
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding laccase/HBT-catalyzed grass delignification at the molecular level†
Roelant
Hilgers
 a,
Gijs
van Erven
a,
Vincent
Boerkamp
a,
Irina
Sulaeva
b,
Antje
Potthast
b,
Mirjam A.
Kabel
a and
Jean-Paul
Vincken
*a
a,
Gijs
van Erven
a,
Vincent
Boerkamp
a,
Irina
Sulaeva
b,
Antje
Potthast
b,
Mirjam A.
Kabel
a and
Jean-Paul
Vincken
*a
aLaboratory of Food Chemistry, Wageningen University & Research, The Netherlands. E-mail: jean-paul.vincken@wur.nl; Tel: +31 (0)317 482234
bDepartment of Chemistry, Division of Chemistry of Renewable Resources, University of Natural Resources and Life Sciences, Austria
First published on 13th February 2020
Abstract
Laccase-mediator systems (LMS) are potential green tools to improve the valorization of lignocellulosic biomass by selective degradation of lignin. Despite extensive attention devoted to lignin degradation by LMS in literature, knowledge on the underlying mechanisms is largely limited to model compound studies. Here, we report a mechanistic study on the delignification of wheat straw (WS) and corn stover (CS) by a laccase/HBT system. Quantitative 13C-IS py-GC-MS analysis revealed that WS and CS were delignified in the range of 28–51% (w/w). Based on a combination of py-GC-MS, 2D NMR, SEC and RP-UHPLC-MS, extensive structural characterization of both residual and solubilized lignin structures was performed, from which we reconstructed the degradation pathway of native lignin by laccase/HBT. For the first time, we show that degradation of native lignin in the plant cell wall matrix by LMS occurs via both Cα–Cβ cleavage and ether cleavage of β-O-4′ aryl ethers, and that the latter primarily occurs via cleavage of the β-O bond. Cγ-Coumaroylated substructures were found to be more recalcitrant towards degradation than non-acylated substructures. In addition to lignin degradation, our results provide evidence for grafting of HBT onto lignin.
Introduction
Agricultural by-products, such as wheat straw (WS) and corn stover (CS), are promising sources of polysaccharides for e.g. biochemical and biofuel production. Nevertheless, the co-occurrence of lignin in these lignocellulosic materials hinders efficient (enzymatic) conversion of the polysaccharides.1 Hence, degradation or removal of lignin prior to polysaccharide conversion is advantageous.2,3 Although multiple chemical and/or physical delignification strategies have been reported, most of these require harsh conditions.2,4 Therefore, green alternatives are receiving increasing attention, such as biological pre-treatments (e.g. by using white-rot fungi) or fractionation with deep eutectic solvents.5–7 Another promising approach, especially with respect to industrial applicability, is enzymatic lignin degradation.8Lignin is an aromatic polymer, built up via polymerization of the phenylpropanoids sinapyl alcohol, coniferyl alcohol and p-coumaryl alcohol. When incorporated in the lignin polymer, these building blocks form syringyl (S), guaiacyl (G) and p-hydroxyphenyl (H) subunits, respectively. Grass lignins also contain acetate, p-coumaric acid and ferulic acid, and also tricin may be incorporated.9,10 Polymerization of the phenylpropanoids results in a variety of interunit linkages, of which the β-O-4 linkage is the most abundant.11 As most interunit linkages are formed via radical coupling of the phenolic hydroxyl groups of the phenylpropanoids, the majority (70–90%) of subunits in the lignin polymer are non-phenolic, while only 10–30% of the subunits are phenolic.12
Several classes of enzymes are known to be active toward lignin, including laccases and various peroxidases (i.e. lignin peroxidases, versatile peroxidases and manganese peroxidases). For industrial delignification of biomass, laccases are preferred to peroxidases, as laccases use molecular O2 rather than H2O2 as electron acceptor, and are, in contrast to peroxidases, not inactivated by their electron acceptor.13 Due to their relatively low redox potential (≤ 800 mV vs. NHE),3 the phenolic subunits in lignin are most prone to direct laccase activity. In contrast, the non-phenolic subunits have higher redox potentials (>1400 mV),14,15 and are, therefore, resistant to oxidation by laccase alone. Nevertheless, when laccase is combined with an appropriate mediator, its oxidizing capacity can be extended to non-phenolic lignin structures. In such a laccase-mediator system (LMS), laccase oxidizes the mediator, which subsequently undergoes a redox reaction with a (non-phenolic) lignin subunit. One of the most effective mediators known is 1-hydroxybenzotriazole (HBT).15–17
After oxidation by the mediator, the formed lignin radical can react further via a variety of pathways. These pathways have, so far, mainly been investigated by using low molecular weight model compounds, resembling substructures of lignin.18–26 Dependent on the structure of the lignin radical, bond cleavage, radical coupling and a number of other modifications have been reported, and it has been suggested that these reaction pathways are in competition with each other. Obviously, this competition determines whether LMS treatment eventually results in overall polymerization or depolymerization. Although these insights from model compound studies are valuable, so far, it remains unclear to what extent these insights can be extrapolated to LMS-induced lignin degradation in situ.
Over the past years, various studies have been published that report successful degradation or removal of lignin from biomass and paper pulp upon LMS treatment.27–39 In most of these studies, however, high laccase and mediator concentrations were used, which is undesirable from both economical and sustainability perspectives. In addition, these studies focused on the effectiveness of the LMS treatments, without performing in-depth structural characterization of the LMS treated lignin. Structural characterization may reveal accumulation of both degraded and recalcitrant substructures, and thereby provide insights into the mechanisms and bottlenecks underlying LMS based delignification. These insights are essential in order to increase the efficiency of LMS based biomass delignification. Although a number of previous studies included structural characterization of LMS treated lignin (e.g. by 2D NMR spectroscopy), limited insights were obtained into the effect of LMS on lignin structure, due to the following reasons: (i) structural characterization was focused on the structure of residual lignin, rather than that of solubilized lignin, even in cases where lignin removal up to 50% was reported, and (ii) LMS treatments were often combined with alkaline peroxide treatments, which are known to induce structural changes in oxidized lignin.40
In this study, our goal was to provide more insights into the mechanisms and bottlenecks underlying LMS-induced delignification of grasses. Wheat straw and corn stover were treated with a laccase/HBT system, after which lignin fractionation and purification was performed to allow detailed characterization of both residual and solubilized lignin populations. Treatments were done at both pH 4 and 6, as the used laccase has been reported to show optimal enzyme activity at pH 4, but optimal lignin model compound oxidation in a LMS at pH 6.41 A combination of py-GC-MS, 2D NMR, SEC and RP-UHPLC-MS was used to provide both quantitative and qualitative information on the structure of LMS treated lignin and enabled us to deduce the underlying ligninolysis mechanisms.
