
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluating lignin valorization via pyrolysis and vapor-phase hydrodeoxygenation for production of aromatics and alkenes†
Alireza
Saraeian
 ab,
Alvina
Aui
c,
Yu
Gao
d,
Mark M.
Wright
c,
Marcus
Foston
d and
Brent H.
Shanks
*ab
ab,
Alvina
Aui
c,
Yu
Gao
d,
Mark M.
Wright
c,
Marcus
Foston
d and
Brent H.
Shanks
*ab
aDepartment of Chemical and Biological Engineering, Iowa State University, Ames, IA 50011, USA. E-mail: bshanks@iastate.edu; Fax: +01 515 294 2689; Tel: +01 515 294 1895
bNSF Engineering Research Center for Biorenewable Chemicals (CBiRC), Ames, IA 50011, USA
cDepartment of Mechanical Engineering, Iowa State University, Ames, Iowa 50014, USA
dDepartment of Energy, Environmental & Chemical Engineering, Washington University in St. Louis, 1 Brookings Drive, Saint Louis, MO 63130, USA
First published on 27th March 2020
Abstract
Lignin valorization to chemicals is an important component of creating economically viable biofuels production from lignocellulosic biomass. Any such strategy should aim at producing chemicals used at scales that can appropriately match lignin availability. Herein, a combined pyrolysis and low-pressure hydrodeoxygenation (HDO) process configuration is proposed to achieve total oxygen removal and obtain hydrocarbon (aromatic and alkene) products. This approach is tested for its robustness for lignin feedstocks obtained from a variety of sources and extracted using different procedures. The experimental results demonstrate that regardless of the lignin source, the HDO process using a MoO3 catalyst was able to funnel the complex mixture of pyrolysis vapors to mono-aromatics (17–29 C%), as well as alkenes and alkanes. The formation of char from lignin pyrolysis retains more than 50% of the feed carbon in the pyrolyzer, allowing only a portion of carbon to volatilize and be converted to products. A partial depolymerization technique is employed on one of the lignin samples prior to pyrolysis as an example of how the amount of char can be drastically reduced leading to an increased yield of aromatics (53–55 C%). Techno-economic analysis based on the experimental results suggest significant economic benefit of this strategy compared to using lignin as simply a boiler feed.
1. Introduction
Among the three biopolymers constituting lignocellulosic biomass, lignin is the most recalcitrant and underutilized component due to its chemical structure and complexity. It is composed of three primary subunits including p-coumaryl, coniferyl and sinapyl alcohols that are bonded together through aryl ether, diaryl ether, dibenzodioxocin, and phenylcoumaran linkages, among others.1,2 Generally, about 15–40% dry weight of lignocellulosic biomass is made of lignin, depending on the biomass source.3 Lignin has traditionally been produced in large quantities as a by-product of pulp and paper processing where it is primarily used as a cheap fuel for supplying heat for boilers; however, the development of next generation biofuels production will likely require a higher value use than boiler fuel. Being a biopolymer composed of aromatic subunits, lignin is an interesting potential feedstock for producing commodity and/or specialty chemicals.Lignin valorization is commonly suggested as producing value-added oxygenated chemicals such as aromatic aldehydes (e.g. vanillin, syringaldehyde, and p-hydroxybenzaldehyde),4–6 organic acids,7 alkyl-catechols,8 phenolic monomers,9–11 and occasionally fuel-like mixtures comprised of fully hydrogenated hydrocarbons.12 While there is value in investigating possible routes for producing specialty oxygenated chemicals, there are limited market sizes for such compounds. For instance, the global market demand for vanillin (largest volume aroma chemical worldwide5) is around 16 thousand tons per year (2016),13 whereas the supply of lignin from potential biorefineries (225 million tons per year of lignin14) could produce about 11 million tons of vanillin per year assuming a conservative vanillin yield of 5%.15,16 In addition, intensive and expensive separation methods will most likely be required for obtaining pure compounds from a lignin fragment mixture, which poses a significant commercial challenge.17 Alternatively, in the case of fuel products, high pressures and temperatures, as well as the added cost of aromatic ring hydrogenation, would worsen the overall economics of the process.
In this work, a process consisting of thermal deconstruction of lignin followed by low-pressure catalytic hydrodeoxygenation (HDO) of the volatilized species is proposed. This process would funnel product species resulting from lignin deconstruction to a product stream primarily consisting of aromatics, alkenes and alkanes, which would minimize the costs of separation. Ideally, the product stream would consist only of aromatics and alkenes, which would allow for direct blending with petroleum-based products downstream of the cracker (into the coldbox) in a petrochemical complex, where existing technology can separate these chemical compounds. This approach would create better match between the amount of lignin available and its downstream utilization.
An important challenge is the lignin deconstruction process used upstream of the HDO reactor. One such process is fast pyrolysis. However, an important issue still to be resolved is the agglomeration of lignin during fast pyrolysis, which would quickly plug the pyrolysis reactor.18 Although some attempts have been made to mitigate this problem in recent years,19 it remains a significant issue. A second challenge with this deconstruction process is the high yield of pyrolytic char, which reduces the volatized carbon yield, leading to a lower amount of products from lignin feedstocks. Pretreatment of lignin has been proposed as a potential approach for mitigating these issues. Several studies have been undertaken to partially deconstruct lignin streams primarily to phenolic monomers and dimers.20–24 Herein, the potential of coupling a pretreatment step (i.e., partial depolymerization of lignin) with pyrolysis and HDO is examined to determine whether enhanced recovery of carbon from a lignin feedstock could be achieved.
Molybdenum trioxide (MoO3) was first discovered by the group of Román-Leshkov to be a promising candidate for low-pressure HDO of biomass-derived model compounds including some lignin-derived oxygenates.25,26 Our group later demonstrated that MoO3 was able to completely deoxygenate the vapors generated from pyrolysis of cellulose, lignin, and corn stover to produce mainly aromatics and alkanes.27 We have recently shown that by optimizing the catalyst to feed ratio, a product stream consisting of aromatics, alkenes, and alkanes could be obtained from a cellulose feed.28 Several studies have investigated the stability of bulk or supported MoO3 for HDO of lignin-derived compounds and lignin.25,29,30 Zhou et al.29 studied the atmospheric HDO of lignin pyrolysis vapors over MoO3 at different temperatures and H2 partial pressures. The highest deoxygenation was achieved at 450 °C; however, deactivation of MoO3 through over-reduction to MoO2 was observed from post-reaction XRD analysis. Based on the tradeoff between catalytic activity and stability, the authors recommended a continuous oxidative catalytic regeneration cycle if MoO3 is to be implemented as a catalyst for HDO of lignin pyrolysis vapors.
In the current work, several lignin samples are used to test our proposed strategy for converting lignin into petrochemical compatible compounds through sequential fast pyrolysis and HDO reactions. Our goal is to evaluate the robustness of the HDO process for a variety of lignin samples, yielding different oxygenated species after pyrolysis. These experimental results are connected with techno-economic analysis (TEA) studies to examine whether such a process scheme could improve the economics of a biorefinery. Additionally, a lignin pretreatment step is performed on one of the feedstocks, as an example, to evaluate the effect of partial depolymerization of lignin prior to pyrolysis on the yields of solid, liquid, and gas products.
