
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-strength cellulose nanofibers produced via swelling pretreatment based on a choline chloride–imidazole deep eutectic solvent†
Juho Antti
Sirviö
 *a,
Kalle
Hyypiö
a,
Shirin
Asaadi
b,
Karoliina
Junka
b and
Henrikki
Liimatainen
a
*a,
Kalle
Hyypiö
a,
Shirin
Asaadi
b,
Karoliina
Junka
b and
Henrikki
Liimatainen
a
aFibre and Particle Engineering Research Unit, University of Oulu, P.O. Box 4300, 90014 Oulu, Finland. E-mail: juho.sirvio@oulu.fi
bBillerudKorsnäs AB, Box 703, 169 27 Solna, Stockholm, Sweden
First published on 18th February 2020
Abstract
A deep eutectic solvent (DES) based on choline chloride and imidazole (CCIMI) was investigated for swelling of cellulose fibers prior to mechanical disintegration into cellulose nanofibers (CNFs). The dimensions of the DES treated and washed fibers were investigated after various treatment conditions (time, temperature, and cellulose consistency) using DES based on choline chloride–urea (CCUrea) and pure imidazole as references. Even mild treatment conditions (15 minutes at 60 °C) with CCIMI increased the diameter of the fibers from 18.1 to 18.9 μm, and a maximum diameter of 19.9 μm was obtained after three hours at 100 °C. Overall, CCIMI resulted in a higher degree of swelling compared to both references. In addition, pure imidazole caused a decrease in the degree of polymerization of cellulose, whereas cellulose degradation in CCIMI was negligible. The mechanical disintegration of CCIMI-treated fibers resulted in the production of CNF films with very good mechanical properties—specific tensile strength and work capacity being over 200 kNm kg−1 and 10 kJ kg−1, respectively—whereas CNFs films produced using choline chloride–urea had notably lower values (182 kNM kg−1 and 7 kJ kg−1, respectively). In addition, CNF films exhibited good oxygen barrier properties, even at an elevated relative humidity level (80%). CCIMI could be recycled without any effect on the mechanical properties of CNF films. The results presented here indicate CCIMI is a highly efficient pretreatment media for swelling and further nanofibrillation of cellulose, even at mild treatment conditions.
Introduction
Nanomaterials are solid components that have one or more dimensions at the nanometric scale (<1000 nm or below 100 nm).1 Compared to their macrosized counterparts, nanomaterials exhibit certain advanced properties, including extreme high surface-to-volume ratio.2 Due to the large surface area, nanomaterials have strong interactions with both their surrounding matrices and themselves. This property allows them to be used in the production of high-strength materials—for example, composites.3 Due to their sizes, nanomaterials uniquely interact with light—that is, nanomaterials can be used to produce visually transparent objects,4 whereas plasmonic nanoparticles can be obtained, for example, from metals.5Of all nanomaterials, cellulose nanofibers (CNFs) are considered sustainable components suitable to be utilized in a variety of ways, such as in composites and self-standing films (e.g., for packaging and flexible electronics).6,7 The sustainability of CNFs originates from the biobased starting material cellulose, which is the most abundant organic polymer on earth. Similarly to natural cellulose fibers, CNFs have several advantage properties compared to the many synthetic counterparts, such as renewability, biodegradability, biocompatibility, and low toxicity.8
Cellulose is widely available as the structural material of plants, where it already exists as nanosized fibers. However, during biosynthesis, these nanometric cellulose fibers (elemental fibrils) aggregate into larger fiber bundles that are held together by strong hydrogen bonds and weak van der Waals forces, and specific methods are requested to break up the natural, recalcitrant fiber structure. The liberation of CNFs from natural cellulose fibers can be achieved using strong mechanical forces. To decrease the large energy demand of mechanical disintegration (nanofibrillation), several chemical methods have been introduced.8 Among the most efficient methods for the production of high-quality CNFs (i.e., individualized nanofibers) are those that generate a strong surface charge on natural fibers.9 Chemical modifications such as (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) mediated oxidation,10,11 carboxymethylation,12,13 and periodate oxidation followed by further derivatization14–18 have been utilized to produce high-quality CNFs. Although these chemical modifications allow the production of CNFs with minimal mechanical forces, they generally utilize hazardous halogenated chemicals and heavily damage cellulose fibers (i.e., cause a decrease in the degree of polymerization [DP] and yield losses). Enzymatic treatments are recognized as sustainable methods to produce CNFs. Enzymes cause the mild hydrolysis of cellulose (i.e., decrease the DP of cellulose)—which, in turn, allows the liberation of CNFs with reduced energy consumption.19,20
Nonmodifying methods (i.e., no or only minimal decrease in DP or alteration of cellulose structure) based on deep eutectic solvents (DESs) have recently been used for the sustainable production of CNFs.21–25 In addition to DESs’ use as nonmodifying pretreatment media, they can be harnessed as solvents for chemical derivatization26,27 and as reagents28 that can even be recycled29 in CNF production. DESs are ionic liquid analogues that are obtained by the simple mixture of two or more components and can often be derived from green and bulk chemicals.30 A noncovalent interaction, such as the hydrogen bonding of DES components, causes the reduction of the melting point of mixtures compared to its individual component. The interaction between DES components allows them to be produced at temperature below melting point of either of components31 The use of mechanical stirring can lower the temperature needed for DES production32 and in some cases DES can be obtained even at room temperature by mild mixing.33 Exclusion of the complicated synthetic routes and any additional chemicals (i.e. solvents and side-products) during their preparation makes DESs promising sustainable alternatives for ionic liquids and other more well-known solvents. Due to the low vapor pressure, DESs do not typically contribute the volatile organic component emissions and are safer to handle than traditional solvents.30
The nonchemical modification of cellulose fibers with DESs is assumed to cause fiber swelling, which is preserved even after the removal of DESs by water washing,23 but no comprehensive studies on the effect of different treatment conditions (e.g., time and temperature) on the swelling of cellulose fibers and the consequent nanofibrillation to produce CNFs have been reported. In this study, we used a DES based on choline chloride and imidazole (CCIMI) as a nonderivatizing pretreatment medium for wood cellulose fibers. Specifically, the role of various conditions in fiber swelling and the consequent nanofibrillation to produce CNFs were addressed. Pure imidazole and previously used DES (CC with urea) were used as references. The effect of different treatment conditions on nanofibrillation and the mechanical and oxygen barrier properties of self-standing films of CNFs were further investigated.
Materials and methods
Materials
Hardwood kraft pulp (BillerudKorsnäs, Sweden) was used as a starting material and was provided as dry sheets. Standard TAPPI methods were used to analyze the composition of the dry sheets: the lignin content with TAPPI T 222 om-02 (<0.1%), acetone-soluble extractives with TAPPI 280 pm-99 (0.14%), NaOH-soluble hemicelluloses, and degraded cellulose portions with TAPPI 212 om-02 (2.9%).Fiber treatment with the DES
The fiber treatment with the DES was performed in a similar manner described previously.21,23 Prior to the treatment, CC and imidazole were mixed at desired temperatures (60 °C, 80 °C, and 100 °C) at a molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 (46.78 g and 53.22 g, respectively). After obtaining a clear liquid, 1 g of cellulose (dry sheets ripped in small pieces by hand) was added to the DES to produce a DES–cellulose suspension with a mass ratio of 1:100. The suspension was then mixed with a magnetic stirrer, and after the desired time (15, 30, 60, 120, and 180 minutes), about 20 ml of water was added. The product was filtrated and further washed under vacuum with 1000 ml of water. The washed fibers were collected and stored at 4 °C until further use. The mass yields of the samples were 90%–100%, and the yield losses were mainly attributed to sample handling (e.g., small amounts of the fibers remained on the filter paper).
:7 (46.78 g and 53.22 g, respectively). After obtaining a clear liquid, 1 g of cellulose (dry sheets ripped in small pieces by hand) was added to the DES to produce a DES–cellulose suspension with a mass ratio of 1:100. The suspension was then mixed with a magnetic stirrer, and after the desired time (15, 30, 60, 120, and 180 minutes), about 20 ml of water was added. The product was filtrated and further washed under vacuum with 1000 ml of water. The washed fibers were collected and stored at 4 °C until further use. The mass yields of the samples were 90%–100%, and the yield losses were mainly attributed to sample handling (e.g., small amounts of the fibers remained on the filter paper).
