
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConversion of birch bark to biofuels†
Ivan
Kumaniaev
a,
Kranti
Navare
 bc,
Natalia
Crespo Mendes
b,
Vincent
Placet
d,
Karel
Van Acker
*be and
Joseph S. M.
Samec
*a
bc,
Natalia
Crespo Mendes
b,
Vincent
Placet
d,
Karel
Van Acker
*be and
Joseph S. M.
Samec
*a
aDepartment of Organic Chemistry, Stockholm University Svante Arrhenius väg 16C, SE 106 91, Stockholm, Sweden. E-mail: joseph.samec@su.se
bDepartment of Materials Engineering, KU Leuven, Kasteelpark Arenberg 44 box 2450, BE-3001 Leuven, Belgium. E-mail: karel.vanacker@kuleuven.be
cUnit Sustainable Materials, VITO, Boeretang 200, 2400 Mol, Belgium
dFEMTO-ST Institute, Applied Mechanics Department, UMR CNRS 6174, University of Franche-Comté, 24 Chemin de l'Epitaphe, F-25000 Besançon, France
eCenter for Economics and Corporate Sustainability (CEDON), KU Leuven, Warmoesberg 26, BE-1000 Brussels, Belgium
First published on 13th March 2020
Abstract
Substitution of fossil energy sources for bio-based ones will require development of efficient processes that can convert inedible and preferably low-value fractions that currently are not used into high-value products. It is desirable that such processes are developed so that both current logistics and infrastructure can be used. Bark, which is the outer layer of woody biomass, is currently burnt in a low-value process or left in the forests to decay and is therefore considered waste. In this work, birch (Betula pendula) bark was converted to hydrocarbons suitable for use in both road and aviation fuels in two efficient steps. Development of an efficient, recyclable, salt- and metal-free solvent-based system to solubilize birch bark under benign reaction conditions was a key outcome. The obtained gum was composed of organosolv lignin and suberin oligomers and was fully characterized. This gum had unique properties and could be directly processed in a conventional hydroprocessing unit set-up to afford hydrocarbons in the road and aviation fuel ranges. Life cycle assessment was applied to evaluate different scenarios for implementing this technology. When using bark generated as a forestry by-product and current infrastructure in a pulp mill, the process had a favorable low carbon dioxide footprint for biofuel generation.
Introduction
According to BP’s latest review of fossil reserves, it is predicted that crude oil reserves will last only 50 years.1 Environmental movements around the world have arisen to stop this usage even before then. At the same time, growing populations will require more energy and food. It is difficult to balance the use of agricultural land for energy and food production.2 In addition, it is not feasible to divert feedstocks for materials such as wood to energy production. One solution is to use low value or unused by-products such as bark. The ability to use current infrastructure to convert these by-products is important for industrialization of such processes. To meet the demand for green fuels it is important to use by-products that are currently not used but can be easily accessed and processes that enable use of current, often Capital Expenses (CAPEX) intensive, infrastructure. Furthermore, it is beneficial if logistics for both handling of the raw material and products are already in place.In the transition from a fossil to a renewable fuel based economy, biomass will play an important role. At the same time, large areas will be required to feed growing populations and this demand is predicted to grow continuously. Thus, it will become vital to use streams that today have little to no value. Bark is the external tissue of plants and currently has very low value. For example, in current biomass processing technologies, such as kraft pulping in the paper industry or in saw mills, bark is separated from trunks and treated as waste. Moreover, the tops, branches and even roots of trees are currently left in forests to decay. Because of their high surface areas, these unused forestry streams are around 40% bark. By comparison, timber and pulp wood is usually less than 5% bark. In Sweden, which only has around 1% of the worlds’ forests, around 30 million tons of wood are harvested annually and around 1.5 million tons of this biomass is bark. In addition to this, around 20 million tons of branches, tops, and roots could be sustainably extracted from Swedish forests. Thus, an additional 8 million tons of bark could potentially be valorized without negatively affecting or competing with current industries or biotopes.3
In any process for conversion of bark into chemicals and/or fuels, polymeric substances must be partially depolymerized and solubilized to make them prone to chemical modifications. Methods of lignin extraction have been widely studied for other types of biomass.4 In particular, the organosolv procedure has been developed with the intention of providing a more environmentally-friendly technology as an alternative to traditional yet problematic kraft pulping process.5 In this type of process, the biomass is treated with organic solvents (MeOH, EtOH, and dioxane) mixed with water in the presence of acids and other additives at 180 °C–200 °C.6 However, even organosolv pulping disrupts the lignin structure and weak C–O bonds are cleaved and recalcitrant C–C bonds are formed. To address this lignin repolymerization, lignin-first methodologies have recently been developed, where the extracted lignin is subjected to an in situ catalytic transformation to form stable products. The lignin-first approach usually applies a metal-catalyzed hydrogenolysis step where lignin is reduced into stable phenolic monomers.7–10
For bark valorization, the focus so far has been to depolymerize suberin. A common method to depolymerize suberin is alkaline methanolysis.11,30,31 With few exceptions, the majority of reports have focused on the analysis of suberin rather than conversion into valuable products. In one of the reported procedures, a typical lignin-first approach was applied to a bark sample to partially depolymerize suberin into a range of fatty acids and alcohols.12 Further depolymerization under alkaline conditions was needed to accomplish the transformation. Another method used FeCl3-catalyzed lignin isolation; however, the transformation of suberin was not described in detail.13 We recently applied a similar procedure (NaOH in MeOH–H2O) to fractionation of Quercus suber bark.14 All these procedures have several shortcomings, including unsustainable use of solvents, the need for stoichiometric neutralization of basic reaction media, problematic product separations, and often the use of transition metals that cannot be recycled because they are affected by the strong base used in the process. The main problem is that suberin is depolymerized at high pH and lignin at low pH, thus the procedures either give high yields of monophenolic compounds from lignin or free fatty acids from suberin.15
We herein report a novel approach to circumvent the shortcomings listed above. By applying an organocatalytic depolymerization of suberin using an inexpensive and easily distilled amine in a benign solvent mixture, suberin can be depolymerized without disrupting lignin (Fig. 1A). To decrease the energy input required in the distillation, the product and solvent mixture was successfully recirculated up to three times before being distilled off. The solubilized mixture of oligomers was then hydrotreated to generate hydrocarbons in the diesel and aviation fuel ranges (Fig. 1A). Life cycle assessment (LCA) of the process was used to evaluate different scenarios with respect to localization of a facility and the energy source (Fig. 1B).
