DOI:
10.1039/C9GC04324A
(Communication)
Green Chem., 2020,
22, 673-677
Photochemical evolution of hydrogen peroxide on lignins†
Received
18th December 2019
, Accepted 21st January 2020
First published on 21st January 2020
Abstract
Means of sustainable on-demand hydrogen peroxide production are sought after for numerous industrial, agricultural, and environmental applications. Herein we present the capacity of lignin and lignin sulfonate to behave as photocatalysts that upon irradiation reduce oxygen to hydrogen peroxide. Water-soluble lignin sulfonate acts as a homogeneous photocatalyst in solution, while lignin in thin-film form behaves as a heterogenous photocatalyst. In both cases, the photochemical cycle is closed via the oxidation of electron donors in solution, a process which competes with the autooxidation of lignin. Therefore, lignins can be destructively photooxidized to produce hydrogen peroxide as well as photochemically oxidizing low-oxidation potential species. These findings enable new photochemistry applications with abundant biopolymers and inform the growing body of knowledge on photochemical evolution of hydrogen peroxide.
Introduction
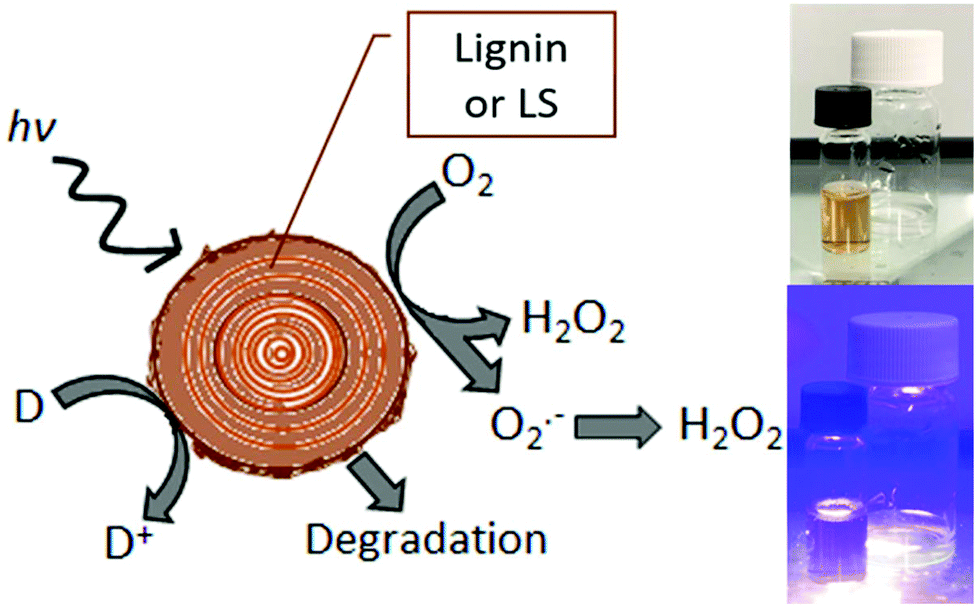
The sustainable production of hydrogen peroxide, H2O2, via reduction of O2 has attracted a great deal of attention in recent years.1,2 This is, in large part, due to the rising use of peroxide as a “green” industrial oxidant, as its only byproducts are oxygen and water.3 For this reason peroxide has been increasingly replacing traditional bleaches such as chlorine-containing reagents.4 Aqueous peroxide solutions also store considerable free energy, and therefore peroxide-based energy conversions schemes have been gaining favor as competitors to hydrogen-based approaches.5,6 In particular, the single-compartment hydrogen peroxide fuel cell has accelerated peroxide to become a chemical fuel with the attractiveness of ease of handling and storage.7,8 For all these applications, sustainable peroxide evolution via photocatalysis,9–11 photoelectrocatalysis,12,13 or direct electrocatalysis14,15 is of great interest. Recently, such catalytic platforms have been applied for on-site generation of peroxide for industrial and agricultural processes.1 These demands motivated us to explore the possibility of peroxide evolution catalyzed by abundant biomaterials. The present study was guided by recent findings that organic carbonyl dyes16 and pigments17 as well as the structurally-related biopolymer eumelanin18 are photocatalysts for selective reduction of oxygen to peroxide. Lignin shares critical structural features with these proven catalytic species – namely aromatic conjugated units with quinone/hydroquinone redox moieties.19,20 These electrochemical properties of lignin have recently been exploited in a number of energy conversion and storage applications such as batteries21 and supercapacitors.22,23 Electrochemical processes are also of major interest for targeted chemical transformations and lignin valorization to useful chemicals.20,24,25 Heterogenous (photo)oxidation of lignins on catalysts is one valorization direction,26,27 and this raises the idea that such processes maybe can be driven by photochemistry alone. The starting question of this work is: Do lignins themselves also afford photo-redox activity, in particular oxygen reduction to hydrogen peroxide? We have tested and confirmed that lignins indeed produce hydrogen peroxide upon irradiation with accompanying autooxidation, or oxidation of available donors in solution (Fig. 1a). Light can therefore be used to degrade lignin with concurrent production of hydrogen peroxide.
