
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScalable synthesis and polymerisation of a β-angelica lactone derived monomer†
Andrea
Dell'Acqua‡
 ,
Bernhard M.
Stadler‡
,
Sarah
Kirchhecker
,
Sergey
Tin
and
Johannes G.
de Vries
*
,
Bernhard M.
Stadler‡
,
Sarah
Kirchhecker
,
Sergey
Tin
and
Johannes G.
de Vries
*
Leibniz Institut für Katalyse, e. V. Albert-Einstein-Strasse 29a, 18059 Rostock, Germany. E-mail: johannes.devries@catalysis.de
First published on 1st May 2020
Abstract
Bio-based levulinic acid is easily ring-closed to α-angelica lactone (α-AL). α-AL can be isomerized to the conjugated β-AL under the influence of base, but since this is an equilibrium mixture it is very hard to devise a scalable process that would give pure β-AL. This problem was circumvented by distilling the equilibrium mixture to obtain a 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 mixture of β- and α-AL in 88% yield. This mixture was used for Diels–Alder reactions on 3 terpenes and on cyclopentadiene in up to 100 g scale. The latter DA adduct was subjected to a ROMP reaction catalysed by the Grubbs II catalyst. The resulting polymer has some similarities to poly-norbornene but is more polar. The polymer can be processed into films with very good transparency.
:10 mixture of β- and α-AL in 88% yield. This mixture was used for Diels–Alder reactions on 3 terpenes and on cyclopentadiene in up to 100 g scale. The latter DA adduct was subjected to a ROMP reaction catalysed by the Grubbs II catalyst. The resulting polymer has some similarities to poly-norbornene but is more polar. The polymer can be processed into films with very good transparency.
Introduction
The debate about when fossil fuels will run out has been ongoing for many years. Technological advancements have made the exploitation of an increasing number of reservoirs profitable. However, there is a consensus that all the natural reservoirs may eventually become depleted, and thus a gradual switch to renewable feedstocks will be necessary. And although it will be impossible to fulfil the total global demand for fuel and energy with biomass-based analogues, there is more than enough biomass available on a yearly basis to serve as the raw material for all of the chemicals we need.1Platform chemicals are small molecules that can be produced in good yields from bio-based raw materials such as lignocellulose or sugars by fermentation or by using thermocatalytic reactions.2,3 Further (preferably catalytic) conversions of these chemicals allows the synthesis of fine chemicals,4–8 monomers,8–13 and fuels.14,15 Especially renewable polymers16–18 and adhesives19 based on them, often have novel properties which are considered an advantage beyond renewability. One very prominent example of a bio-based platform chemical is levulinic acid (LA) which can be efficiently obtained by the acid-catalysed decomposition of the C-6 sugars in lignocellulose.2,20–23 LA itself can be converted into a wide range of useful compounds.24 The most studied and used ones are aminolevulinic acid (herbicide), 2-methyl tetrahydrofuran (solvent and fuel),25,26 γ-valerolactone27 (solvent), ester derivatives (plasticisers, fragrances and fuels),28–30 and the nylon intermediates adipic acid and caprolactam.31–37 LA can be converted into methyl vinyl ketone (monomer, vitamin A precursor) under oxidative conditions,38 or via decarbonylation of the intermediate angelica lactone.39,40
There are three isomers of angelica lactone (α-AL, β-AL, and γ-AL) which are shown in Scheme 1a. α-AL is conveniently obtained by reactive distillation of LA in high yields with water being the sole side product.41–43 This makes α-AL an interesting bio-based building block, which has been used as a monomer in UV-light induced, cationic and ring-opening polymerisations.44 The UV-light induced and cationic polymerisations tend to afford only sticky colourful oligomers.45,46 The product obtained by anionic ring-opening polymerisation has a higher molecular weight but consists of a mixture of C–C and ester connected monomer units.47 Such an irregular structure would make it rather challenging to use these polymers in applications where an exact control of