 (tentative) | 8 | |
| DBDO-α | 4 | |
| Phenylcoumaran | 10 | |
| Others (4-O-5′, α-O-4 etc.) | 3 |
We suggest that these structures are Alk-O-Alk in general (α- and γ-) as well as various β-O-4 units, potentially including minor amounts of non-cyclic α-O-4 units. The material balance in the specific positions of lignin units (Table 4) shows that maximal 0.16/100 Ar and 0.18/100 Ar of the unknown Alk–O moieties can be accommodated at the γ- and α-positions, respectively.
This is supported by detailed analysis of 2D NMR spectra of spruce MWL. There are some signals which surprisingly have not been considered until now in spite of the extensive work on the characterization of native lignins. We assign these signals based on the data available in the literature33–40 and ChemDraw modeling of some hypothetical moieties when the literature data are lacking. This analysis indicates that significant signal overlap is plausible even in the 2D spectrum (Fig. 8). γ-Methylene (CH2) signals of potential γ-ether moieties have definitely different response factors as compared to the CH signals in the region of 79–90 ppm that are used as the quantification reference (Table 5). Thus, detailed quantitative HSQC analysis of Alk-O-Alk moieties is not reliable so far and the integral values reported (Table 5) can just be used for an approximate discussion.
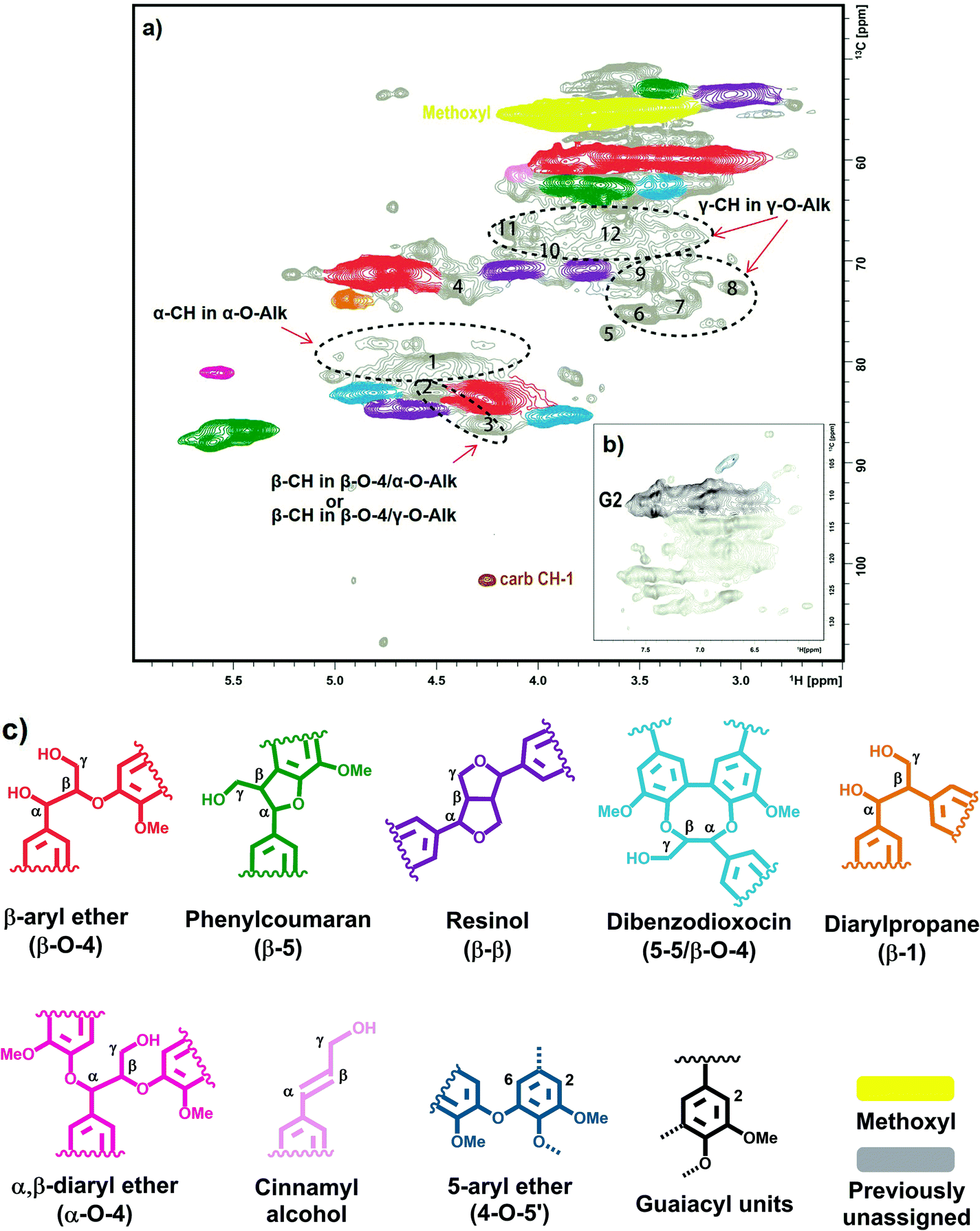 | ||
| Fig. 8 HSQC NMR spectrum of spruce milled wood lignin in DMSO-d6: (a), side-chain region; (b), aromatic region; (c), identified structural units of lignin. | ||
| HSQC peaks | Integration range, ppm | Integral | Possible assignment | |
|---|---|---|---|---|
| δ C | δ H | |||
| a The value is set based on 13C NMR quantification. | ||||
| Cluster 1 | 89.6–78.9 | 6.0–3.7 | 83.0 | Reference clustera |
| carb-CH1 | 103.1–99.4 | 4.3–4.2 | 0.6 | Carbohydrate (CH-1) |
| 1 | 81.4–78.5 | 4.8–4.3 | 8.2 | α-O-Alk (α-CH) |
| 2 | 83.7–81.7 | 4.7–4.4 | 9.5 | Unknown; α-O-Alk (α-CH) or/and β-O-4/γ-O-Alk (β-CH) |
| 3 | 87.7–85.1 | 4.4–4.1 | 2.9 | α-O-Alk/β-O-4 (β-CH) |
| Aβ | 84.9–81.7 | 4.4–4.0 | 20.0 | α-OH/β-O-4/γ-OH (β-CH) |
| Aα | 72.9–68.9 | 5.1–4.5 | 32.5 | α-OH/β-O-4 (α-CH) |
| I2/6 | 107.5–100.7 | 6.9–6.6 | 2.1 | 4-O-5′ |
| Cγ | 72.1–69.2 | 4.3–4.0 | 6.6 | β-β (γ-CH2)-1 |