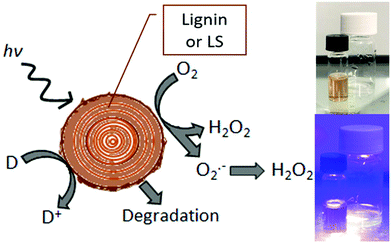 |
| | Fig. 1 Schematic representation of photochemical reduction of dissolved oxygen on lignin or LS (left). Reduction can proceed via a single-electron over the superoxide intermediate or two-electron mechanism. Photogenerated cationic species lead to autooxidation and degradative breakdown of lignin as well as oxidation of donors (D to D+). Photographs on the right show the simple photochemical experiment with lignin deposited on PET and LS in solution, non-irradiated and irradiated with a violet light source. | |
Results and discussion
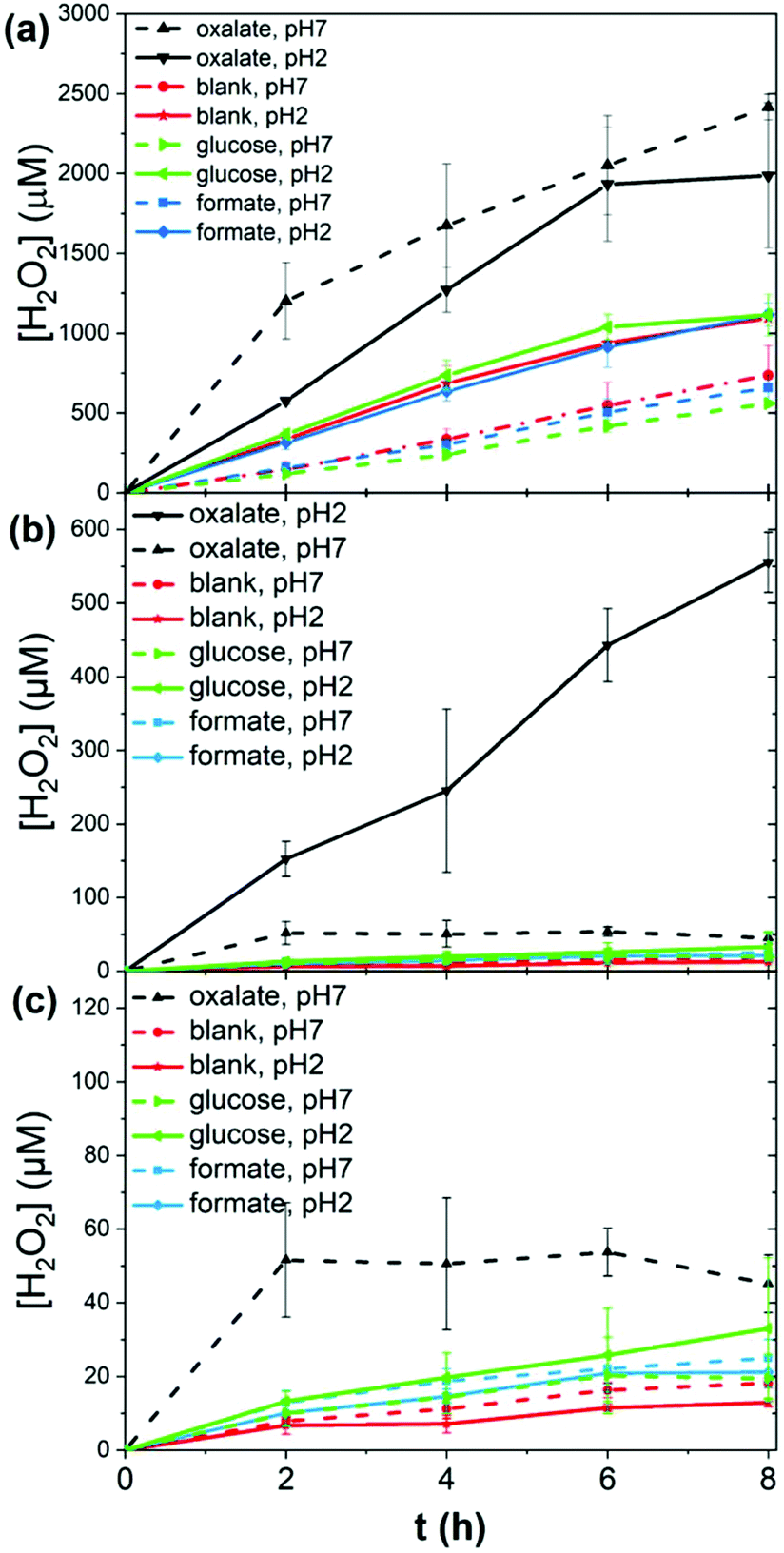
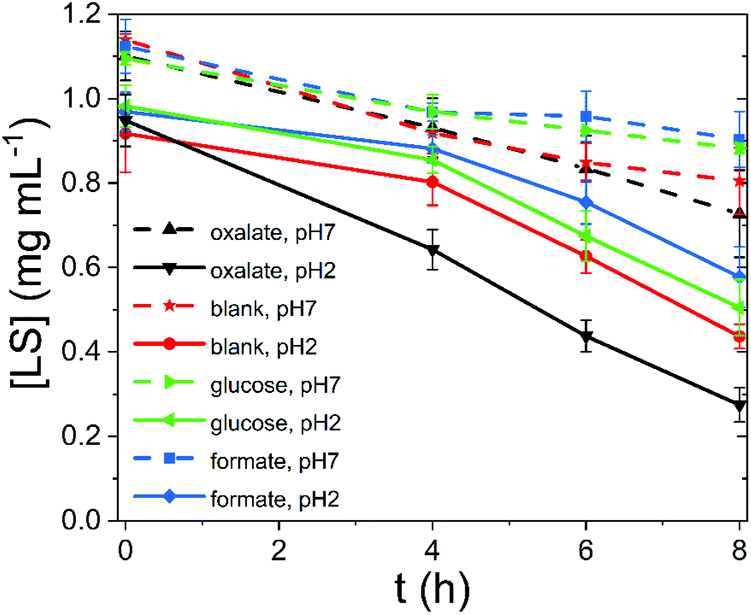
To test the photocatalytic performance of lignin, as a heterogeneous catalyst, and lignin sulfonate (LS), as a homogeneous analog, we employed procedures developed recently by our group.17 In these experiments, we used a given mass of lignin or LS placed in a transparent glass vial together with oxygenated aqueous electrolyte. The pH of the solution was varied from acidic to neutral. Basic conditions were not considered due to known lignin instability. Three different potential electron-donors (compounds which may be sacrificially oxidized) were tested, namely glucose, formate, and oxalate. These were compared always with a “blank” situation with just lignin (or LS) and water. Different concentrations of LS were evaluated. In the case of lignin, solid films were spincoated onto polyethylene terephthalate (PET) plastic foils from a dioxane solution to yield a known quantity of lignin immobilized on the PET carrier. The PET/lignin could conveniently be placed at the bottom of the reactor vial. In the case of LS, aqueous solutions were prepared directly in the vials. The vials with lignin/PET or LS solution were placed onto a violet LED source (λmax = 400 nm, 120 mW cm−2 intensity) and irradiated for up to 8 hours. For determination of the hydrogen peroxide concentration over time, aliquots were removed and the horseradish peroxidase/3,3′,5,5′-tetramethylbenzidine (HRP/TMB) assay was applied to quantify H2O2.12 We found that under all studied