Analysis of the fiber dimensions
Two milligrams of the fibers (either original cellulose or DES-treated fibers) were manually mixed with water to obtain a 0.004 wt% suspension. A 50 mL aliquot of the suspension was prepared and then analyzed using a Fiber Lab image-based analyzer (Fiber Lab 2000, Metso Automation, Jyväskylä, Finland). The fiber width and cell wall thickness (CWT) were obtained as average values of over 5000 individual qualified fibers captured by the scanning camera in the instrument.Limiting viscosity
The limiting viscosity of the cellulose samples was measured using the ISO 5351 standard. At first, 200 mg of an absolutely dry sample was shredded and placed into small bottles. Then five small copper sticks and 25 ml of water were added, and the suspensions were mixed in a shaker for about 30 minutes to properly disperse the fibers. After that, 25 ml of cupriethylenediamine solution was added into each bottle, and the resulting mixture was re-placed in the shaker for another 30 minutes. Finally, the sample bottles were placed in an oil bath under mixing, which was maintained at a constant temperature of 25 °C. Each sample was sucked into the capillary viscometer above the upper marker line with a pileusball. Then the solution was allowed to drain out of the tube, and the time interval between the two marker lines was measured with a stopwatch. The analysis was conducted in triplicates, and the results were averaged. The limiting viscosity values were converted to DP values using the following equation: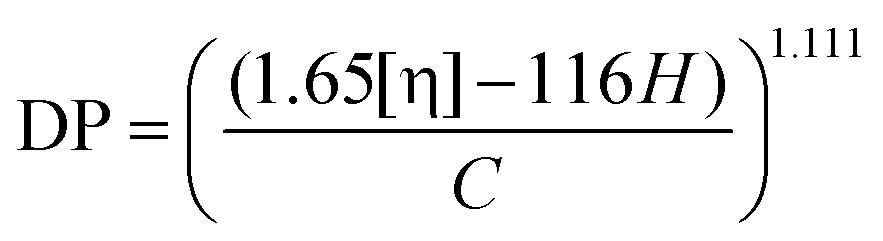 | (1) |
This calculation corrects the contribution of hemicellulose to the limiting viscosity value and DP of cellulose, assuming that the average DP of hemicellulose is 140.
Molar mass measurements
The number average molar mass (Mn), weight average molar mass (Mw), and polydispersity (PD) were determined by size exclusion chromatography (SEC) in DMAc/0.8% LiCI eluent using pullulan calibration from original pulp and pulp after treatment with CCIMI at 100 °C for 180 minutes and CCUrea at 100 °C for 120 minutes. The freeze-fried samples DMAc/8% LiCl according to the solvent exchange method.34 After dissolution, the samples were diluted with DMAc providing final LiCl concentration of 0.8% as in the eluent. In all cases the samples were filtered (0.45 μm) before the measurement. The SEC measurements were performed using 2× PL gel MiniMixed A columns with a pre-column in DMAc/0.8% LiCl eluent (0.36 ml min−1, T = 80 °C). The elution curves were detected using Waters 2414 refractive index detector. The molar mass distributions (MMD) were calculated against 8× pullulan (6100–624000 g mol−1) standards, using Waters Empower 3 software.
X-ray diffraction
The crystalline structure of the original and DES-treated pulp was scanned with a wide-angle X-ray diffraction device. Measurements were conducted on a Rigaku SmartLab 9 kW rotating anode diffractometer (Japan) using Co Kα radiation (40 kV, 135 mA; λ = 1.79030 nm). Freeze-dried CNFs were pressed to around 1 mm thick tablets. Scanning was performed over a 2θ (Bragg angle) range from 5° to 50° at a speed of 10° s−1 and using a step of 0.5°. The degree of crystallinity in terms of the crystallinity index (CrI) was calculated from the peak intensity of the main crystalline plane (200) diffraction (I200) at 26.2° and the peak intensity at 22.0° associated with the amorphous fraction of cellulose (Iam) according to eqn (2)35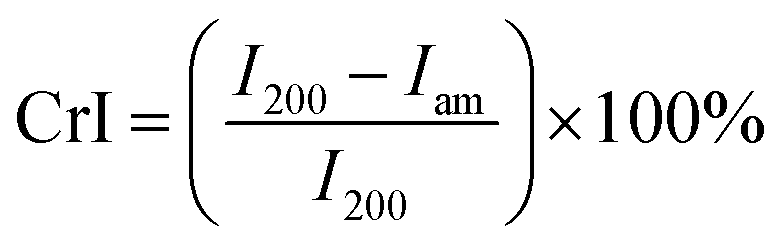 | (2) |
Field emission scanning electron microscope
The DES-treated sample was weighed, to match 2.0 mg of the absolutely dry mass, into a decanting glass and then diluted with 200 ml of water. Then the sample was dried via vacuum filtering on a polyvinylidene fluoride membrane with a pore size of 0.65 μm, which was then dipped in liquid nitrogen and dried with a freeze-dryer. When the samples were dry, a double-sided sticker was glued on the bottom of the weight, and a small piece of the film sample was glued on the sticker. Prior to the measurement, the samples were coated with platinum. Images were acquired with the Zeiss Ultra Plus (Carl Zeiss SMT AG, Germany) field emission scanning electron microscope (FESEM).Nanofibrillation of cellulose
Before nanofibrillation, the DES pretreated pulp was diluted to 0.5 wt% and premixed with an Ultra-Turrax IKA T25 mixer at 10000 rpm for 5 minutes. This was performed to create a suspension as homogenous as possible, which was then nanofibrillated with Microfluidics’ M-110EH-30-microfluidizer. Each sample first passed the system thrice through 400 and 200 μm chambers at 1000 bar and then twice through 400 and 100 μm chambers at 1500 bar.
Transmission electron microscope
To analyze the size and degree of fibrillation of the CNFs, the samples were imaged with a transmission electron microscope (TEM, JEOL JEM-2200FS, Japan). The samples were prepared by adding 7 μl of a diluted poly-L-lysine solution, 7 μl of a diluted CNF suspension, and 7 μl of 2% uranyl acetate on a carbon-coated copper grid. Between adding the chemicals, each of these solutions was allowed to remain on the grid for approximately 30 seconds and, after that, were dried with filter paper. The diameters of the nanofibers were measured using ImageJ's software 1.50i. The average was calculated using 70 individual nanofibers.Thermogravimetric analysis
Thermogravimetric (TG) analyses of the CNFs were carried out using a thermal analyzer (STA 449F3, Netzsch) under the air flow (dynamic air) using a constant flow rate of 60 mL min−1. Approximate 5 mg of the freeze-dried sample was weighed in an aluminum oxide pan, and the sample was heated from 30 °C to 900 °C with a heating rate of 10 °C min−1.Preparation of the CNF films
The CNFs were dosed to match 0.265 g of absolutely dry matter and placed in a decanting glass. The suspension was then diluted with water to a total weight of 100 g, and the sample was degassed for 10 minutes with an ultrasonic bath. After degassing, the sample was vacuum filtrated on top of a membrane (Durapore DVPP 0.65 μm, Merck Millipore Ltd, Ireland) with a negative pressure of approximately 800 mbar, and, after that, the sample dried with a vacuum drier (Karl Schröder KG, Germany) to obtain a film with a grammage of 60 g m−2. Drying was executed at 93 °C and with a negative pressure of 930 mbar for 10 minutes.Tensile strength measurement for CNF films
The films were tested at 23 °C and relative humidity (RH) 50% with a universal testing device (Instron 5544, USA) using the ISO 1924-2 standard. Prior to the measurement, the films were conditioned at above mentioned measurement conditions for 48 hours. The films were then cut to sample strips with a width of 5 mm thickness and length of 70 mm, and their average thickness was calculated by selecting five random points, which were measured with a thickness gauge (Precision Thickness Gauge FT3, Hanatek Instrument, UK). The tensile testing device was equipped with a 2 kN force sensor, and the gauge length was set to 40 mm. The samples were strained at a speed of 5 mm min−1 until they snapped. The average and standard deviation of the samples from each film were calculated.Recycling and reuse of the DES
Cellulose pulp (2.5 g) was treated with CCIMI DES at 60 °C for 15 minutes using cellulose:DES mass ration of 1:100. After treatment, 50 ml of deionized water was added followed by filtration and washing with 250 ml of deionized water. DES-water solution was collected and cellulose pulp was further washed with 900 ml of deionized water to remove any trace of DES components. Nanofibrillation, film preparation, and tensile test measurement were conducted in similar manner as mentioned above.
Water was removed from the DES-water solution with Buchi rotavapor using approximately vacuum of −0.9 mbar and water bath at 95 °C. Evaporation was conducted until no water was observed to drop into collection flack. After evaporation, the mass of the DES was measured and DES was directly used for treatment of fresh batch of cellulose fibers.
Oxygen barrier properties
The oxygen transmission rate (OTR) of the films was measured using a MOCON OxTran 2/20 (Minneapolis, MN). The film was exposed to 100% oxygen on one side and to oxygen-free nitrogen on the other side. The oxygen permeability (OP) was calculated by multiplying the OTR by the thickness of the film and dividing it by the difference in the partial pressure of the oxygen gas between the two sides of the film. The measurements were made at 23 °C, a normal atmospheric pressure, and a 50% and 80% RH with a specimen area of 5 cm2.Results and discussion
Swelling of the cellulose fibers by DES treatment
Previously, the eutectic point of the CC and IMI mixture was achieved with a molar ratio of 3:7, resulting in the formation of a DES with a melting point of around 56 °C.36 Therefore, the minimum temperature of 60 °C was chosen here for fiber treatment with CCIMI. All treatments were conducted at normal atmosphere pressure without any protection gas, and after treatment, DES components were removed by water washing. The diameters of the fibers were directly analyzed from never-dried samples to prevent the collapse of the swollen fibers.