the polymer structure is necessary. For instance, the oligomers that are typically used in the production of thermoplastic polyurethanes require well-defined end groups.48 Higher molecular weight polyesters on the other hand are typically used in extrusion and spinning processes49 for which high crystallinity and melting points are desirable traits, something poly α-AL may not provide due to the high stereo- and regio-irregularity. Other studies which were initiated by Mascal and co-workers focused on the utilisation of α-AL as a precursor for jet-fuels. Here α-AL is converted in the presence of K2CO3 to its di- and trimers, which can then be hydrodeoxygenated to the branched alkanes typically used in gasoline (Scheme 1b).50,51 The dimerisation step caught our attention as this reaction is known to occur via the β-isomer which we deem to be an interesting building block. β-AL itself has been dimerised to jet-fuels precursor and, for the first time, polymerized to acrylic-type polymers by the group of Hong.52
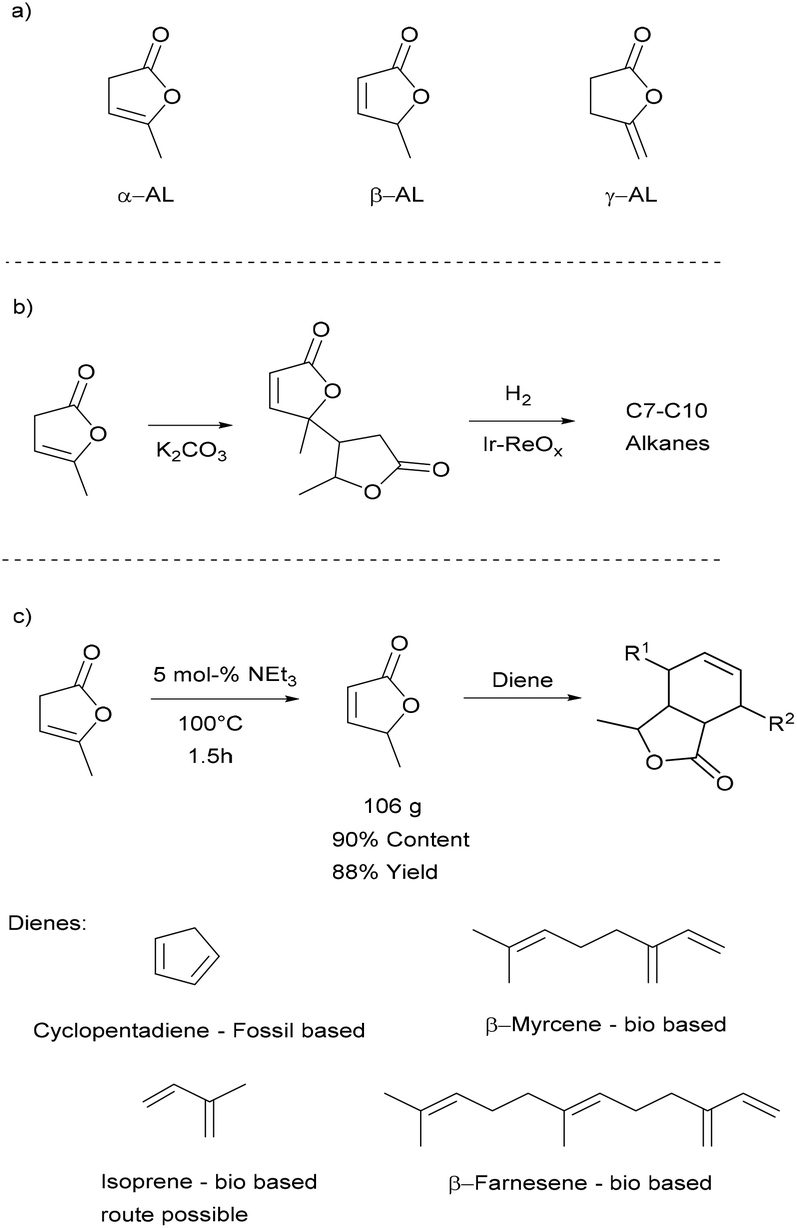 | ||
| Scheme 1 a) Different isomers of angelica lactone. (b) The pioneering work of Mascal and co-workers regarding the conversion of Al to fuels. (c) Our approach to upgrade α-AL to functional monomers for ROMP. | ||
We have explored the synthesis of bio-based monomers via Diels–Alder reactions (Scheme 1c) to obtain lactone functionalised derivatives. Diels–Alder reactions for the preparation of bio-based monomers recently received much attention53–56 as they are very atom-efficient and usually can be conducted in the absence of a solvent. Such functionalised norbornenes can then be used for example as monomer for adhesives,57 shape memory materials58 and polymer based electrolytes in batteries.59 Adducts containing the angelica lactone moiety have been prepared using this approach, but thus far the β-AL that was used in these reactions was not made from α-AL, likely due to the necessity to have pure β-AL.60–62
We wanted to use these Diels–Alder adducts in ring-opening metathesis reactions (ROMP) to obtain bio-based norbornene polymers. They can be easily functionalised as it is known that the γ-valerolactone moiety readily undergoes reactions with primary amines.63
Results and discussion
α-AL and β-AL are in equilibrium with each other in the presence of base but also undergo dimerisation under the reaction conditions necessary for isomerisation, a fact that makes the highly selective synthesis of pure β-AL from α-AL at high conversion a very challenging reaction that has not been achieved up to this date. Usually yields between 40–60% of β-AL can be achieved.64–66 This and the fact that in the existing protocols product separation needs to be carried out by column chromatography negatively affects the sustainability and scalability. Additionally, the formation of various azeotropes (see ESI†) of the isomers under vacuum distillation conditions results in a further yield penalty.However, as shown in Scheme 2, we found that it is possible to obtain a high content of β-AL in the product fraction (90 mol%), if the reaction time is kept rather short and no solvent is used. The usage of triethylamine allows its easy separation during vacuum distillation preventing further dimerisation. The mixture of the two angelica lactones was thus obtained in 88% yield after distillation on a 100 g scale. A small forerun was obtained, containing mostly α-AL, which can be reused. The only side products that remain are the di- and trimers which might be interesting raw materials for fuels.50,67
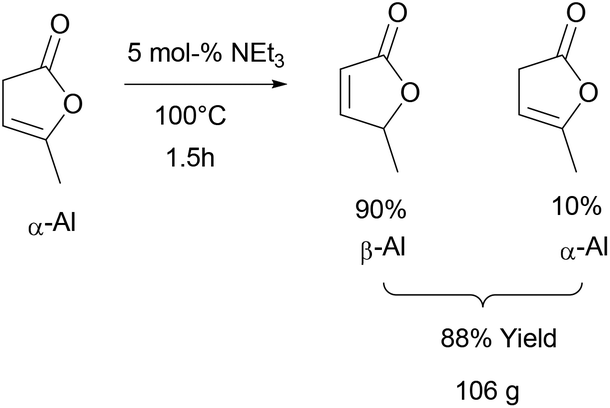 | ||
| Scheme 2 Synthesis of a mixture enriched with β-AL. | ||
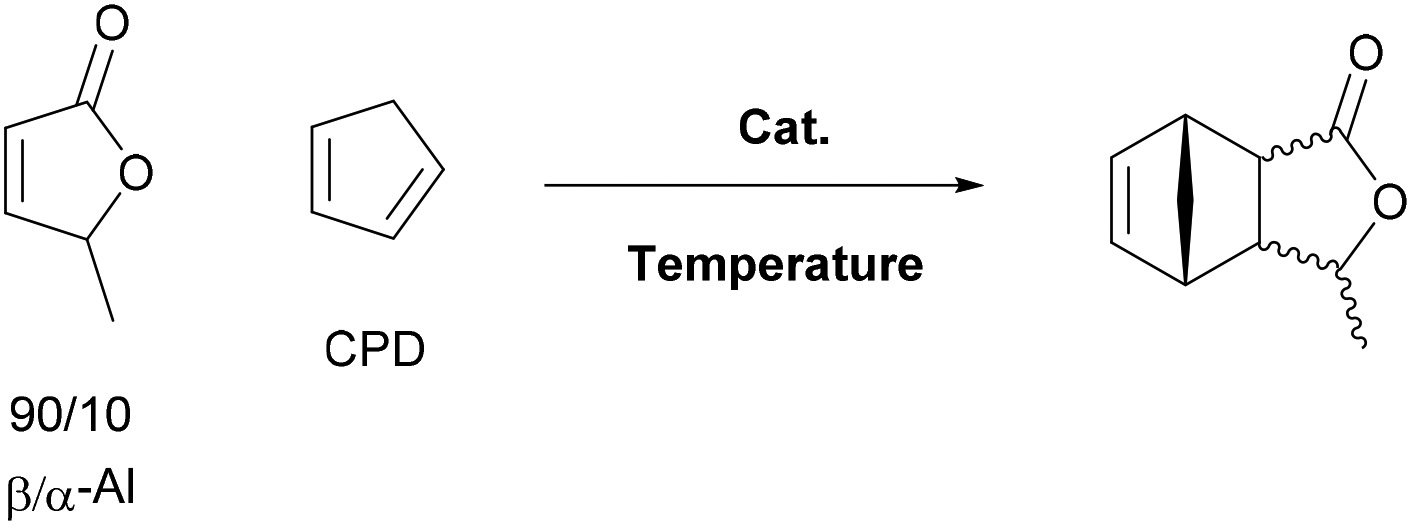