conditions, aqueous solutions with lignin or LS produced measurable amounts of hydrogen peroxide upon irradiation. The peroxide concentration over time under different conditions is plotted in Fig. 2. In the case of LS (Fig. 2a), 1–2.4 mM [H2O2] was produced within a few hours of irradiation. We tested peroxide yield as a function of LS concentration and found that a LS loading of 1 mg mL−1 in water was optimal, since it showed the highest LS amount to hydrogen peroxide concentration ratio (ESI, Fig. S1†). Generally, acidic conditions promoted more peroxide evolution than neutral conditions. The addition of oxalate turned out to boost peroxide yield in both acidic and neutral media with a produced amount of peroxide 2000 μM and 2400 μM, respectively. Formate and glucose do not act as efficient donors since similar concentrations of 650 (neutral pH) to 1100 μM (acidic pH) peroxide compared to samples without additional electron donor (blank) were achieved. All show an almost linear increase in peroxide concentration. Qualitatively, lignin performed in the same way as LS, albeit peroxide yields were roughly ten times lower (Fig. 2b and c). Based on our previously published results on peroxide-evolving dyes16 in solution we would maintain that it is most probable that both for lignin and LS the reaction mechanism proceeds via single-electron reduction of oxygen to superoxide, which subsequently disproportionates to form stable hydrogen peroxide. Such a mechanism is kinetically most facile, however a contribution from concerted two-electron/two-proton reduction of oxygen to peroxide cannot be ruled out. In either case, decreasing pH would be expected to enhance peroxide production, which is experimentally confirmed. Higher pH mechanisms could not be tested due to breakdown of lignins at high pH. The question is whether lignins are purely photosubstrates in the reaction, where photochemical reduction of oxygen is accompanied by autooxidation of the material, or if photogenerated cationic species can oxidize different species. Over the course of irradiation, the lignin and LS samples bleached, which