2. Materials and methods
2.1. Lignin samples
Lignin samples were obtained from a variety of sources. Organosolv corn stover lignin (OCL) extracted from corn stover by acetic acid was obtained from Archer Daniel Midland Company and was further washed with 0.1 M nitric acid before use. DuPont lignin (DPL) extracted from corn stover using a proprietary mild ammonia-based extraction method was obtained from DuPont and used with no further treatment. Renmatix lignin (RL) extracted from mixed hardwoods using a two-stage supercritical water extraction method31 was obtained from Renmatix and used without any treatment. Organosolv poplar lignin (OPL) was obtained by extraction from poplar wood chips using methanol and concentrated HCl according to a method described previously.21 All lignin samples were crushed and sieved to a size range of 38–106 μm.OPL oils were obtained using a method described previously.21 Briefly, a stainless steel bomb reactor with an internal volume of ∼10 mL was charged with 100 mg of lignin and 100 mg of a copper-doped porous metal oxide (CuPMO) catalyst (synthesis method described elsewhere).32 Methanol (3 mL) and n-decane (1.76 μL) were added as the solvent and internal standard, respectively. The reactors were heated in an isothermal muffle furnace (Thermo Fisher Scientific) at 300 °C for reaction times of 3 and 6 h (OPL-3 h and OPL-6 h) and then quenched in an ice water bath. The contents were washed with methanol and the combined liquids filtered using a 10 mL syringe fitted with a 0.2 μm nylon membrane filter. The collected liquid phase was placed under vacuum and the methanol removed via rotary evaporation to collect the oils.
All samples were characterized by thermal gravimetric analysis (TGA) and elemental analysis (CHNS/O). Proximate analysis was performed using a Mettler Toledo TG/DSC1. Approximately 30 mg of each sample was loaded in an alumina crucible, which was then placed on a platinum plate attached to a microbalance inside the instrument. The furnace was moved over the sample and the modified ASTM method described previously was used.33 Moisture was determined by a temperature ramp from 40 °C to 105 °C at a rate of 10 °C min−1 under flowing nitrogen (100 mL min−1) and holding at 105 °C for 40 min. Using the same nitrogen flow rate, the temperature was ramped from 120 °C to 900 °C at a rate of 10 °C min−1 and held at 900 °C for 20 min to determine the volatile carbon. Finally, the fixed carbon was determined by switching the atmosphere to air (100 mL min−1) while the temperature was held at 900 °C for 30 min. The remaining mass measured was considered ash content. Ultimate analysis was conducted using an Elementar vario MICRO cube elemental analyzer in CHNS mode. About 4–5 mg of each sample along with 4–5 mg tungsten oxide (to improve heat transfer) were loaded into flexible nickel boats. The boats were folded to avoid sample spills and were dropped into a combustion tube held at 1050 °C. CO2, H2O and N2 were measured with a thermal conductivity detector (TCD) and SO2 was measured using an infrared detector. C, H, N and S content of the samples were determined based on the dry, ash-free mass of each sample, whereas oxygen content was calculated by the difference from 100%. Data from ultimate and proximate analyses of the lignin and oil samples are shown in Table 1.
| Proximate (wt%) | Ultimatea (wt%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Moistureb | Volatiles | Fixed carbon | Ash | C | H | N | S | Oc | |
| a Dry ash-free basis. b OPL oil samples weight loss started at low temperatures due to their volatility, thus water could not be determined (see Fig. S1 vs. Fig. S2†). c Oxygen was calculated by difference. | |||||||||
| DPL | 4.7 | 61.1 | 20.1 | 14.1 | 56.81 | 4.61 | 4.39 | 1.21 | 32.98 |
| RL | 2.4 | 67.6 | 29.4 | 0.6 | 62.59 | 4.99 | 0.36 | 0.02 | 32.04 |
| OCL | 1.8 | 63.1 | 34.6 | 0.6 | 64.23 | 4.77 | 1.90 | 0.21 | 28.89 |
| OPL | 1.1 | 70.9 | 27.2 | 0.8 | 61.26 | 5.40 | 0.29 | 0.05 | 33.00 |
| OPL-3 h | — | 88.0 | 6.8 | 5.3 | 68.13 | 6.57 | 0.30 | 0.01 | 24.99 |
| OPL-6 h | — | 98.5 | 0 | 1.5 | 67.91 | 7.33 | 0.27 | 0.01 | 24.48 |
2.2. Pyrolysis and HDO reactions
The reactions were performed in a single-shot tandem micro-pyrolyzer/reactor system (RX-3050TR, Frontier Labs, Japan) described previously (Fig. S3†).27,28 The samples were added into stainless steel cups and loaded into the tandem system. A sample mass of ∼500 μg was used for the reaction studies, whereas for char measurements 600–800 μg lignin samples were used for higher accuracy. At the start of a run, the sample cup was dropped into the pyrolyzer and the pyrolysis vapors were swept by 120 sccm H2 into the HDO reactor. The products were then swept into the GC for analysis. For all runs in this study, the pyrolyzer and the reactor were kept at 500 and 400 °C, respectively. All interfaces including the GC inlet were kept at 300 °C to minimize condensation and repolymerization of reactive species. In the case of non-catalytic pyrolysis, the second reactor was also kept at 300 °C to avoid reactions occurring on the inner wall of the stainless steel tubing. Additionally, the reactor tube was deactivated using SilcoNert® 1000 technology provided by SilicoTek® Corporation.MoO3 (Sigma, ACS reagent, ≥99.5%) was pelletized, calcined in a muffle furnace at 550 °C for 4 h, crushed, sieved to a size fraction of 38–106 μm, and stored in a desiccator until further use. The MoO3 catalyst (5 or 10 mg) was mixed with ∼60 mg acid-washed glass beads (Sigma, 150–212 μm), fixed between two layers of quartz wool (Micromeritics), and reduced at 400 °C for 1 h in the reactor prior to reaction. The glass beads and quartz wool were tested and found to be completely inactive for HDO under the reaction conditions.
2.3. Product identification and quantification
The pyrolysis or HDO products were analyzed using a gas chromatogram (GC – Agilent 7890B) coupled with a TCD, a flame ionization detector (FID), and a mass spectrometer (MS – Agilent 5977A). The GC oven was programmed to initiate at 30 °C and hold for 7 min and then ramp to 300 °C at 10 °C min−1. Non-condensable species and hydrocarbons were separated using a GasPro column (Agilent GS-GasPro) and quantified by TCD. Condensable species were separated using two medium polarity columns (Agilent VF-1701 MS) and identified and quantified by MS and FID, respectively. The TCD and FID were calibrated for a variety of compounds, a list of which can be found in the ESI (Table S1†). All calibration curves were attained using 5 calibration standards with R2 higher than 0.99. For compounds not available for calibration, standard compounds with the most similar chemical structure were used instead. Results are reported as an average of at least three measurements.The product yields were reported as:34

2.4. Techno-economic analysis
The techno-economic analysis was based on a 2000 dry metric tonne per day (MTPD) of corn stover in an ethanol and chemicals biorefinery. Ethanol was generated from biochemical upgrading of the corn stover cellulose and hemicellulose fractions, and the chemicals derived from the thermochemical conversion of lignin. The biochemical conversion was based on work by Humbird et al.38 The thermochemical conversion built upon previous work by Bbosa et al. for the upgrading of lignin to chemicals.39 In this analysis, vapor-phase products from lignin fast pyrolysis were catalytically upgraded into aromatics and other hydrocarbons. The aromatics were valued based on bulk market prices, and the minimum ethanol-selling price (MESP) was estimated using a discounted cash flow rate of return analysis.The lignin fast pyrolysis and vapor-phase catalytic upgrading process was modeled using Aspen Plus 10™ to obtain mass and energy balances for the unit operations. The process model consisted of a hierarchy unit (A1000) as shown in Fig. S4.†Fig. 1 shows the main unit operations within the fast pyrolysis and vapor-phase upgrading hierarchy. Lignin recovered from the biochemical process was diverted from the steam generation unit to the fast pyrolysis process. The pyrolysis vapors were catalytically upgraded into hydrocarbons. The catalytic upgrading process employed hydrogen from a natural gas steam methane reformer (SMR). Low molecular weight gases from the upgrading unit consisting of CO, CH4, and C2–C6 alkanes were recycled to the SMR to reduce natural gas demand. The ex situ catalytic upgrading unit also included a combustor as the continuous catalyst regenerator. Complete configurations of the process are illustrated in a simplified block diagram in Fig. S5.† Pyrolysis char was recovered from the cyclone and combusted in the biorefinery steam generation unit to provide process heat for the system.