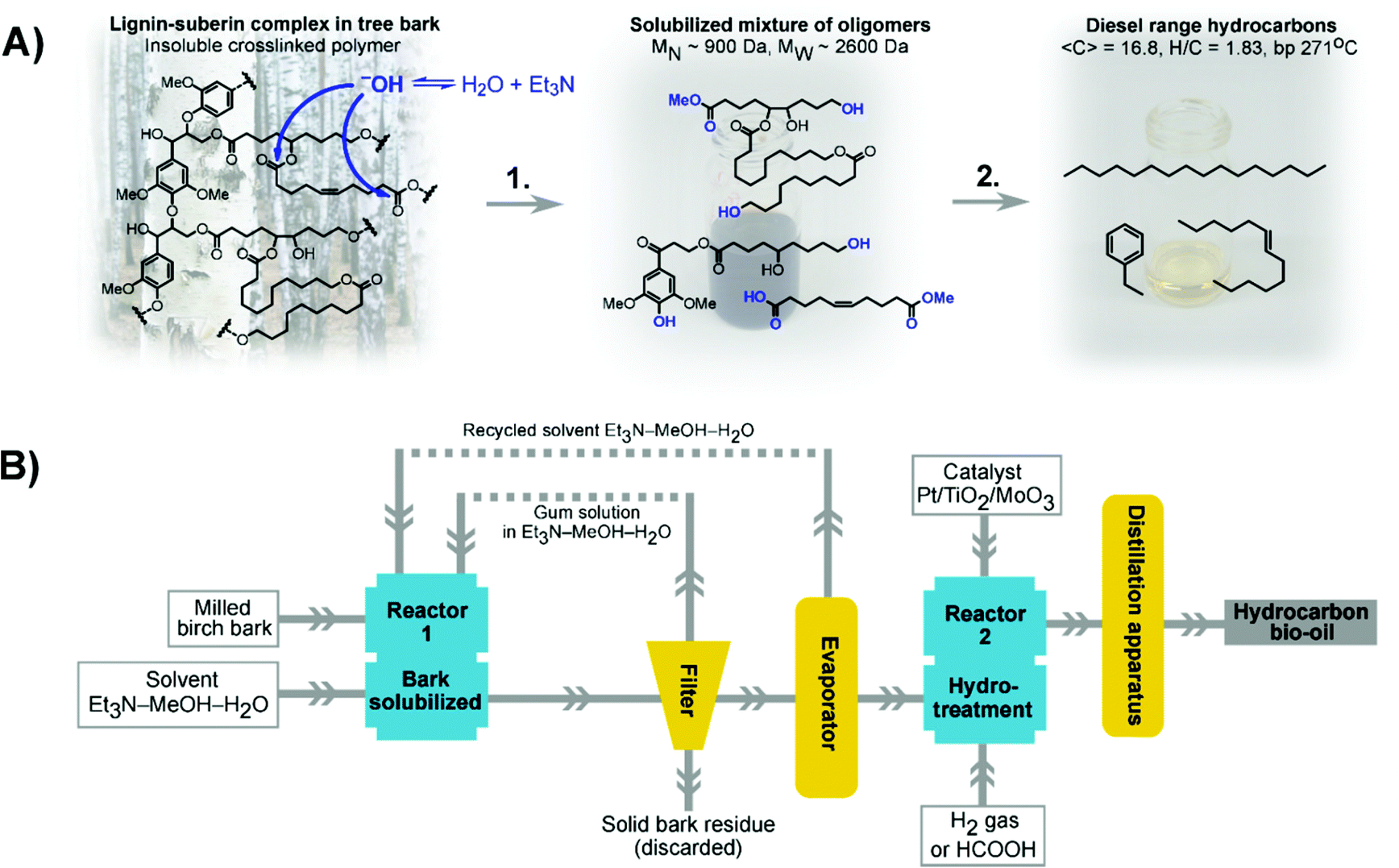 | ||
| Fig. 1 (A) Chemistry of the bark treatment process. (B) Principal process schematic. | ||
Results and discussion
Part A: experimental results
Bark has a high energy content because it is mainly composed of fatty acid esters and lignin (21–24 MJ kg−1).16 The elemental composition of the birch in this study was C, 70.1%; H, 9.2%; N, 0.3%; and O, 19.5% (ESI section 1.6†). Besides low-molecular-weight extractives and water (29% for the EtOH extractives plus water content in birch bark, ESI section 1†), the two major substances in birch bark were suberin (33%) and lignin (15%), which are both hydrophobic polymers. In addition, hydrophilic suberin-associated fragments, presumably polyols, were present (11%). This is in line with previously reported analyses (Table 1).32,33 Although lignin is abundant in other tissues as well, suberin is a unique component of bark and serves as a protective barrier of the plant. Suberin is an aliphatic polyester composed of hydroxylated fatty acids (Fig. 1A). The lignin and suberin domains are highly crosslinked and form an insoluble rigid network.17| Bark type | Extractives | Total suberin (alkaline extraction) | Lignin (acid-insoluble) | Cellulose | Hemicellulose |
|---|---|---|---|---|---|
| a Average for several species. | |||||
| Birch (Betula Pendula) (this work) | 26 | 44 | 15 | 0 | 0 |
| Birch (Betula Pendula)11 (dewaxed) | — | 73 | 15 | 1 | |
| Pine (Pinus sylvestris)28 | 19.1 | 44.9 | 25.4 | 14.7 | |
| Oak (Quercus suber)a29 | 14 | 33 | 24 | 26 | |
Amines are the most common and obvious candidatures of volatile recyclable bases. However, ammonia, primary and secondary amines cannot be used because they form amides when reacting with esters. Pyridine and other heterocyclic amines could be used, however, their high boiling points would make the recycling more energy demanding. In addition, their basicity is lower than the aliphatic amines.
For the purpose described herein, a simple tertiary aliphatic amine Et3N (pKa 10.7) seemed to be the best option. In the presence of Et3N in organosolv pulping conditions, suberin would undergo alkaline hydrolysis (Fig. 1A). After the solubilization is accomplished, Et3N can easily be removed by distillation (bp 89 °C) together with the other components of the solvent.