| Cγ | 72.1–69.8 | 3.9–3.6 | 5.9 | β-β (γ-CH2)-2 |
| Cluster 2 | 78.6–70.0 | 3.6–3.0 | 16.1 | Unknown, γ-O-Alk (cluster) |
| Cluster 3 | 69.1–64.8 | 4.2–3.4 | 13.8 | Unknown, γ-O-Alk (cluster) |
| 6 | 77.2–73.9 | 3.6–3.4 | 3.2 | Unknown, γ-O-Alk |
| 7 | 76.1–72.9 | 3.4–3.2 | 2.2 | Unknown, γ-O-Alk |
| 8 | 74.1–71.4 | 3.1–3.0 | 0.9 | Unknown, γ-O-Alk |
| 9 | 73.0–70.3 | 3.6–3.4 | 3.8 | Unknown, γ-O-Alk |
| 10 | 70.9–67.8 | 4.0–3.9 | 2.0 | Unknown, γ-O-Alk |
| 11 | 68.4–65.5 | 4.2–4.1 | 1.2 | Unknown, γ-O-Alk |
| 12 | 69.8–65.6 | 3.8–3.5 | 6.7 | Unknown, γ-O-Alk |
Some weak signals at δC/δH 70–65/3.5–4.0, which can be attributed to γ-ether moieties,33–40 were detected in the HMQC12 and HSQC spectra (Fig. 8, Table 5). Thus, it is likely that a large variety of γ-ether moieties exists, resulting in many resonances and a consequent spreading of resonance intensities in this region. About 7 different potential signals can be observed in the spectrum (Fig. 8, Table 5).
The broad signal from CH-α in α-O-Alk moieties has been detected at δC/δH ∼ 81/4.6 ppm.33–40 As the amount of carbohydrates in the MWL preparation was too low to consider some significant contribution from lignin carbohydrate complex (LCC) benzyl ethers,39 the signal observed was assigned to some (but not all) α-O-Alk linkages between lignin units. Importantly, the amount of α-OH/β-O-4 moieties derived from α-CH is significantly higher than that from the corresponding β-CH (Table 5). It can be hypothesized that some of α-OH/β-O-4 moieties contain a substituent (such as -O-Alk) at the γ-position of the side chain; this would shift the resonance of the β-CH but have a very subtle effect on α-CH, which fits well the hypothesis of the presence of α-OH/β-O-4/γ-O-Alk moieties. ChemDraw simulations support this hypothesis. Therefore, the cross-peak at ca. 83.1/4.53 ppm (Fig. 8, Table 5, peak 2) might be tentatively assigned to α-OH/β-O-4/γ-O-Alk moieties.
The rather high content of γ- and α-ether moieties estimated by 13C NMR is consistent with NMR analysis of ginko MWL which was selectively 13C-enriched at the γ- and α-positions.4113C NMR showed an appreciable resonance intensity gain in the regions of 70–65 ppm and 82–78 ppm, respectively, although no individual peaks were assigned.