 | ||
| Fig. 1 Simplified process block diagram of lignin pyrolysis and catalytic vapor-phase upgrading (A1000). | ||
Process equipment was selected and sized using the operating conditions, and mass and energy balances of the unit operations. Equipment costs for most units were gathered from Aspen Process Economic Analyzer. Costs of custom engineering equipment such as the fast pyrolysis and ex situ catalytic upgrading unit were based on the work of Dutta et al.40 Installation factors were also based on the same study with an overall LANG factor of 4.1.40
The integrated biorefinery had a project lifetime of 30 years with an annual operating capacity of 96% equivalent to 8410 hours per year. The economic feasibility of this process was assessed using a multi-year discounted cash flow rate of return analysis. Key financial assumptions included 40% equity with a 10 year loan at 8% interest. A double-decline balance (DDB) depreciation method was applied to the general plant (7 years) and steam generation unit (10 years). The plant salvage value was set at $0. The facility construction period was 3 years, and its startup time was 0.25 years. During the startup period, the facility incurred 100% and 75% of fixed and variable costs, respectively; and generated 50% of its nameplate capacity revenue. The income tax rate was 35%. The objective used for the calculation was the minimum ethanol-selling price at an internal rate of return of 10%. All costs presented in this study were on a 2007$ basis to provide a direct comparison to Humbird's enzymatic hydrolysis study.38 This plant was assumed to have an “n-th” plant design. A summary of the financial assumptions used for such plants are given in Table 2. Other assumptions and limitations to the TEA model from having a state-of-the-art process is described in the ESI (List S1†).
| Parameter | Assumptions |
|---|---|
| Equity | 40% |
| Plant life | 20 years |
| Construction period | 3 years |
| Depreciation period | 7/10 years, 200 DDB |
| Working capital | 5% of FCI |
| Plant salvage value | 0 |
| Start-up time | 0.25 years |
| Revenue & cost during startup (% of normal) | Revenue: 50% |
| Variable costs: 75% | |
| Fixed costs: 100% | |
| Construction costs | Year 1: 32% |
| Year 2: 60% | |
| Year 3: 8% | |
| Interest rate for financing | 8% annually |
| Internal rate of return | 10% |
| Income tax rate | 35% |
The primary operating cost assumptions are tabulated in Table 3. These assumptions were employed in estimating the variable operating costs such as feedstock, by-product and waste disposal. There have been many estimates of plant gate feedstock costs. The feedstock cost was estimated by NREL to be $30 MT−1 for corn stover,41 which was then updated to $46 MT−1 by Foust et al.42 Oak Ridge National Laboratory assumed feedstock costs to be from $38 MT−1 to $55 MT−1 of stover43 and Pacific Northwest National Lab employed feedstock costs for wood chips of $60 MT−1.44 Hence, we assumed the feedstock cost to be $58.50 MT−1 in this study, which was within the range provided by previous studies and to provide a direct comparison to the work by Humbird et al.38 Other raw materials included natural gas and aromatics. The cost of aromatics varies from $905 MT−1 to $1800 MT−1 [https://www.alibaba.com/showroom/chemicals-price-list.html, accessed 2019-08-28]. For this work, the assumed price of aromatics was $1335 MT−1 based on a previous study.45 A sensitivity analysis was employed to evaluate the impact of the wide range in aromatics pricing to the MESP. The cost of olefins was estimated based on the price of ethylene on Alibaba ranging from $300–1000 MT−1 [https://www.alibaba.com/showroom/ethylene-price.html, accessed 2019-08-28]. In this study, the average cost of olefins was assumed to be $650 MT−1 as higher value olefins were also included in the output. The cost of natural gas was assumed to be $5 MMBtu−1, which was based on an updated study by NREL.46 Other operating costs such as cost of corn steep liquor, diammonium phosphate, sulfuric acid and more were assumed similarly as those in Humbird's study.38 Fixed variable costs consisted of annual salaries, maintenance costs, taxes and insurance. The annual wages were estimated based on the number of workers and their salary rates from publicly available information in the NREL report.38 Additionally, overhead costs, insurance and maintenance costs were based on NREL's assumptions of 90% of the total labor cost, 0.7% of fixed capital investments and 3% of inside battery limits costs.38
Seven scenarios were compared for this analysis: power, DPL, RL, OCL, OPL, OPL-3 h, and OPL-6 h. In the power scenario, all the process lignin was combusted for heat and power generation. In the case of DPL, RL, OCL, and OPL scenarios, lignin recovered from lignocellulosic biomass via various methods was upgraded into chemicals. In OPL-3 h and OPL-6 h scenarios, lignin was initially pretreated into an oil-like liquid prior to fast pyrolysis. No capital or operating costs associated with the partial depolymerization process were considered in the techno-economic model, which means that the MESPs reported for these two samples were based on the same costs for the pyrolysis/HDO sections similar to all lignin cases. Therefore, the difference between these two scenarios and the OPL (parent lignin) case can be considered as a potential margin for guiding future efforts to create cost effective pretreatment processes.