Solubilization of the bark using a mixture of MeOH–H2O–triethylamine (Et3N) was optimized with respect to minimizing the mass of solid residue remaining after filtration (ESI section 2.2†). The degrees of solubilization (%) are reported in relation to the mass of extractive-free bark (EtOH extractives plus water content of 29%, ESI section 1.1†). As a starting point, the bark was treated with MeOH–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v or 46% volume fraction of H2O) at 220 °C for 1 h in the absence of Et3N. Without a catalyst, only 27% of the bark was solubilized (Fig. 2). Addition of Et3N (4% volume fraction) improved the results and 69% of the bark was solubilized. Increasing the volume fraction of Et3N to 7% gave 91% solubilization of the bark. A further increase in the Et3N volume fraction (12%) caused a decrease in the solubilization degree (73%).
:1 v/v or 46% volume fraction of H2O) at 220 °C for 1 h in the absence of Et3N. Without a catalyst, only 27% of the bark was solubilized (Fig. 2). Addition of Et3N (4% volume fraction) improved the results and 69% of the bark was solubilized. Increasing the volume fraction of Et3N to 7% gave 91% solubilization of the bark. A further increase in the Et3N volume fraction (12%) caused a decrease in the solubilization degree (73%).
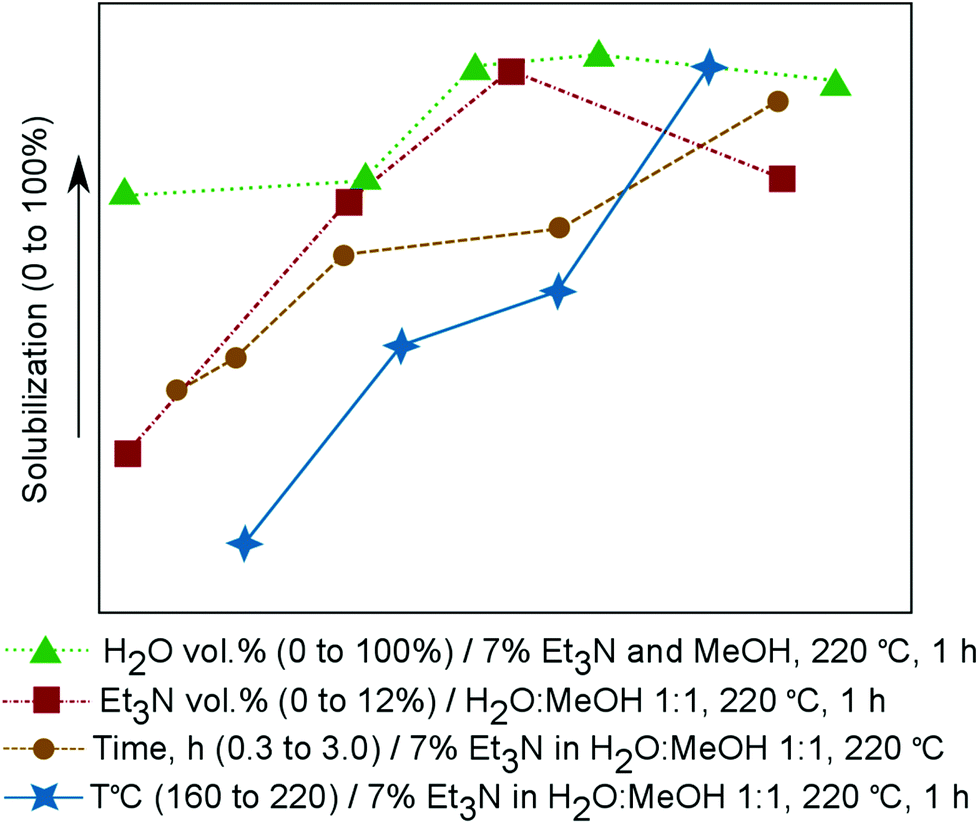 | ||
| Fig. 2 Influences of time, temperature, and volume fractions of water and triethylamine on bark solubilization. | ||
Using the optimized Et3N volume fraction (7%), the role of water in the solvent mixture was explored. If no water was added (i.e., a Et3N–MeOH mixture was used), the solubilization degree was lower than that obtained with MeOH–H2O 1:1 v/v, but still relatively high (70%). Addition of water (30% volume fraction) did not greatly improve the solubilization (72%). When water became the major component at a volume fraction of 60%, the degree of solubilization reached a maximum (93%). Use of the H2O–Et3N system without MeOH led to a small decrease in the degree of solubilization (89%); however, the resulting mixture was difficult to handle during filtration. For optimal solubilization and separation, a MeOH volume fraction of 46% was used.
The effect of temperature was also investigated. When the process was carried out for 1 h with the optimized solvent system (MeOH–H2O 1:1 v/v, 7% volume fraction of Et3N) at 160 °C, poor solubilization was observed (13%). Increasing the temperature afforded better results with degrees of solubilization of 45% at 180 °C, 54% at 200 °C, and 91% at 220 °C.
Because of the low loading of bark in the reactor (150 kg m−3), the lower limit of the solvent to bark ratio (V) was approximately 7 L kg−1. Up to that value, the solubilization degree did not depend on this parameter and values of 91%, 92%, 91%, and 90% were obtained for V = 20, 15, 10, and 7 L kg−1, respectively (ESI section 2.2†). However, evaporation of the solvent required an energy input of approximately 1.7 MJ per liter of solvent (ESI section 4†). Because solvent recycling by evaporation caused a slight decline in the product yield (Table 2), it would be beneficial to decrease V by reusing the solvent several times before evaporation, that is, use the solution for processing additional portions of bark in a looped system. Importantly, the presence of solubilized bark components in the solution did not affect the efficiency of solubilization of new portions of bark. In three consecutive experiments with V = 10 L kg−1, the degrees of solubilization were 91%, 90%, and 90%. Thus, the solvent to bark ratio was reduced to 3.3 L kg−1.