Experimental
Materials
Wheat straw (Triticum aestivum, WS) and corn stover (Zea mays, CS) were kindly provided by CNC (Milsbeek, The Netherlands). Uniformly 13C-labeled wheat straw lignin isolate (13C-IS, 97.7 atom% 13C) was available in our laboratory from previous research,42 as well as 1-(3,4-dimethoxyphenyl)-3-hydroxypropan-1-one and 1-(3,4-dimethoxyphenyl)-2,3-dihydroxypropan-1-one.43 Laccase from Trametes versicolor was purchased from Sigma Aldrich (St Louis, MO, USA) and was used without any further purification. Laccase activity was measured spectrophotometrically by oxidation of ABTS (1 U = 1 μmol oxidized ABTS min−1, see ESI†). Rovabio® Advance was obtained from Adisseo (Antony, France) and ViscoStar 150L was purchased from Dyadic (Jupiter, FL, USA). All other chemicals were of analytical grade, purchased from commercial suppliers, and used without further purification. Water was purified using a MilliQ system (Millipore, Billerica, MA, USA).Methods
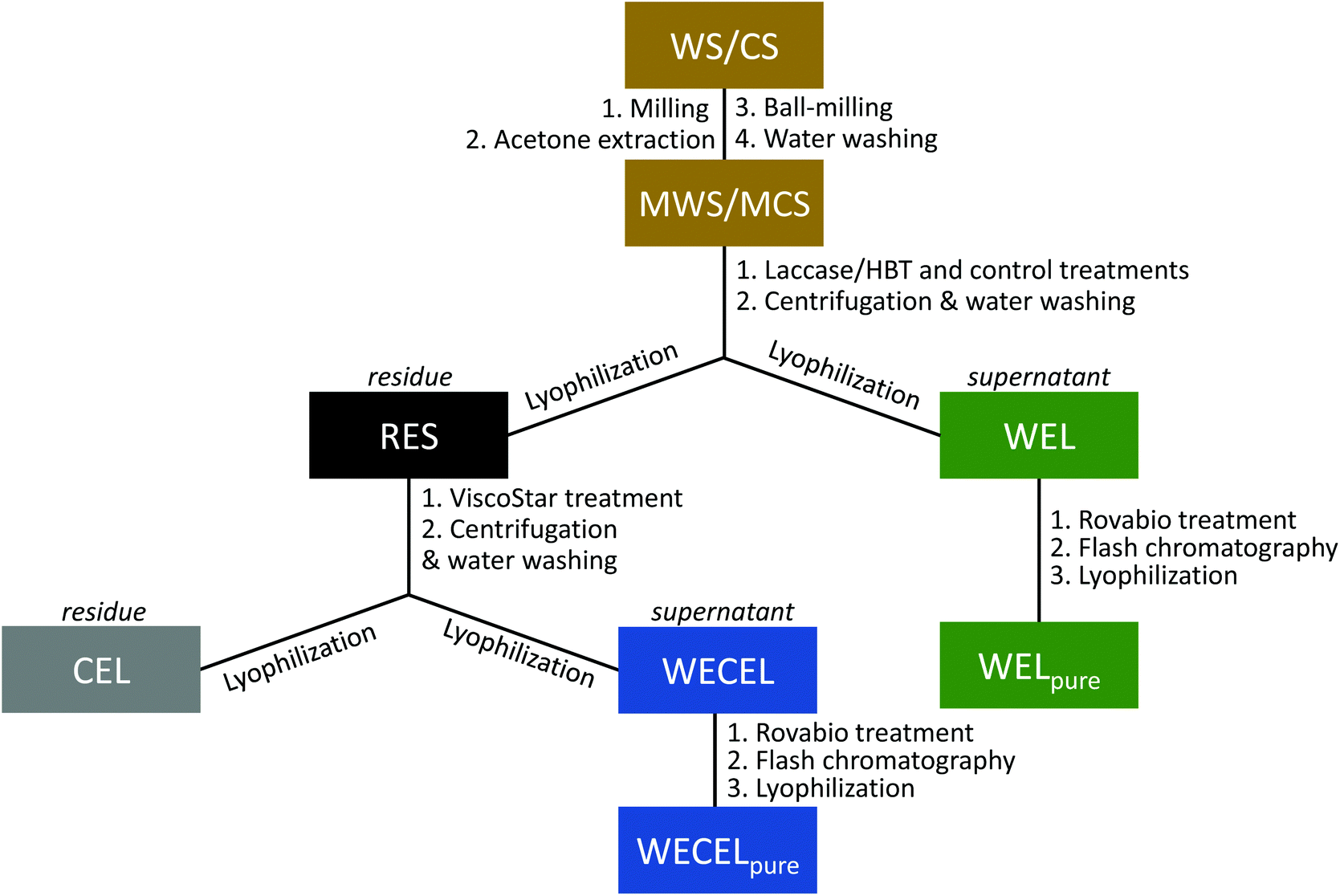 | ||
| Fig. 1 Schematic overview of the incubations, fractionations and purifications in this study. (M)WS = (Milled) wheat straw; (M)CS = (Milled) corn stover; RES = residue; WEL = water-extractable lignin; CEL = cellulolytic enzyme lignin; WECEL = water-extractable cellulolytic enzyme lignin. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 15 min, 20 °C). The residue was incubated for a second time, using fresh ViscoStar 150L and fresh buffer. The supernatant of the first incubation was stored at 4 °C. After incubation and centrifugation, the residue was washed three times with water at ∼0.5% (w/w) solid loading, and lyophilized to yield cellulolytic enzyme lignin (CEL). The supernatants of both ViscoStar treatments and the supernatant from the three wash steps were combined and lyophilized to obtain water extractable cellulolytic enzyme lignin (WECEL).
000g, 15 min, 20 °C). The residue was incubated for a second time, using fresh ViscoStar 150L and fresh buffer. The supernatant of the first incubation was stored at 4 °C. After incubation and centrifugation, the residue was washed three times with water at ∼0.5% (w/w) solid loading, and lyophilized to yield cellulolytic enzyme lignin (CEL). The supernatants of both ViscoStar treatments and the supernatant from the three wash steps were combined and lyophilized to obtain water extractable cellulolytic enzyme lignin (WECEL).
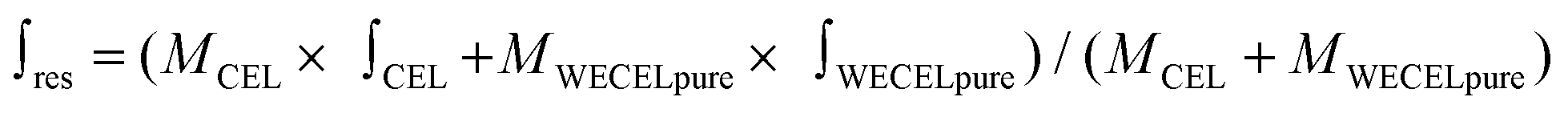 | (1) |
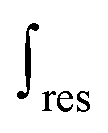 is the mass-weighted averaged HSQC integral for residual lignin; MCEL and MWECELpure are the masses of lignin in the CEL and WECELpure fraction, respectively;
is the mass-weighted averaged HSQC integral for residual lignin; MCEL and MWECELpure are the masses of lignin in the CEL and WECELpure fraction, respectively; 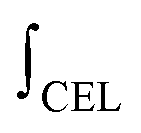 and
and 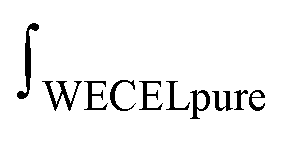 are the HSQC integrals obtained for the CEL and WECELpure fraction, respectively.
500g, 5 min, 20 °C) and analyzed using RP-UHPLC-PDA-MS. A detailed description of the method can be found in the ESI.†
are the HSQC integrals obtained for the CEL and WECELpure fraction, respectively.
500g, 5 min, 20 °C) and analyzed using RP-UHPLC-PDA-MS. A detailed description of the method can be found in the ESI.†
Results and discussion
Delignification of MWS and MCS
After laccase/HBT treatment of MWS and MCS, the water-washed and lyophilized residues (i.e. WS and CS-RES fractions) were weighed and analyzed by using quantitative py-GC-MS analysis with 13C wheat straw lignin as internal standard. The analysis indicated that the laccase/HBT treatment resulted in a delignification of 44–51% and 36–44% for MWS and MCS, respectively, whereas only 0–5% delignification was observed in control incubations without laccase/HBT (Fig. 2). These values are in a similar range as those reported in a previous study on laccase/HBT based wheat straw delignification and confirm that grasses can be effectively delignified by laccase/HBT treatments.28 The used py-GC-MS based lignin quantification method has been validated for quantification of both wheat straw and corn stover lignin,42,44 but this was the first time the method was used for lignin quantification in LMS treated biomass. Therefore, for comparison, a Klason lignin determination was performed on the WS-RES fractions incubated at pH 4. Although the latter method suggested a MWS delignification of 32% at pH 4 (Table S5†), which is, admittedly, lower than the values obtained by using py-GC-MS, both methods show a significant decrease in lignin content after laccase/HBT treatment. As both lignin quantification methods have their drawbacks (see ESI† for a more detailed discussion), and only py-GC-MS provides additional structural information, we used this method for lignin quantification on all other samples in this study.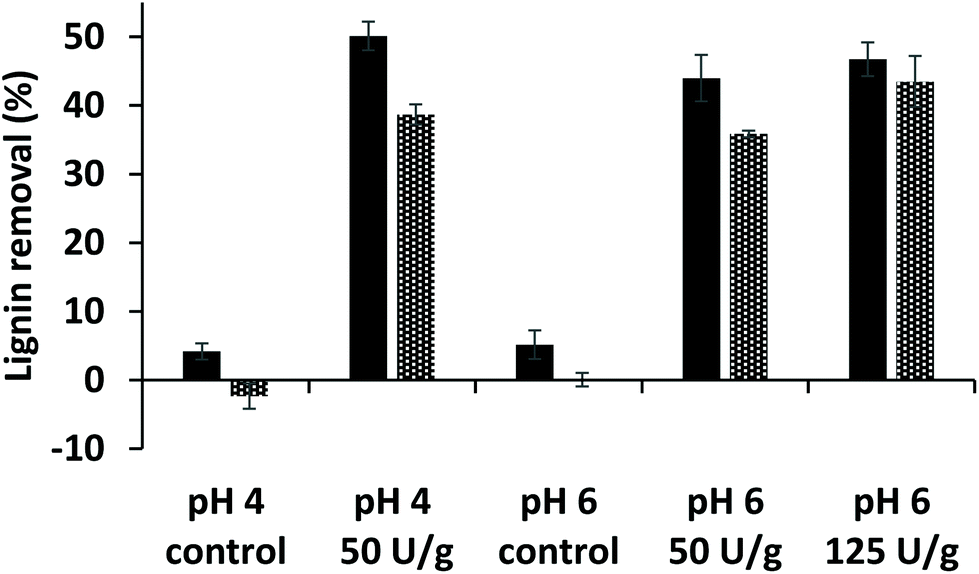 | ||
| Fig. 2 Estimated lignin removal from milled wheat straw (solid bars) and milled corn stover (dotted bars) by laccase/HBT treatments based on quantitative 13C-IS py-GC-MS analysis (see ESI† for details). | ||
Although no lignin quantification could be performed for the unpurified WEL fractions,48 it was assumed that all removed lignin ended up in the WEL fractions (see Fig. 1), as no volatile reaction products were expected to be formed.