Next the solvent free Diels–Alder reaction of the AL-mixture containing 90% β-AL with cyclopentadiene (CPD) was investigated in the presence and absence of a catalyst (Table 1). When aluminium triflate was used as the catalyst (entries 1 + 2) the contents were converted to a black charred residue and no desired product formation was observed. When no catalyst was used it was possible to obtain the product in 19 to 25% at 80 °C to 90 °C respectively (Table 1, entries 3 and 5). At a higher temperature (100 °C) it was possible to isolate 40% of the DA-adduct. A similar yield was obtained at 80 °C in 30 minutes when anhydrous zinc chloride was used as catalyst. Increasing the reaction time from 30 minutes to 16 hours only led to a yield of 63%. As it was apparent that the low yields are caused by the competing di- and trimerisation of the CPD with itself, the equivalents of CPD with respect to β-AL were increased up to 10. This resulted in 90% yield of the desired product. Since a process that uses 10 eq. of CPD is not very efficient and sustainable, we decided to perform the reaction under semi-batch conditions on a 50 g scale (see Scheme 3). After dissolving zinc chloride in the β-AL enriched mixture 2 equivalents of CPD were slowly added over 10 hours. The ZnCl2 could be easily separated by adding acetone and any formed di- or trimers of CPD could be precipitated by simple dissolution of the product mixture in methanol. No column chromatography was necessary to obtain the product as a mixture of isomers in 82% yield. This is very fortunate as purification by distillation of the adduct is not an option since the retro-Diels–Alder reaction to β-AL commences at 136 °C (see ESI† for a DSC/TGA). Aiming for a higher renewable atom content, dienes other than cyclopentadiene have also been investigated. Use of furans was not successful under both catalysed and thermal conditions, due to rapid formation of humines (see ESI†). On the other hand, use of isoprene and terpenes (β-myrcene and farnesene) were more promising (Table 2). Upon reaction with β-AL at high temperature, full conversion was achieved. The diene oligomers formed as by-product were removed by a simple filtration through a short silica path, affording the Diels–Alder products in yields of up to 60%.
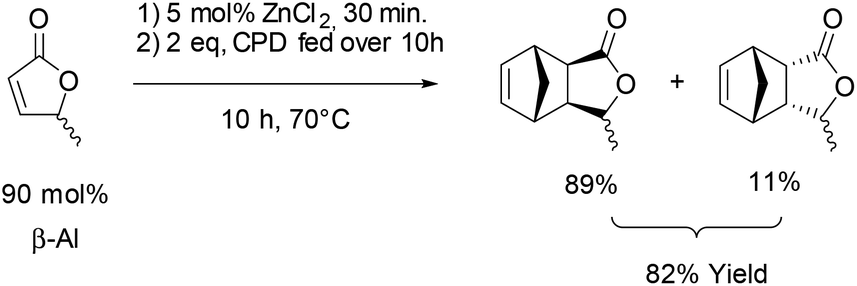 | ||
| Scheme 3 Synthesis of the DA-adduct with CPD on 50 g scale. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Eq. (CPD) | T [°C] | t [h] | Yielda [%] | Endo/exoa |
| General conditions: Reactions were carried out in closed reaction tubes and heated with microwave irradiation.a Determined by 1HNMR spectroscopy.b Isolated by column chromatography. | ||||||
| 1 | 5 mol% Al(OTf)3 | 3 | RT | 16 | — | — |
| 2 | 5 mol% Al(OTf)3 | 3 | 100 | 2 | — | — |
| 3 | — | 3 | 80 | 0.5 | 19 | 91/9 |
| 4 | — | 3 | 100 | 0.5 | 40b | 70/30 |
| 5 | — | 3 | 60 | 0.5 | 25 | 95/5 |
| 6 | 5 mol% ZnCl2 | 3 | 80 | 0.5 | 40 | 84/16 |
| 7 | 5 mol% ZnCl2 | 3 | RT | 16 | 63 | 89/11 |
| 8 | 5 mol% ZnCl2 | 10 | RT | 16 | 90 (86)b | 90/10 |
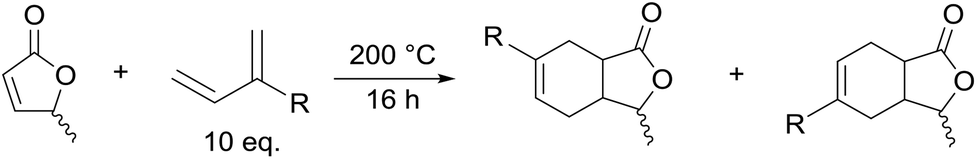
The ring-opening metathesis polymerisation (ROMP) of the DA adducts was next investigated.68–70 Grubbs’ 2nd generation catalyst was chosen for this (Fig. 1). Dichloromethane (DCM) is the solvent of choice for this reaction, since the polymeric products normally dissolve in it, thus allowing the formation of higher molecular weight polymers. The fully bio-based adducts shown in Table 2 were not polymerizable under these conditions, probably due to the absence of the ring strain that is present in the norbornene-type structure. On the other hand, the polymerization of the Cp/β-AL adduct was successful. Interestingly, at the initial monomer concentration of 1 mol l−1 after the addition of the initiator (0.5 mol% w.r.t. Cp/β-AL) the reaction mixture formed a gel within one minute indicating a great reactivity of this monomer in ROMP reactions (see Scheme 4). To further fine tune the polymerisation conditions different solvents, commonly considered as “greener” or safer choices, beside DCM were investigated at a lower catalyst loading. These solvents and their influence on the ROMP of the CP adduct are shown in Table 3. 2-Methyl-tetrahydrofuran was chosen due to its similar polarity to DCM but lower toxicity71 and potential renewability. Ethyl acetate (EtOAc) was included as it is one of the safest solvents regarding flammability and toxicity,72,73 which is also applicable for methyl isobutyl ketone (MIBK). Although not a “green” compound in general we also included tert-butyl methyl ether (MTBE) since it remains still a common solvent in industry and does not form peroxides.