As a general trend, both fiber width and CWT increased when the treatment time with CCIMI increased. For example, fiber width increased from 18.1 μm of original fibers to 18.9 μm after DES treatment of 15 minutes at 60 °C (Table 1, entry 2). The prolonged treatment time caused a further increase of the fiber width to 19.3 μm at 60 °C (Table 1, entry 3). At shorter treatment times, the increase in the temperature only showed a minimal effect on the fiber width. However, at longer treatment times, the increase of the temperature had a more notable effect and the maximum width of 19.9 μm was observed after three hours of treatment at 100 °C (Table 1, entry 16). The increase in the fiber width in function of temperature is mostly likely due to the increased molecular motion at elevated temperature, which enables DES to penetrate deeper into the cellulose fiber structure, in turn increasing the swelling of fibers. Similarly, the increase of treatment time allows a longer interaction between cellulose fibers and DES.
:100. Samples used for the production of CNFs are in bold print
| Entry | Solvent | Temperature (°C) | Time (min) | Fiber properties | DP | CrI (%) | CNF sample | Average diameter of CNFs (nm) | |
|---|---|---|---|---|---|---|---|---|---|
| Width (μm) | CWT (μm) | ||||||||
| a Original cellulose fibers. b Not measured. | |||||||||
| 1a | — | — | — | 18.1 | 5.6 | 3960 | 69 | — | — |
| 2 | CCIMI | 60 | 15 | 18.9 | 5.8 | 3740 | 67 | 1 | 17 ± 18 |
| 3 | CCIMI | 60 | 30 | 19.1 | 6.1 | 3910 | —b | — | — |
| 4 | CCIMI | 60 | 60 | 19.3 | 6.0 | 4010 | —b | — | — |
| 5 | CCIMI | 60 | 120 | 19.6 | 6.4 | 3950 | —b | — | — |
| 6 | CCIMI | 60 | 180 | 19.3 | 6.1 | 3890 | 67 | 2 | 10 ± 8 |
| 7 | CCIMI | 80 | 15 | 19.0 | 6.1 | 3860 | —b | — | — |
| 8 | CCIMI | 80 | 30 | 19.0 | 6.1 | 3760 | —b | — | — |
| 9 | CCIMI | 80 | 60 | 19.7 | 6.4 | 3910 | —b | — | — |
| 10 | CCIMI | 80 | 120 | 19.7 | 6.2 | 3890 | —b | — | — |
| 11 | CCIMI | 80 | 180 | 19.5 | 6.1 | 3830 | —b | — | — |
| 12 | CCIMI | 100 | 15 | 19.0 | 6.2 | 3790 | 66 | 3 | 18 ± 23 |
| 13 | CCIMI | 100 | 30 | 19.0 | 6.2 | 3860 | —b | — | — |
| 14 | CCIMI | 100 | 60 | 19.3 | 6.2 | 3870 | —b | — | — |
| 15 | CCIMI | 100 | 120 | 19.5 | 6.2 | 3820 | —b | — | — |
| 16 | CCIMI | 100 | 180 | 19.9 | 6.0 | 3880 | 64 | 4 | 14 ± 10 |
| 17 | Imidazole | 100 | 180 | 19.0 | 5.9 | 3450 | —b | — | — |
| 18 | CCurea | 100 | 120 | 19.0 | 5.9 | 3640 | 66 | 5 | 17 ± 21 |
The CWT of the fibers mostly followed a similar trend, rather than fiber width. That is, CWT values mostly increased when treatment time and temperature were increased. However, a small drop in the CWT values was noted at the longest treatment time (180 min), which may indicate cell walls were partly collapsed when the overall width of the fibers increased.
The CCUrea DES, which has been previously used as pretreatment media for cellulose to improve nanofibrillation,21 also had a notable effect on fiber width, and a fiber diameter of 19.0 μm was obtained (Table 1, entry 18) with two hours of treatment at 100 °C. Nevertheless, the swelling of the fibers with CCUrea was lower compared to that of CCIMI at the same conditions (fiber width of 19.0 μm vs. 19.5 μm, respectively) (Table 1, entry 15). The higher swelling ability of CCIMI may be attributed to the higher alkalinity of IMI compared to that of urea. It is well known that cellulose swells in aqueous alkaline solutions.37
Pure imidazole has been previously used for the dissolution of starch38 (similarly to CCIMI36) and, thus, is assumed to also interact with cellulose fibers, possibly leading to the swelling of the fibers. In addition, pure imidazole has been used for the production of CNFs after a long treatment time (24 h) at a high temperature (120 °C) followed by ultrasonic treatment39 and in lignocellulose fractionation (i.e. delignification).40,41 Here, we compared the effects of pure imidazole and DES CCIMI on fiber swelling and the possible benefits of DES formation. The treatment with imidazole was conducted for three hours at 100 °C, and fiber width was observed to increase to 19.0 μm. This result is notably lower compared to that obtained by CCIMI (19.9 μm) at the same conditions and proves the advantageous features of DES in fiber swelling. In addition, CCIMI resulted in similar fiber swelling already after a half hour of treatment at 60 °C. It is noteworthy that imidazole has a melting point range of 89 °C–91 °C and cannot be used at 60 °C.
The decrease in the mass ratio between cellulose fibers and CCIMI had no, or only, minimal effect on fiber swelling—that is, similar fiber widths were obtained at cellulose:CCIMI weight ratios of 1:100, 1:50, 1:33, and 1:25 after one hour of treatment at all the studied temperatures (Table S1†). Therefore, CCIMI is a potential swelling medium, even at a higher cellulose consistency. However, it should be noted that at a weight ratio of 1:25, a highly viscous suspension was observed, and more intensive mixing than can be obtained using a magnetic stirrer is recommended, especially if the aim is to further decrease the weight ratio.
Characterization of DES-treated cellulose fiber
Different solvent treatments can cause various chemical and physical transformations of the cellulose fibers. The DP values of CCIMI-treated cellulose were similar (94% or higher) to that of original pulp, indicating the hydrolysis of cellulose was minimal (Table 1). However, when pure imidazole was used, a notable drop in the DP of cellulose was noted (from 3960 to 3450) (Table 1, entry 17). A decrease in the DP with pure imidazole might originate from a higher imidazole concentration compared to CCIMI. Imidazole is amphoteric molecule with base-dominating characteristic, being thus mild degradation agent for cellulose as was reported in previous study.39In addition to the different concentrations of imidazole, the presence of CC in a DES may protect cellulose molecules from hydrolysis. However, the role of individual DES components in the hydrolysis of cellulose during treatment with a DES is partly unknown, and previous results have also shown severe hydrolysis of cellulose occurs when carboxylic or sulfonic acids are used, as in CC-based DESs.42 CCUrea, used here as a reference, was not previously reported to cause cellulose hydrolysis.21 However, a small decrease in the DP was observed (Table 1, entry 18), which was more significant compared to that of CCIMI and similar to that previously reported with ammonium thiocyanate–urea and guanidine hydrochloride–urea DESs.23
In addition to the determination of DP using limiting viscosity, molar mass of cellulose before and after treatment with CCIMI at 100 °C for 180 min and CCUrea 100 °C for 120 min was analyzed using SEC. It was observed that molar mass and the PD of CCIMI remained in a similar level compared to the original values of cellulose pulp (Table S2†). This indicates that no cellulose degradation or dissolution of hemicelluloses took place when CCIMI was used in at most severe conditions studied. On the other hand, changes in the Mw values after CCUrea treatment indicates that some changes in the constitution of pulp took place, and the Mw was noticed to increase. This phenomenon was likely due to dissolution of hemicelluloses.
The DRIFT spectra of original cellulose pulp and the fibers treated with CCIMI for 180 min at 100 °C and with CCUrea for 120 min at 100 °C are presented in Fig. 1. Other spectra are presented in ESI (Fig. S1†). It is apparent that no alteration of the cellulose chemical structure took place during fiber treatment. Previously, a small carbonyl peak at 1715 cm−1 was observed when cellulose was treated with CCUrea, indicating the formation of a minor amount of cellulose carbamate.21 In the current study, no carbamate peak was observed, demonstrating both CCUrea and CCIMI are nonderivatizing treatment media for cellulose in the used conditions.