The number of etherified phenolic units (Table 4) is: 1–0.31 (PhOH) – 0.02 (SD) = 0.67/Ar, with 0.56/Ar of them having been identified so far. By wet chemistry methods, the amount of total β-O-4 units is 0.50/Ar,5,7 so we add 0.08/Ar to previously identified 0.42/Ar (Table 1). The remaining 0.03/Ar etherified units at the 4-position could be some minor moieties such as α-O-4, 4-O-5′ etc., as well as the experimental error.
The unidentified 0.13 units/Ar at the β-position (Table 4) likely belong to non-conjugated carbonyl groups (0.04/Ar)12 and to the saturated aliphatic type (0.09/Ar). The latter is very difficult to quantify accurately as two very strong resonances – from OMe groups and especially from the solvent (DMSO) – may overlap with the resonances of these saturated moieties and may also affect the baseline in this region. Again, the diversity should be rather high because no distinct cross-peaks in the HSQC spectra are observed.
If we consider the side chain as the “tail” and the aromatic ring as the “head” of the lignin monomeric unit, it is noteworthy that the proportion of terminal “heads” is significantly higher than that of “tails” (26 vs. 18/100 Ar). This is due to a significant number of interunit connections of “tail-to-tail” type (e.g., β–β type). At the same time, the DBDO moiety (0.04/Ar) would eventually produce two terminal side chains (tails) per one phenolic OH (head).
Tentative model of spruce MWL
On the basis of the extensive quantification discussed so far, we are now in the position to propose a possible structure of spruce MWL and a plausible model (Fig. 9). It is important to emphasize that this model does not correspond to the lignin in situ, due to much less structural information available for the latter. Moreover, we note that many of the analytical methods that are feasible for MWL, such as high resolution NMR, cannot be applied for lignin in situ and that MWL has undergone an isolation procedure which certainly impacted its structure.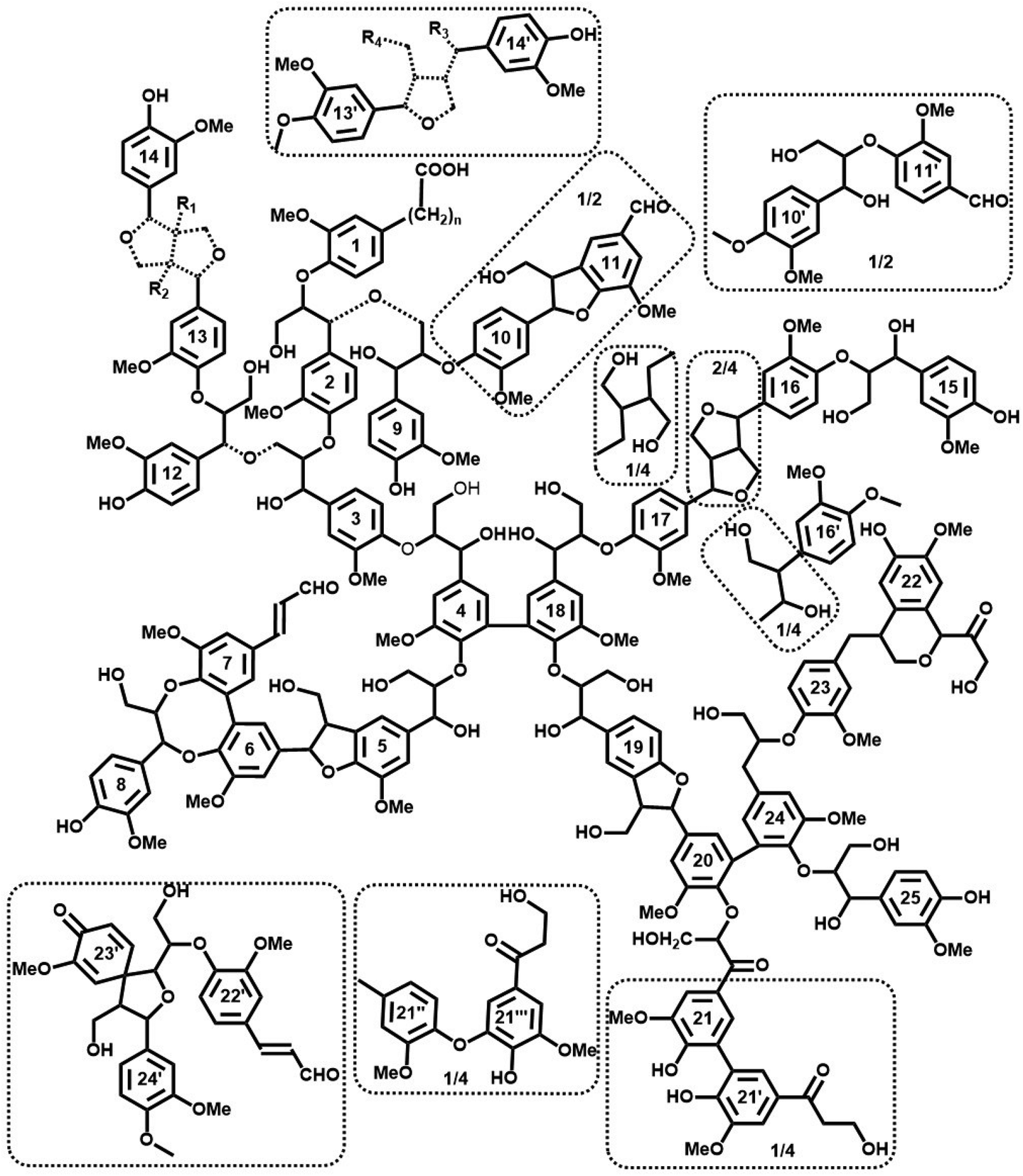 | ||
| Fig. 9 Tentative quantitative structural model of spruce MWL, in agreement with currently available structural data on lignin moieties and their occurrence frequencies. Dashed bond: Different structures agreeing with available information; the exact structure of the Alk-O-Alk moieties (units 2–9, 3–12, 13–14) is not well defined, therefore the model indicates a general type of units rather than insisting on a precise structure. Minor structures (below 4/100 Ar) are given in the dotted boxes. | ||
Starting with the premise that despite the vast amount of information available, a structural image can only be a representation, a “snapshot”, of a single lignin molecule: it is impossible to draw an exact chemical structure of lignin per se. There is infinite structural diversity between individual lignin molecules (and in addition still incomplete information on the exact chemical structures of all lignin structural units and their linkage modes). As an example, a single guaiacyl glycerol (G-β-O-4) unit can be erythro- or threo-configured, phenolic or etherified, condensed (of different types) or non-condensed, giving rise to a vast variety of important structural differences that are not defined. More importantly, the sequence of lignin subunits in the macromolecule is unknown and there is no appropriate analytical method so far to define it in a polymer (only in some oligomeric fractions). Thus, even the most simplified structural lignin models3,42 must remain speculative.