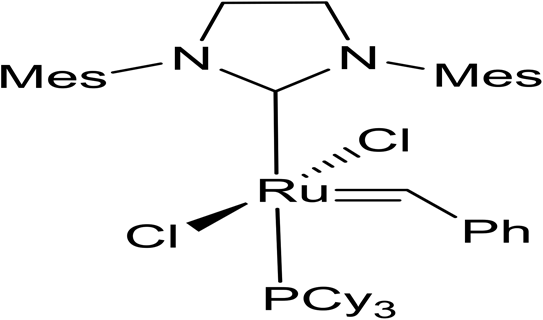 | ||
| Fig. 1 Grubbs II metathesis catalyst. | ||
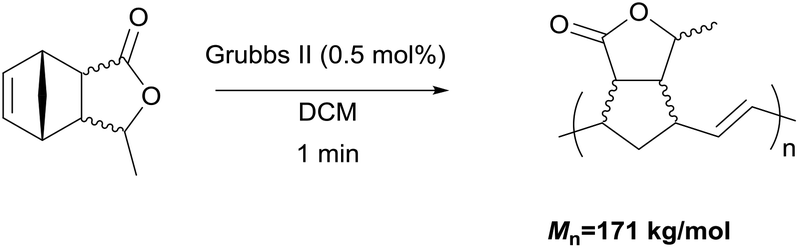 | ||
| Scheme 4 Initial ROMP experiment with Cp/β-AL. | ||
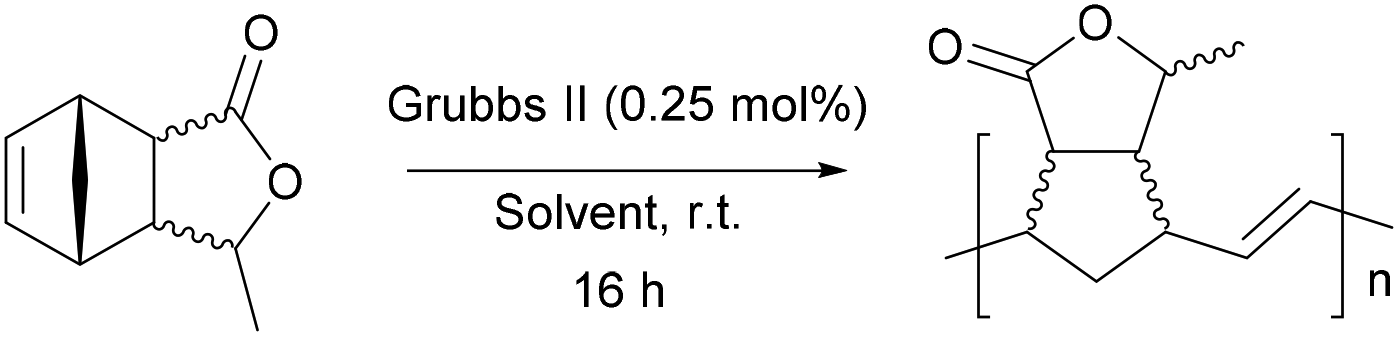
Notably, in all solvents expect DCM a white precipitate appeared after a few minutes of reaction time. In all the attempts the DA adduct did polymerise, yielding white gummy materials after precipitation with cold methanol.
As expected, the polymer obtained in DCM had a substantially higher molecular weight than the one predicted assuming a living polymerization (Table 3, entry 1). When using 2-MeTHF the obtained molecular weight was reduced to 80.6 kg mol−1. Ethyl acetate (EtOAc) and MTBE on the other hand afforded molecular weights of 64.1 kg mol−1 and 67.8 kg mol−1 respectively which are relatively close to the theoretical molecular weight of 65.6 kg mol−1. These findings indicate that the very safe and potentially renewable solvent EtOAc does not lead to catalyst deactivation/inhibition compared to DCM.
Aiming to assess the relation between substrate to catalyst ratio and molecular weight of the polymer, different catalyst loadings were investigated. A substantial linearity in the range between 100:1 and 600:1 (mol of substrate per mol of catalyst) was achieved, showing that the final molecular weight can be controlled by varying the amount of catalyst (Fig. 2).
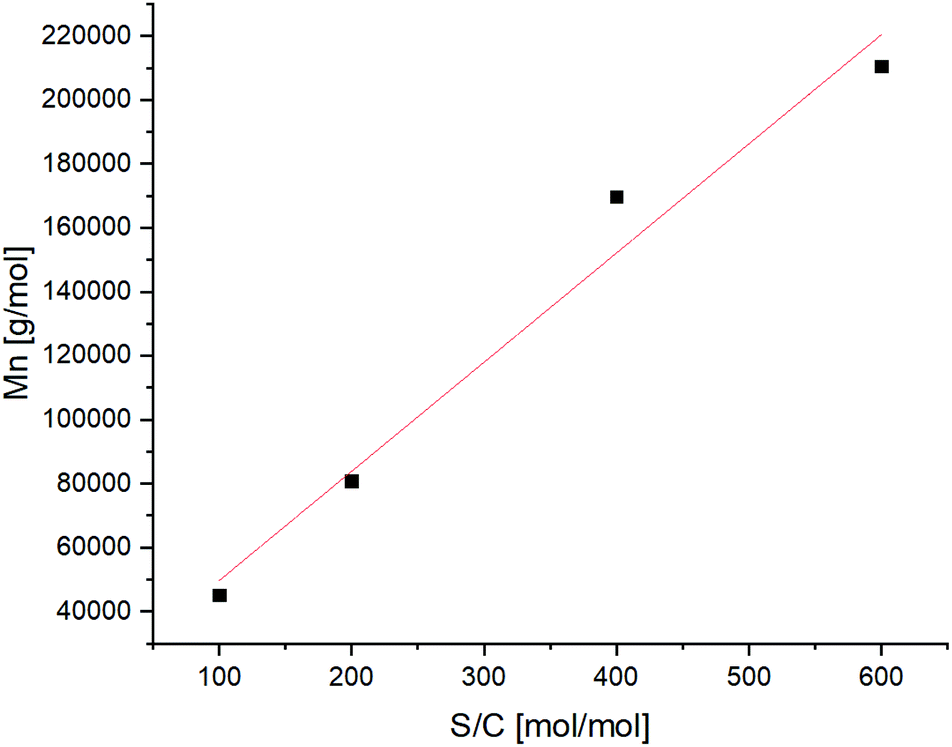 | ||
| Fig. 2 Obtained Mn values via ROMP of Cp/β-AL performed at different substrate to catalyst ratios (S/C). | ||
The obtained polymers tend to decompose at about 378 °C in a nitrogen atmosphere. No melting points were detected in the DSC analysis (as expected) confirming the amorphous nature of the polymer. In addition, a glass transition could also not be observed, which is an indication that the Tg occurs at or above the decomposition temperature (Fig. S8 and S9†). In contrast, poly-norbornene has a Tg at 35 °C. It is not easy to account for the huge difference in Tg between the two polymers. The high Tg of our polymer can possible be explained by the stereoregular nature. For steric reasons, the polymer has to be all trans.