could be easily followed spectrophotometrically (Fig. 3, ESI Fig. S2–S10†). Since the addition of electron donors such as oxalate increases markedly the peroxide yield, it may be that autooxidation competes with oxidation of the donor molecules. Typically, the catalytic performance and stability is expressed in terms of a turnover number (TON) as number of moles of product divided by the amount of catalyst which degrades over a given time. Since we cannot assign a precise molecular mass for the lignin samples, we express TON as μmols of H2O2 g−1 consumed lignin or LS (Table 1). The starting and final catalyst mass of LS and lignin were determined via UV-Vis spectroscopy (ESI Fig. S10†). In this way of data evaluation, we can see that addition of electron donors in all cases serves to increase the TON, clearly evidencing that the photooxidative process of electron donors competes with lignin autooxidation. Excepting kinetic considerations, oxalate is the strongest electron donor (thermodynamically), and is by far the most effective in increasing peroxide yield, though the faster rate of overall peroxide evolution leads to more rapid bleaching. For lignin with oxalate, a max TON of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 254 μmol g−1 was achieved, which is comparable to peroxide productivity from synthetic pigments.17 We note that to understand the redox processes in lignin, we attempted cyclic voltammetry measurements, however photocurrents were found to be very low, in the nA cm−2 range, and we could not make any useful conclusions based on this data. The low conductivity and heterogeneity of lignin samples makes photoelectrochemistry difficult.
254 μmol g−1 was achieved, which is comparable to peroxide productivity from synthetic pigments.17 We note that to understand the redox processes in lignin, we attempted cyclic voltammetry measurements, however photocurrents were found to be very low, in the nA cm−2 range, and we could not make any useful conclusions based on this data. The low conductivity and heterogeneity of lignin samples makes photoelectrochemistry difficult.