Purification of lignin fractions
After LMS treatment, the residue (RES) and supernatant (WEL) were further fractionated and purified to facilitate structural lignin analysis by 2D NMR. Hereto, cellulolytic enzyme lignin (CEL) was prepared from RES, which resulted in a 2.0 to 2.7-fold increase in lignin content (Tables S9–S12†). The resulting supernatant (WECEL) contained the released carbohydrates, but also a significant amount of lignin (i.e. 31–64% of the lignin originating from RES), indicating that also WECEL lignin should be analyzed for a complete overview the residual lignin structure. To facilitate analysis, WECEL was purified by treatment with a xylanase-enriched enzyme cocktail containing additional ferulic acid esterase activity, and subsequent Flash chromatography. Based on py-GC-MS analysis it was estimated that this purification increased the lignin content from ≤10.6% (w/w) in WECEL to ≥50.4% (w/w) in WECELpure (Tables S13–S16†). The same strategy was used to remove sugars and mediator from WEL fractions, which resulted in WELpure fractions with estimated lignin contents of 6.6–23.2% (w/w) (Table S17†). The summed lignin yields of the three final fractions (i.e. CEL, WECELpure and WELpure) were estimated to be 93–100% for control incubations and 39–67% for laccase/HBT incubations (see Tables S9–S17†), though it should be noted that the yields of the laccase/HBT treated fractions are expected to be underestimations (see discussion on page S13–14).Overall extent of lignin oxidation and degradation in CEL, WECELpure and WELpure fractions
As a measure for the extent of lignin oxidation in the purified WS and CS fractions (i.e. CEL, WECELpure and WELpure), the relative abundance of Cα-oxidized structures was determined based on semi-quantitative processing of 2D HSQC NMR spectra (see Fig. S1† for spectra and Table S3† for all assignments). It was observed that laccase/HBT treatment increased the relative content of Cα-oxidized subunits in all fractions, due to oxidation of both S and G-units (Fig. 3A and Fig. S2†). The extent of Cα-oxidation was also estimated based on the abundance of Cα-oxidized products in py-GC-MS analysis of the same fractions (Fig. 3B). Although absolute values differed from the HSQC analysis, a very similar trend was observed: Cα-oxidation increased in all fractions, but the extent was vastly different for each fraction. Whereas CEL fractions of laccase/HBT treated MWS only showed 3–18% Cα-oxidation (based on HSQC NMR), for WECELpure and WELpure fractions, Cα-oxidation was 24–40% and 69–75%, respectively.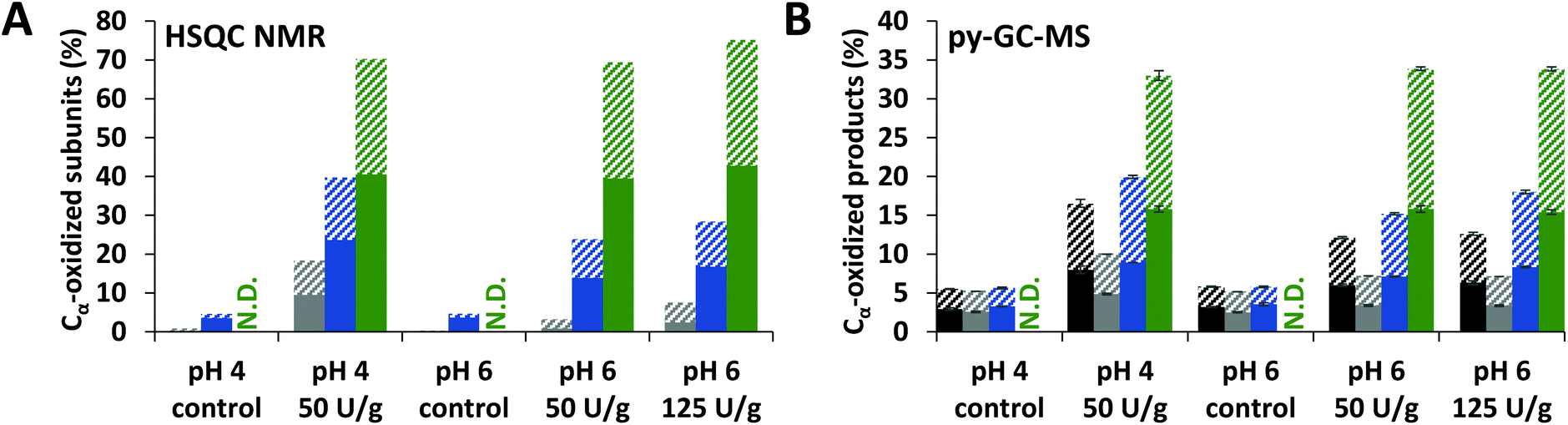 | ||
| Fig. 3 Relative abundance of Cα-oxidized subunits as determined by using HSQC NMR (A) and estimated based on py-GC-MS (B) in RES (black), CEL (grey), WECELpure (blue) and WELpure (green) fractions of laccase/HBT treated MWS and controls. RES was only analyzed by using py-GC-MS. Solid and striped bars correspond to G and S units, respectively. Because of their low abundance, H-units are not included. Error bars in B represent the standard deviation of two treatment duplicates and two analytical duplicates. N.D. = Not determined. Cα-Oxidation upon treatment of MCS showed similar trends and can be found in Fig. S2.† | ||
To verify that delignification of MWS and MCS was truly caused by cleavage of interunit linkages, the amount of intact interunit linkages per 100 aromatic subunits (S, G, H and oxidized analogues) was determined from the HSQC spectra. Indeed, in all fractions of laccase/HBT treated samples a decrease (up to 51%) in intact interunit linkages was observed (Fig. 4A and Fig. S3†). We also estimated the amount of interunit linkages based on the py-GC-MS data. Hereto, the relative amount of pyrolysis products with intact 3-carbon side chains (PhCγ) were taken, after correction for cleavage markers (i.e. diketones and vinylketones, as shown previously).5,43 Although the absolute values differed from those obtained using HSQC NMR, similar trends were observed (Fig. 4B). Interestingly, the amount of interunit linkages in WELpure was 2–4 times lower than in CEL and WECELpure.