After recirculation, the solvent mixture eventually needs to be distilled off from the product and recycled back. The efficiency of distillation was studied by evaporating the solvent mixture after the catalytic solubilization and measuring the losses of MeOH and Et3N. The solvent was recycled three times by distillation under vacuum (Table 2). The concentration of Et3N slightly decreased after each distillation. The recycled solvent was used for solubilization of new samples of bark without any deviation in the degree of solubilization. As major loss was observed in the first distillation, we assumed that the equipment might be saturated with volatiles. It should be noted that a small-scale set-up was used in these experiments and that an industrial process with a closed system should be even more efficient.
According to size exclusion chromatography results (ESI section 2.3†), the gum obtained by bark solubilization contained a variety of oligomeric products of suberin and lignin resulting from partial depolymerization, especially of suberin, with MW = 2630 Da and MN = 932 Da (PD = 2.8). This meant that an average dissolved molecule was composed of 4–5 monomeric units of lignin and/or suberin. The elemental composition of the material was similar to that of bark (ESI section 2.4†). The material was insoluble in hexane, moderately soluble in toluene (28% of the gum mass), and readily soluble in methanol (87% of the gum mass) (ESI section 2.5†). Notably, the gum became miscible with tall oil fatty acid at 120 °C, and the suspension remained stable at room temperature. Therefore, tall oil fatty acid which is produced during kraft pulping34 could be used as a carrier liquid in industrial gum hydrotreatment processes, as shown previously for kraft lignin acylated with fatty acids.18 The viscosity of the suspension at room temperature was 15–500 mPa s for the mass fraction range of 7%–33% and temperature range of 25 °C–70 °C (ESI section 2.6†). These parameters are important for applicability in a green hydrotreater, where co-processing in carrier liquids such as feeds for hydrogenated vegetable oils (HVO) is a feasible option.
HSQC NMR results (Fig. 3A) demonstrated the presence of typical structural motifs of suberin and were in accordance with previous data obtained for birch bark.11 To analyze the monomeric fatty acids, the gum was subjected to alkaline methanolysis and the extract was studied by GC (Fig. 3B). A variety of C16–C22 hydroxylated carboxylic acids and diacids were identified (ESI section 2.7†),11,19,20 with the main components being 22-hydroxydocosanoic acid (26% total ion current analysis was performed on silylated derivatives) and 1,18-octadec-9-enedioic acid (14%). In addition, ferulic acid (3%) was detected.
 | ||
| Fig. 3 Analyses of bark-derived gum by (A) HSQC NMR and (B) GC-MS of methanolysate. | ||
The gum was subjected to hydrotreatment in the presence of a Pt/MoO3/TiO2 catalyst at 360 °C (ESI section 3.2†).21 Hydrotreatment includes hydrodeoxygenation, hydrodenitrogenation and hydrodesulfurization to generate a hydrocarbon. This catalyst was used because it provides very efficient hydrotreatment without the need for a carrier liquid. Compared with using a carrier liquid, this enables more accurate analysis of the products that are formed. However, as the gum is miscible in fatty acids, it could conveniently be used as a feed in a hydrotreater for hydrogenated vegetable oil production (Fig. S10†). Simulated distillation of the obtained bio-oil showed that it contained hydrocarbons within the aviation and road fuel ranges (ESI section 3.3†). The lightest components had boiling points of 70 °C and 90% of the mixture boiled at below 350 °C (Fig. 4A). Thus, this proof of concept trial showed that the majority of the feed ended up as high-value products.
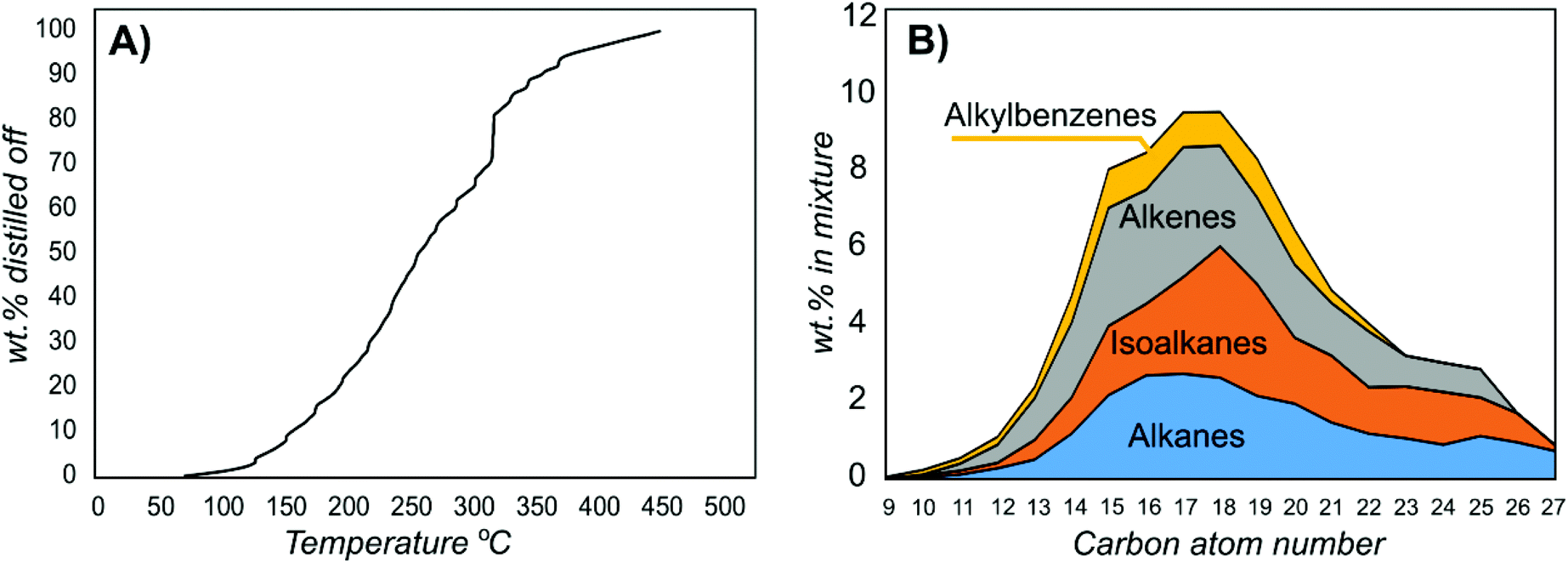 | ||
| Fig. 4 Analysis of the bio-oil in (A) a simulated distillation and (B) by two-dimensional GC for distribution of the components by carbon atom number. | ||
Analysis of the mixture from hydrotreatment by two-dimensional GC showed the different types of components in the mixture (ESI section 3.4,†Fig. 4B, Table 3). The most abundant molecules were C15–C19 hydrocarbons. In natural suberin, only fatty acids with even carbon atom numbers are present. Hydrocarbons with uneven chain lengths emerge because of cracking and/or decarboxylation processes. Higher molecular weight compounds such as naphthalenes (20% mass fraction) are probably also products of cracking since their carbon atom numbers are generally lower than the ones of other observed hydrocarbons (average of 14.4 versus 16.8 for the whole mixture). Unsaturated and monounsaturated hydrocarbons account for up to 73% of the mixture by mass; however, because of the presence of aromatic compounds, the average number of double bonds and/or cycles per molecule for the whole mixture is 2.4 and the H/C ratio is 1.83. The theoretically estimated higher heating value of the generated biofuel is 45.4–48.2 MJ kg−1 (average of 46.5 MJ kg−1). It should be noted that straight chains could be isomerized to generate a larger fraction of aviation fuels.22 The yield of the obtained bio-oil was 40% of the initial bark mass (56% of the extractive-free bark mass), and a major fraction of the extractives could be used.23