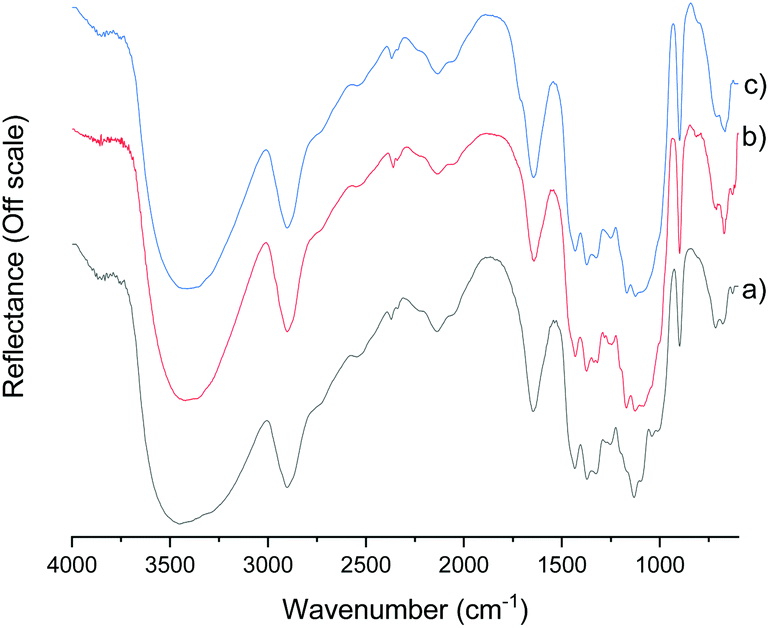 | ||
| Fig. 1 DRIFT spectra of original cellulose fibers (a), fibers treated with CCIMI at 100 °C for 180 min (b), and fibers treated with CCUrea at 100 °C for 120 min (c). | ||
DESs are generally considered solvents with a high dissolution capacity regarding many organic and inorganic components,43 and as stated above, CCIMI has previously been used to dissolve starch. In addition, some DESs are known to dissolve cellulose,44 especially low molecular weight microcrystalline cellulose.45,46 From the XRD crystallography results, it can be seen that the crystalline structure of cellulose remained similar to the original cellulose pulp—that is, cellulose I (Fig. 2). The absence of evidence of other cellulose crystalline allomorphs indicates there was no cellulose dissolution and regeneration during the pretreatment with CCIMI and CCUrea, which is in line with previous studies with CCUrea21 and triethylmethylammonium chloride–imidazole DESs.26,33 In addition, the amount of crystalline fraction in cellulose remained at its original level (Table 1, entry 1) when either CCIMI (Table 1, entries 2, 6, 12, and 16) or CCUrea (Table 1, entry 18) was used. Therefore, the swelling of the cellulose fibers was concluded to be caused by the weakening of the hydrogen bonds between the crystalline plates and nonordered region of cellulose, and solvents do not penetrate the crystalline region of cellulose. The inability of CCIMI and CCUrea to infiltrate the crystalline part of cellulose might be the reason why these particular DESs do not dissolve cellulose.
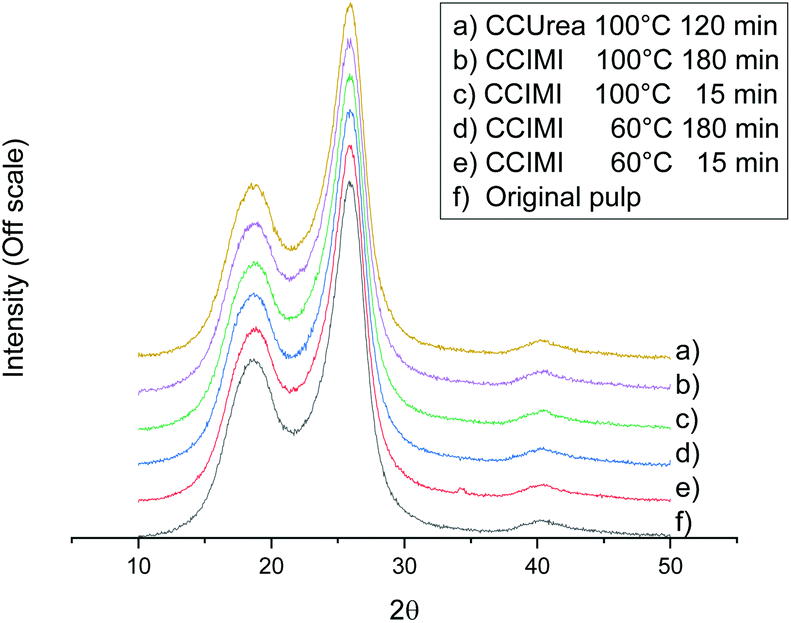 | ||
| Fig. 2 XRD diffraction patterns of original cellulose and DES-treated fibers. | ||
After DES treatment, the surface structure of cellulose fibers appeared slightly rougher compared to original fibers (Fig. 3). Changes in the surface are especially evident in Fig. 3e and f. In addition, surface microfibers were partly loosened and protruded out from the interface. However, it is clear there were not large visual changes in the fiber morphology caused by DES treatments in FESEM (dried samples) despite clear swelling, as indicated by image analysis (never-dried samples) (Table 1).
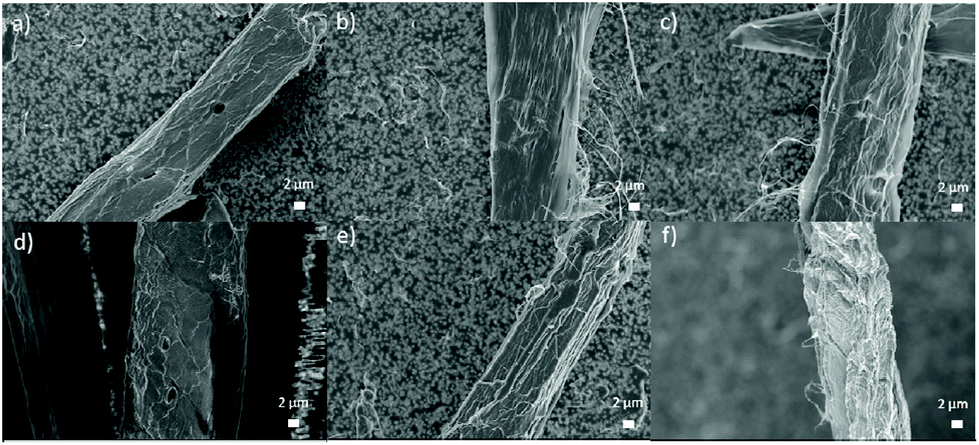 | ||
| Fig. 3 FESEM images of original (a) and CCIMI-treated fibers at 60 °C for 15 minutes (b) and 180 minutes (c) and at 100 °C for 15 min (d) and 180 min (e) and CCUrea-treated fiber at 100 °C for 120 min (f). | ||
Nanofibrillation of DES-treated cellulose fibers
Selected CCIMI-treated and CCUrea-treated cellulose fibers (Table 1, entries 2, 6, 12, 16, and 18) were mechanically disintegrated with a microfluidizer to produce CNFs. All the samples passed easily through the microfluidizer without blocking it—indicating that swelling treatment improved the nanofibrillation, as nontreated fibers tend to block the narrow chambers of the apparatus.47 The samples existed as slightly gel-like opaque dispersions (Fig. 4a), which is common for nonchemically modified CNFs. After freeze-drying, all the samples formed self-standing, white aerogels (Fig. S2†) that can be used, for example, in oil–water separation.48 | ||
| Fig. 4 (a) Dispersion of CNFs (concentration around 0.5 wt%) and (b) self-standing films prepared from CNFs. CNFs obtained from fibers treated with CCUrea (1) and CCIMI at 60 °C for 15 minutes (2) and 180 minutes (3) and at 100 °C for 15 min (4) and 180 min (5). (c) Demonstration of opaque characteristic of CNF film 1. | ||
TEM images of CNFs indicate all the samples disintegrated into nanofibers with heterogeneous size distribution ranging from diameters of around 5 nm (individual nanofibrils) to tens of nanometers (nanofibril aggregates) (Fig. 5). The average diameters ranged from 10 to 18 nm (histograms of the fiber diameters are presented in Fig. S3†). High temperature during CCIMI treatment seemed to result in slightly thinner and more uniform nanofibers; however, due to the large variations in the size of the fibers, no direct conclusion can be drawn. The lateral dimensions of CNFs produced with CCIMI are in line with previous studies with CNFs produced using nonderivatizing DES treatment21,23 and pure mechanical disintegration.49
 | ||
| Fig. 5 TEM images of CNFs obtained from fibers treated with CCUrea (a) and CCIMI at 60 °C for 15 minutes (b) and 180 minutes (c) and at 100 °C for 15 min (d) and 180 min (e). Examples of the diameters of the nanofibers are indicated with arrows. | ||
Thermal properties of CNFs
TG analysis showed CNFs produced using CCIMI and CCUrea exhibited a slightly lower onset temperature compared to the original cellulose pulp (288 °C of original pulp vs. 273 °C–278 °C of CNFs), indicating marginally lower thermal stability (Fig. S4†). The decrease in the thermal stability mainly originates from the damage caused by high mechanical forces applied during nanofibrillation.50There were minor differences between CCIMI-treated samples, as those produced with longer treatment times exhibited slightly higher onset temperature (onset temperature 277 °C and 278 °C of samples produced at 60 °C and 100 °C, respectively, for 180 min) compared to those produced using 15 min treatment (onset temperature 273 and 270 °C of samples produced at 60 and 100 °C, respectively). The onset temperature of the sample produced with CCUrea was in-between long- and short-time CCIMI-treated samples (275 °C). Small differences might originate from minor changes in the composition of the pulp during the pulp treatment: longer treatment times result in the removal of some of the hemicelluloses (2.9% of alkaline soluble hemicelluloses) still present in the pulp. Previous studies have demonstrated the thermal stability of the pulp increases by DES treatment due to the removal of hemicelluloses, and the thermal properties of CNFs produced from DES treatment of birch pulp are similar to those of the original pulp.23 The difference between the thermal properties of the CNFs produced in previous studies and the current study mostly originates from the hemicellulose content of the pulps. As in previous studies, the birch pulp contained 24% of residual hemicelluloses.