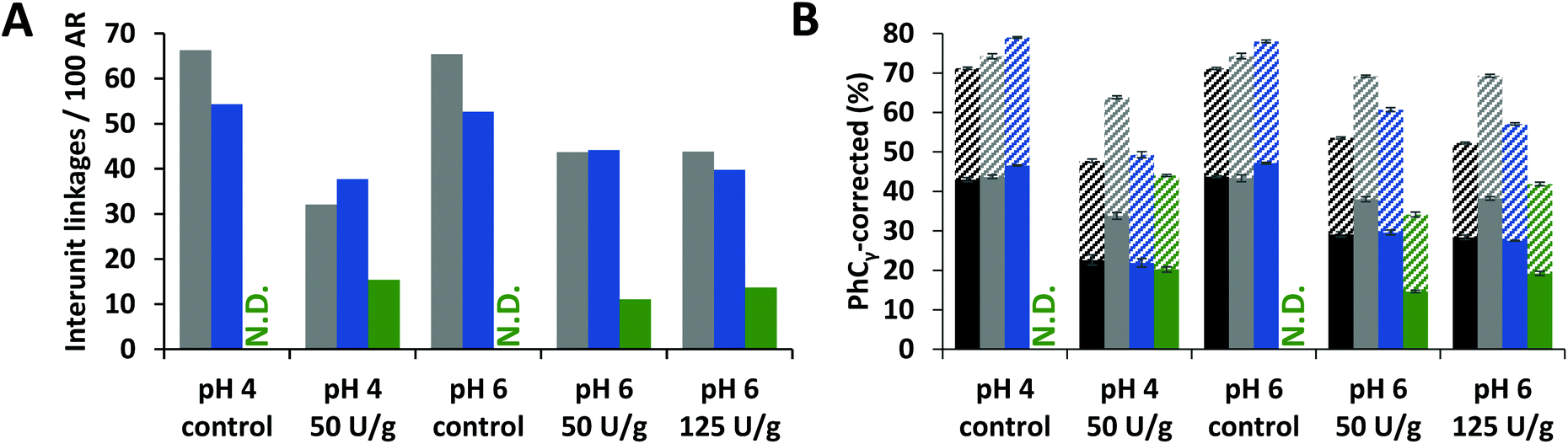 | ||
| Fig. 4 HSQC NMR based (A) and py-GC-MS based (B) estimation of intact interunit linkages in RES (black), CEL (grey), WECELpure (blue) and WELpure (green) fractions of laccase/HBT treated WS and controls. RES was only analyzed by using py-GC-MS. Data corresponding to CS showed similar trends (Fig. S3†). In B, solid and striped bars refer to G and S units, respectively. Error bars in B represent the standard deviation of two treatment duplicates and two analytical duplicates. N.D. = Not determined. | ||
Overall, the results shown in Fig. 3 and 4 indicated that oxidized and degraded substructures accumulated in WELpure (i.e. solubilized lignin) and to a lower extent in WECELpure. Consequently, analysis of only residues, as often performed in literature,28,29,38,49 would heavily underestimate the overall effect of the laccase/HBT treatment on the structure of lignin, especially since a considerable amount of lignin is solubilized upon laccase/HBT treatment (Fig. 2).
Detailed insights into laccase/HBT induced reactions of lignin
To shed more light on the degradation pathways underlying the observed delignification, we zoomed in on the structural features of the most oxidized lignin fractions of laccase/HBT-treated MWS and MCS (i.e. WECELpure and WELpure), and compared those to the structural features of the WECELpure fraction of the corresponding control (Fig. 5). Although the spectra of laccase/HBT fractions showed multiple correlations that could not be annotated (grey correlations in Fig. 5), the vast majority of these correlations also occurred in spectra from the Rovabio enzyme cocktail and the control incubation containing only laccase and HBT (Fig. S5†). Thus, these correlations were mostly unrelated to lignin and were, therefore, not included in further processing of the spectra.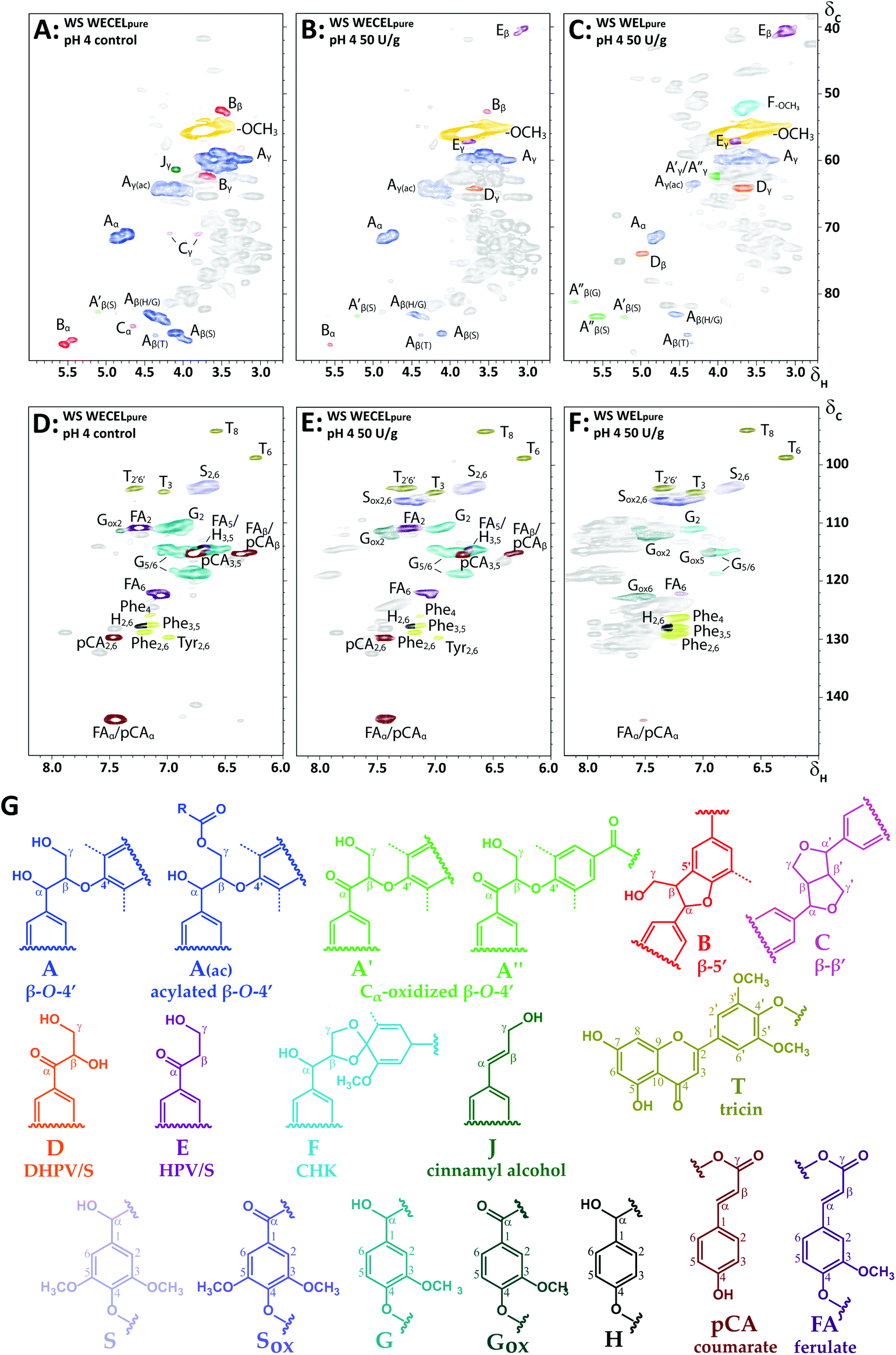 | ||
| Fig. 5 Aliphatic (A–C) and aromatic (D–F) regions of HSQC spectra of (un)treated WS at pH 4: WECELpure of control incubation (A & D), WECELpure of laccase/HBT treatment (B & E) and WELpure of laccase/HBT treatment (C & F). Lignin substructures corresponding to the colored annotations are shown in (G). Dashed lines correspond to –H or –OCH3. Annotations Phe and Tyr correspond to phenylalanine and tyrosine residues, respectively. Grey correlations are unassigned signals, mainly originating from Rovabio Advance and (reaction products of) HBT. Annotation was done based on literature and by comparison with purified degradation products of a lignin model compound (see Fig. S5†).43,56–59 Note that F is a tentative annotation, and that the displayed structure is only one of the possible cyclohexadienone structures (see van Erven et al. for other possible structures).43 A complete list of annotations can be found in Table S3.† HSQC spectra of other incubations (pH 6 and CS) showed similar correlations (data not shown). | ||
From the lignin-related correlations in the aliphatic region it was observed that the decreased abundance of interunit linkages upon laccase/HBT treatment (Fig. 4) was caused by a decrease in β-O-4′ linkages, as well as phenylcoumaran (β-5′) and resinol (β–β′) linkages (Fig. 5A–C). Concurrently, several new C–H correlations appeared, which were annotated based on comparison to HSQC spectra of purified degradation products of a lignin model dimer, and additional HMBC experiments (Fig. S6†). Based on these annotations, it was concluded that three new structural features appeared: (i) Cα-oxidized intact β-O-4′ linkages, (ii) hydroxypropiovanillone/syringone residues (HPV/HPS), and (iii) dihydroxy-propiovanillone/syringone residues (DHPV/S) (Fig. 5). HPV/S and DHPV/S resulted from cleavage of the β-O and O-4′ bonds of β-O-4′ linkages, respectively, and have been reported in studies on LMS-induced degradation of dimeric lignin model compounds.19,23,25 To the best of our knowledge, our research is the first to show evidence for such degradation pathways in a LMS treatment of actual lignocellulosic biomass.