| Component | wt% | Number-average carbon atom number |
|---|---|---|
| Summary of two-dimensional GC to evaluate the hydrocarbon composition of the biofuel. | ||
| n-Alkanes | 24.1 | 18.2 |
| Branched alkanes | 23.4 | 18.5 |
| Alkenes and cycloalkanes | 25.1 | 17.2 |
| Alkylbenzenes | 7.7 | 16.3 |
| Higher aromatics | 19.7 | 14.4 |
| Whole mixture | 100.0 | 16.8 |
Part B: environmental assessment
To critically assess the process, a theoretical evaluation was performed. The energy demand to process 1 kg of the biofuel product was calculated considering the following parameters: yield of the biofuel, solvent volume to bark mass ratio, energy of solvent evaporation, heat capacity of the solvent, heat capacity of the reactor material, and reaction time. Four principal summands were considered: heating of the reaction mixture in both stages, and distillation of the mixture in both stages. Calculation of the energy demand is described in detail in the ESI (section 4†).The temperature range of the process is crucial. Some studies24–26,35 have shown that water and alcohols at 300 °C–400 °C are able to solubilize biomass of a similar nature without addition of a catalyst. The necessity to heat the solvent to these temperatures would create a prohibitive additional demand of 10–20 MJ per kilogram of the biofuel product. In addition, CAPEX intensive facilities would be required. Introduction of Et3N as a benign catalyst facilitates suberin depolymerization and allows the first stage to run at a lower temperature, as presented in this study. This means additional expenditures associated with high pressure equipment could be avoided.
The most energy-consuming part of the process is heating and evaporation of the MeOH–H2O–Et3N solvent in the first stage. Consequently, the energy demand is predominantly influenced by the ratio of the solvent volume to the mass of the feedstock. To be efficient, the process should meet the requirement V < 4 L kg−1. At this point, the energy demand to distill the solvent will be equal to the energy content of the fuel. Other important factors are the bark loading, which affects the surface to volume ratio and thus influences heat loss and reactor heating, and the coefficient of heat exchange between the reactor material and air. Under the initial laboratory conditions used in this study (bark loading = 1 g, V = 10–20 L kg−1), the process was extremely inefficient and required >100 MJ for production of 1 kg of biofuel. The energy demand dropped substantially when the bark loading was increased and the solvent to bark ratio was decreased by recirculating the solvent. An energy demand of 31 MJ kg−1 is expected when industry-applicable conditions are used (bark loading = 104 kg, effective V = 3 L kg−1, overall heat loss coefficient = 10−3 MJ m−2 K−1 min−1). Under industrial conditions with efficient heat exchangers there is virtually no heat loss, and if the optimized conditions used (V = 2.3 L kg−1, initial volume = 7 L kg−1, recirculation three times without evaporation) the energy demand should decrease to 23 MJ kg−1.
Further environmental benefits and drawbacks of the optimized two-stage process for birch bark conversion into biofuel were evaluated by LCA.27 LCA is a tool that quantifies the potential environmental impacts of products throughout their entire life cycle from raw material extraction to the end of life. The applied methodology follows the four-phases framework standardized by ISO 14040 [ISO]. Further details and the data and assumptions used in the analysis are given in ESI section 5.† It should be noted that the technology should not be implemented at areas where it could compete with people who rely on birch bark for living, for example the Sami people in north of Scandinavia.
The environmental performance of biodiesel production was compared with that of fossil-based diesel production (from crude oil). The functional unit chosen for this study was 46.5 MJ of energy produced by the fuel. The reference flow was developed by comparing the calorific values of biofuel and a fossil-derived fuel (ESI section 5.1†). Four scenarios were examined for the environmental performance assessment and used to evaluate improvements to the developed process (Fig. 5):
- Scenario 1 (Baseline): Optimized two-stage continuous process at an industrial scale with natural gas as the fuel for heat energy [geographical scope: Europe]
- Scenario 2: Electricity as the input for heat energy using the European electricity mix [geographical scope: Europe]
- Scenario 3: Electricity as the input for heat energy using the Swedish electricity mix [geographical scope: Sweden]
- Scenario 4: Heat energy and methanol as waste streams from the paper and pulp industry [geographical scope: Sweden].