Mechanical properties of CNF films
CNF films were produced by the filtration and vacuum drying method, and all the films exhibited a typical opaque visual appearance (Fig. 4b and c). Tensile test results showed films had significantly high strength properties: all the films had tensile strength values above 200 MPa, and samples 1 and 3 recorded specific tensile strength values above 200 kNM kg−1 (Fig. 6a). The high specific strength values of samples 1 and 3 are interesting, as they were produced using the shortest treatment time (15 min). However, all tensile strength values were similar when taking measurement error into account.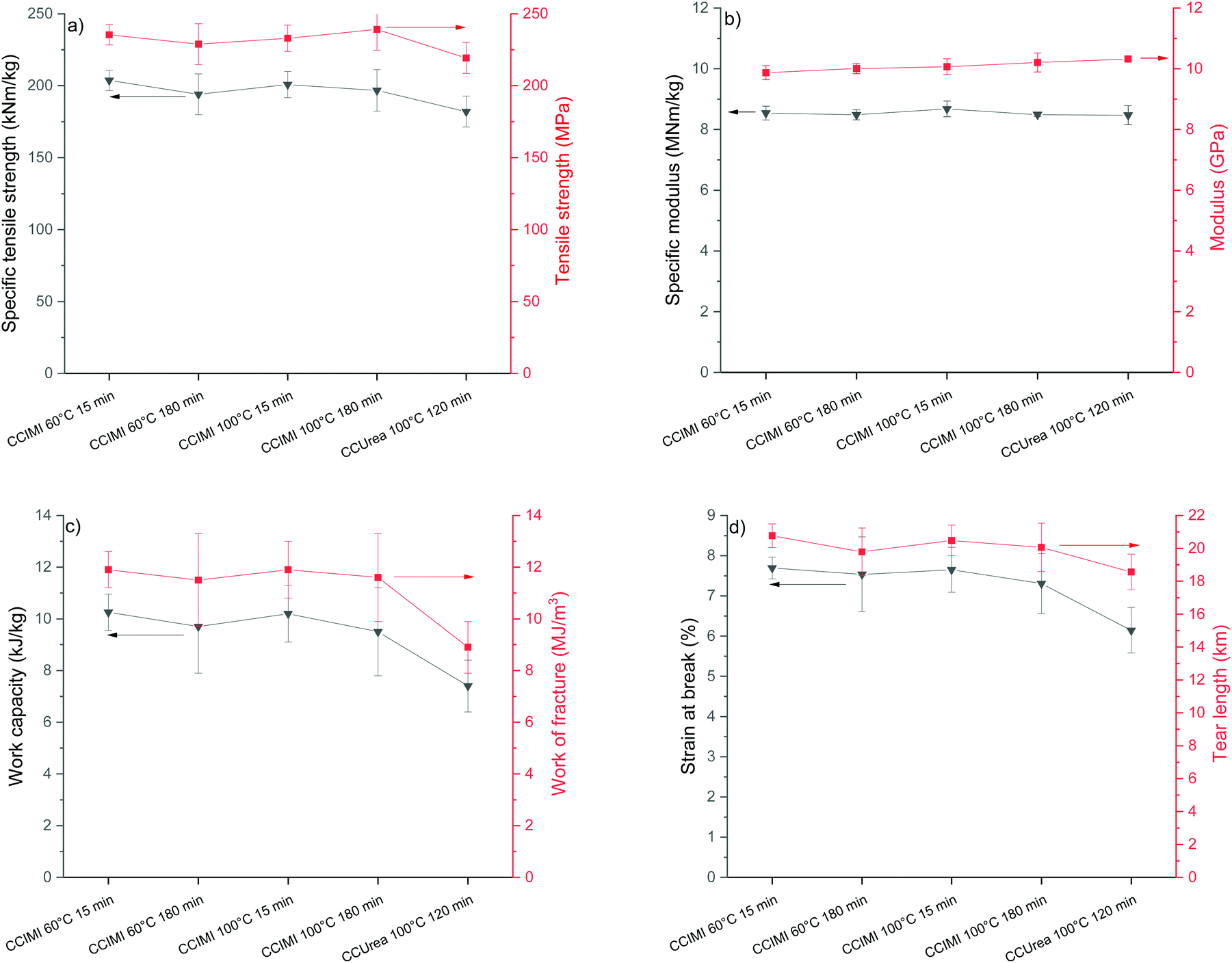 | ||
| Fig. 6 Mechanical properties of CNF films prepared from DES-treated fibers: (a) Tensile and specific strength, (b) modulus and specific modulus, (c) work capacity and work of fracture, and (d) strain at break and tear length. | ||
It is noteworthy that all the CNF films produced from CCIMI-treated fibers showed better mechanical properties compared to those produced using CCUrea. Particularly, a high difference in value was observed in the toughness of the film. For example, sample 1 had a toughness of 11.9 MJ m−3, whereas the toughness of film produced using CCUrea was 8.9 MJ m−3 (Fig. 6c). On the other hand, the moduli of all the films were similar.
The mechanical properties of the film produced using CCIMI DES (especially tensile strength [235 MPa] and specific tensile strength [204 kNm kg−1]) are interesting, as they are among the highest reported for cellulose nanomaterials (see Table 2, which compares mechanical properties of CNFs reported in the literature). Tensile strength of 233 MPa was reported for CNFs produced using TEMPO-mediate oxidation (Table 2, entry 6).51 However, when taking into account the density of the film, the specific tensile strength was lower than that reported in this study (160 vs. 204 kNm kg−1). Nevertheless, in a separate study, a specific tensile strength of 268 kNm kg−1 was obtained for a CNF film produced using TEMPO-mediate oxidation (Table 2, entry 5).
| Entry | Method | Density (g cm−2) | Specific modulus (MNm kg−1) | Modulus (GPa) | Specific tensile strength (kNm kg−1) | Tensile strength (MPa) | Work capacity (kJ kg−1) | Work of fracture (MJ m−3) | Strain (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CCIMI DES pretreatment (60 °C, 15 min) | 1.2 | 8.5 | 9.9 | 204 | 235 | 10.2 | 11.9 | 7.7 | This work |
| 2 | CCUrea DES pretreatment (100 °C, 120 min) | 1.2 | 8.7 | 10.2 | 182 | 219 | 7.4 | 8.9 | 6.1 | This work |
| 3 | CCIMI DES pretreatment (100 °C, 240 min) | — | — | 12.1 | — | 142 | — | — | 2.4 | 25 |
| 4 | CCUrea DES pretreatment (100 °C, 120 min) | — | — | 12.9 | — | 119 | — | — | 2.1 | 25 |
| 5 | TEMPO-mediated oxidation | 0.89 | 10.5 | 9.3 | 268 | 239 | — | — | 6.2 | 53 |
| 6 | TEMPO-mediated oxidation | 1.46 | 4.7 | 6.9 | 160 | 233 | — | — | 7.6 | 51 |
| 7 | Microfluidization | 1.08 | 12.2 | 13.2 | 198 | 214 | 14.0 | 15.1 | 10.1 | 54 |
| 8 | Microfluidization | 1.33 | 5.4 | 7.2 | 214 | 284 | 12.0 | 16 | 8.7 | 55 |
| 9 | Enzymatic pretreatment | — | — | 10 | — | 196 | — | — | 6.5 | 56 |
| 10 | TEMPO-mediated oxidation | — | — | 9 | — | 210 | — | 10 | 8 | 57 |
| 11 | Masuko grinding | 1.34 | 5.1 | 6.8 | 127 | 170 | — | — | 8.7 | 58 |
| 12 | Periodate/chlorite oxidation | 1.47 | 8.2 | 12 | 143 | 210 | — | — | 5.3 | 12 |
| 13 | Periodate oxidation/sulfonation | 1.07 | 19.7 | 21.1 | 152 | 162 | — | — | 1.6 | 15 |
| 14 | GHClUrea DES pretreatment (100 °C, 120 min) | — | — | 7.7 | — | 189 | — | — | 9.4 | 23 |
| 15 | Enzymatic pretreatment | >1.4 | — | 13.4 | — | 232 | — | — | 5.0 | 59 |
For nonchemically modified CNF films, tensile strength of 284 MPa has been reported (Table 2, entry 8), whereas the specific tensile strength (214 kNm kg−1) was still in line with the highest values obtained in the current study (Table 2, entry 1). A similar tensile strength (232 MPa) compared to films produced using CCIMI-treated fibers was reported for CNF film obtained from enzyme-treated fibers (Table 2, entry 15). In addition, previous results with CCIMI (treatment time of 4 h at 100 °C) showed a significantly lower tensile strength compared to the current study (Table 2, entry 3).25
It should be noted that in Table 2, the mechanical properties of films with randomly oriented nanofibers are reported. Orientation—for example, by drawing—can significantly improve the mechanical properties of the films. Cold drawing with a draw ratio of 1:4 increased the tensile strength of CNF films from 185 MPa of nondrawn films to 428 MPa.52
The work of fracture or work capacity (toughness) is less frequently reported in the literature. However, these values are important in many applications and represent the overall mechanical performance of materials, as a high toughness request balances strength and ductility (strain). In the literature, a significantly higher work of fracture (and also work capacity) compared to the current study has been reported for CNF films produced using only mechanical disintegration with a microfluidizator (Table 2, entries 7 and 8), whereas a slightly lower work capacity has been reported for CNFs produced using TEMPO-mediate oxidation (Table 2, entry 10).