3. Results and discussion
3.1. Lignin pyrolysis and HDO
As shown in Fig. 2, lignin pyrolysis produced considerable amounts of char (>51 C%) regardless of the feedstock source and extraction procedure. As discussed previously, this result was found to be due to the formation of reactive species through cleavage of ether bonds in the lignin structure followed by coupling reactions to form polymerization products, under pyrolysis conditions.47 Among the lignin samples, OPL had the lowest char yield; however, the char still represented over 51 C% of the pyrolysis products, allowing only 48% of the carbon to be volatilized. Other lignin samples yielded higher char amounts with DPL being the highest at 58.3 C%. The higher yield of char obtained from DPL pyrolysis could be due to its high ash content (ca. 14%) as determined by TGA (Table 1). Alkali and alkaline earth metals present in the ash fraction have been reported to increase char formation during pyrolysis of lignocellulosic biomass.48,49 Detailed yields of the pyrolysis products and the HDO products are presented in Tables S2–S5† for all four lignin samples. | ||
| Fig. 2 Carbon yields from fast pyrolysis at 500 °C and for pyrolysis followed by HDO at 400 °C over 5 mg MoO3 (average of four injections). | ||
The major products from fast pyrolysis of the lignin samples were expectedly oxygenates, mainly including phenolic monomers such as phenol, guaiacol, syringol, and their alkylated forms. These compounds were previously found to be produced from thermal deconstruction of various lignin samples.18,50–52 Other phenolic oxygenates included trimethoxybenzenes, coniferyl aldehyde, sinapaldehyde, and hydroxylated and methoxylated benzaldehydes (e.g., syringaldehyde and vanillin), acetophenones (e.g., 3′,5′-dimethoxyacetophenone and acetosyringone), and benzoic acids (e.g., 4-hydroxybenzoic acid). Non-phenolic oxygenates, represented by red bars in Fig. 2, included different compounds depending on the lignin sample. In the case of DPL, they were a variety of C1–C6 oxygenates such as alcohols, acids, aldehydes, ketones, esters, and furans summing to ∼4.6 C%. Observing these species in the pyrolysis vapors indicated the presence of carbohydrate residues after ammonia extraction of corn stover. Pyrolysis of RL yielded about 10.5 C% non-phenolic oxygenates with over 70% selectivity to levoglucosan among non-phenolics (ca. 7.4 C% yield). This six-carbon anhydrosugar is known to be produced exclusively as a primary product of cellulose pyrolysis,50,53,54 clearly suggesting the presence of a considerable cellulose fraction in the RL sample. Non-phenolics from OCL pyrolysis were about 3.8 C%, mainly containing acetic acid, which could be due to the extraction process of corn stover lignin by acetic acid in these samples. Finally, from OPL pyrolysis, methanol was produced almost exclusively with 96% selectivity among the non-phenolic oxygenates. The rest of the compounds in this category only composed ∼0.2 C% clearly indicating the absence of carbohydrate residues in this sample.
Despite the numerous oxygenated species in the complex mixture of pyrolysis vapors and regardless of the differences in variety and abundance of these species from the four lignin samples, MoO3 was able to funnel the complex mixture into a product stream mainly composed of aromatics, alkenes, and alkanes (see Fig. 2 and Tables S2–S5†). A carbon balance of over 90% was achieved for all samples. In addition to coke and unidentified products, the missing carbon was most likely due to condensation of reactive species on the interfaces in the experimental system where temperature fell below 400 °C as evidenced from a photo of the pyrolysis quartz tube after ∼60 lignin pyrolysis shots (Fig. S6a†).
It should be noted that when the catalyst was placed in the second reactor, oxygenated compounds would deoxygenate fully and thus would not react further downstream of the catalyst bed. However, when no catalyst was used in the second reactor, oligomerization reactions could take place from the bottom of the pyrolyzer all the way to the GC inlet, which were all at 300 °C. Oligomerization of reactive species after rapid condensation of pyrolysis vapors of a corn stover lignin (and even some phenolic monomers) has been evidenced in a micropyrolyzer similar to the one used in this work.55 This could partly explain the reason for lower carbon yields for lignin pyrolysis experiments compared to the HDO tests since phenolic oligomers cannot be detected and quantified in the GC. Moreover, these oligomers could stick to the inner surface of tubing walls as verified by the brown/black residues in the GC inlet liner, the first 5 cm of the guard column, and the waste after cleaning the GC inlet with ethanol and acetone as shown in Fig. S6.† There were also numerous minor peaks in the GC spectra from pyrolysis of lignin samples that could not be identified using the MS system because they were either too small or overlapped with other compounds (Fig. S7–S10†). It is reported that over 500 compounds were identified from pyrolysis of lignin using a photoionization high resolution mass spectrometry technique (although after condensation),55 whereas in this study the relatively high overall carbon balance was based on only about 80 compounds that were identified and quantified. In contrast, after HDO fewer peaks/compounds were observed on FID chromatograms, making it possible to identify and quantify a large amount of the final products (Fig. S7–S10†).
Of the carbon volatized in the pyrolyzer, a significant portion was converted to aromatic hydrocarbons in the second reactor. DPL, RL, OCL, and OPL, respectively, yielded 16.9, 19.1, 22.6, and 29.4 C% aromatics with a selectivity of 81–91% to benzene, toluene, ethylbenzene, and xylenes (BTEX). Nine-carbon aromatics accounted for most of the rest of the carbon in aromatics with a selectivity of 8–14%. Interestingly, though RL had the lowest yield of phenolics in its pyrolysis vapors the yield of aromatics after HDO was comparable to the other lignin samples. This result could have been due to the aromatization activity of MoO3 with oxygenated molecules as observed in the pyrolysis and HDO of cellulose over MoO3.28
DPL had the highest yield of alkene/alkane hydrocarbons at 13.3 C% and OPL had the lowest yield at 3.4 C%. The selectivity to alkenes among non-aromatic hydrocarbons was in the range of 15–33% depending on the feedstock. Alkene species are desired over alkanes as their production would consume less hydrogen during HDO. In addition, alkenes are more valuable since they can be used after separation without the need for further processing.56 The annual USA production amount of ethylene and propylene are about 24 and 11 million tonne per year, respectively. C4 alkenes are produced at about 3 million tonne per year, whereas the market for C5 and C6 alkenes is smaller, although their market could be expanded through making alkylate. More importantly, the price of all of these molecules is >$800 per tonne, which is greater than two times that of fuels. The sum of hydrocarbons was comparable across all four samples from 28.3 C% (RL) to 32.8 C% (OPL).
In addition to non-phenolic oxygenates, nitrogen containing compounds were observed from pyrolysis of DPL and OCL in agreement with elemental analysis results showing 3.56 wt% and 1.85 wt% nitrogen (dry ash-free basis) for these samples, respectively. MoO3 was found to be an effective catalyst for removing nitrogen under the reaction conditions since no nitrogen containing compounds were detected after upgrading. No sulfur containing compounds were identified after pyrolysis of lignin samples even in the case of DPL with 1.2 wt% sulfur. This is likely due to overshadowing any peaks for sulfur containing compounds with large peaks corresponding to a variety of oxygenated species. Unfortunately, our experimental setup did not allow for oil collection and its further characterization via elemental analysis.
CO and CO2 contributed a measurable portion of the pyrolysis products (i.e., 2.9–3.3 C% in sum) for different lignin samples due to decarboxylation and decarbonylation reactions, whereas methane was sufficiently low as to not be distinguishable from CO in the TCD chromatogram. Decarboxylation is generally preferred over decarbonylation because it removes two oxygen atoms with each atom of carbon leading to higher carbon retention in the useful products. Comparing the yields of CO and CO2 before and after upgrading over MoO3 catalyst suggested that decarboxylation was not significantly catalyzed by MoO3, whereas decarbonylation was enhanced. CO2 yields of about 0.6–2 C% were obtained from pyrolysis of the four lignin samples, while these yields only increased slightly to 0.7–2.4 C% after HDO. Furthermore, methane was produced in relatively large quantities (ca. 3–9 C%) after HDO indicating methanation of CO and CO2, and cracking reactions, in addition to conversion of methanol to methane.