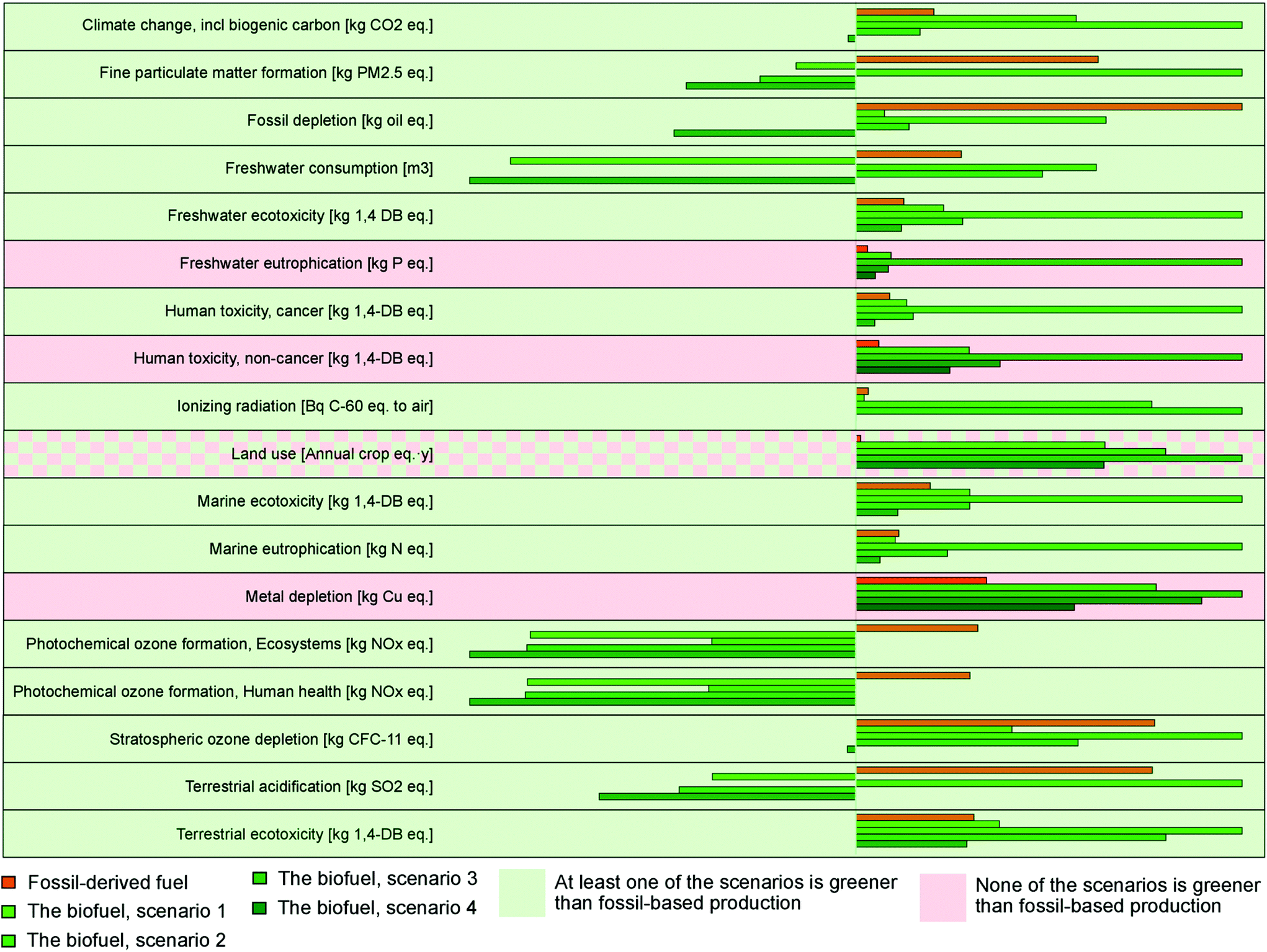 | ||
| Fig. 5 Comparative assessment of the environmental impacts of different scenarios. Within each impact category, the impacts are depicted as a percentage of the maximum impact. | ||
The baseline scenario (Scenario 1) assumed a stand-alone continuous process, where the solvent was recycled and new solvent was continuously added to compensate for solvent loss occurring during the distillation process. The energy supplied for the process was from natural gas. In this scenario, biofuel performed better than fossil-based diesel in the following environmental impact categories: fine particulate matter formation, fossil depletion, freshwater consumption, ionizing radiation, marine eutrophication, photochemical ozone formation, stratospheric ozone depletion, and terrestrial acidification (Fig. 5, ESI section 5.3†). Climate change, freshwater eutrophication, land use, human toxicity, metal depletion, and freshwater, marine, and terrestrial ecotoxicity were identified as impact categories where the biofuel performed worse than fossil-based diesel. The main hotspots in the biofuel production life cycle for these impact categories were catalyst production, bark chip production, flue gas emissions (containing mainly carbon dioxide), and heat energy production (ESI section 5.3†).
For the climate change impact category, the fossil-based diesel emitted 0.603 kg of CO2 eq. compared with the biodiesel, which emitted 1.7 kg of CO2 eq. corresponding to the functional unit. The main contributors to the global warming potential of biodiesel were the flue gas emissions (containing mainly carbon dioxide) and energy input. The flue gas emissions contributed to 65% of the global warming potential of biodiesel, mainly during the HDO process (3.03 kg of CO2 eq.) and the bark residue incineration (0.30 kg of CO2 eq.). Additionally, 34% of the climate change impact was caused by heat energy production (0.76 kg of CO2 eq. for bark solubilization, 0.76 kg of CO2 eq. for filtration and distillation, and 0.23 kg of CO2 eq. for the HDO process). About 67% of the total negative impacts were compensated for by positive impacts of bark chip production because of carbon sequestration and avoidance of impacts of hydrogen production from the HDO process (ESI section 7†) and heat energy production (heat produced by bark residue incineration).
The main cause for the impacts related to the land-use change was the use of bark chips (0.45 annual crop eq. per year). However, the bark chips that can be used as a feedstock for biofuel production are currently either left behind in forests or burnt for bioenergy in the paper and pulp industry. Hence, use of the bark does not require use of additional land. However, the results of the LCA show the impact on land use. This is because bark that was used in the paper and pulp industry is now redirected to making biofuel, and some of the impact that was allocated to the paper and pulp industry is reallocated to biofuel. If the system boundaries of this study were to include the paper and pulp industry, the results would show zero impact on the land-use change impact category. It is important to note that the bark that is diverted from the paper and pulp industry to biofuel production will have to be substituted for by some other source of energy in the paper and pulp industry. The other available sources of energy vary in different European countries and hence are not included in the system boundaries of this study. The different sources of energy could have very different environmental impacts, especially for climate change. However, it should be noted that all paper and pulp mills generate an energy surplus. Thus, the pulp mill itself would not require energy compensation.