Recycling and reuse of DES
The decrease of the solvent and chemical consumption in cellulose processing is important to improve the sustainability of the DES use in terms of environmental friendliness and cost efficiency. The recycling of solvent is highly important step to minimize the waste emissions. Similar to the ionic liquids, DESs are known to be recyclable when used in many applications,60,61 including cellulose treatment.29 It was observed that during the recycling of CCIMI by evaporation of water used in the washing, mass of the solvents slightly increased, indicating that small amount of water (approximately 4–5%) (Table S1†) remained in DES even after prolonged evaporation. It has been shown that water can take part in the structure of DESs,62,63 and the removal of residual water can be cumbersome process requesting severe drying conditions.64 Previous studies of pretreatment of paper and board cellulose pulp with DES were successfully conducted in presence of small amount of water (i.e., by using wet biomass)24 and therefore the recycling experiments were studied without any further drying steps than evaporation.Fig. 7 shows that the specific tensile strength and modulus of the films produced using recycled DES were similar to the film produced using pristine DES. Furthermore, the DRIFT analysis indicated that the chemical structure of cellulose remained intact also in the recycled DES (Fig. S5†). Therefore, it can be concluded that CCIMI can be recycled using simple water evaporation and the mechanical properties of produced CNFs can still be retained. However, it should be noted that no optimization of the recycling process was performed. One critical point in the recycling is the water consumption. The evaporated water can be used e.g. directly for next washing step, thus creating a closed water cycle. However, the amount of used water should be minimized as the evaporation of water request high amount of energy. In addition, comprehensive studies should be conducted where DES pre-treatment is compared to more common cellulose pre-treatment. Typically, chemical pretreatments such as oxidation and carboxymethylation utilize hazardous halogenated chemicals and long reaction times at elevated temperature. The enzymatic pre-treatments can be conducted at slightly lower temperature (typically at 50 °C) compared to CCIMI-treatment, but longer treatment times are requested (ranging from few hours to several days).19,20,65 However, it should be noted that various pre-treatments can have different effect on the nanofibrillation efficiency (i.e., different amount of mechanical energy is requested to disintegrate cellulose fibers into nanofibers). Therefore, direct comparison of pre-treatments reported in literature might not be entirely meaningful without using similar systems and conditions (e.g. by using same raw material and mechanical disintegration process).
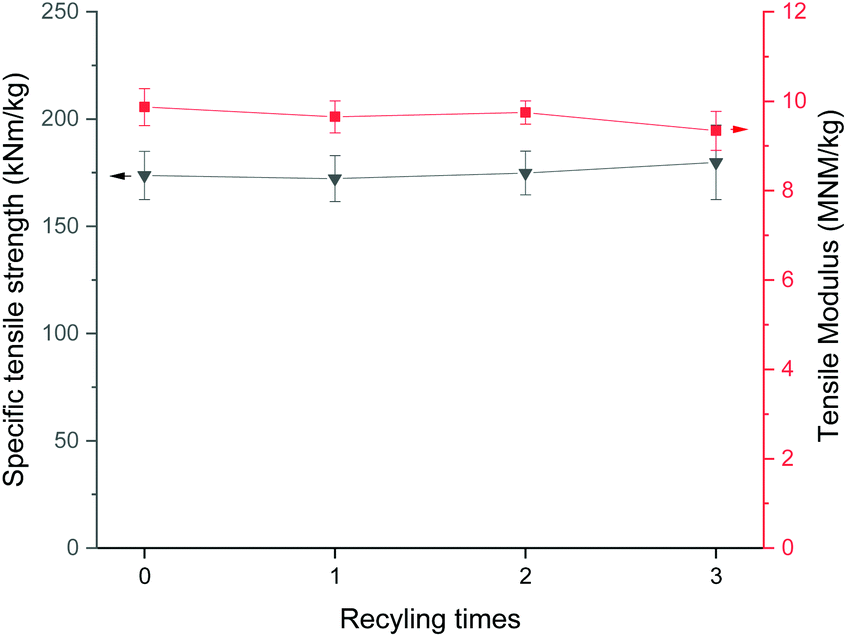 | ||
| Fig. 7 Specific tensile strength and modulus of CNF films produced using pristine and recycled CCIMI. | ||
Oxygen barrier properties of CNF films
Surprisingly, the trend in the oxygen barrier properties of CNF films did not follow that of the mechanical properties—that is, the best barrier properties were observed with films produced using CCUrea (Table 3). At an RH of 50%, the oxygen permeability of sample 5 was four to seven times lower than those produced using CCIMI. When the RH was elevated from 50% to 80%, the OP value of film produced using CCUrea-treated fibers increased more radically compared to those produced with CCIMI and the difference in oxygen permeability was only 1.2 to 1.7 times in favor of sample 5. In addition, of the CNF films produced using CCIMI, films obtained by short treatment times exhibited notably higher OP values at elevated humidity levels (Table 3, entries 1 and 3).| Entry | Sample | 50% RH | 80% RH | ||
|---|---|---|---|---|---|
| OTR (cm3 [m2 d]−1) | OP (cm3 μm [m2 d atm]−1) | OTR (cm3 [m2 d]−1) | OP (cm3 μm [m2 d atm]−1) | ||
| 1 | CCIMI 60 °C 15 min | 2.8 | 176 | 9.2 | 580 |
| 2 | CCIMI 60 °C 180 min | 2.4 | 162 | 7.2 | 486 |
| 3 | CCIMI 100 °C 15 min | 1.6 | 105 | 10.4 | 681 |
| 4 | CCIMI 100 °C 180 min | 1.6 | 100 | 7.8 | 486 |
| 5 | CCUrea 60 °C 180 min | 0.4 | 25 | 6.2 | 393 |
The excellent oxygen barrier property of CNFs is generally thought to be a combination of high polarity (i.e., hydrophilicity), crystallinity, and a tight hydrogen bond network of nanofibers, resulting in minimal cavities between nanofibers. As shown above, the CrI of all the samples were similar, and the mechanical properties of the film produced using the CCUrea-treated fibers were even slightly lower compared to those of the CCIMI films, indicating similar or even slightly worse bonding between nanofibers. Therefore, the difference between the barrier properties of CNFs from CCUrea and CCIMI treatments may be ascribed to some molecular level properties. As the hydrogen bond acceptor in both DESs was the same (CC), the hydrogen bond donor, can presumably have a significant effect, in particular, on the gas barrier of the CNF films.