Although optimization of the pyrolysis conditions was not the aim of this study, it was previously shown that 500 °C was the optimal temperature for maximizing liquid products from an organosolv lignin.57 Additionally, it was found that at this temperature the yield of methoxylated phenols decreased simultaneous with an increase in alkylated phenols, which could explain the production of methanol from all lignin samples in this study.57 Note that dehydration is also a major reaction that takes place under pyrolysis conditions, but since carbon atoms were tracked from the feedstock to products, it was not necessary to quantify water produced during pyrolysis.34
3.2. Pyrolysis and HDO of pretreated lignin
OPL-3 h and OPL-6 h samples were merely used as two examples to evaluate the influence of lignin partial depolymerization before pyrolysis and HDO on the yield of final products. Note that we are not claiming OPL as an extraordinary sample nor are we advocating this specific lignin pretreatment technique. Fig. 3 compares the pyrolysis results of OPL and pretreated OPL oils, and the deoxygenated products after HDO over the MoO3 catalyst. The pretreatment step successfully reduced the amount of pyrolytic char from ∼51 C% to ∼18 C% for both the 3 h and 6 h treated oil samples. The use of CuPMO catalyst in supercritical methanol (sc-MeOH) for hydrogenolysis of α-O-4 and β-O-4 aryl ether linkages and depolymerization of lignin and other biomass components has been extensively studied.21,58–63 These studies showed that the reactive intermediates formed during lignin solvolysis were stabilized and further depolymerized and altered through hydrogenation, hydrodeoxygenation, and methylation reactions in the presence of sc-MeOH and the CuPMO catalyst, preventing repolymerization into char and leading to essentially no char formation.21,61,62 Consequently, most of the ether bonds in the lignin structure were cleaved before pyrolysis, which would reduce the formation of reactive species that could potentially repolymerize and form char. Although previous studies of lignin depolymerization with CuPMO showed that nearly all lignin was converted into an oily product, that product included some proportion of oligomeric species.59 The presence of recalcitrant lignin oligomers remaining after treatment with CuPMO could partly explain the formation of 18 C% char during pyrolysis. The formation of this char could also be attributed to dehydration of phenolic alcohols to unsaturated species that were reactive enough for repolymerization.55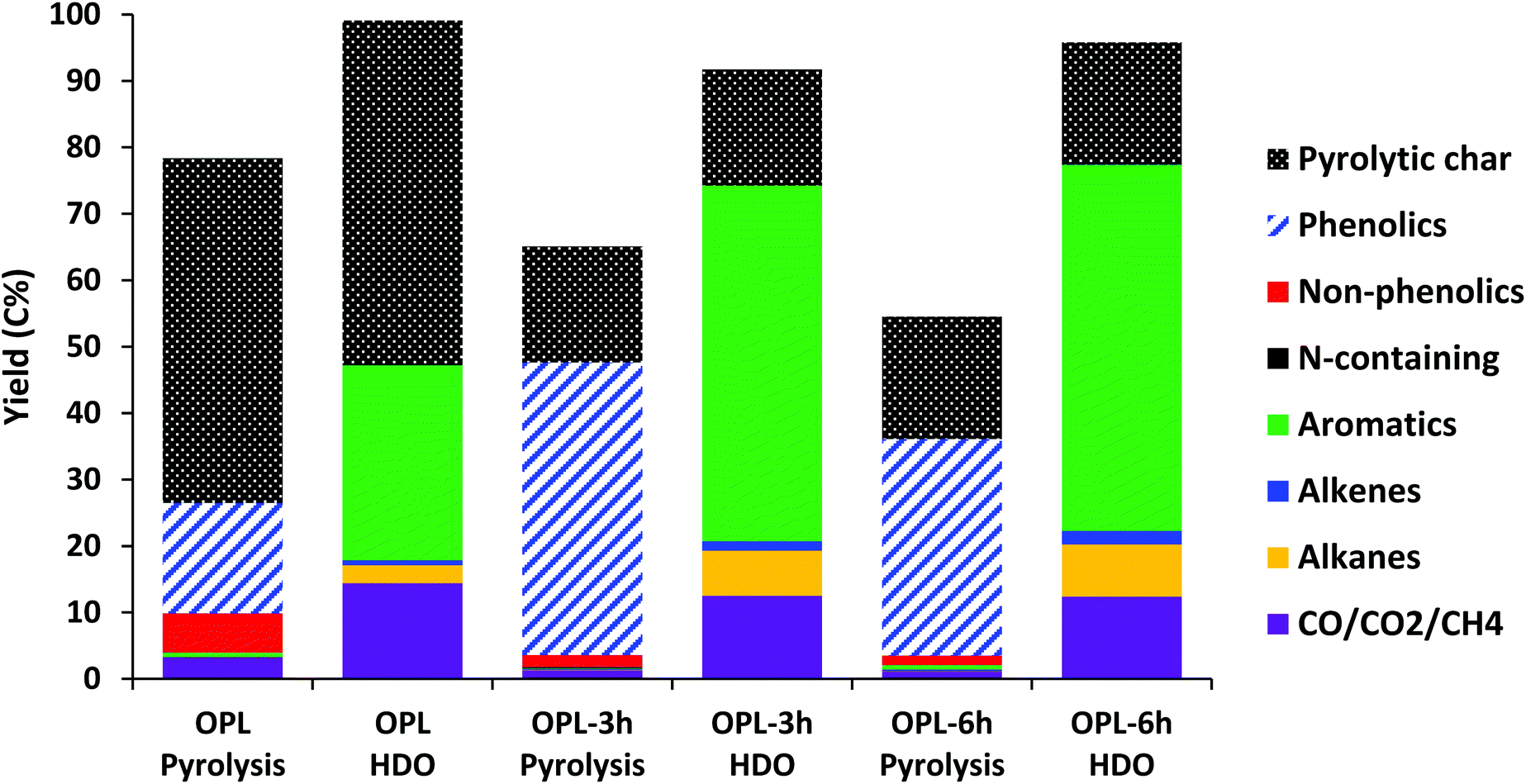 | ||
| Fig. 3 Carbon yields from fast pyrolysis of OPL and OPL oils at 500 °C and for pyrolysis followed by HDO at 400 °C over 5 and 10 mg MoO3, respectively (average of four injections). | ||
Given that little to no char was formed during the pretreatment step, the lowered char yields could be claimed as lower overall solid yields from the parent OPL. However, the incorporation of methanol into the structure of the depolymerized species should also be accounted for if one wishes to calculate carbon yields based on the initial lignin sample. The carbon yields displayed in Fig. 3 were calculated based on the carbon in each sample (not the parent lignin, except in the case of OPL).
Phenolic oxygenates were significantly increased from 16.6 C% (OPL) to 44 and 32.6 C% for OPL-3 h and OPL-6 h, respectively. Lower phenolic yields from pyrolysis of OPL-6 h was likely due to the number of products drastically increasing, making it much more difficult to identify specific products among the extensively diversified product mixture by the GC-MS system. Additionally, the large number of products would make it impossible to find standard compounds by which the FID could be calibrated making quantification inaccurate. Nevertheless, it was generally observed that oxygenated molecules produced from pyrolysis of OPL-6 h contained more cyclohexanol and cyclohexanediol-type compounds, indicating substantial arene hydrogenation after 3 hours of pretreatment. Furthermore, more methyl ester compounds and less carboxylic acid compounds (e.g., homovanillic acid) were observed compared with OPL-3 h, which could have been from esterification of the organic acids by methanol over the CuPMO catalyst. Reactions occurring during fast pyrolysis could also play a significant role in product diversification as to change the slate of compounds found in the product stream. A detailed list of pyrolysis and HDO products can be found for the OPL oils in the ESI (Tables S6, S7 and Fig. S11, S12†).