In addition to the new structural features mentioned above, one of the formed correlations (F in Fig. 5) was tentatively annotated as a cyclohexadienone ketal (CHK).43 Such structures have been reported as products from one-electron oxidation of β-O-4′ linked model compounds,50 but could in our study only be tentatively annotated, as no other evidence for CHK formation was found than the HSQC correlation at δC/δH 51.6/3.60.
The aromatic region also showed new C–H correlations after laccase/HBT treatment (Fig. 5D–F). In addition to Cα-oxidized S and G units, several new correlations appeared that showed overlap with HSQC correlations of HBT or its degradation products (Fig. S5†). The presence of HBT-related correlations in WECELpure and WELpure fractions could be explained by the fact that, even after purification, some residual (free) HBT and its degradation product benzotriazole (BT) were present (Table S6†). Nevertheless, as these HSQC correlations were, with lower intensity, also visible in the laccase/HBT treated CEL fractions (Fig. S1†), and because the CEL fractions were extensively washed with water, it indicated that grafting of HBT on WS and CS lignin occurred during incubation, as has been suggested more often in literature.51,52 Corroborating evidence for HBT grafting was found in RP-UHPLC-MS analysis of WECELpure fractions, which revealed the formation of reaction products that were tentatively annotated as lignin-HBT coupling products, based on their molecular formula with 3 nitrogen and ≫6 carbon atoms (Table S6†). Therefore, the grey peaks in Fig. 5E and F, likely, predominantly correspond to a combination of free, degraded and lignin-grafted HBT.
In addition to HSQC NMR, SEC-MALS(IR) was performed on the CEL and WECELpure fractions to investigate the effect of the LMS treatment on the molecular weight distribution. No significant effect was observed in the case of CEL fractions. Remarkably, WECELpure fractions showed a slightly increased molecular weight after LMS treatment (Fig. S4†). This suggests that degradation coincided with (re)polymerization.
As lignin model compound studies have shown that, in addition to ether cleavage products, also benzoic acids could be formed (via Cα–Cβ cleavage) upon LMS treatment,23,25 and such structures would lack diagnostic correlations in HSQC spectra, we compared HMBC spectra of the WECELpure and WELpure fractions. The HMBC spectra indeed showed that, in addition to phenylketones, benzoic acids were formed upon laccase/HBT treatment, and that they were absent in the control sample (Fig. 6). In WELpure a small peak was also observed at δC 192 ppm, that was annotated as an aldehyde based on the presence of a correlation at δC/δH 192/9.8 in the corresponding HSQC spectrum (data not shown).53 Nevertheless, as this HSQC correlation was also observed in WECELpure control samples, and laccase/HBT treatment did not result in more than 1 aldehyde group per 100 aromatic rings, we concluded that aldehyde formation was not a main degradation pathway during LMS treatment. Remarkably, very clear correlations were observed that corresponded to S-ketones and S-acids (δH 7.1–7.3), whereas the intensity of their G-analogues was very low. Possibly, G-units selectively undergo (re)polymerization and grafting, and that, via C–O coupling at C5, this results in aromatic rings with HSQC correlations that overlap with those of S-units.54,55 Nevertheless, this hypothesis does not match with HSQC spectra, which showed formation of both Cα-oxidized G and S units, and py-GC-MS data, which showed that the cleavage markers (i.e. vinyl ketones and diketones)43 are formed from both G and S units (Tables S9–S17†). Therefore, the reason behind this discrepancy remains to be elucidated.
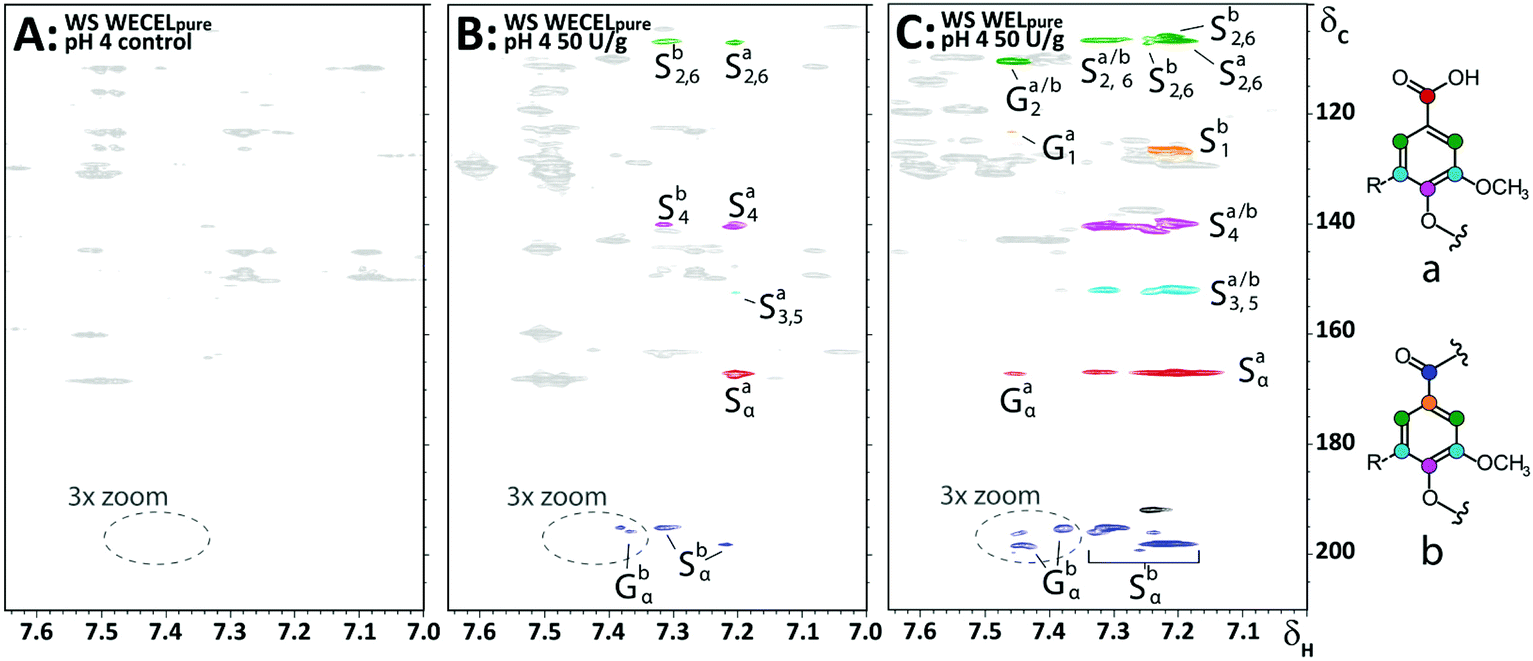 | ||
| Fig. 6 Aromatic regions of 1H–13C HMBC spectra obtained from WECELpure (A & B) and WELpure (C) fractions of WS incubated at pH 4. A = control, B & C = laccase/HBT treated. Annotation was done based on comparison with reported chemical shifts of lignin model compounds.60 The black correlation at δC 192 ppm in C most likely corresponds to an aldehyde, but is only present in small amounts (see main text). Substructures corresponding to the annotated correlations are shown on the right. A full 3× zoomed version of the WELpure fraction, showing more ketone correlations, can be found in Fig. S7.† | ||
Relative susceptibility of lignin substructures to laccase/HBT activity
To get more insights into the relative susceptibility of lignin substructures towards degradation by laccase/HBT, lignin mass-weighted averages of the HSQC integrals of the residual lignin fractions (i.e. CEL and WECELpure) were determined (see Methods section for calculation). Based on these integrals, relative abundances of structural features in the residual lignin were calculated (Table 1). The most apparent observations are discussed below.| Wheat straw | Corn stover | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pH 4 | pH 6 | pH 4 | pH 6 | |||||||
| a Amount of linkages per 100 aromatic units (S + Sox + G + Gox + H). b Numbers between brackets refer to relative abundance. | ||||||||||
| Laccase (U g−1) | 0 | 50 | 0 | 50 | 125 | 0 | 50 | 0 | 50 | 125 |
| Lignin subunits (%) | ||||||||||
| H | 3 | 3 | 3 | 3 | 3 | 3 | 5 | 3 | 4 | 4 |
| G | 64 | 37 | 65 | 51 | 48 | 58 | 40 | 59 | 50 | 49 |
| Gox | 1 | 19 | 1 | 8 | 11 | 5 | 13 | 4 | 7 | 9 |
| S | 31 | 28 | 31 | 31 | 30 | 34 | 32 | 34 | 32 | 32 |
| Sox | 1 | 13 | 1 | 7 | 9 | 1 | 10 | 0 | 7 | 6 |
| S/G | 0.48 | 0.75 | 0.47 | 0.63 | 0.66 | 0.57 | 0.81 | 0.57 | 0.68 | 0.67 |
| Hydroxycinnamates | ||||||||||
| pCA | 10 | 11 | 10 | 11 | 11 | 69 | 98 | 68 | 87 | 86 |