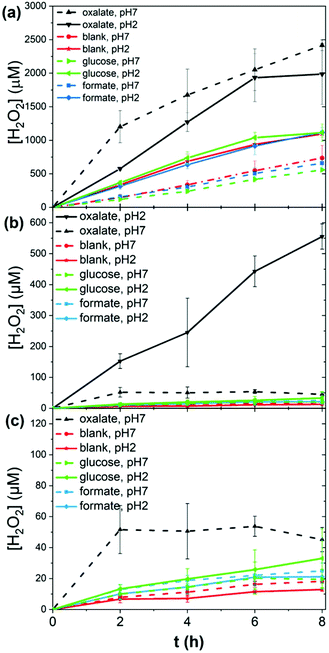 |
| | Fig. 2 Photochemical evolution of peroxide by lignins. (a) 1 mg ml−1 LS solutions irradiated over eight hours, under acidic and neutral conditions, with different added electron donors. (b and c) Peroxide over time measured for spin-coated lignin films under the same set of conditions. (c) shows a magnified view of values below 120 μM [H2O2]. | |
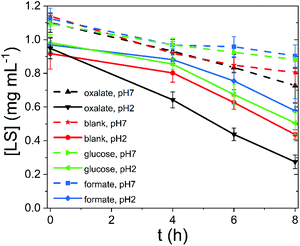 |
| | Fig. 3 LS concentration over irradiation time, calculated spectrophotometrically. | |
Table 1 Calculated “TON” values (i.e. mol H2O2 per g of degraded lignin) for LS and lignin in the various electron-donor/pH solutions after 8 hours of irradiation
| Material |
Units |
Blank, pH2 |
Blank, pH7 |
Oxalate, pH2 |
Oxalate, pH7 |
Formate, pH2 |
Formate, pH7 |
Glucose, pH2 |
Glucose, pH7 |
| LS |
μmol H2O2 g−1 LS |
2280 |
2199 |
2948 |
6466 |
2852 |
2979 |
2043 |
2640 |
| Lignin |
μmol H2O2 g−1 LS |
389 |
548 |
17254 |
1358 |
638 |
750 |
995 |
587 |
In conclusion, we have identified reproducible photochemical evolution of hydrogen peroxide using lignin or lignin sulfonate. The process is primarily a destructive photochemical process, consuming the lignin material, however other substrates can also be photochemically oxidized concurrently. From the point of view of catalytic peroxide evolution, lignins do not appear to be promising candidates for supporting a true catalytic technology. However, in numerous applications the photochemical evolution of peroxide by lignins may be highly advantageous. One potential perspective is using lignin-mediated peroxide evolution as an initiation to some chemical process. Recent reports concerning spontaneous oxidative polymerization inside of living plants implicates reactive oxygen species as the initiator.28 Light stimulation of lignin can be used to such effect intentionally and locally. Based on our results, lignin as a mass-produced material can be photochemically degraded to generate hydrogen peroxide under solar illumination, producing vats of relatively low concentration peroxide which can be used as for chemical initiation, oxidation, or antimicrobial purposes. On the other hand, the photooxidative side of this reaction may be of great interest: considering the extensive application of oxidation of lignin to yield added-value products,26,27 the direct photooxidation byproducts of the photochemical reaction we present here could be of interest. Overall our findings represent a new green chemical approach to peroxide evolution using an abundant biomaterial.
Experimental methods
Preparation of lignin samples
100 μL of 10 mg mL−1 solution of lignin in dioxane:H2O 9:1 were spin coated (1500 rpm, 1 min) on a PET foil (∅ = 22 mm), which was previously cleaned with IPA, detergent, and 18 MΩ water. Afterwards, the samples were dried at room temperature. Next, the samples were placed on the bottom of a 20 mL vial and filled with 2 mL of 10 mM donor aqueous solution (blank, oxalate, formate, glucose) with different pH (2, 7). The vials were closed with a lid with two holes for O2 inlet and outlet.
Preparation of LS solutions
A solution of 10 mg mL−1 LS in water was prepared and sonicated for 10 min. The appropriate amount for reaching the desired concentration of LS in water was pipetted to a 5 mL volume flask, which was equipped with a lid with two holes for O2 inlet and outlet. In total, 2 mL of reaction solution with 1 mg mL−1 LS in water and 10 mM of electron donor (without or blank, oxalate, formate and glucose) with different pH (2 and 7) were prepared. Samples for irradiation and non-irradiation for control comparison were prepared.