However, accumulation and visualization of all currently available information in a plausible lignin structural model is certainly useful for understanding structural features of lignin. This is even more relevant as several previous visualization attempts have become contradictory, in view of the ever-growing amounts of experimental data that are becoming available. The most recent quantitative model of spruce MWL has been developed by Sakakibara.7 Analytical developments, especially 2D NMR methods, have significantly changed our image of lignin structure since then, leading to a new structural model suggested by Brunow in 1998.42 However, as admitted by Brunow, such model did not quantitatively correlate the amount of suggested subunits and functional groups with their experimental numbers obtained from analyses. Similarly, a very recent model of Ralph et al.3 is based on the current ligninfication theory, which includes some approximative HSQC quantification.
In our proposed model, Fig. 9, we combine all available analytical data, both from our own work and the literature, with recent findings on the absolute molecular weights of MWL. Consequently, we offer an up-to-date structural model of spruce MWLs. The high degree of branching/cross-linking is derived from the combination of new results on lignin average DP10 and well-known and accepted data on the amount of various terminal units.4–7,9,11–14,26,27,32 The model depicts 7 formal individual chains with one formal branching point containing one unit in the branching chain (units 8, DBDO phenolic) and 4 cross-linking points. Half of the cross-links are of the 5–5′ etherified type (units 4–18 and 20–24) while the other half is of different Alk-O-Alk type (units 2–9, 3–12). As mentioned above, the exact structure of these Alk-O-Alk moieties is not well defined, therefore the model indicates a general type of units rather than a precise structure.
Differences between MWL and lignin in situ
The discussion above related to spruce MWL, not lignin in situ. The latter is much more difficult to analyze. Therefore, we can discuss only some specific features, as follows. It is well known that β-O-4 linkages are cleaved during ball milling in an extent proportional to milling intensity.26,43,44 This disadvantageous structural change is almost inherent in lignin isolation: also “whole cell wall” lignin analyzed with solution HSQC NMR method45 has undergone ball milling and therefore is no longer native. However, the structure of MWL (i.e., the extracted and purified fraction of the total wood lignin) is rather independent of the milling intensity and quite constant in a wide range of MWL yield.26,44,46 The difference in the amount of β-O-4 structures has not been well defined yet. Hu et al. did not find any difference between MWL and lignin in situ by ozonolysis,44 while Gellerstedt et al.47 reported a 40% decrease in β-O-4 in MWL vs. biomass lignin according to TA analysis. Also, as the results of the cleavage of β-O-4 structures during ball milling, the amount of phenolic OH in MWL is significantly higher than in biomass lignin, ca. 0.3 vs. 0.1 per C9-unit, respectively.4–7,48 It is also likely that the amount of carbonyl groups increases during ball milling through oxidation;49 although some carbonyl moieties, such as vanillin and coniferyl aldehyde are already present in in situ lignin.50Remarkably, softwood lignin in situ is even more condensed than the corresponding MWLs (DC of 41% vs. 35%, correspondingly) and all condensed moieties in lignin in situ are etherified.24 The authors suggested that phenolic condensed moieties of MWL were formed from cleavage of the corresponding etherified units during ball milling. In addition, lignin in biomass is linked to polysaccharides forming so-called lignin-carbohydrate complexes (LCCs).4–7,51,52 This would result in additional crosslinking. The main LCC linkages are of benzyl ether, phenyl glycoside and γ-ester types; their total amounts is about 0.07/C9-unit.39,52 Obviously, the molecular weight is significantly decreasing during ball milling,46,49 but so far it is not possible to determine the decrease quantitatively due to challenges in detection of the starting value, the molecular weight of lignin in situ.