| FA | 26 | 22 | 27 | 19 | 19 | 74 | 28 | 72 | 22 | 26 |
| Flavonoids | ||||||||||
| Tricin | 10 | 10 | 15 | 7 | 6 | 6 | 6 | 8 | 4 | 4 |
| Interunit linkages , | ||||||||||
| β-O-4′ (A) | 52 (82) | 28 (82) | 51 (82) | 42 (85) | 38 (84) | 46 (91) | 27 (92) | 40 (91) | 30 (88) | 23 (88) |
| β-O-4′ox (A′/A′′) | 1 (2) | 4 (12) | 2 (3) | 2 (5) | 2 (4) | 1 (1) | 1 (2) | 1 (1) | 3 (7) | 1 (5) |
| β-5′ (B) | 8 (13) | 2 (6) | 8 (12) | 4 (9) | 5 (10) | 4 (7) | 2 (6) | 3 (8) | 2 (5) | 2 (8) |
| β–β′ (C) | 2 (3) | 0 (0) | 2 (3) | 1 (2) | 1 (2) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Total (100%) | 63 | 35 | 63 | 50 | 46 | 50 | 29 | 44 | 35 | 26 |
| Cleavage products | ||||||||||
| DHPV/DHPS (D) | 0.0 | 0.7 | 0.0 | 0.0 | 0.2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| HPV/HPS (E) | 0.3 | 3.4 | 0.0 | 1.0 | 1.4 | 0.0 | 0.6 | 0.0 | 0.5 | 0.8 |
Firstly, Table 1 shows that, although the overall decrease in intact linkages was mainly caused by degradation of β-O-4′ linkages, the relative abundances of the interunit linkages suggested a preferential degradation of phenylcoumaran and resinol linkages. Although it is unknown whether these linkages are truly degraded or only modified, at least these findings show that not only β-O-4′ linkages, but also β-5′ and β–β′ linkages are targets of the LMS treatment.
Secondly, in all laccase/HBT treated samples the S/G ratio of the residue increased, and this has previously been suggested to result from a preferential removal of G-units.28,29 At first sight, the increased S/G ratio indeed hints at a preferential removal of G-units, and this seems to be confirmed by the results shown in Fig. 4B and Fig. S3B,† which indicates that mainly linkages are degraded that have their Cα atom connected to a G-unit (G-X linkage). Nevertheless, in case of preferential removal of G-units from the residue, an accumulation of G-units (low S/G ratio) is expected in the solubilized lignin. This is clearly not the case, as the S/G ratios in WELpure fractions are all higher than the S/G ratios of the residual lignin (Table S7†). Another explanation for the observed increase in S/G ratio could be a higher degree of side-reactions of G-units (e.g. grafting and repolymerization at the C5 position), after which they may be excluded from data processing. A preferential removal of G-units and degradation of G-X linkages is therefore not proven.
Thirdly, the results shown in Table 1 indicate that pCA moieties accumulate in the residual lignin, in particular in CS. This accumulation is confirmed by an increased abundance of 4-vinylphenol in py-GC-MS analysis of laccase/HBT treated samples (Table S18†).61 Hence, it was concluded that pCA-esterified linkages were more recalcitrant toward degradation by laccase/HBT than other linkages. This may explain the somewhat lower degree of delignification in incubations of MCS (Fig. 2), since the pCA content of CS is much higher than that of WS (Table 1). Extrapolating this observation to other lignocellulosic materials, it could be expected that materials with a low degree of pCA-acylation, such as barley straw and rye straw,62 are better targets for LMS based delignification strategies than highly pCA-acylated materials like sugarcane bagasse.58
Lastly, a remarkable pH effect was observed for the degradation of tricin after laccase/HBT treatment. Whereas the relative abundance of tricin remained unaltered at pH 4, a decrease was observed at pH 6. As no accumulation of tricin was observed in the WELpure fraction, tricin was truly degraded, rather than only solubilized.
In summary, these insights show that the laccase/HBT acts on a broad range of lignin substructures, and that the relative susceptibility is dependent on both the local structure of lignin and the reaction conditions.
Mechanisms underlying grass delignification by laccase/HBT
Overall, our results indicate that the β-O-4′ linkages of MWS and MCS lignin undergo four types of reactions in laccase/HBT treatments: (i) Cα–Cβ cleavage, yielding carboxylic acid residues (route A in Fig. 7); (ii) O-4′ cleavage, yielding DHPV/S residues (route B in Fig. 7); (iii) β-O cleavage, yielding HPV/S residues (route C in Fig. 7); and Cα oxidation of intact linkages (route D in Fig. 7). As HPV/S residues and Cα-oxidized β-O-4′ linkages are formed to a much larger extent than DHPV/S residues (Table 1 and Table S7†), we concluded that Cα-oxidation and β-O cleavage are the major reactions induced by laccase/HBT treatments and that O-4′ cleavage only occurs to a minor extent. A quantitative comparison between these pathways and Cα–Cβ cleavage was not possible, as benzoic acids (formed via Cα–Cβ cleavage) lacked diagnostic HSQC correlations.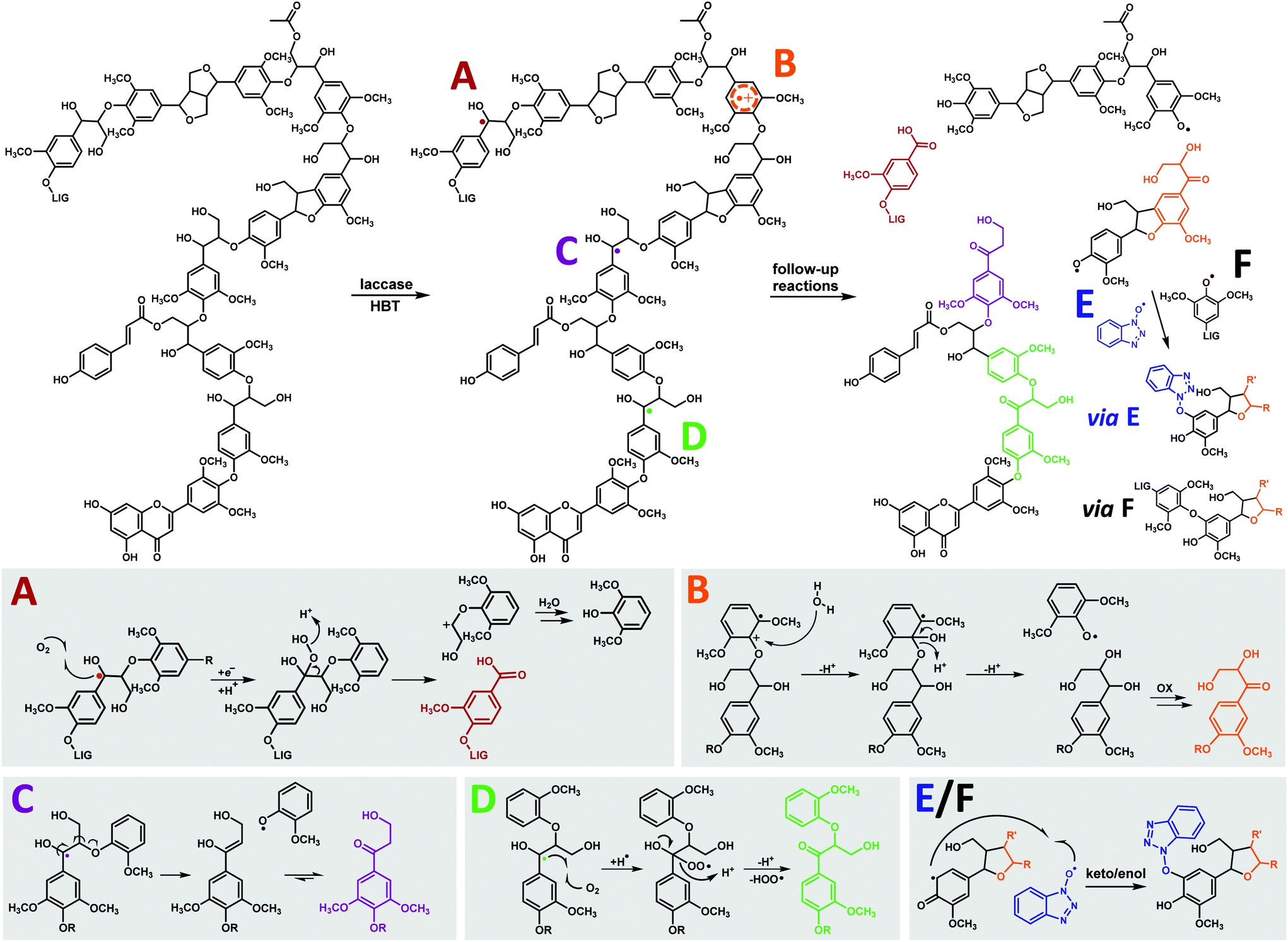 | ||
| Fig. 7 General overview of the observed structural changes upon laccase/HBT treatment of MWS and MCS and suggested corresponding reactions mechanisms, shown on substructures of the lignin structure in the upper panel. A = Cα–Cβ cleavage, B = O-4′ cleavage, C = β-O cleavage, D = Cα oxidation, E = HBT grafting and F = repolymerization. As E and F are expected to follow similar mechanisms, only reaction E is completely shown. Mechanisms are based on model compound studies in literature.18,23,63,64 Degradation pathways of β-5′ and β–β′ linkages are not distinctly shown due to their low abundance. | ||