Other than methanol (which was used in multiple steps for preparation of pretreated OPL samples), non-phenolic oxygenates from OPL-3 h and OPL-6 h mostly included higher alcohols and diols, which could be made by C–C scission of phenolic monomer side chains and C–C addition reactions.64
The sum of CO and CO2 yields was lower in the case of pretreated samples from about 3.3 C% in OPL to about 1.1–1.3 C%. This could be attributed to a lower number of C–O bonds available for decarbonylation or decarboxylation. No particular pattern was observed with regards to the yield of aromatics among the three samples, especially since the yields were very little, but alkenes and alkanes were produced in larger quantities from pyrolysis of pretreated OPL samples than that of OPL.
Fig. 3 also presents the carbon yields obtained from HDO of raw and pretreated OPL pyrolysis vapors. Since nearly twice the amount of carbon was volatilized during pyrolysis, 10 mg of MoO3 was used for the HDO step instead of the 5 mg used for the other lignin feedstocks. The amount of aromatics obtained from HDO of the OPL oils was significantly higher than that of the base OPL. OPL-3 h and OPL-6 h yielded about 53.5 and 55.0 C% aromatics, respectively, while OPL gave approximately 29.0 C% aromatics. Additionally, the composition of the aromatic products changed for the pretreated OPL materials.
The yields of alkenes and alkanes were higher after pretreatment, but all had similar selectivity to alkenes (ca. 20%) among non-aromatic hydrocarbons. The yields of CO and CO2 decreased while methane yields were increased, which was likely due to the higher relative abundance of methyl ether and methyl ester groups in the pyrolysis vapors of OPL oils.
It was apparent that lignin pretreatment prior to pyrolysis could reduce the overall yield of char and increase the amount of volatilized carbon as to increase the yield of valuable products (see Fig. 4). As there was little to no char formed during the pretreatment step21,61,62 and that it was unlikely that much of the methanol incorporated during pretreatment ended up in the final aromatic yield, it appeared that this particular lignin pretreatment did lead to improved carbon atom efficiency from the lignin feedstock into valuable products. However, analysis of methanol losses to gas and CuPMO stability as well as accurate carbon tracking from the parent lignin and methanol into pretreated samples (including any carbon loss to char) and then into deoxygenated products must be done before any quantitative conclusions about the value of this, or any, pretreatment can be drawn. While reporting detailed product yields from a carbon balance perspective would allow for evaluation of a particular pretreatment system, it is ultimately the overall cost of such systems that would determine their industrial relevance. However, the results presented suggest, from a technical standpoint, that a cost-effective pretreatment process preserving most of the carbon in the deconstructed lignin would be an ideal strategy for lignin valorization through the proposed commodity chemical route.
 | ||
| Fig. 4 Lignin valorization overview via fast pyrolysis-HDO with and without pretreatment. | ||
3.3. Economic evaluation
The basis biorefinery used for the analysis would produce 61 million gallons per year of ethanol with an estimated fixed capital cost of $401 million (MM) including equipment costs of $214 MM. This is comparable to previous studies, which reported total project investments for a biorefinery with a pyrolysis/upgrading section of approximately $220–657 MM.65,66 A similar capital cost was assumed for all scenarios except for the power scenario in which the pyrolysis/upgrading section was excluded as lignin was directly combusted to heat and power. The wastewater section (A600) was the most expensive area, which contributed 22% of the total capital cost ($49 MM), followed by the pyrolysis/upgrading section (A1000) with a 21% contribution ($45 MM). The pyrolysis/upgrading section includes various processes – fast pyrolysis, steam methane reforming, and upgrading, as illustrated in Fig. 1. Cost contributions of various equipment used in area 1000 are shown in Fig. S13.† As observed in this figure, the most expensive equipment is the fast pyrolysis reactor, followed by the steam methane reformer and catalytic reactor. Detailed contributions of different sections to the capital cost can also be found in the ESI (Fig. S14†).Annual fixed variable cost for the biorefinery was estimated at $21.4 MM, which included the cost of labor, land, insurance, maintenance and overhead costs. The variable operating cost for the seven different scenarios varied from $26–68 MM annually. Both OPL-3 h and 6 h cases had the lowest variable operating costs compared to all other scenarios. This result was mainly due to the credits obtained from the additional aromatics and olefins.
Fig. 5 compares the contribution of operating costs to the MESP in dollars per gallon ($ gal−1) for all scenarios analyzed in this study. The minimum selling price of ethanol varied between $0.75–2.02 gal−1, with OPL-6 h being the lowest. While the cost of partial depolymerization was not included in the model for OPL-3 h and OPL-6 h cases, the difference between MESP of OPL and OPL-3 h/OPL-6 h scenarios indicate the economic margins that partial depolymerization procedures could operate in to be considered cost effective (assuming similar product yields). In all scenarios, the feedstock cost contributed the most to the MESP with an approximate average of 40% and 95% for the 4 lignin samples and 2 pretreated lignin cases, respectively. However, the difference in the MESP was primarily due to the credits from aromatics and olefins, which reduced the MESP by approximately 20–34% for the DPL, RL, OCL and OPL cases, while it reduced the MESP by an average of 70% for the OPL oils. Note that the ‘Other’ category in Fig. 5 included estimates of the costs associated with process chemicals and utilities such as sulfuric acid, natural gas, ammonia, corn steep liquor, diammonium phosphate, sorbitol, host nutrients, sulfur dioxide, caustic, boiler chemicals, flue gas desulfurization lime, cooling tower chemicals, and makeup water, as well as costs of ash and catalyst disposal.
 | ||
| Fig. 5 Contribution of different operating cost parameters to the MESP ($ gal−1) for all seven lignin utilization scenarios – power, DPL, RL, OCL, OPL and OPL-3 h and OPL-6 h. | ||
A complete list of different parameters that contributed to the costs associated with the MESP is available in the ESI in Table S8.† Additionally, in the power scenario, there was a $0.2 gal−1 difference in fixed capital costs compared to other scenarios, which was simply due to the exclusion of the pyrolysis and HDO equipment. Scenarios in which pretreated lignin (i.e., OPL-3 h and OPL-6 h) were used reduced the MESP by an average of $1.36 gal−1 when compared to Humbird's38 cost of ethanol from enzymatic hydrolysis and fermentation.