With regard to other key impact categories, freshwater eutrophication impacts were mainly caused by catalyst production (1.01 × 10−4 kg of P eq.) and heat energy production (9.18 × 10−5 kg of P eq.). Catalyst production was also the main contributor to human toxicity (non-cancer) impacts (0.84 kg of 1,4-DB eq.) and metal depletion (1.62 × 10−3 kg of 1,4-DB eq.) This is mainly because the catalyst consumption rate in biofuel production is four times that of fossil-based diesel production. Development of targeted guard beds for these feeds to recover precious catalysts in refineries could decrease the catalyst deactivation dramatically and increase the lifetime of the catalyst.
LCA was performed for two additional scenarios to assess the environmental impact when heat was supplied by electricity instead of combustion of natural gas. The two scenarios under consideration used the average electricity mixes of Europe (Scenario 2) and Sweden (Scenario 3) as the sources of electricity. Comparing the three scenarios, Scenario 2, which was heavily dependent on fossil fuel for electricity generation, had higher environmental impacts than the baseline scenario (Scenario 1) and Scenario 3 for all categories except ionizing radiation and land use. Scenario 3, which mostly used nuclear and renewable electricity, had the highest impacts for ionizing radiation and land use. Scenario 1 performed better than the other two scenarios for all impact categories except climate change, fine particulate matter formation, and terrestrial acidification, where Scenario 3 had the best results. For freshwater eutrophication, marine ecotoxicity, and photochemical ozone formation there was slight variation in terms of environmental impacts between Scenarios 1 and 3. When compared with fossil-based diesel, both Scenarios 2 and 3 performed better for photochemical ozone formation and terrestrial ecotoxicity. Scenario 3 also had better results for climate change, fine particulate matter formation, fossil depletion, stratospheric ozone depletion, and terrestrial acidification. For the remaining categories, fossil-based diesel had the lowest environmental impacts (Fig. 5).
Scenario 4 represents the bark valorization process in conjunction with the paper and pulp industry. It is a best-case scenario and possible wherever there is pulp production and excess heat and methanol from paper and pulp industries can be used in the bark valorization process. Hence, the impacts of these processes are assumed as null. Among all the biofuel production scenarios, Scenario 4 had the best performance for all environmental impact categories. Compared with fossil-based diesel, Scenario 4 still performed better for all categories except freshwater eutrophication, human toxicity, and metal depletion, for which none of the biofuel scenarios performed better than fossil-based diesel.
Fig. 5 summarizes the environmental impact of fossil-based diesel production and biodiesel production in the four scenarios. The results suggest that the fossil-based diesel is environmentally beneficial compared with biodiesel only in the following four categories: freshwater eutrophication, human toxicity, land use, and metal depletion. When considering land use, biofuel performed worse than fossil fuel in the strict LCA. However, as stated above, since bark is waste from the forestry industry and pulp and paper production, the land-use value is null and the biofuel performs better than fossil fuel. For the remaining categories biofuel performed better than fossil-based fuel in all of the scenarios. Therefore, even if biofuel might not be beneficial compared with fossil diesel in the base scenario, the environmental benefits of biofuel can be enhanced by supplying energy from renewable sources, operating the process in conjunction with other industries (e.g., paper and pulp), and utilizing the by-products of these industries.
The sensitivity analysis performed for this study provides an overview of the parameters (material and energy inputs) that greatly influence the LCA results. Among the parameters, hydrogen produced in the HDO process affects the results the most. This is followed by heat energy and bark chips. The volume of solvent consumed has little influence on the results. These results suggest that the values for the amount of hydrogen (produced and consumed), energy, and bark chips in the process should be accurately known before designing the process.
Conclusions
We developed a two-stage process for conversion of birch bark into a green biofuel. The first stage uses the recyclable, salt- and metal-free solvent system of MeOH–H2O–Et3N in which triethylamine plays the role of a benign catalyst for hydrolysis of ester bonds in the bark tissue. Suberin and lignin in the bark undergo partial cleavage and become accessible for hydrotreatment (the second stage of the process), which affords a mixture of hydrocarbons in the gasoline–aviation–diesel fuel range with a 40% yield (from the initial bark mass). On an industrial scale, the process would require 23 MJ of energy to yield 1 kg of diesel product. The potential environmental impact of implementing the technology has been evaluated using a LCA of the two-stage process and considering different scenarios. It is important to choose the right location for implementation of the technology. The life cycle greenhouse gases (GHG) emitted during the process amount to 1.7 kg of CO2 eq. per kilogram of biofuel produced from the bark. This base scenario assumes a stand-alone facility at an industrial scale using heat and electricity from natural gas. If using electricity as a source of heat energy, the life cycle GHG increases to 2.98 kg of CO2 eq. for the European electricity mix. The GHG emissions decrease to 0.495 kg of CO2 eq. for the Swedish electricity mix. If the facility is integrated with a pulp mill and uses excess heat and methanol that is generated by the pulp mill, the life cycle GHG further decreases to −0.06 kg of CO2 eq. per kilogram of biofuel produced from bark and the scenario becomes very attractive. Considering that most bark is currently left in forests or burnt in a low-value process in a pulp mill, the de facto land use is null. Implementing this technology close to existing infrastructure and using downstream logistics would be a very appealing approach to produce a biofuel with minimum environmental impact.Conflicts of interest
JSM Samec is a co-owner of RenFuel and a Professor at Stockholm University.Acknowledgements
This project received funding from the Bio-based Industries Joint Undertaking under the European Union's Horizon 2020 research and innovation program (Grant No. 744349). We thank the Swedish Energy Agency for financial support.References
- BP Statistical Review of World Energy 2019.
- D. H. Meadows, D. L. Meadows, J. Randers and W. W. Behrens III, in The Limits to Growth; A Report for the Club of Rome's Project on the Predicament of Mankind, Universe Books, 1972 Search PubMed.
- C.-A. Helander, in Forests and Forestry in Sweden, The Royal Swedish Academy of Agriculture and Forestry, 2015 Search PubMed.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678, DOI:10.1021/acs.chemrev.7b00588.
- A. Tejado, C. Peña, J. Labidi, J. M. Echeverria and I. Mondragon, Bioresour. Technol., 2007, 98, 1655, DOI:10.1016/j.biortech.2006.05.042.