Analytical data and current lignification theory
Current lignification theory3 cannot completely explain all structural varieties that are derived directly from analytical results or indirectly from different experimental methods. This is obviously no reason to reject either the analytical findings or the theory as a whole. On the contrary, the latter should be adapted to reconcile it with the analytical data.It is worth noting that the presence of α-O-Alk moieties in MWL has been considered by a few researchers.4,7,33,53 Glasser et al.54 carried out computational modeling of lignin and concluded that about 20% of lignin units should come from a “non-hydrogenative polymerization” route. The exact biosynthetic route for these units is not clear yet and needs further investigation.
There are two main points of disagreement between the lignification theory and the experimental data:
1. The high amounts of 5–5′ etherified units (not DBDO). Corroborated by different analytical methods, this fact is analytically well proven, however, not accounted for by the lignification theory.3 A possible explanation was suggested very recently by Chang and Jiang.9
2. High amounts of unidentified Alk-O-Alk moieties, very likely of α-O-Alk and γ-O-Alk types. Their exact structures are not known yet and need both further analytical studies and integration into lignification theory.
Some differences between the experimental data on MWL and lignification theory (implying lignin in situ) might originate from lignin changes during ball milling. In particular, a high diversity of structural units is typical for degradation processes of native lignin.40 Similarly, this could be a result of very complex (and not known yet) lignin transformation in ball milling, in particular the aliphatic α-O-Alk and γ-O-Alk moieties. With regard to aromatic structures, by contrast, it has been shown24 that the amount of 5–5′ and 4-O-5′ etherified units in lignin in situ is even higher than in MWL, and that their high number is therefore not an artifact of the isolation process. Thus, the degree of branching/crosslinking through aromatic rings is even higher in the true native lignin than it is in MWL.
Impact of native lignin structure on lignin valorization
The structure of native lignin is significantly altered upon processing, and obviously has an influence on the structures generated that make up the technical lignins. On the one hand, understanding the structure of native lignins – and also of close-to native milled wood lignins – is the basis of comprehending the structural changes that occur in the large variety of biorefinery processes, e.g. different chemical pulping methods, and different pre-treatments in bioethanol production. It is obvious that chemical degradation of branched and crosslinked lignin is more difficult than depolymerization of linear lignin, requiring more drastical conditions and higher energy input. On the other hand, the higher reluctance in delignification of certain biomass types, specifically softwood, would mean indirect support of the branched lignin structure.A better understanding of the structure of lignins in original biomass and better knowledge of the resulting reaction pathways allow for a prediction of the main characteristics of technical lignins and related biorefinery products. In particular, inter-unit C–C bonds (such as in 5–5′ units) are very stable in most biorefinery processes and are therefore expected to accumulate in the corresponding technical lignins. In contrast, alkyl ethers, especially those of the benzylic type, are more reactive, and most of them should be cleaved during biorefinery treatments, depending of course on process severity. Therefore, a higher degree of branching revealed in softwood native lignins will be conveyed to the corresponding technical lignins and have significant influence on their follow-up chemistry and application potential.
Supramolecular properties of lignin
We end our discussion with a subsidiary but yet relevant evidence found in the formation of supramolecular structures based on lignin, which are always far from being one-dimensional. Some of the earlier indications in such context is the case of Kraft lignin in solution, which was reported to form fractal associations.55 Indeed, it was suggested that Kraft lignin clusters “exhibit a supramolecular structure similar to what earlier has been proposed for native softwood lignin”. Moreover, lignins typically display spherical morphologies after removal from the cell wall or middle lamella. For example, lignin is extruded from the cell wall into beads during hydrolysis of biomass.56,57 Furthermore, upon extrusion of extracted lignins dissolved in an appropriated solvent in an anti-solvent (solvent shifting) the lignins form spherical particles.58 Related morphologies are also evident when lignocellulosic biomass is deconstructed into nanocellulose.59 Similar to these observations, when a lignin solution is carried by a gas under laminar flow in an aerosol system, the lignin dries into perfectly spherical particles.60 The tendency for lignin to bead up can be partially explained by the role of the high surface tension of water. However, the effect is very notorious across the different processes, as presented above, which hints to the possibility that the contribution of molecular interactions and shape may be prime factors.Another piece of evidence can be taken from attempts to extrude lignin by wet or dry spinning, e.g., into filaments. Such processes are quite challenging unless a material that helps to align the structure under the flow field, such as nanocellulose.61 It is clear that there is a general reluctance of lignin to align in linear fashion. In fact, alignment of secondary components (for example to assist filament formation) is clearly reduced by the presence of lignin.