Hydrogen peroxide evolution
The closed vials were equipped with steel needles as inlet and outlet and the headspace over the solution was purged with oxygen (120 s, oxygen flow 100 ml min−1). Afterwards the samples were placed on the light source (LED, λmax = 400 nm, 120 mW cm−2 intensity) and illuminated for 8 h. The sample vials were maintained approximately at room temperature over this time due to installed cooling fans.
HRP assay
After every 2 h samples of 50 to 1 μL were taken and mixed with 250 to 299 μL of freshly prepared HRP assay mixture consisting of 993 μL phosphate-citric acid buffer solution, 5 μL 3,3′,5,5′-tetramethylbenzidine (TMB) and 2 μL horseradish peroxidase (HRP). Then the samples with a total volume of 300 μL were spectrophotometrically measured at 653 nm with a Synergy H1 Microplate reader (BioTek® Instruments, Inc.). With a previously determined calibration line the concentration could be calculated based on the measured absorbance. In the case of LS, due to interaction of HRP with LS,29 a specific calibration had to be done. An HRP assay mixture was prepared as mentioned above, except that a certain amount of 1 mg mL−1 LS was added mimicking the dilution concentration of the aliquots taken from the reaction mixture. Afterwards, the respective amount of H2O2 to gain concentrations of 40, 30, 20, 10 and 0 μM was added. With the obtained calibration lines (ESI Fig. S11†), the H2O2 content was accurately calculated. The effects from LS degradation during evolution reaction was not considered for calibration, thus providing a conservative measurement of H2O2 concentration.
Calibration of LS concentration
Solutions with a concentration of 0.2, 0.4, 0.6, 0.8, and 1 mg mL−1 LS in water were prepared. 300 μL of each solution were placed in a plate-reader plate and spectra from 300 to 700 nm with a Synergy H1 Microplate reader (BioTek® Instruments, Inc.) were recorded. For the calibration line the absorbance at 450 nm was chosen.
Calibration of lignin concentration
Solutions with a concentration of 0.1, 0.025, 0.5, 0.075, and 0.1 mg mL−1 lignin in dioxane:H2O 9:1 were prepared. 3 mL of each solution were placed in a QS high precision cell (Quartz Suprasil, 10 × 10 mm light path, Hellma Analytics) and spectra from 300 to 700 nm were recorded with an absorption spectrometer PerkinElmer Lambda 900. For the calibration line, the absorbance at 324 nm was chosen.
UV-Vis spectroscopy – degradation
For LS: Spectra from 300 to 700 nm of the irradiated reaction solution were recorded with a Synergy H1 Microplate reader (BioTek® Instruments, Inc.) at 0, 2, 4, 6 and 8 h. For comparison spectra of non-irradiated (dark) samples were measured in parallel. For Lignin: PET foils with spin coated lignin (used and unused samples) were placed in 3 mL of dioxane:H2O 9:1 and sonicated for 10 min. Afterwards, the PET foils were removed. Each solution was placed in a QS high precision cell (Quartz Suprasil, 10 × 10 mm light path, Hellma Analytics) and spectra from 300 to 700 nm were recorded with an absorption spectrometer PerkinElmer Lambda 900.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors are grateful to the Knut and Alice Wallenberg Foundation, especially to the Wallenberg Wood Science Centre 2.0 and the Wallenberg Centre for Molecular Medicine at Linköping University, for financial support. Support from Vinnova within the framework of treesearch.se is likewise gratefully acknowledged.
References
- S. C. Perry, D. Pangotra, L. Vieira, L.-I. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef CAS.
- S. Fukuzumi, Y. Yamada and K. D. Karlin, Electrochim. Acta, 2012, 82, 493–511 CrossRef CAS PubMed.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed. Engl., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- G. Goor, J. Glenneberg and S. Jacobi, Ullmann's Encycl. Ind. Chem., 2012, 18, 393–427 Search PubMed.
- R. S. Disselkamp, Int. J. Hydrogen Energy, 2010, 35, 1049–1053 CrossRef CAS.