Lignin oxidation by HBT radicals is generally suggested to occur via hydrogen atom transfer (HAT), which results in the formation of benzylic radicals.14 The formation of carboxylic acids, HPV/S and Cα-oxidized linkages can, indeed, be explained from benzylic radical intermediates (Fig. 7). The formation of DHPV/S, however, is more likely to occur via a radical cation intermediate. The latter is formed via direct electron transfer (ET), rather than HAT. Although the redox potentials of non-phenolic lignin subunits (>1.4 V vs. NHE)14,15 are higher than that of HBT (1.08 V vs. NHE),15 a slow ET between HBT radicals and non-phenolic lignin subunits with relatively low redox potentials seems plausible, and has been suggested more often.23 Since S-units have a lower redox potential than G-units, due to the presence of an extra methoxyl group, it is expected that O-4′ cleavage preferentially occurs in β-O-4′ linkages with a 4′-linked S-unit. Route B in Fig. 7 could, in theory, also occur via one-electron oxidation of Cα-oxidized structures and subsequent cleavage. Since we found that a Cα-oxidized lignin dimer (veratrone) was inert to the laccase/HBT system (data not shown), we consider the mechanism displayed in Fig. 7 more plausible. Considering that we observed that Cα-oxidation and β-O cleavage were more predominant than O-4′ cleavage, our results confirm that HAT is the main oxidation mechanism underlying lignin degradation by laccase/HBT.
Upon cleavage of β-O-4′ linkages, phenoxyl radicals are expected to be formed.23 We suggest that these radicals undergo coupling (grafting) to HBT radicals present in solution. This is further evidenced by model compound studies, in which HBT-coupling was observed with phenoxyl radicals, and not with benzylic radicals.18,19 In addition to HBT grafting, phenoxyl radicals may also undergo radical coupling to other lignin-derived radicals (repolymerization).18,63 Although no NMR based evidence could be obtained for this, SEC results indeed suggested that (re)polymerization of lignin occurred (Fig. S4†).
The insights presented in this study can be used as a starting point for further optimization of lignin degradation by LMS. Our results indicated that the presence of esterified pCA decreases the susceptibility of linkages toward degradation by LMS. Therefore, removal of pCA esters, either during a pretreatment step or simultaneously with LMS treatment, is expected to increase LMS-induced lignin degradation. A green approach for pCA removal could be the addition of feruloyl or coumaroyl esterases.65,66
Conclusions
By fractionation, purification and comprehensive analysis of laccase/HBT treated milled wheat straw (MWS) and milled corn stover (MCS), we were able to elucidate laccase/HBT induced delignification pathways. We have shown that laccase/HBT treatment successfully degrades lignin in MWS and MCS, via both ether cleavage and Cα–Cβ cleavage. For the first time, we showed that β-O-4′ ether cleavage predominantly occurs via cleavage of the β-O bond, rather than the O-4′ bond. Furthermore, our findings suggested that pCA-esterification decreased the susceptibility of linkage degradation in lignin and that HBT grafts onto lignin. These mechanistic insights contribute to the fundamental understanding of enzymatic lignin degradation and can be used to optimize lignin degradation by LMS or comparable treatments in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Pieter de Waard (MAGNEFY, WUR) is greatly acknowledged for performing the NMR analyses. Collaboration was facilitated by COST Action CA17128 “Establishment of a Pan-European Network on the Sustainable Valorisation of Lignin (LignoCOST)”, supported by COST (European Cooperation in Science and Technology).Notes and references
- S. D. Mansfield, C. Mooney and J. N. Saddler, Biotechnol. Prog., 1999, 15, 804–816 CrossRef CAS PubMed
.
- L. P. Christopher, B. Yao and Y. Ji, Front. Energy Res., 2014, 2, 12 Search PubMed
- L. Munk, A. K. Sitarz, D. C. Kalyani, J. D. Mikkelsen and A. S. Meyer, Biotechnol. Adv., 2015, 33, 13–24 CrossRef CAS PubMed
- S. Behera, R. Arora, N. Nandhagopal and S. Kumar, Renewable Sustainable Energy Rev., 2014, 36, 91–106 CrossRef CAS
- G. Van Erven, N. Nayan, A. S. M. Sonnenberg, W. H. Hendriks, J. W. Cone and M. A. Kabel, Biotechnol. Biofuels, 2018, 11, 262 CrossRef PubMed
- J. L. Mamilla, U. Novak, M. Grilc and B. Likozar, Biomass Bioenergy, 2019, 120, 417–425 CrossRef CAS
- D. L. Gall, J. Ralph, T. J. Donohue and D. R. Noguera, Curr. Opin. Biotechnol., 2017, 45, 120–126 CrossRef CAS PubMed
- A. T. Martinez, F. J. Ruiz-Duenas, M. J. Martinez, J. C. del Río and A. Gutierrez, Curr. Opin. Biotechnol., 2009, 20, 348–357 CrossRef CAS PubMed
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Marita, R. D. Hatfield, S. A. Ralph and J. H. Christensen, Phytochem. Rev., 2004, 3, 29–60 CrossRef CAS
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed
- K. Lundquist and J. Parkås, BioResources, 2011, 6, 920–926 CAS
- D. M. Mate and M. Alcalde, Microb. Biotechnol., 2017, 10, 1457–1467 CrossRef CAS PubMed
- P. Baiocco, A. M. Barreca, M. Fabbrini, C. Galli and P. Gentili, Org. Biomol. Chem., 2003, 1, 191–197 RSC
- M. Fabbrini, C. Galli and P. Gentili, J. Mol. Catal. B: Enzym., 2002, 16, 231–240 CrossRef CAS
- F. d'Acunzo and C. Galli, Eur. J. Biochem., 2003, 270, 3634–3640 CrossRef PubMed
- P. Astolfi, P. Brandi, C. Galli, P. Gentili, M. F. Gerini, L. Greci and O. Lanzalunga, New J. Chem., 2005, 29, 1308–1317 RSC
- R. J. Hilgers, J.-P. Vincken, H. Gruppen and M. A. Kabel, ACS Sustainable Chem. Eng., 2018, 6, 2037–2046 CrossRef CAS PubMed
- R. Hilgers, M. Twentyman-Jones, A. van Dam, H. Gruppen, H. Zuilhof, M. A. Kabel and J.-P. Vincken, Catal. Sci. Technol., 2019, 9, 1535–1542 RSC
- S. Kawai, M. Asukai, N. Ohya, K. Okita, T. Ito and H. Ohashi, FEMS Microbiol. Lett., 1999, 170, 51–57 CAS
- S. Kawai, T. Umezawa and T. Higuchi, Arch. Biochem. Biophys., 1988, 262, 99–110 CrossRef CAS PubMed
- S. Kawai, M. Nakagawa and H. Ohashi, FEBS Lett., 1999, 446, 355–358 CrossRef CAS PubMed