Uniform one-dimensional supramolecular structures assembled from irregular, highly branched molecules are rarely reported. Special exceptions have been discussed in the literature were unique matching of directional intermolecular interactions and steric constraints are at play (e.g., in the case of some dendritic structures).62 Otherwise, dendritic molecules typically possess globular or close to globular shapes in solution. Similar observations can be translated to other molecules in solution, including lignin. This is in fact a main issue in the field of colloids, which is not fully discussed in the literature and is particularly relevant to lyophobic colloidal particles such as extracted lignins.
The nature of the interactions between the single molecule to form supramolecular structures are determined by electrodynamic interactions (van der Waals and dispersion forces), steric and depletion forces but also connected to particle/medium interfaces, which is an important consideration for any conclusion to be made, namely, the media used in any observation (interactions between the particle and the surrounding medium). For biopolymers, such as lignins, there are many possible conformations and interactions in solution, much more than the more widely studied synthetic polymers. Moreover, the rules of thumb that apply to typical polymers may not be applicable to complex macromolecules such as lignins. It is with no doubt that a debate exists whether lignin is branched or linear. While a definite conclusion pointing to a branched lignin structure is not possible based on the “beading” evidence, an assumption of strict linearity would rather challenge all experimental observations. Overall, supramolecular shape provide indirect evidence supporting the direct chemical evidence discussed in this paper, it is nevertheless interesting to note the correlation between macromolecular branching and structure with supramolecular assembly, a topic that we hope to expand in our future inquiries.
Conclusions
Based on the absolute molecular weight of MWL, our suggested lignin molecular model consists on an average of 25 C9-units. The analyses of terminal groups and inter-unit linkages agree with spruce MWL being rather branched and cross-linked (about 1 branching and 4 cross-linking points per 25 C9-units) with short branching chains. The results of four independent analytical methods (CuO-PO, TA-31P NMR, 13C NMR of NaBH4 reduced lignin) indicate the presence of significant amounts of 5–5′ etherified units. The amount of branching/cross-linking units in the aromatic ring (5–5′ and 4-O-5′ etherified) is about 20/100 Ar. Quantitative 13C NMR and semi-quantitative HSQC analysis indicates that also aliphatic ether (Alk–O–Alk) linkages at the α- and γ-positions of the side chain, with intact β-O-4 bonds, significantly contribute to lignin cross-linking.The suggested up-to-date structural model of spruce MWL is thus in agreement with the current structural and molecular weight data. The example formula depicts one out of a plethora of molecules that accurately match the available quantitative data; yet it is precisely this agreement that distinguishes it from previous, qualitative structural proposals. An assertion of the randomness of the model would be illogical here: lignin itself shows exactly this randomness in its structure, and any reasonable attempt to grasp this structural arbitrariness and diversity, which is deeply rooted in its biosynthesis, must be limited only to snapshots. This is what our structural model affords – it puts together the experimental analytical puzzle pieces to an entire image, in one possible combination. Not the only one, but one that considers and incorporates all contributing elements. We sincerely hope that the model will serve as a helpful and useful tool for further contemplation of lignin structure, reactivity and properties. The agreement with all the available experimental data will be certainly a plus. Despite the accordance with the current status of experimentally determined frequency of lignin structural units, we are aware that lignin research is constantly adding information that might change our rationalization of this fascinating macromolecule.
Conflicts of interest
There are no conflicts to declareAcknowledgements
The authors are grateful to Dr J. Ralph for valuable discussion. We are also grateful for support by the Academy of Finland's Flagship Programme under Projects No. 318890 and 318891 (Competence Center for Materials Bioeconomy, FinnCERES) and the Austrian Biorefinery Center Tulln (ABCT).References
- D. A. I. Goring, in Lignins: Occurrence, formation, structure and reaction, ed. K. V. Sarkanen and C. H. Ludwig, Wiley-Interscience, New York, 1971, pp. 695–768 Search PubMed.
- C. Crestini, F. Melone, M. Sette and R. Saladino, Biomacromolecules, 2011, 12, 3928–3935 CrossRef CAS PubMed.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- K. Freudenberg, in Molecular Biology Biochemistry and Biophysics, ed. A. Kleinszeller, G. F. Springer and H. G. Wittmann, Springer-Verlag, Berlin, 1968, vol. 2, pp. 47–122 Search PubMed.
- E. Adler, Wood Sci. Technol., 1977, 11, 169–218 CrossRef CAS.
- C. L. Chen, in Lignins: Occurrence in wood tissues, isolation, reactions and structure, ed. M. Lewis and I. S. Goldstein, Marcel Dekker Inc., New York, 1991, pp. 183–261 Search PubMed.
- A. Sakakibara, in Wood and Cellulose Chemistry, ed. D. N. S. Hon and N. Shiraishi, Marcel Dekker Inc., New York, 1991, pp. 113–175 Search PubMed.