- S. Fukuzumi, Joule, 2017, 1, 689–738 CrossRef CAS.
- S. Yamazaki, Z. Siroma, H. Senoh, T. Ioroi, N. Fujiwara and K. Yasuda, J. Power Sources, 2008, 178, 20–25 CrossRef CAS.
- E. Miglbauer, P. J. Wójcik and E. D. Głowacki, Chem. Commun., 2018, 54, 11873–11876 RSC.
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed. Engl., 2014, 53, 13454–13459 CrossRef CAS PubMed.
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 774–780 CrossRef CAS.
- Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa and T. Hirai, Nat. Mater., 2019, 18–21 Search PubMed.
- M. Jakešová, D. H. Apaydin, M. Sytnyk, K. Oppelt, W. Heiss, N. S. Sariciftci and E. D. Głowacki, Adv. Funct. Mater., 2016, 26, 5248–5254 CrossRef.
- M. Gryszel, A. Markov, M. Vagin and E. D. Głowacki, J. Mater. Chem. A, 2018, 6, 24709–24716 RSC.
- Y. Liu, X. Quan, X. Fan, H. Wang and S. Chen, Angew. Chem., Int. Ed. Engl., 2015, 54, 6837–6841 CrossRef CAS PubMed.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, Nat. Catal., 2018, 1, 156–162 CrossRef CAS.
- M. Gryszel, R. Rybakiewicz and E. D. Głowacki, Adv. Sustainable Syst., 2019, 3, 1900027 CrossRef.
- M. Gryszel, M. Sytnyk, M. Jakesova, G. Romanazzi, R. Gabrielsson, W. Heiss and E. D. Głowacki, ACS Appl. Mater. Interfaces, 2018, 10, 13253–13257 CrossRef CAS PubMed.
- L. Migliaccio, M. Gryszel, V. Đerek, A. Pezzella and E. D. Głowacki, Mater. Horiz., 2018, 5, 984–990 RSC.
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC.
- D. Rochefort, D. Leech and R. Bourbonnais, Green Chem., 2004, 6, 14–24 RSC.
- W. E. Tenhaeff, O. Rios, K. More and M. A. McGuire, Adv. Funct. Mater., 2014, 24, 86–94 CrossRef CAS.
- P. Schlee, S. Herou, R. Jervis, P. R. Shearing, D. J. L. Brett, D. Baker, O. Hosseinaei, P. Tomani, M. M. Murshed, Y. Li, M. J. Mostazo-López, D. Cazorla-Amorós, A. B. Jorge Sobrido and M. M. Titirici, Chem. Sci., 2019, 10, 2980–2988 RSC.
- N. Guo, M. Li, X. Sun, F. Wang and R. Yang, Green Chem., 2017, 19, 2595–2602 RSC.
- Y. Liu, Y. Nie, X. Lu, X. Zhang, H. He, F. Pan, L. Zhou, X. Liu, X. Ji and S. Zhang, Green Chem., 2019, 3499–3535 RSC.
- H. Guo, D. M. Miles-Barrett, B. Zhang, A. Wang, T. Zhang, N. J. Westwood and C. Li, Green Chem., 2019, 21, 803–811 RSC.
- J. C. Colmenares and R. Luque, Chem. Soc. Rev., 2014, 43, 765–778 RSC.
- R. Behling, S. Valange and G. Chatel, Green Chem., 2016, 18, 1839–1854 RSC.
- E. Stavrinidou, R. Gabrielsson, K. P. R. Nilsson, S. Kumar, J. F. Franco-gonzalez, A. V. Volkov, M. P. Jonsson, A. Grimoldi, M. Elgland, I. V. Zozoulenko and D. T. Simon, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2807–2812 CrossRef CAS PubMed.
- D. Yang, Y. Chang, X. Wu, X. Qiu and H. Lou, RSC Adv., 2014, 4, 53855–53863 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Optimization of lignin sulfonate concentration, UV-Vis spectra for lignin and lignin sulfonate over the course of irradiation, optical analysis calibration curves. See DOI: 10.1039/c9gc04324a |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab and
Eric Daniel
Głowacki
ab and
Eric Daniel
Głowacki