Sensitivity analysis of key process parameters was conducted to determine their influence on the MESP. The results of the sensitivity analysis to selected operating parameters for the OPL-6 h scenario are shown in Fig. 6. It was assumed that these operating parameters were similar for all cases – for instance, in all cases, the amount of feedstock and operating hours were assumed the same. As shown in Fig. 6, ±20% variations in the aromatics price had a greater impact than a similar change in the fixed capital, operating hours and fuel output, while the char output had negligible impact on the MESP. Hence, the three most significant parameters affecting the MESP were aromatics price, operating hours and fuel output and the least significant parameter was char output. A 20% increase in the price of aromatics from $1335 MT−1 to $1602 MT−1, decreased the MESP by 48% from $0.75 gal−1 to $0.39 gal−1. Similarly, an increase in 20% of operating hours decreased the MESP by 32%, while a 20% increase in the fixed capital increased the MESP by 35%. On the contrary, the char output only affected the MESP by approximately 1%. The same trend was observed for OPL-3 h case as well, but for the other lignin scenarios – the three parameters that affected the MESP most significantly were fuel output, fixed capital and operating hours. The price of aromatics did not affect the MESP as significantly in these cases because aromatic yields were lower compared to the pretreated lignin cases. Fig. S15† includes the sensitivity analysis plots for all other scenarios.
 | ||
| Fig. 6 Sensitivity analysis results for the OPL-6 h scenario on the MESP to key economic parameters (parameters were varied by ±20%). | ||
4. Conclusion
Evaluated was the concept of HDO of lignin pyrolysis vapors as a method to valorize lignin into drop-in chemicals (i.e., aromatics and alkenes) both from carbon recovery and techno-economic perspectives. While more than half of the feedstock carbon converted to solid char during pyrolysis, the majority of the volatilized carbon was converted to mono-aromatics over a MoO3 catalyst at 400 °C and ∼1.7 bar H2. Importantly, this approach allowed for funneling of the complex pyrolysis vapor stream of both the more quantifiable oxygenated species (phenolics and non-phenolics) and ∼20% carbon in the unidentified species into the final products (i.e., aromatics, alkenes, and alkanes). Despite, the loss of carbon to char, TEA based on the experimental results suggested lignin valorization via the pyrolysis and HDO strategy to be economically attractive for incorporation into a lignocellullosic biorefinery as evidenced by an 8–24% reduction of the MESP from $2.2 gal−1 (base case – power) to $1.67–2.02 gal−1 when hydrocarbons were generated from lignin. To address the carbon loss issue, an example pretreatment method was also examined. As demonstrated, lignin partial depolymerization prior to pyrolysis can improve carbon volatilization and recovery of desirable products by reducing char from 52 to 18 C%. Such an improvement would offer a large margin with respect to carbon recovery and, therefore, a considerable economic margin for implementing cost effective pretreatment processes for lignin valorization via combined pyrolysis and HDO.The proposed strategy for valorizing lignin by funneling lignin fragments into a diminished number of non-oxygenated aromatics and alkenes that could be directly integrated into the existing petrochemical infrastructure has significant economic potential. However, further technological advancements are still required to best achieve this potential. One such is an inexpensive lignin pretreatment that would partially deconstruct the lignin so as to maximize the amount of carbon that can be volatilized rather than being lost as char. While the MoO3 catalyst used for low-pressure HDO is remarkably effective at deoxygenating a broad range of molecules to petrochemical relevant hydrocarbons, further improvement in catalyst selectivity such that only mono-aromatics and alkenes are generated without alkanes would be desirable.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is sponsored in part by the Iowa Energy Center, Iowa Economic Development Authority and its utility partners under the grant number 17-IEC-002. In addition, we would like to acknowledge funding from the Mike and Jean Steffenson Chair. Partial funding for this research was provided by the National Science Foundation Catalysis Program under award number CBET-1603692.References
- J. C. Del Río, J. Rencoret, P. Prinsen, A. n. T. Martínez, J. Ralph and A. Gutiérrez, J. Agric. Food Chem., 2012, 60, 5922–5935 CrossRef PubMed.
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna and M. Keller, Science, 2014, 344, 1246843 CrossRef PubMed.
- J. D. P. Araújo, C. A. Grande and A. E. Rodrigues, Chem. Eng. Res. Des., 2010, 88, 1024–1032 CrossRef.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2015, 4, 35–46 CrossRef.
- P. C. Rodrigues Pinto, E. A. Borges da Silva and A. E. Rodrigues, Ind. Eng. Chem. Res., 2010, 50, 741–748 CrossRef.
- I. Hasegawa, Y. Inoue, Y. Muranaka, T. Yasukawa and K. Mae, Energy Fuels, 2011, 25, 791–796 CrossRef CAS.
- M. L. Stone, E. M. Anderson, K. M. Meek, M. Reed, R. Katahira, F. Chen, R. A. Dixon, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2018, 6, 11211–11218 CrossRef CAS.
- H. Guo, B. Zhang, Z. Qi, C. Li, J. Ji, T. Dai, A. Wang and T. Zhang, ChemSusChem, 2017, 10, 523–532 CrossRef PubMed.
- W. Wanmolee, N. Laosiripojana, P. Daorattanachai, L. Moghaddam, J. Rencoret, J. C. del Río and W. O. S. Doherty, ACS Sustainable Chem. Eng., 2018, 6, 3010–3018 CrossRef.
- S. Van den Bosch, T. Renders, S. Kennis, S. F. Koelewijn, G. Van den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. M. Courtin and J. M. Thevelein, Green Chem., 2017, 19, 3313–3326 RSC.
- P. De Wild, R. Van der Laan, A. Kloekhorst and E. Heeres, Environ. Prog. Sustainable Energy, 2009, 28, 461–469 CrossRef.
- C. S. Lancefield, B. M. Weckhuysen and P. C. A. Bruijnincx, in Lignin Valorization Emerging Approaches, ed. G. T. Beckham, Royal Society of Chemistry, Cambridge, ch. 7, 2018, pp. 159–198 Search PubMed.
- J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, Top value-added chemicals from biomass-Volume II—Results of screening for potential candidates from biorefinery lignin, Pacific Northwest National Laboratory, Richland, WA, US, 2007 Search PubMed.
- I. A. Pearl, J. Am. Chem. Soc., 1942, 64, 1429–1431 CrossRef.
- R. Behling, S. Valange and G. Chatel, Green Chem., 2016, 18, 1839–1854 RSC.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef PubMed.
- D. J. Nowakowski, A. V. Bridgwater, D. C. Elliott, D. Meier and P. de Wild, J. Anal. Appl. Pyrolysis, 2010, 88, 53–72 CrossRef.
- S. Zhou, R. C. Brown and X. Bai, Green Chem., 2015, 17, 4748–4759 RSC.
- J. Zakzeski, A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602–1609 CrossRef.
- J. A. Barrett, Y. Gao, C. M. Bernt, M. Chui, A. T. Tran, M. B. Foston and P. C. Ford, ACS Sustainable Chem. Eng., 2016, 4, 6877–6886 CrossRef.
- R. Shu, Y. Xu, L. Ma, Q. Zhang, C. Wang and Y. Chen, Chem. Eng. J., 2018, 338, 457–464 CrossRef.
- H. Ma, H. Li, W. Zhao, L. Li, S. Liu, J. Long and X. Li, Green Chem., 2019, 21, 658–668 RSC.
- K. Y. Nandiwale, A. M. Danby, A. Ramanathan, R. V. Chaudhari and B. Subramaniam, ACS Sustainable Chem. Eng., 2018, 7, 1362–1371 CrossRef.
- T. Prasomsri, M. Shetty, K. Murugappan and Y. Román-Leshkov, Energy Environ. Sci., 2014, 7, 2660–2669 RSC.
- T. Prasomsri, T. Nimmanwudipong and Y. Román-Leshkov, Energy Environ. Sci., 2013, 6, 1732–1738 RSC.
- M. W. Nolte, J. Zhang and B. H. Shanks, Green Chem., 2016, 18, 134–138 RSC.
- M. W. Nolte, A. Saraeian and B. H. Shanks, Green Chem., 2017, 19, 3654–3664 RSC.