- M. Y. Balakshin, E. A. Capanema, B. Goldfarb, J. Frampton and J. F. Kadla, Holzforschung, 2005, 59(5), 488–496 CAS.
- H. M. Chang and X. Jiang, J. Wood Chem. Technol., 2019, 1–10 Search PubMed.
- G. Zinovyev, I. Sulaeva, S. Podzimek, D. Rössner, I. Kilpeläinen, I. Sumerskii, T. Rosenau and A. Potthast, ChemSusChem, 2018, 11(18), 3259–3268 CrossRef CAS PubMed.
- L. Zhang and G. Gellerstedt, in Proceedings of the 6th European Workshop on Lignocellulosics and Pulp, Bordeaux, 2000, pp. 7–10.
- E. A. Capanema, M. Y. Balakshin and J. F. Kadla, J. Agric. Food Chem., 2004, 52, 1850–1860 CrossRef CAS PubMed.
- M. Y. Balakshin and E. A. Capanema, J. Wood Chem. Technol., 2015, 35(3), 220–237 CrossRef CAS.
- G. F. Zakis, in Functional analysis of lignins and their derivatives, Tappi Press, Atlanta, 1994 Search PubMed.
- I. A. Pearl, J. Am. Chem. Soc., 1942, 64(6), 1429–1431 CrossRef CAS.
- F. Lu and J. Ralph, J. Agric. Food Chem., 1997, 45, 2590–2592 CrossRef CAS.
- C. Rolando, B. Monties and C. Lapierre, in Methods in Lignin Chemistry, ed. S. Y. Lin and C. W. Dence, Springer, Berlin, 1992, pp. 334–349 Search PubMed.
- M. Erickson, S. Larsson, G. E. Miksche, A. C. Wiehager, B. O. Lindgren and C. G. Swahn, Acta Chem. Scand., 1973, 27, 903–914 CrossRef CAS.
- R. Smit, I. D. Suckling and R. M. Ede, in Proceedings of 9th International Symposium on Wood and Pulping Chemistry, Montreal, 1997, pp. L4–1.
- S. Tohmura and D. S. Argyropoulos, J. Agric. Food Chem., 2001, 49, 536–542 CrossRef CAS.
- M. Y. Balakshin and E. A. Capanema, in 59th Appita Annual Conference and Exhibition: Incorporating the 13th ISWFPC (International Symposium on Wood, Fibre and Pulping Chemistry), Auckland, 2005, vol. II., pp. 353–360.
- S. Larsson and G. E. Miksche, Acta Chem. Scand., 1971, 25, 647–662 CrossRef CAS.
- G. Gellerstedt, in Methods in Lignin Chemistry, ed. S. Y. Lin and C. W. Dence, Springer, Berlin, 1992, pp. 322–333 Search PubMed.
- B. Nanayakkara, M. Manley-Harris and I. D. Suckling, J. Agric. Food Chem., 2011, 59, 12514–12519 CrossRef CAS.
- K. M. Holtman, H. M. Chang, H. Jameel and J. F. Kadla, J. Agric. Food Chem., 2003, 51, 3535–3540 CrossRef CAS PubMed.
- M. Y. Balakshin, E. A. Capanema and H. M. Chang, in Characterization of Lignocellulosics, ed. T. Hu, Oxford, UK, 2008, pp. 148–166 Search PubMed.
- L. Zhang and G. Gellerstedt, Magn. Reson. Chem., 2007, 45, 37–45 CrossRef CAS PubMed.
- Y. Li, T. Akiyama, T. Yokoyama and Y. Matsumoto, Biomacromolecules, 2016, 17, 1921–1929 CrossRef CAS PubMed.
- F. Yue, F. Lu, S. Ralph and J. Ralph, Biomacromolecules, 2016, 17, 1909–1920 CrossRef CAS.
- M. Y. Balakshin, E. A. Capanema, C. L. Chen and H. S. Gracz, J. Agric. Food Chem., 2003, 51, 6116–6127 CrossRef CAS.
- M. Y. Balakshin, E. A. Capanema, R. B. Santos, H. M. Chang and H. Jameel, Holzforschung, 2016, 70, 95–108 CAS.
- K. Lundquist, Acta Chem. Scand., 1980, B34, 497–501 Search PubMed.
- G. J. Leary, D. A. Sawtell and H. Wong, Holzforschung, 1983, 37(4), 213–215 CrossRef CAS.
- G. Brunow, J. Sipilä and T. Mäkelä, Holzforschung, 1989, 43(1), 55–59 CrossRef CAS.
- F. Lu and J. Ralph, Org. Biomol. Chem., 2008, 6(20), 3681–3694 RSC.
- S. A. Ralph, J. Ralph and L. L. Landucci, in NMR database of lignin and cell wall model compounds, US Forest Prod. Lab., Madison, 2004 (http://ars.usda.gov/Services/docs.htm) Search PubMed.