At an RH of 50%, the oxygen barrier properties (OP of 25 cm3 μm [m2 d atm]−1) of sample 5 can be described as very high (<40 cm3 μm [m2 d atm]−1),66 and it is generally better than those obtained using carboxymethylation (37,67 5268 and 8669 cm3 μm [m2 d atm]−1) or TEMPO-mediated oxidations (35 cm3 μm [m2 d atm]−1).70 A lower OP value (12 cm3 μm [m2 d atm]−1) than that of a CNF film produced using CCUrea was reported for films obtained using sequential periodate and chlorite oxidation.15 It should be noted that the oxygen barrier properties of carboxylated CNFs can be improved by changing the sodium cation to multivalent aluminum or calcium ions, and an OP of 3.6 cm3 μm (m2 d atm)−1 was obtained with CNFs produced by TEMPO-mediated oxidation with calcium as a counter-ion (amounts to a 70 times decrease compared to that with sodium).71
Generally, the barrier properties of CNF films produced from nonchemically modified fibers (e.g., those produced from enzymes or solvent treated or produced purely by mechanical disintegration) are reported to be lower than those with chemical modification.67,72 At an RH of 50%, OP values similar to the values obtained using CCIMI at an RH of 80% (350–500 cm3 μm [m2 d atm]−1) have been observed for CNF films produced by the homogenization of cellulose pulp,73 whereas OP values as low as 48 cm3 μm (m2 d atm)−1 were obtained with CNFs produced from enzyme-treated fibers.74 In addition, hot pressing has been observed to significantly increase the barrier properties of nonmodified CNF films, and an OP value of 20 cm3 μm (m2 d atm)−1 has been reported after two hours of pressing at about 100 °C and 1800 Pa.72
Highly hydrophilic CNF films are known to drastically lose their oxygen barrier properties at elevated humidity levels (>65% RH) due to the adsorption of water on the film structure and loosening of the bonding between nanofibers.66 The decrease in the oxygen barrier properties is especially prominent in CNFs containing chemical groups such as carboxylates, as the natural hydroxyl groups of cellulose have been replaced with more hydrophilic carboxylic acid and its salt. For example, in the case of carboxymethylated CNFs, the OP values increased from 52 to 45400 cm3 μm (m2 d atm)−1 when the RH changed from 50 to 80%.68 The OP value of cellulose nanocrystals (containing highly hydrophilic sulfate groups on their surfaces) from bacterial cellulose was observed to increase from 6.1 to 52264 μm (m2 d atm)−1 when the RH increased from 0% to 80%.75 A relatively low OP value (899 cm3 μm [m2 d atm]−1) at an RH of 80% has been observed with CNF produced using phosphorylation.76 Among the lowest OP values at RH of 80% for nanocellulosic materials reported are OP values obtained through using periodate oxidation followed by the reduction of aldehydes to alcohols to produce CNFs (550 cm3 μm [m2 d atm]−1) and reductive amination to produce butylamine-modified cellulose nanocrystals (590 cm3 μm [m2 d atm]−1).77 The above-mentioned results are still higher compared to results obtained by CCIMI-treated fibers (180 min at 60 °C and 100 °C) and CCUrea (Table 3, entries 2, 4, and 5). Of the oxygen barrier properties of the current study, the one obtained with CCUrea still falls under the category of a high oxygen barrier, whereas those produced using CCIMI can be classified as a medium barrier, even at an elevated RH.66
Discussion about the effect of different DES on the properties of CNFs
Although no direct conclusion can be made and more research (e.g., molecular dynamic simulation) is recommended, it appears that CCIMI and CCUrea DESs have somewhat different behaviors when used as treatment media for cellulose fibers. Urea is a well-known additive for the dissolution of cellulose in an aqueous alkaline solution.78 It has been demonstrated that in an aqueous solution, urea has a weak interaction with the carbon ring plane of cellulose. The interaction is attributed to either van der Waals forces or electrostatic interactions between urea and the carbon ring atoms with partially positive charges.79 In addition, urea has two NH2 groups with partially positive charges and an oxygen atom with a partially negative charge, which can all contribute to the hydrogen bond interaction with cellulose. It has been shown that urea has a similar affinity to the hydroxyl groups of cellulose compared to that of water.79On the other hand, glucose (cellulose monomer) ring plane and imidazole ring stacking was observed with molecular simulation studies in water solution.80 This interaction was attributed to van der Waals interactions, similar to the interaction between cellulose and urea. However, only the N3 nitrogen atom was observed to take part in significant hydrogen bonding with glucose due to the low polarity of the NH group in N1.80 Therefore, it might be that although both urea and imidazole can interact with the hydrophobic carbon ring plate of cellulose, a different ability for hydrogen bonding might cause variations when these chemicals are used together with CC in cellulose treatment. Furthermore, imidazole has a higher possibility for interaction with the hydrophobic planes of cellulose—which might expose the planes toward the surface of cellulose, making CCIMI-treated fibers slightly more hydrophobic at the molecular level. On the other hand, urea disturbs, to a higher degree, the hydrogen bonding of cellulose, which might result in slightly decreased mechanical properties. In an oxygen barrier test, more hydrophilic (polar) CNFs produced from CCUrea-treated fiber exhibited less interactive nonpolar oxygen atoms at a moderate humidity level (50%). However, at higher humidity levels, these fibers suffer from higher polarity, as they have interact, to a higher degree, with water molecules—which, in turn, results in a decrease of barrier properties in to a significantly higher extent compared to CNFs produced from CCIMI-treated fibers. The lowest relative increase in OP values of those CNF films produced using CCIMI-treated fibers from longer treatment times suggest the exposure of the hydrophobic plate is a kinetically controlled reaction and favors long treatment times.
Conclusions
DESs based on CC and IMI showed excellent capacity for cellulose fiber swelling at mild conditions without causing detectable chemical alterations of the cellulose structure. In addition, neither the DP nor crystallinity of cellulose changed during DES treatment, whereas pure IMI caused slight hydrolysis of cellulose. Furthermore, recycling experiments indicated that CCIMI can be directly used for cellulose fiber treatment after removal of water used in fiber washing. Compared to previously used DESs, fibers treated with CCIMI provided CNF films with better mechanical properties. On the other hand, the oxygen barrier properties of CNF films produced using CCUrea-treated fibers were notably better compared to those produced using CCIMI at an RH of 50%, whereas the relative difference was notably smaller at higher humidity levels. Specifically, the different behaviors of the films in the oxygen barrier test indicate there are molecular level differences in the actions of CCIMI and CCUrea on the cellulose fibers. Therefore, more atomic-level studies are needed to fully understand and utilize the different DESs in the production of nanosized cellulose materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Mr Sami Saukko, Mr Tommi Kokkonen, and Mr Marcin Selent for their assistance in TEM, TG, and XRD measurements, respectively. Mr Jani Österlund is acknowledged for the film preparation and tensile test, adn BillerudKorsnäs Produktlab Skärblacka for OTR measurements. BillerudKorsnäs AB is gratefully acknowledged for its financial support of this work. The facilities at the Center of Microscopy and Nanotechnology at the University of Oulu were used in this research.References
- V. Prakash Sharma, U. Sharma, M. Chattopadhyay and V. N. Shukla, Mater. Today: Proc., 2018, 5, 6376–6380 CAS.
- P. N. Navya and H. K. Daima, Nano Convergence, 2016, 3, 1 CrossRef CAS PubMed.
- J. Jordan, K. I. Jacob, R. Tannenbaum, M. A. Sharaf and I. Jasiuk, Mater. Sci. Eng., A, 2005, 393, 1–11 CrossRef.
- X. Ho and J. Wei, Materials, 2013, 6, 2155–2181 CrossRef CAS PubMed.
- S. Khatua, P. Manna, W.-S. Chang, A. Tcherniak, E. Friedlander, E. R. Zubarev and S. Link, J. Phys. Chem. C, 2010, 114, 7251–7257 CrossRef CAS.
- D. Klemm, E. D. Cranston, D. Fischer, M. Gama, S. A. Kedzior, D. Kralisch, F. Kramer, T. Kondo, T. Lindström, S. Nietzsche, K. Petzold-Welcke and F. Rauchfuß, Mater. Today, 2018, 21, 720–748 CrossRef CAS.
- E. Kontturi, P. Laaksonen, M. B. Linder, Nonappa, A. H. Gröschel, O. J. Rojas and O. Ikkala, Adv. Mater., 2018, 30, 1703779 CrossRef PubMed.
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS PubMed.
- F. Rol, M. N. Belgacem, A. Gandini and J. Bras, Prog. Polym. Sci., 2019, 88, 241–264 CrossRef CAS.
- T. Saito and A. Isogai, Biomacromolecules, 2004, 5, 1983–1989 CrossRef CAS PubMed.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- L. Wågberg, G. Decher, M. Norgren, T. Lindström, M. Ankerfors and K. Axnäs, Langmuir, 2008, 24, 784–795 CrossRef PubMed.
- Ch. Eyholzer, N. Bordeanu, F. Lopez-Suevos, D. Rentsch, T. Zimmermann and K. Oksman, Cellulose, 2010, 17, 19–30 CrossRef CAS.
- A. Tejado, M. N. Alam, M. Antal, H. Yang and T. G. M. van de Ven, Cellulose, 2012, 19, 831–842 CrossRef CAS.
- J. A. Sirviö, A. Kolehmainen, M. Visanko, H. Liimatainen, J. Niinimäki and O. E. O. Hormi, ACS Appl. Mater. Interfaces, 2014, 6, 14384–14390 CrossRef PubMed.
- H. Liimatainen, M. Visanko, J. A. Sirviö, O. E. O. Hormi and J. Niinimaki, Biomacromolecules, 2012, 13, 1592–1597 CrossRef CAS PubMed.
- H. Liimatainen, T. Suopajärvi, J. Sirviö, O. Hormi and J. Niinimäki, Carbohydr. Polym., 2014, 103, 187–192 CrossRef CAS PubMed.
- S. Pan and A. J. Ragauskas, Carbohydr. Polym., 2014, 111, 514–523 CrossRef CAS PubMed.
- M. Henriksson, G. Henriksson, L. A. Berglund and T. Lindström, Eur. Polym. J., 2007, 43, 3434–3441 CrossRef CAS.
- M. Pääkkö, M. Ankerfors, H. Kosonen, A. Nykänen, S. Ahola, M. Österberg, J. Ruokolainen, J. Laine, P. T. Larsson, O. Ikkala and T. Lindström, Biomacromolecules, 2007, 8, 1934–1941 CrossRef PubMed.
- J. A. Sirviö, M. Visanko and H. Liimatainen, Green Chem., 2015, 17, 3401–3406 RSC.
- A. Hosseinmardi, P. K. Annamalai, L. Wang, D. Martin and N. Amiralian, Nanoscale, 2017, 9, 9510–9519 RSC.
- P. Li, J. A. Sirviö, A. Haapala and H. Liimatainen, ACS Appl. Mater. Interfaces, 2017, 9, 2846–2855 CrossRef CAS PubMed.