- G. Zhou, P. A. Jensen, D. M. Le, N. O. Knudsen and A. D. Jensen, ACS Sustainable Chem. Eng., 2016, 4, 5432–5440 CrossRef.
- M. Shetty, K. Murugappan, T. Prasomsri, W. H. Green and Y. Román-Leshkov, J. Catal., 2015, 331, 86–97 CrossRef.
- S. Kilambi and K. L. Kadam, US Pat, US9359651B2, 2016 Search PubMed.
- G. S. Macala, T. D. Matson, C. L. Johnson, R. S. Lewis, A. V. Iretskii and P. C. Ford, ChemSusChem, 2009, 2, 215–217 CrossRef.
- P. A. Johnston, PhD thesis, Iowa State University, 2017.
- A. Saraeian, M. W. Nolte and B. H. Shanks, Renewable Sustainable Energy Rev., 2019, 104, 262–280 CrossRef.
- R. K. Sharma, J. B. Wooten, V. L. Baliga, X. Lin, W. G. Chan and M. R. Hajaligol, Fuel, 2004, 83, 1469–1482 CrossRef.
- R. Lou and S.-b. Wu, Appl. Energy, 2011, 88, 316–322 CrossRef.
- A. Demirbas, J. Anal. Appl. Pyrolysis, 2004, 72, 243–248 CrossRef.
- D. Humbird, R. Davis, L. Tao, C. Kinchin, D. Hsu, A. Aden, P. Schoen, J. Lukas, B. Olthof and M. Worley, Process Design and economics for biochemical conversion of lignocellulosic biomass to ethanol: Dilute-acid pretreatment and enzymatic hydrolysis of corn stover, National Renewable Energy Laboratory, Golden, CO, US, 2011 Search PubMed.
- D. Bbosa, M. Mba-Wright and R. C. Brown, Biofuels, Bioprod. Biorefin., 2018, 12, 497–509 CrossRef.
- A. Dutta, A. H. Sahir, E. Tan, D. Humbird, L. J. Snowden-Swan, P. A. Meyer, J. Ross, D. Sexton, R. Yap and J. Lukas, Process design and economics for the conversion of lignocellulosic biomass to hydrocarbon fuels: Thermochemical research pathways with in situ and ex situ upgrading of fast pyrolysis vapors, Pacific Northwest National Laboratory, Richland, WA, US, 2015 Search PubMed.
- A. Aden, M. Ruth, K. Ibsen, J. Jechura, K. Neeves, J. Sheehan, B. Wallace, L. Montague, A. Slayton and J. Lukas, Lignocellulosic biomass to ethanol process design and economics utilizing co-current dilute acid prehydrolysis and enzymatic hydrolysis for corn stover, National Renewable Energy Laboratory, Golden, CO, US, 2002 Search PubMed.
- T. D. Foust, A. Aden, A. Dutta and S. Phillips, Cellulose, 2009, 16, 547–565 CrossRef.
- M. E. Walsh, R. L. Perlack, A. Turhollow, D. de la Torre Ugarte, D. A. Becker, R. L. Graham, S. E. Slinsky and D. E. Ray, Biomass feedstock availability in the United States: 1999 state level analysis, EERE Publication and Product Library, 2000 Search PubMed.
- Y. Zhu and S. B. Jones, Techno-economic analysis for the thermochemical conversion of lignocellulosic biomass to ethanol via acetic acid synthesis, Pacific Northwest National Laboratory, Richland, WA, US, 2009 Search PubMed.
- W. Hu, Q. Dang, M. Rover, R. C. Brown and M. M. Wright, Biofuels, 2016, 7, 57–67 CrossRef.
- R. E. Davis, N. J. Grundl, L. Tao, M. J. Biddy, E. C. Tan, G. T. Beckham, D. Humbird, D. Thompson and M. S. Roni, Process Design and economics for the conversion of lignocellulosic biomass to hydrocarbon fuels and coproducts: 2018 biochemical design case update; biochemical deconstruction and conversion of biomass to fuels and products via integrated biorefinery pathways, National Renewable Energy Laboratory, Golden, CO, US, 2018 Search PubMed.
- H. Kawamoto, J. Wood Sci., 2017, 63, 117 CrossRef.
- R. Mahadevan, S. Adhikari, R. Shakya, K. Wang, D. Dayton, M. Lehrich and S. E. Taylor, Energy Fuels, 2016, 30, 3045–3056 CrossRef.
- K. Wang, J. Zhang, B. H. Shanks and R. C. Brown, Appl. Energy, 2015, 148, 115–120 CrossRef.
- P. R. Patwardhan, R. C. Brown and B. H. Shanks, ChemSusChem, 2011, 4, 1629–1636 CrossRef CAS.
- M. Garcia-Perez, S. Wang, J. Shen, M. Rhodes, W. J. Lee and C.-Z. Li, Energy Fuels, 2008, 22, 2022–2032 CrossRef CAS.
- Y. Yu, X. Li, L. Su, Y. Zhang, Y. Wang and H. Zhang, Appl. Catal., A, 2012, 447, 115–123 CrossRef.
- P. R. Patwardhan, D. L. Dalluge, B. H. Shanks and R. C. Brown, Bioresour. Technol., 2011, 102, 5265–5269 CrossRef CAS.
- P. R. Patwardhan, R. C. Brown and B. H. Shanks, ChemSusChem, 2011, 4, 636–643 CrossRef CAS.
- X. Bai, K. H. Kim, R. C. Brown, E. Dalluge, C. Hutchinson, Y. J. Lee and D. Dalluge, Fuel, 2014, 128, 170–179 CrossRef CAS.
- M. W. Nolte and B. H. Shanks, Energy Technol., 2017, 5, 7–18 CrossRef CAS.
- S. Zhou, M. Garcia-Perez, B. Pecha, S. R. A. Kersten, A. G. McDonald and R. J. M. Westerhof, Energy Fuels, 2013, 27, 5867–5877 CrossRef CAS.
- M. Chui, G. Metzker, C. M. Bernt, A. T. Tran, A. C. B. Burtoloso and P. C. Ford, ACS Sustainable Chem. Eng., 2017, 5, 3158–3169 CrossRef CAS.
- D. J. McClelland, P. H. Galebach, A. H. Motagamwala, A. M. Wittrig, S. D. Karlen, J. S. Buchanan, J. A. Dumesic and G. W. Huber, Green Chem., 2019, 21, 2988–3005 RSC.
- C. M. Bernt, G. Bottari, J. A. Barrett, S. L. Scott, K. Barta and P. C. Ford, Catal. Sci. Technol., 2016, 6, 2984–2994 RSC.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640–1647 RSC.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef CAS PubMed.
- A. J. Kumalaputri, G. Bottari, P. M. Erne, H. J. Heeres and K. Barta, ChemSusChem, 2014, 7, 2266–2275 CrossRef CAS PubMed.
- P. H. Galebach, S. Thompson, A. M. Wittrig, J. S. Buchanan and G. W. Huber, ChemSusChem, 2018, 11, 4007–4017 CrossRef CAS PubMed.
- R. Thilakaratne, T. Brown, Y. Li, G. Hu and R. Brown, Green Chem., 2014, 16, 627–636 RSC.
- T. R. Brown, Bioresour. Technol., 2015, 178, 166–176 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc04245h |
| This journal is © The Royal Society of Chemistry 2020 |