- P. Sannigrahi and A. J. Ragauskas, in Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals, ed. C. E. Wyman, John Wiley & Sons, Ltd, Chichester, 2013, pp. 201–214. DOI:10.1002/9780470975831.
- R. Rinaldi, R. Jastrzebski, M. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215, DOI:10.1002/anie.201510351.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558, DOI:10.1002/cssc.201600237.
- T. Renders, S. Van den Bosch, S.-F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557, 10.1039/C7EE01298E.
- W. Schutyser, T. Renders, S. Van den Bosch, S.-F. Koelewijn, G. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908, 10.1039/C7CS00566K.
- A. Karnaouri, H. Lange, C. Crestini, U. Rova and P. Christakopoulos, ACS Sustainable Chem. Eng., 2016, 4(10), 5289–5302, DOI:10.1021/acssuschemeng.6b01204.
- M. D. Garrett, S. C. Bennett, C. Hardacre, R. Patrick and G. N. Sheldrake, RSC Adv., 2013, 3, 21552, 10.1039/C3RA44382E.
- G. Koumba-Yoya and T. Stevanovic, Catalysts, 2017, 7, 294, DOI:10.3390/catal7100294.
- I. Kumaniaev and J. S. M. Samec, ACS Sustainable Chem. Eng., 2018, 6, 5737–5742, DOI:10.1021/acssuschemeng.8b00537.
- T. Vangeel, T. Renders, K. Van Aelst, E. Cooreman, S. Van den Bosch, G. Van den Bossche, S.-F. Koelewijn, C. M. Courtinb and B. F. Sels, Green Chem., 2019, 21, 5841–5851, 10.1039/C9GC02139F.
- A. Rossi, in Progress in Biomass Conversion, ed. A. D. Tillman and E. C. Jahn, Academic Press, 1984, pp. 69–99 Search PubMed.
- J. Graça, Front. Chem., 2015, 3, 62, DOI:10.3389/fchem.2015.00062.
- J. Löfstedt, C. Dahlstrand, A. Orebom, G. Meuzelaar, S. Sawadjoon, M. V. Galkin, P. Agback, M. Wimby, E. Corresa, Y. Mathieu, L. Sauvanaud, S. Eriksson, A. Corma and J. S. Samec, ChemSusChem, 2016, 9, 1392–1396, DOI:10.1002/cssc.201600172.
- R. Järvinen, A. Silvestre, U. Holopainen, M. Kaimainen, A. Nyyssölä, A. M. Gil, N. C. Pascoal, P. Lehtinen, J. Buchert and H. Kallio, J. Agric. Food Chem., 2007, 55, 1327–1336, DOI:10.1021/jf9008907.
- R. Järvinen, A. J. D. Silvestre, U. Holopainen, M. Kaimainen, A. Nyyssölä, A. M. Gil, C. P. Neto, P. Lehtinen, J. Buchert and H. Kallio, J. Agric. Food Chem., 2009, 57, 9016–9027, DOI:10.1021/jf9008907.
- K. Kon, T. Toyao, W. Onodera, S. M. A. H. Siddiki and K. Shimizu, ChemCatChem, 2017, 14, 2822–2827, DOI:10.1002/cctc.201700219.
- P. Vozka, P. Šimáček and G. Kilaz, Energy Fuels, 2018, 32, 11595–11606, DOI:10.1021/acs.energyfuels.8b02787.
- P. Šiman, A. Filipová, A. Tichá, M. Niang, A. Bezrouk and R. Havelek, PLoS One, 2016, 11, e0154933, DOI:10.1371/journal.pone.0154933.
- L. Yang, Y. Li and P. E. Savage, Bioresour. Technol., 2016, 211, 779–782, DOI:10.1016/j.biortech.2016.03.151.
- D. Kusdiana and S. Saka, Bioresour. Technol., 2004, 91, 289–295, DOI:10.1016/s0960-8524(03)00201-3.
- Y. Wu, Y. Chen and K. Wu, in Near-critical and Supercritical Water and Their Applications for Biorefineries, ed. Z. Fang and C. Xu, Springer, 2014, ch. 3. DOI: DOI:10.1007/978-94-017-8923-3.
- ISO 14040:2006 Environmental management – Life cycle assessment – Principles and framework.
- L. Valentín, B. Kluczek-Turpeinen, S. Willför, J. Hemming, A. Hatakka, K. Steffen and M. Tuomela, Bioresour. Technol., 2010, 101, 2203–2209, DOI:10.1016/j.biortech.2009.11.052.
- A. Sen, M. Zhianski, M. Glushkova, K. Petkova, J. Ferreira and H. Pereira, Wood Sci. Technol., 2016, 50, 1261–1276, DOI:10.1007/s00226-016-0836-y.
- P. C. R. O. Pinto, A. F. Sousa, A. J. D. Silvestre, C. P. Neto, A. Gandini, C. Eckerman and B. Holmbom, Ind. Crops Prod., 2009, 29, 126–132, DOI:10.1016/j.indcrop.2008.04.015.
- I. Miranda, J. Gominho, I. Mirra and H. Pereira, Ind. Crops Prod., 2013, 41, 299–305, DOI:10.1016/j.indcrop.2012.04.024.
- R. Alén, in Pulp Mills and Wood-Based Biorefineries, ed. A. Pandey, R. Höfer, M. Taherzadeh, K. M. Nampoothiri and C. Larroche, Elsevier, Amsterdam, Oxford, Waltham, 2015, pp. 91–126 Search PubMed.
- B. Holmbom, in Biorefining of Forest Resources, ed. R. Alén, Paperi ja Puu Oy, 2011, pp. 178–224 Search PubMed.
- R. Höfer, in Pulp Mills and Wood-Based Biorefineries, ed. A. Pandey, R. Höfer, M. Taherzadeh, K. M. Nampoothiri and C. Larroche, Elsevier, Amsterdam, Oxford, Waltham, 2015, pp. 127–155 Search PubMed.
- E. Dinjus, Ul. Arnold, N. Dahmen, R. Höfer and W. Wach, in Sustainable Solutions for Modern Economies, ed. R. Höfer, RSC Publishing, London, 2009, pp. 125–158 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc00405g |
| This journal is © The Royal Society of Chemistry 2020 |