
A practical strategy for construction and regulation of multi-functional triazepinium salts via highly efficient I2-catalyzed cyclization†

Abstract
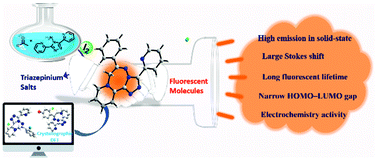
Synthesis of new functional organic molecules is a critical path that may greatly accelerate the evolution of organic optoelectronic materials. We have achieved a facile synthesis strategy to produce unique saddle-shaped multi-functional triazepinium salts that exhibit excellent solid-state fluorescence. Their fluorescence performance could be easily regulated by adjusting the dihedral angle between the main skeleton and the substituted moiety, giving a large Stokes shift, non-aggregation quenching, long lifetime, and multilevel-redox characteristics.
 Please wait while we load your content...
Please wait while we load your content...