- T. Suopajärvi, J. A. Sirviö and H. Liimatainen, Carbohydr. Polym., 2017, 169, 167–175 CrossRef PubMed.
- S. Jonasson, A. Bünder, T. Niittylä and K. Oksman, Cellulose, 2020, 27, 185–203 CrossRef CAS.
- J. A. Sirviö, Carbohydr. Polym., 2018, 198, 34–40 CrossRef PubMed.
- T. Selkälä, J. A. Sirviö, G. S. Lorite and H. Liimatainen, ChemSusChem, 2016, 9, 3074–3083 CrossRef PubMed.
- J. A. Sirviö, J. Ukkola and H. Liimatainen, Cellulose, 2019, 26, 2303–2316 CrossRef.
- P. Li, J. A. Sirviö, B. Asante and H. Liimatainen, Carbohydr. Polym., 2018, 199, 219–227 CrossRef CAS PubMed.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 RSC.
- D. E. Crawford, L. A. Wright, S. L. James and A. P. Abbott, Chem. Commun., 2016, 52, 4215–4218 RSC.
- J. A. Sirviö and M. Visanko, J. Mater. Chem. A, 2017, 5, 21828–21835 RSC.
- R. Berggren, F. Berthold, E. Sjöholm and M. Lindström, J. Appl. Polym. Sci., 2003, 88, 1170–1179 CrossRef CAS.
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef CAS.
- M. Zdanowicz, T. Spychaj and H. Mąka, Carbohydr. Polym., 2016, 140, 416–423 CrossRef CAS PubMed.
- S. M. Neale, Trans. Faraday Soc., 1933, 29, 228–234 RSC.
- T. Jordan, S. Schmidt, T. Liebert and T. Heinze, Green Chem., 2014, 16, 1967–1973 RSC.
- J. Mao, H. Abushammala, H. Hettegger, T. Rosenau and M.-P. Laborie, Polymers, 2017, 9, 473 CrossRef PubMed.
- A. R. C. Morais, J. V. Pinto, D. Nunes, L. B. Roseiro, M. C. Oliveira, E. Fortunato and R. Bogel-Łukasik, ACS Sustainable Chem. Eng., 2016, 4, 1643–1652 CrossRef CAS.
- A. Toscan, A. R. C. Morais, S. M. Paixão, L. Alves, J. Andreaus, M. Camassola, A. J. P. Dillon and R. M. Lukasik, Ind. Eng. Chem. Res., 2017, 56, 5138–5145 CrossRef CAS.
- J. A. Sirviö, M. Visanko and H. Liimatainen, Biomacromolecules, 2016, 17, 3025–3032 CrossRef PubMed.
- H. Qin, X. Hu, J. Wang, H. Cheng, L. Chen and Z. Qi, Green Energy Environ. DOI:10.1016/j.gee.2019.03.002.
- J. A. Sirviö, J. Mater. Chem. A, 2019, 7, 755–763 RSC.
- M. Sharma, C. Mukesh, D. Mondal and K. Prasad, RSC Adv., 2013, 3, 18149–18155 RSC.
- H. Fu, X. Wang, H. Sang, Y. Hou, X. Chen and X. Feng, J. Mol. Liq., 2019, 112140 Search PubMed.
- K. Missoum, M. N. Belgacem and J. Bras, Materials, 2013, 6, 1745–1766 CrossRef CAS PubMed.
- O. Laitinen, T. Suopajärvi, M. Österberg and H. Liimatainen, ACS Appl. Mater. Interfaces, 2017, 9, 25029–25037 CrossRef CAS PubMed.
- M. Visanko, J. A. Sirviö, P. Piltonen, R. Sliz, H. Liimatainen and M. Illikainen, Cellulose, 2017, 24, 4173–4187 CrossRef CAS.
- J. Li, X. Wei, Q. Wang, J. Chen, G. Chang, L. Kong, J. Su and Y. Liu, Carbohydr. Polym., 2012, 90, 1609–1613 CrossRef CAS PubMed.
- H. Fukuzumi, T. Saito, T. Iwata, Y. Kumamoto and A. Isogai, Biomacromolecules, 2009, 10, 162–165 CrossRef CAS PubMed.
- H. Sehaqui, N. Ezekiel Mushi, S. Morimune, M. Salajkova, T. Nishino and L. A. Berglund, ACS Appl. Mater. Interfaces, 2012, 4, 1043–1049 CrossRef CAS PubMed.
- M. Salajkova, L. Valentini, Q. Zhou and L. A. Berglund, Compos. Sci. Technol., 2013, 87, 103–110 CrossRef CAS.
- M. Henriksson, L. A. Berglund, P. Isaksson, T. Lindström and T. Nishino, Biomacromolecules, 2008, 9, 1579–1585 CrossRef CAS PubMed.
- G. Reyes, M. Borghei, A. W. T. King, J. Lahti and O. J. Rojas, Biomacromolecules, 2019, 20, 502–514 CrossRef CAS PubMed.
- R. Mao, N. Meng, W. Tu and T. Peijs, Cellulose, 2017, 24, 4627–4639 CrossRef CAS.
- F. Lossada, J. Guo, D. Jiao, S. Groeer, E. Bourgeat-Lami, D. Montarnal and A. Walther, Biomacromolecules, 2019, 20, 1045–1055 CrossRef CAS PubMed.
- J. Sethi, M. Farooq, S. Sain, M. Sain, J. A. Sirviö, M. Illikainen and K. Oksman, Cellulose, 2018, 25, 259–268 CrossRef CAS.
- H. Sehaqui, A. Liu, Q. Zhou and L. A. Berglund, Biomacromolecules, 2010, 11, 2195–2198 CrossRef CAS PubMed.
- A. K. Sanap and G. S. Shankarling, Catal. Commun., 2014, 49, 58–62 CrossRef CAS.
- C. Mukesh, D. Mondal, M. Sharma and K. Prasad, Carbohydr. Polym., 2014, 103, 466–471 CrossRef CAS PubMed.
- T. El Achkar, S. Fourmentin and H. Greige-Gerges, J. Mol. Liq., 2019, 288, 111028 CrossRef CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- C. Du, B. Zhao, X.-B. Chen, N. Birbilis and H. Yang, Sci. Rep., 2016, 6, 1–14 CrossRef PubMed.
- J. Y. Zhu, R. Sabo and X. Luo, Green Chem., 2011, 13, 1339–1344 RSC.
- J. Wang, D. J. Gardner, N. M. Stark, D. W. Bousfield, M. Tajvidi and Z. Cai, ACS Sustainable Chem. Eng., 2018, 6, 49–70 CrossRef CAS.
- C. Aulin, M. Gällstedt and T. Lindström, Cellulose, 2010, 17, 559–574 CrossRef CAS.
- C. Aulin, G. Salazar-Alvarez and T. Lindström, Nanoscale, 2012, 4, 6622 RSC.
- I. Siró, D. Plackett, M. Hedenqvist, M. Ankerfors and T. Lindström, J. Appl. Polym. Sci., 2011, 119, 2652–2660 CrossRef.
- V. Kumar, R. Bollström, A. Yang, Q. Chen, G. Chen, P. Salminen, D. Bousfield and M. Toivakka, Cellulose, 2014, 21, 3443–3456 CrossRef CAS.
- M. Shimizu, T. Saito and A. Isogai, J. Membr. Sci., 2016, 500, 1–7 CrossRef CAS.
- M. Österberg, J. Vartiainen, J. Lucenius, U. Hippi, J. Seppälä, R. Serimaa and J. Laine, ACS Appl. Mater. Interfaces, 2013, 5, 4640–4647 CrossRef PubMed.
- K. Syverud and P. Stenius, Cellulose, 2009, 16, 75 CrossRef CAS.
- A. Liu, A. Walther, O. Ikkala, L. Belova and L. A. Berglund, Biomacromolecules, 2011, 12, 633–641 CrossRef CAS PubMed.
- M. Martínez-Sanz, A. Lopez-Rubio and J. M. Lagaron, Carbohydr. Polym., 2013, 98, 1072–1082 CrossRef PubMed.
- A. Naderi, T. Lindström, G. Flodberg, J. Sundström, K. Junel, A. Runebjörk, C. F. Weise and J. Erlandsson, Nord. Pulp Pap. Res. J., 2016, 31, 20–29 CAS.
- M. Visanko, H. Liimatainen, J. A. Sirviö, K. S. Mikkonen, M. Tenkanen, R. Sliz, O. Hormi and J. Niinimäki, RSC Adv., 2015, 5, 15140–15146 RSC.
- T. Budtova and P. Navard, Cellulose, 2015, 1–51 Search PubMed.
- E. Wernersson, B. Stenqvist and M. Lund, Cellulose, 2015, 22, 991–1001 CrossRef CAS.
- M. Chen, Y. J. Bomble, M. E. Himmel and J. W. Brady, Carbohydr. Res., 2012, 349, 73–77 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fiber width and CWT at various concentrations, DRIFT spectra, photograph of CNF aerogels, and TGA curves. See DOI: 10.1039/c9gc04119b |
| This journal is © The Royal Society of Chemistry 2020 |