Contact angle measurement revealed that the presence of the lactone moiety in poly-Cp/β-AL is increasing the hydrophilicity compared to poly-norbornene (θ(Cp/β-AL) = 75.7 ± 1.9°; θ(Cp/poly-norbornene) = 83.9 ± 2.3°).
As shown in Fig. 2 the polymer obtained via the ROMP of Cp/β-AL can be cast into clear transparent films. Comparison with poly-norbornene (prepared with the same catalyst in DCM) shows there is no negative effect of the lactone group on transparency (Fig. 3). Therefore it might be possible to find similar applications in optical wave guides,74 transparent coatings75 or other applications where high transparency in a large range of wavelengths is necessary.76
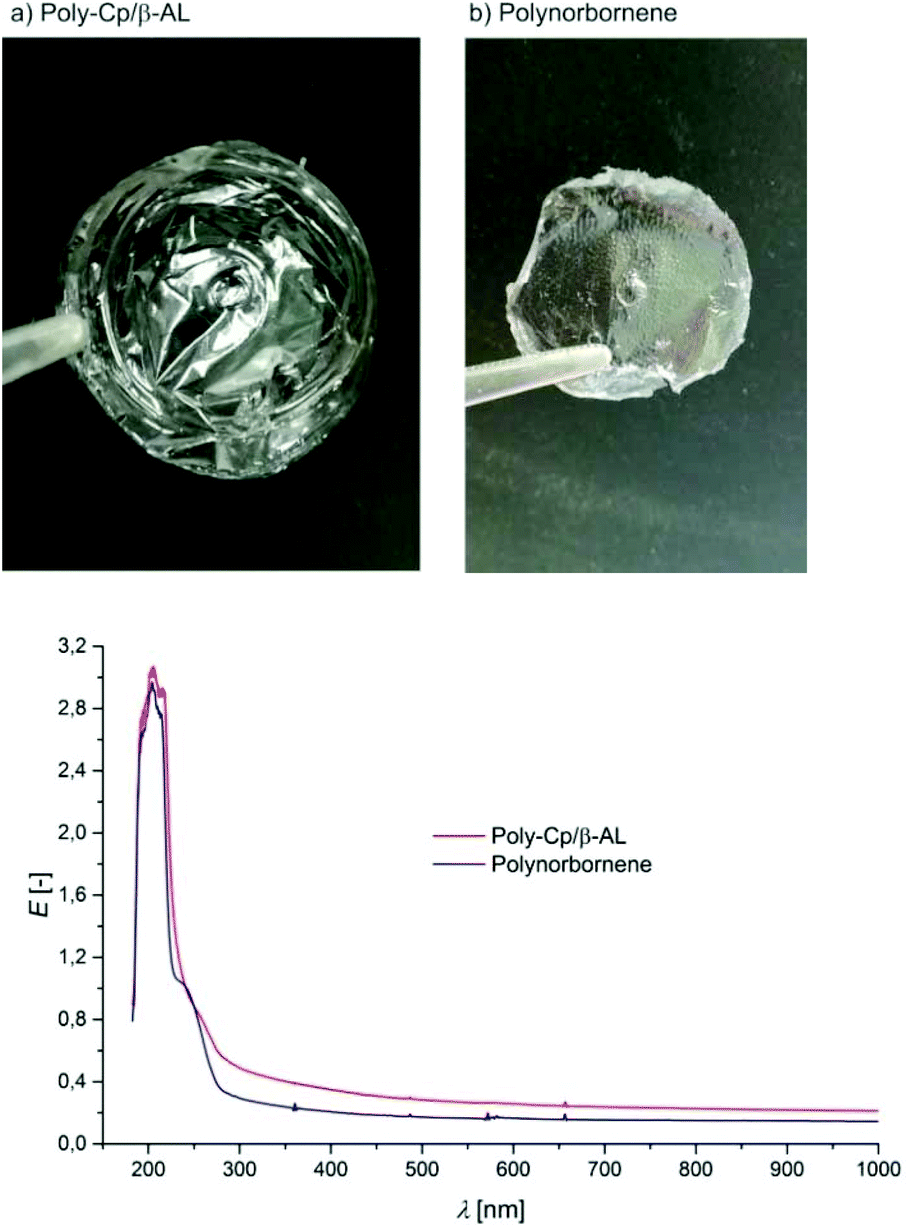 | ||
| Fig. 3 Films obtained from (a) poly-Cp/β-AL and (b) polynorbornene obtained by solution castings. Below: absorbance spectra of these films in the range from 150–1000 nm. E = extinction. | ||
Conclusions
It is still challenging to obtain pure β-AL from renewable α-AL in a scalable and sustainable procedure. In this work we show that a 90:10 mixture of the two can be prepared in a scalable procedure. This mixture is rich enough in β-AL to be used efficiently as dienophile in Diels–Alder reactions with cyclopentadiene, isoprene, myrcene and β-farnesene. The adduct obtained with cyclopentadiene can be efficiently polymerized in a ROMP reaction catalysed by the Grubbs II catalyst, either in DCM or alternatively using the renewable solvents 2-MeTHF or ethyl acetate. A linear relationship between Mn and the amount of catalyst is observed between a monomer to catalyst ratio of 100 to 600, thus allowing control of the molecular weight. The resulting polymers can be processed into films that have high transparency over a wide range of wavelengths. Compared with poly-norbornene the lactone moiety increases (surface) hydrophilicity, which could give rise to various benefits; for instance, higher miscibility with polar additives or better interaction with certain (wet) surfaces in coating applications. The lactone moiety does not only increase the bio-based carbon content of the materials, but it also enables further derivatization which gives easy access to a plethora of different polymers.