- S. Kawai, M. Nakagawa and H. Ohashi, Enzyme Microb. Technol., 2002, 30, 482–489 CrossRef CAS
- A. Potthast, T. Rosenau, H. Koch and K. Fischer, Holzforschung, 1999, 53, 175–180 CAS
- E. Srebotnik and K. E. Hammel, J. Biotechnol., 2000, 81, 179–188 CrossRef CAS PubMed
- R. Bourbonnais, M. Paice, B. Freiermuth, E. Bodie and S. Borneman, Appl. Environ. Microbiol., 1997, 63, 4627–4632 CrossRef CAS PubMed
- A. Rico, J. Rencoret, J. C. Del Río, A. T. Martínez and A. Gutiérrez, BioEnergy Res., 2015, 8, 211–230 CrossRef CAS
- J. Rencoret, A. Pereira, J. C. Del Río, A. T. Martínez and A. Gutiérrez, BioEnergy Res., 2016, 9, 917–930 CrossRef CAS
- A. Rico, J. Rencoret, J. C. Del Río, A. T. Martínez and A. Gutiérrez, Biotechnol. Biofuels, 2014, 7, 6 CrossRef PubMed
- S. Camarero, D. Ibarra, Á. T. Martínez, J. Romero, A. Gutiérrez and J. C. Del Río, Enzyme Microb. Technol., 2007, 40, 1264–1271 CrossRef CAS
- X. Du, J. Li, G. Gellerstedt, J. Rencoret, J. C. Del Río, A. T. Martínez and A. Gutiérrez, Biomacromolecules, 2013, 14, 3073–3080 CrossRef CAS PubMed
- Q. Chen, M. N. Marshall, S. M. Geib, M. Tien and T. L. Richard, Bioresour. Technol., 2012, 117, 186–192 CrossRef CAS PubMed
- Z. Zheng, H. Li, L. Li and W. Shao, Biotechnol. Lett., 2012, 34, 541–547 CrossRef CAS PubMed
- E. D. Babot, A. Rico, J. Rencoret, L. Kalum, H. Lund, J. Romero, J. C. Del Rio, Á. T. Martínez and A. Gutiérrez, Bioresour. Technol., 2011, 102, 6717–6722 CrossRef CAS PubMed
- A. G. Barneto, E. Aracri, G. Andreu and T. Vidal, Bioresour. Technol., 2012, 112, 327–335 CrossRef CAS PubMed
- D. Moldes, M. Díaz, T. Tzanov and T. Vidal, Bioresour. Technol., 2008, 99, 7959–7965 CrossRef CAS PubMed
- D. Moldes, E. Cadena and T. Vidal, Bioresour. Technol., 2010, 101, 6924–6929 CrossRef CAS PubMed
- J. Rencoret, A. Pereira, J. C. Del Río, A. T. Martínez and A. Gutiérrez, ACS Sustainable Chem. Eng., 2017, 5, 7145–7154 CrossRef CAS
- J. Rencoret, A. Pereira, G. Marques, J. C. Del Río, Á. T. Martínez and A. Gutiérrez, Holzforschung, 2018, 73, 45–54 Search PubMed
- J. Gierer, F. Imsgard and I. Noren, Acta Chem. Scand., Ser. B, 1977, 31, 561–572 CrossRef
- T. Rosado, P. Bernardo, K. Koci, A. V. Coelho, M. P. Robalo and L. O. Martins, Bioresour. Technol., 2012, 124, 371–378 CrossRef CAS PubMed
- G. Van Erven, R. De Visser, D. W. H. Merkx, W. Strolenberg, P. De Gijsel, H. Gruppen and M. A. Kabel, Anal. Chem., 2017, 89, 10907–10916 CrossRef CAS PubMed
- G. Van Erven, R. J. Hilgers, P. De Waard, E.-J. Gladbeek, W. J. H. Van Berkel and M. A. Kabel, ACS Sustainable Chem. Eng., 2019, 7, 16757–16764 CrossRef CAS
- G. Van Erven, R. De Visser, P. De Waard, W. J. H. Van Berkel and M. A. Kabel, ACS Sustainable Chem. Eng., 2019, 7, 20070–20076 CrossRef CAS
- E. Jurak, A. M. Punt, W. Arts, M. A. Kabel and H. Gruppen, PLoS One, 2015, 10, e0138909 CrossRef PubMed
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Nat. Protoc., 2012, 7, 1579 CrossRef CAS PubMed
- G. Zinovyev, I. Sulaeva, S. Podzimek, D. Rössner, I. Kilpeläinen, I. Sumerskii, T. Rosenau and A. Potthast, ChemSusChem, 2018, 11, 3259–3268 CrossRef CAS PubMed
- Unpurified WEL fractions were expected to have a very high content of HBT, which is explosive in unhydrated form, and therefore unsuitable for pyrolysis.
- R. Martin-Sampedro, E. A. Capanema, I. Hoeger, J. C. Villar and O. J. Rojas, J. Agric. Food Chem., 2011, 59, 8761–8769 CrossRef CAS PubMed
- S. Kawai, T. Umezawa and T. Higuchi, FEBS Lett., 1987, 210, 61–65 CrossRef CAS
- T. Jestel, S. Roth, D. Heesel, A. Kress, R. Fischer and A. C. Spiess, Biocatal. Biotransform., 2019, 37, 66–76 CrossRef CAS
- L. Munk, A. M. Punt, M. A. Kabel and A. S. Meyer, RSC Adv., 2017, 7, 3358–3368 RSC
- A. F. Martin, Y. Tobimatsu, R. Kusumi, N. Matsumoto, T. Miyamoto, P. Y. Lam, M. Yamamura, T. Koshiba, M. Sakamoto and T. Umezawa, Sci. Rep., 2019, 9, 1–14 CrossRef PubMed
- Y. Li, T. Akiyama, T. Yokoyama and Y. Matsumoto, Biomacromolecules, 2016, 17, 1921–1929 CrossRef CAS PubMed
- F. Yue, F. Lu, S. Ralph and J. Ralph, Biomacromolecules, 2016, 17, 1909–1920 CrossRef CAS PubMed
- J. Zeng, G. L. Helms, X. Gao and S. Chen, J. Agric. Food Chem., 2013, 61, 10848–10857 CrossRef CAS PubMed
- J. C. Del Río, J. Rencoret, P. Prinsen, A. T. Martínez, J. Ralph and A. Gutiérrez, J. Agric. Food Chem., 2012, 60, 5922–5935 CrossRef PubMed
- J. C. Del Río, A. G. Lino, J. L. Colodette, C. F. Lima, A. Gutiérrez, Á. T. Martínez, F. Lu, J. Ralph and J. Rencoret, Biomass Bioenergy, 2015, 81, 322–338 CrossRef
- H. Guo, D. M. Miles-Barrett, A. R. Neal, T. Zhang, C. Li and N. J. Westwood, Chem. Sci., 2018, 9, 702–711 RSC
- S. A. Ralph, J. Ralph and L. Landucci, 2009, available at URL http://www.glbrc.org/databases_and_software/nmrdatabase/.
- In the py-GC-MS analysis, 4-vinylphenol was used as a marker for the abundance of p-coumaric acid. Although also pyrolysis of H-units may contribute to the formation of 4-vinylphenol, this contribution is expected to be minor as the abundance of H-units in the WS and CS fractions was very low (≤5%).
- R. Sun, X. Sun, S. Wang, W. Zhu and X. Wang, Ind. Crops Prod., 2002, 15, 179–188 CrossRef CAS
- B. Ramalingam, B. Sana, J. Seayad, F. Ghadessy and M. Sullivan, RSC Adv., 2017, 7, 11951–11958 RSC
- R. Ten Have and P. J. Teunissen, Chem. Rev., 2001, 101, 3397–3414 CrossRef CAS PubMed
- I. Benoit, D. Navarro, N. Marnet, N. Rakotomanomana, L. Lesage-Meessen, J.-C. Sigoillot, M. Asther and M. Asther, Carbohydr. Res., 2006, 341, 1820–1827 CrossRef CAS PubMed
- W. S. Borneman, L. Ljungdahl, R. Hartley and D. Akin, Appl. Environ. Microbiol., 1991, 57, 2337–2344 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc04341a |
| This journal is © The Royal Society of Chemistry 2020 |