- J. Sipilä and G. Brunow, Holzforschung, 1991, 45, 275–278 CrossRef.
- H. H. Nimz and H. D. Lüdemann, Holzforschung, 1976, 30(2), 33–40 CrossRef CAS.
- M. Y. Balakshin, E. A. Capanema, H. Gracz, H. M. Chang and H. Jameel, Planta, 2011, 233(6), 1097–1110 CrossRef CAS PubMed.
- M. Y. Balakshin and E. A. Capanema, RSC Adv., 2015, 5(106), 87187–87199 RSC.
- N. Terashima, D. Evtuguin, C. Pascoal Neto, J. Parkas, M. Paulsson, U. Westermark, S. Ralph and J. Ralph, in Proceedings of the 12th International Symposium on Wood and Pulping Chemistry, Madison, 2003, vol. I, pp. 175–178.
- G. Brunow, I. Kilpeläinen, J. Sipilä, K. Syrjänan, P. Karhunen, H. Setälä and P. Rummakko, in Lignin and Lignan Biosynthesis, ed. N. G. Lewis and S. Sarkanen, American Chemical Society, Washington, DC, 1998, pp. 131–147 Search PubMed.
- A. Fujimoto, Y. Matsumoto, H. M. Chang and G. Meshitsuka, J. Wood Sci., 2005, 51, 89–91 CrossRef CAS.
- Z. Hu, T. F. Yeh, H. M. Chang, Y. Matsumoto and J. F. Kadla, Holzforschung, 2006, 60, 389–397 CAS.
- H. Kim, J. Ralph and T. Akiyama, BioEnergy Res., 2008, 1(1), 56–66 CrossRef.
- G. Zinovyev, I. Sumerskii, T. Rosenau, M. Balakshin and A. Potthast, Molecules, 2018, 23(9), 2223 CrossRef PubMed.
- H. Önnerud and G. Gellerstedt, Holzforschung, 2003, 57, 165–170 Search PubMed.
- J. M. Yang and D. A. I. Goring, Can. J. Chem., 1979, 58, 2411–2414 CrossRef.
- H. M. Chang, E. B. Cowling, W. Brown, E. Adler and G. Miksche, Holzforschung, 1975, 29, 153–159 CrossRef CAS.
- D. V. Evtuguin, M. Y. Balakshin, N. Terashima, C. Pascoal Neto and A. M. S. Silva, in Proceedings of the 12th International Symposium on Wood and Pulping Chemistry, Madison, 2003, pp. 177–180.
- T. Koshijima and T. Watanabe, in Association between lignin and carbohydrates in wood and other plant tissues, Springer Science & Business Media, Berlin, 2013 Search PubMed.
- M. Y. Balakshin, E. A. Capanema and A. Berlin, in Studies natural products chemistry, Elsevier, Amsterdam, 2014, vol. 42, pp. 83–115 Search PubMed.
- G. J. Leary, Wood Sci. Technol., 1982, 16, 67–70 CrossRef CAS.
- W. G. Glasser and H. R. Glasser, Holzforschung, 1974, 28, 5–11 CrossRef CAS.
- M. Norgren, H. Edlund and L. Wågberg, Langmuir, 2002, 18(7), 2859–2865 CrossRef CAS.
- B. S. Donohoe, S. R. Decker, M. P. Tucker, M. E. Himmel and T. B. Vinzant, Biotechnol. Bioeng., 2008, 101(5), 913–925 CrossRef CAS PubMed.
- M. Ago, B. L. Tardy, L. Wang, J. Guo, A. Khakalo and O. J. Rojas, MRS Bull., 2017, 42(5), 371–378 CrossRef CAS.
- A. P. Richter, B. Bharti, H. B. Armstrong, J. S. Brown, D. Plemmons, V. N. Paunov and O. D. Velev, Langmuir, 2016, 32(25), 6468–6477 CrossRef CAS PubMed.
- A. Ferrer, E. Quintana, I. Filpponen, I. Solala, T. Vidal, A. Rodríguez and O. J. Rojas, Cellulose, 2012, 19(6), 2179–2193 CrossRef CAS.
- M. Ago, S. Huan, M. Borghei, J. Raula, E. I. Kauppinen and O. J. Rojas, ACS Appl. Mater. Interfaces, 2016, 8(35), 23302–23310 CrossRef CAS PubMed.
- L. Wang, M. Ago, M. Borghei, A. Ishaq, A. C. Papageorgiou, M. Lundahl and O. J. Rojas, ACS Sustainable Chem. Eng., 2019, 7(6), 6013–6022 CrossRef CAS PubMed.
- M. Ornatska, S. Peleshanko, K. L. Genson, B. Rybak, K. N. Bergman and V. V. Tsukruk, J. Am. Chem. Soc., 2004, 126(31), 9675–9684 CrossRef CAS.
Footnote |
| † Current address: State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 110623, Liaoning, China. |
| This journal is © The Royal Society of Chemistry 2020 |