Experimental section
Preparation of a mixture of angelica lactones with 90 mol% content of the β-isomer
α-Angelica lactone (98%, 120 g, 1.2 mol) was added to a 250 ml two neck flask equipped with a condenser and a magnetic stirrer followed by the addition of triethyl amine (5 mol%, 8.5 ml). The mixture was heated to 100 °C under an argon atmosphere and monitored by 1H-NMR. After 1.5 hours a ratio of β/α-angelica lactone of 90–95/10–5 was reached and the condenser exchanged with a distillation head. Subsequent vacuum distillation at 6 × 10−2 mbar yielded two fractions: 38–42 °C containing mainly the α-isomer (this fraction can be reused for the next isomerisation run) and a fraction at 45–50 °C containing a mixture of angelica lactones with 90 mol% content of the β-isomer (106 g, 88% of theory).1H NMR (400 MHz, CD2Cl2, signals of β-isomer are reported) δ 7.46 (dd, J = 5.7, 1.5 Hz, 1H), 6.04 (dd, J = 5.7, 2.0 Hz, 1H), 5.11 (qt, J = 6.9, 2.0, 1.5 Hz, 1H), 1.41 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CD2Cl2) δ 173.3, 158.0, 121.3, 80.0, 19.0.
Diels–Alder reaction between β-angelica lactone and cyclopentadiene
In a dry and argon purged 10 ml reaction tube the angelica lactone mixture (β-AL = 90%, 10 mmol, 900 μl) and the desired catalyst were mixed. Freshly prepared CPD (3.0–10 eq. 2.5–8.3 ml) was added, the tube was sealed and heated with a microwave oven to the desired reaction temperature where it is kept for the indicated time. The 2 diastereomers of the product were separated by flash column chromatography (heptane/ethyl acetate 8:2), affording the endo and exo isomers (each of them is a racemic mixture of 2 stereoisomers) adduct as colourless oils (endo: 949 mg, 5.9 mmol, 30%; exo: 119 mg, 0.7 mmol, 7%; endo/exo 89/11).
Exo: 1H NMR (300 MHz, CD2Cl2) δ 6.21–6.04 (m, 2H), 4.17 (qd, J = 6.4, 3.2 Hz, 1H), 3.24–3.03 (m, 1H), 2.82 (dtq, J = 3.1, 1.5, 0.8 Hz, 1H), 2.59 (dt, J = 8.3, 1.3 Hz, 1H), 2.08–1.97 (m, 1H), 1.50–1.35 (m, 7H), 1.30 (d, J = 6.4 Hz, 3H). 13C NMR (75 MHz, CD2Cl2) δ 177.4, 136.4, 134.9, 78.9, 51.6, 48.5, 48.2, 46.0, 45.6, 22.8.
Endo: 1H NMR (300 MHz, CD2Cl2) δ 6.22–6.09 (m, 2H), 3.95 (qd, J = 6.5, 3.1 Hz, 1H), 3.26–2.97 (m, 3H), 2.67–2.54 (m, 1H), 1.57–1.46 (m, 1H), 1.39–1.29 (m, 1H), 1.25 (d, J = 6.5 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 177.3, 137.6, 137.5, 80.7, 50.0, 48.9, 47.6, 46.3, 43.1, 23.2.
Semi continuous synthesis of the β-angelica lactone CPD adduct
A 500 mL 2-neck round bottom flask was filled with dicyclopentadiene (100 mL) and iron(0) powder and equipped with a distillation setup. Cyclopentadiene was obtained by thermal cracking of its corresponding dimer at 180 °C and then condensed into a dropping funnel. 2 equivalents (84 mL, 67 g, 1.0 mol) were dropped over 10 hours into a 1 liter 3-necks round bottom flask containing the angelica lactone mixture (β-AL = 90%; 50 g, 0.5 mol) and zinc(II) chloride (3.5 g, 0.03 mol, 0.05 eq.) while heating up to 70 °C. Once the addition was complete, the reaction was stirred for another 10 hours and monitored by GC. After cooling down to room temperature, acetone (80–150 ml) was added to precipitate the Lewis acid catalyst. The remaining cyclopentadiene and α-angelica lactone were removed by vacuum distillation (50 °C/0.06 mbar) and the remaining cyclopentadiene dimers were precipitated by addition of ice-cold methanol (100 ml) followed by filtration. Solvent removal afforded the product as an orange liquid (69 g, 0.4 mol, 82% yield, endo:exo 89/11).
Ring-opening metathesis polymerization of the Cp/βAL adduct
To a stirred solution of Grubbs II catalyst (4.7 mg, 0.006 mmol, 0.25 mol%) in the desired solvent (Table 3) the Cp/βAL adduct (0.2 mL, 2.2 mmol, 1 eq.) was added under argon atmosphere. The reaction was stirred overnight. In few minutes after the addition, all the solutions turned opalescent and a whiteish precipitate appeared, except for the reaction in DCM. The reaction mixtures were then concentrated in vacuo and washed several times with methanol (using DCM to re-dissolve the polymer). All the reaction afforded a whiteish, gummy solid. A small portion of each sample was dissolved in a DMF/LiBr solution and analyzed by GPC. 1H-NMR spectra were recorded in CDCl3.
1H NMR (300 MHz, CDCl3) δ 5.84–5.12 (m, 2H, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 4.53–4.09 (m, 1H, O–CH–CH3), 3.33–2.38 (m, 4H, ring junction- and allylic-CH-), 1.89 (m, 2H, bridged-CH2-), 1.62–0.92 (m, 3H, CH3).
), 4.53–4.09 (m, 1H, O–CH–CH3), 3.33–2.38 (m, 4H, ring junction- and allylic-CH-), 1.89 (m, 2H, bridged-CH2-), 1.62–0.92 (m, 3H, CH3).
Film formation procedure
Films were obtained by casting a saturated solution of the polymer in DCM into a Teflon mold (depth 1 mm), letting the solvent evaporate over 4 hours.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors greatly appreciate the help of C. Wulf with the GPC and DSC measurements and to A. Wotzka for further DSC and TGA measurements. AD and SK are highly thankful to Henkel AG & Co for the financial support. The authors are grateful to Andreas Taden, Kenji Ito, Adrian Brandt and Horst Beck (Henkel AG & Co) for the useful discussions.This research was funded by the Bio-Based Industries Joint Undertaking under the European Union's Horizon 2020 research and innovation program, grant agreement no. 720695 (GreenSolRes).
Notes and references
- P. N. R. Vennestrøm, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–10509 CrossRef PubMed.
- Top Value-Added Chemicals from Biomass Vol. I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas, ed. T. Werpy and G. Petersen, U. S. Department of Energy (DOE) by the National Renewable Energy Laboratory a DOE national Laboratory, 2004 Search PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- W. G. Fan, C. Verrier, Y. Queneau and F. Popowycz, Curr. Org. Synth., 2019, 16, 583–614 CrossRef CAS PubMed.
- C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588–5617 RSC.
- B. Wozniak, S. Tin and J. G. de Vries, Chem. Sci., 2019, 10, 6024–6034 RSC.
- P. Gallezot, Catal. Today, 2007, 121, 76–91 CrossRef CAS.
- J. G. de Vries, Chem. Rec., 2016, 16, 2787–2800 CrossRef PubMed.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS.
- O. R. Schade, P.-K. Dannecker, K. F. Kalz, D. Steinbach, M. A. R. Meier and J.-D. Grunwaldt, ACS Omega, 2019, 4, 16972–16979 CrossRef CAS PubMed.
- T. Asano, H. Takagi, Y. Nakagawa, M. Tamura and K. Tomishige, Green Chem., 2019, 21, 6133–6145 RSC.
- W. Leitner, J. Klankermayer, S. Pischinger, H. Pitsch and K. Kohse-Höinghaus, Angew. Chem., Int. Ed., 2017, 56, 5412–5452 CrossRef CAS PubMed.
- K. I. Galkin and V. P. Ananikov, ChemSusChem, 2019, 12, 185–189 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354 CrossRef CAS PubMed.
- J. A. Galbis, M. d. G. García-Martín, M. V. de Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS PubMed.
- A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637–1669 CrossRef CAS PubMed.
- L. A. Heinrich, Green Chem., 2019, 21, 1866–1888 RSC.
- D. J. Hayes, S. W. Fitzpatrick, M. H. B. Hayes and J. R. H. Ross, in Biorefineries - Industrial Processes and Products: Status Quo and Future Directions, ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley-VCH, Weinheim, 2008, vol. 1, pp. 139–164 Search PubMed.
- F. D. Pileidis and M.-M. Titirici, ChemSusChem, 2016, 9, 562–582 CrossRef CAS PubMed.
- F. Yu, J. Thomas, M. Smet, W. Dehaen and B. F. Sels, Green Chem., 2016, 18, 1694–1705 RSC.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef.
- A. T. Adeleye, H. Louis, O. U. Akakuru, I. Joseph, O. C. Enudi and D. P. Michael, AIMS Energy, 2019, 7, 165–185 CAS.
- D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156–159 CrossRef CAS.
- V. Pace, P. Hoyos, L. Castoldi, P. Dominguez de Maria and A. R. Alcantara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- P. Bloom, To Archer-Daniels-Midland Company, WO2007094922A2, 2007.
- A. Démolis, N. Essayem and F. Rataboul, ACS Sustainable Chem. Eng., 2014, 2, 1338–1352 CrossRef.
- X. Yi, M. G. Al-Shaal, W. Ciptonugroho, I. Delidovich, X. Wang and R. Palkovits, ChemSusChem, 2017, 10, 1494–1500 CrossRef CAS PubMed.
- J.-P. Lange, J. Z. Vestering and R. J. Haan, Chem. Commun., 2007, 3488–3490 RSC.
- J. G. de Vries, N. Sereinig, E. W. M. van de Vondervoort and M. C. C. Janssen, To DSM IP Assets BV, WO2012131028A1, 2012.
- P. K. Wong, C. Li, L. Stubbs, M. van Meurs, D. G. Anak Kumbang, S. C. Y. Lim and E. Drent, To Agency for Science, Technology and Research, WO2012134397A1, 2012.
- Y. Yang, X. R. Wei, F. X. Zeng and L. Deng, Green Chem., 2016, 18, 691–694 RSC.
- J. D. Nobbs, N. Z. B. Zainal, J. Tan, E. Drent, L. P. Stubbs, C. Li, S. C. Y. Lim, D. G. A. Kumbang and M. van Meurs, ChemistrySelect, 2016, 1, 539–544 CrossRef CAS.
- A. Marckwordt, F. El Ouahabi, H. Amani, S. Tin, N. V. Kalevaru, P. C. J. Kamer, S. Wohlrab and J. G. de Vries, Angew. Chem., Int. Ed., 2019, 58, 3486–3490 CrossRef CAS PubMed.
- J. Lin, H. Song, X. Shen, B. Wang, S. Xie, W. Deng, D. Wu, Q. Zhang and Y. Wang, Chem. Commun., 2019, 55, 11017–11020 RSC.
- Y. Gong, L. Lin and B. Zhang, Chin. J. Chem., 2012, 30, 327–332 CrossRef CAS.
- J. A. Dumesic and R. M. West, To Wisconsi Alumni Research Foundation, WO002011087962A1, 2011.
- W. Skorianetz and G. Ohloff, Helv. Chim. Acta, 1975, 58, 1272–1275 CrossRef CAS.
- J. H. Helberger, S. Ulubay and H. C. Civelekoglu, Justus Liebigs Ann. Chem., 1949, 561, 215–220 CrossRef CAS.
- D. Sun, Y. Takahashi, Y. Yamada and S. Sato, Appl. Catal., A, 2016, 526, 62–69 CrossRef CAS.
- C. G. S. Lima, J. L. Monteiro, T. de Melo Lima, M. Weber Paixao and A. G. Correa, ChemSusChem, 2018, 11, 25–47 CrossRef CAS PubMed.
- V. E. Tarabanko and K. L. Kaygorodov, Chem. Sustainable Dev., 2010, 321–328 Search PubMed.
- C. S. Marvel and C. L. Levesque, J. Am. Chem. Soc., 1939, 61, 1682–1684 CrossRef CAS.
- Y. Yokoyama, M. Okada and H. Sumitomo, Makromol. Chem., 1975, 176, 3537–3550 CrossRef CAS.
- K. L. Kaygorodov, V. E. Tarabanko and N. Tarabanko, Cogent Chem., 2018, 4, 1443689 Search PubMed.
- N. Adam, G. Avar, H. Blankenheim, W. Friederichs, M. Giersig, E. Weigand, M. Halfmann, F.-W. Wittbecker, D. Larimer, U. Maier, S. Meyer-Ahrens, K.-L. Noble and H.-G. Wussow, in Ullmann's Encyclopedia of Industrial Chemistry, 2005, DOI:10.1002/14356007.a21_665.pub2.
- E. Gubbels, T. Heitz, M. Yamamoto, V. Chilekar, S. Zarbakhsh, M. Gepraegs, H. Köpnick, M. Schmidt, W. Brügging, J. Rüter and W. Kaminsky, in Ullmann's Encyclopedia of Industrial Chemistry, 2018, pp. 1–30, DOI:10.1002/14356007.a21_227.pub2.
- M. Mascal, S. Dutta and I. Gandarias, Angew. Chem., 2014, 126, 1885–1888 CrossRef.
- J. Xu, N. Li, X. Yang, G. Li, A. Wang, Y. Cong, X. Wang and T. Zhang, ACS Catal., 2017, 7, 5880–5886 CrossRef.
- X.-J. Wang and M. Hong, Angew. Chem., Int. Ed., 2020, 59, 2664–2668 CrossRef CAS PubMed.
- A. D. Pehere, S. Xu, S. K. Thompson, M. A. Hillmyer and T. R. Hoye, Org. Lett., 2016, 18, 2584–2587 CrossRef CAS PubMed.
- Y. Bai, M. De bruyn, J. H. Clark, J. R. Dodson, T. J. Farmer, M. Honoré, I. D. V. Ingram, M. Naguib, A. C. Whitwood and M. North, Green Chem., 2016, 18, 3945–3948 RSC.
- M. Iqbal, R. A. Knigge, H. J. Heeres, A. A. Broekhuis and F. Picchioni, Polymers, 2018, 10, 1177 CrossRef PubMed.
- A. Blanpain, J. H. Clark, T. J. Farmer, Y. Guo, I. D. V. Ingram, J. E. Kendrick, S. B. Lawrenson, M. North, G. Rodgers and A. C. Whitwood, ChemSusChem, 2019, 12, 2393–2401 CAS.
- N. R. Grove, P. A. Kohl, S. A. Bidstrup Allen, S. Jayaraman and R. Shick, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 3003–3010 CrossRef CAS.
- D. Yang, W. Huang, J. Yu, J. Jiang, L. Zhang and M. Xie, Polymer, 2010, 51, 5100–5106 CrossRef CAS.
- D. Rosenbach, N. Mödl, M. Hahn, J. Petry, M. A. Danzer and M. Thelakkat, ACS Appl. Energy Mater., 2019, 2, 3373–3388 CrossRef CAS.
- Z. Chen and R. M. Ortuño, Tetrahedron: Asymmetry, 1994, 5, 371–376 CrossRef CAS.
- T. Řezanka and K. Sigler, Eur. J. Org. Chem., 2006, 4277–4284 CrossRef.
- H. Yanai, A. Takahashi and T. Taguchi, Tetrahedron, 2007, 63, 12149–12159 CrossRef CAS.
- M. Chalid, H. J. Heeres and A. A. Broekhuis, J. Appl. Polym. Sci., 2012, 123, 3556–3564 CrossRef CAS.
- Y. Wu, R. P. Singh and L. Deng, J. Am. Chem. Soc., 2011, 133, 12458–12461 CrossRef CAS PubMed.
- L. Zhou, L. Lin, J. Ji, M. Xie, X. Liu and X. Feng, Org. Lett., 2011, 13, 3056–3059 CrossRef CAS PubMed.
- C. R. Jones, M. D. Greenhalgh, J. R. Bame, T. J. Simpson, R. J. Cox, J. W. Marshall and C. P. Butts, Chem. Commun., 2016, 52, 2920–2923 RSC.
- B. Lu, J. Li, G. Lv, Y. Qi, Y. Wang, T. Deng, X. Hou and Y. Yang, RSC Adv., 2016, 6, 93956–93962 RSC.
- I. Choinopoulos, Polymers, 2019, 11, 298 CrossRef PubMed.
- A. K. Pearce, J. C. Foster and R. K. O'Reilly, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1621–1634 CrossRef CAS.
- J.-A. Song, B. Park, S. Kim, C. Kang, D. Lee, M.-H. Baik, R. H. Grubbs and T.-L. Choi, J. Am. Chem. Soc., 2019, 141, 10039–10047 CrossRef CAS PubMed.
- V. Antonucci, J. Coleman, J. B. Ferry, N. Johnson, M. Mathe, J. P. Scott and J. Xu, Org. Process Res. Dev., 2011, 15, 939–941 CrossRef CAS.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- M. Singh and K. Weidner, in Optical Interconnects for Data Centers, ed. T. Tekin, R. Pitwon, A. Håkansson and N. Pleros, Woodhead Publishing, 2017, pp. 157–170, DOI:10.1016/B978-0-08-100512-5.00006-1.
- G. Sung, M.-C. Choi, S. Nagappan, W.-K. Lee, M. Han and C.-S. Ha, Polym. Bull., 2012, 70, 619–630 CrossRef.
- M.-C. Choi, J.-C. Hwang, C. Kim, S. Ando and C.-S. Ha, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1806–1814 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc00338g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |